Back to Journals » Pharmacogenomics and Personalized Medicine » Volume 11
CYP2C9 polymorphisms in epilepsy: influence on phenytoin treatment
Authors Silvado CE ![]() , Terra VC, Twardowschy CA
, Terra VC, Twardowschy CA ![]()
Received 18 October 2017
Accepted for publication 1 February 2018
Published 29 March 2018 Volume 2018:11 Pages 51—58
DOI https://doi.org/10.2147/PGPM.S108113
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Martin H Bluth
Carlos Eduardo Silvado,1 Vera Cristina Terra,1 Carlos Alexandre Twardowschy2
1Comprehensive Epilepsy Program, Hospital de Clinicas, Federal University of Parana (UFPR), Curitiba, Brazil; 2Department of Neurology, Catholic University of Parana (PUCPR), Curitiba, Brazil
Abstract: Phenytoin (PHT) is an antiepileptic drug widely used in the treatment of focal epilepsy and status epilepticus, and effective in controlling focal seizures with and without tonic–clonic generalization and status epilepticus. The metabolization of PHT is carried out by two oxidative cytochrome P450 enzymes CYP2C9 and CYP2C19; 90% of this metabolization is done by CYP2C9 and the remaining 10% by CYP2C19. Genetic polymorphism of CYP2C9 may reduce the metabolism of PHT by 25–50% in patients with variants *2 and *3 compared to those with wild-type variant *1. The frequency distribution of CYP2C9 polymorphism alleles in patients with epilepsy around the world ranges from 4.5 to 13.6%, being less frequent in African-Americans and Asians. PHT has a narrow therapeutic range and a nonlinear pharmacokinetic profile; hence, its poor metabolization has significant clinical implications as it causes more frequent and more serious adverse effects requiring discontinuation of treatment, even if it had been effective. There is evidence that polymorphisms of CYP2C9 and the use of PHT are associated with an increase in the frequency of some side effects, such as cerebellar atrophy, gingival hypertrophy or acute cutaneous reactions. The presence of HLA-B*15:02 and CYP2C9 *2 or *3 in the same patient increases the risk of Stevens–Johnson syndrome and toxic epidermal necrolysis; hence, PHT should not be prescribed in these patients. In patients with CYP2C9 *1/*2 or *1/*3 alleles (intermediate metabolizers), the usual PHT maintenance dose (5–10 mg/kg/day) must be reduced by 25%, and in those with CYP2C9 *2/*2, *2/*3 or *3/*3 alleles (poor metabolizers), the dose must be reduced by 50%. It is controversial whether CYP2C9 genotyping should be done before starting PHT treatment. In this paper, we aim to review the influence of CYP2C9 polymorphism on the metabolization of PHT and the clinical implications of poor metabolization in the treatment of epilepsies.
Keywords: phenytoin, antiepileptics, CYP2C9, cytochrome P450, epilepsy, polymorphisms, adverse effects
Introduction
Phenytoin (PHT) was introduced about 80 years ago for use in epilepsy1 and still remains one of the most prescribed antiepileptic drugs (AEDs) to control partial seizures, either with or without secondary generalization,2 and for the treatment of status epilepticus. It is available worldwide for use in outpatient setting, as well as in almost all emergency services.
The main mechanism of action of PHT is the selective blockage of neuronal voltage-dependent sodium channels in their inactive state.3 Fosphenytoin is the prodrug (a water-soluble phosphate ester) of PHT, which is hydrolyzed to PHT by phosphatases.
The metabolism of PHT is rather complex, and 95% of PHT is eliminated by biotransformation though a number of pathways. The main step is the conversion to (R,S)-5-(p-hydroxyphenyl)-5-phenyl-hydantoin (p-HPPH), leading to the formation of a reactive arene oxide intermediate, which could play a role in the pathogenesis of some of the idiosyncratic adverse reactions, including serious cutaneous reactions, hepatotoxicity and teratogenicity.4 Secondary hydroxylated metabolites include m-HPPH and a dihydrodiol, which is further metabolized to a glucuronide and to a catechol derivate. Approximately 98% of circulating p-HPPH is in levo isoform “S”.5 Overall, p-HPPH accounts for 67–88% and dihydrodiol for 7–11% of the human urinary metabolites of PHT.6
PHT exhibits a nonlinear pharmacokinetic profile, due to saturation of the oxidation route by two hepatic oxidative cytochrome P450 enzymes (CYPs) CYP2C9 and CYP2C19.7–9 This metabolism is reduced in persons with genetic polymorphisms and also by the interaction of these systems with other drugs.8
PHT is metabolized by both CYP2C9 and CYP2C19, which are responsible for the conversion of inactive PHT to its main metabolite, p-HPPH; CYP2C9 is responsible for 90% of this metabolization,8 whereas CYP2C19 metabolizes the remaining 10%.10
Genetic polymorphism of CYP2C9 may reduce the metabolism of PHT by 25–50% in patients who have the genetic variants *2 and *3, as compared to those with variant *1 (normal metabolizers), named the “wild types” or the “extensive metabolizers”.11
PHT has a nonlinear pharmacokinetic profile,11 and its therapeutic range is rather narrow and may be even more pronounced in patients who have a genetic polymorphism that may cause poor metabolization which might have significant clinical implications as it causes more frequent and more serious adverse effects requiring discontinuation of treatment, even if it had been effective in seizure control.
In this review, we discuss the influence of genetic polymorphism of the CYP2C9 on the metabolism of PHT and the clinical implications of poor metabolization.
Pharmacogenetics of CYP2C9
Pharmacogenetic is the study of the relationship between variations in a single gene and the action of drugs, and that these genetic variations help to explain the reason for either treatment failure or toxic effects of the administration of various pharmaceutical compounds. Knowledge of these variations may help to predict which patients are at an increased risk of toxicity and identify those who are most likely to achieve the desired drug therapeutic effect.12
Additionally, response variability depends on differences in metabolism, usually caused by genetic polymorphisms. Genetic polymorphisms occurring as a result of single-nucleotide exchange in the DNA sequence are more frequent, whereas others occurring as a result of either insertions or deletions of multiple-nucleotide sequences are less frequent. Currently, it is quite clear that genetic polymorphisms play a key role in the variability in pharmacokinetics and pharmacodynamics of AEDs, and can affect their efficacy, tolerability, safety and duration of action.13–17
Elimination of drugs usually occurs by either hepatic (metabolism) or renal (excretion) routes. Metabolism is the most important route both quantitatively and qualitatively, and can be further divided into Phase I, in which oxidative reactions are catalyzed by many CYPs, and Phase II, in which conjugation reactions occur as a result of glucuronidation.18
The CYPs are encoded by 57 human genes, whose products take part in the oxidative drug metabolism of approximately 90% of all clinically prescribed drugs, and genetic polymorphisms of CYPs are responsible for the interindividual variability in the pharmacokinetics of AEDs. CYP3A4 is the most important hepatic CYP and accounts for more than a third of all hepatic CYPs. For many AEDs (PHT, phenobarbital, carbamazepine, ethosuximide, felbamate, oxcarbazepine, eslicarbazepine, clobazam), oxidation by CYPs (3A4, 2C9, 2C19) is the main metabolization route.18–22 Therefore, changes in this enzyme system have important implications in the management of epileptic patients, particularly side effects and drug interactions.23–25
Depending on DNA variability, drug metabolism may be altered in various locations with potential changes in absorption, distribution, transport, metabolism, elimination and sites of action. The AEDs are not only mere substrates but can also induce or inhibit genes involved in their own and other drugs’ metabolism.26
The action of CYP2C9 is induced by rifampicin, increasing the clearance of drugs eliminated by CYP2C9, such as losartan, PHT, tolbutamide, warfarin and others, by almost 50%. Inhibition of CYP2C9 metabolization, reducing the clearance of drugs eliminated by CYP2C9, could be caused by antidepressants (fluoxetine, fluvoxamine, imipramine, sertraline, trazodone, viloxazine), antimicrobials (chloramphenicol, fluconazole, isoniazid, miconazole, sulfaphenazole) and other drugs (allopurinol, amiodarone, cimetidine, dextropropoxyphene, diltiazem, disulfiram, doxifluridine, 5-fluorouracil, omeprazole, phenylbutazone, sulfinpyrazone, tacrolimus, tamoxifen, ticlopidine, tolbutamide). The inhibitory effect is more relevant in patients with poor metabolization due to the presence of CYP2C9 variants *2 or *3, increasing the incidence and severity of side effects.4,8,27
The gene CYP2C9 and its variants
CYP2C9 is one of the most abundant CYPs in the human liver (∼20% of total hepatic CYP content), responsible for the metabolization of approximately 15% of all clinical drugs (>100 drugs) and is highly polymorphic, with >35 genetic variations being reported.27
It is an important member of the CYP2C subfamily, has been mapped to chromosome 10q24.2 and is a highly variable gene; nucleotide sequencing has already identified almost 60 alleles.28 However, only three alleles, namely CYP2C9 *1 (the wild-type allele, as it is the most common in the population), CYP2C9 *2 and CYP2C9 *3 (both with reduced enzyme activities), are often identified in ethnic population studies.29
The CYP2C9 *2 variant has a C>T transition at position 432 of exon 3 encoding arginine, causing an exchange of cysteine at position 144 (Arg144Cys) of CYP2C9 protein, while the analysis of the variant CYP2C9 *3 demonstrated A>T1077 transversion in exon 7 which encodes an amino acid exchanging isoleucine to leucine at position 359 (Ile359Leu).30
Ethnicity should be taken into account when pharmacogenetic information is used to make a clinical decision, as the frequency of CYP2C9 alleles varies among populations according to the race and ethnic background. A large number of studies have demonstrated a marked variation in the frequency distribution of CYP2C9 alleles around the world, with the wild-type allele frequency ranging from 76.5 to 96.7%.29–46 This difference occurs among white and black Africans,30 white and black Americans,30 Indian ethnic groups,44 Japanese,29 Middle East and North African populations,47 Swedish population,38 and Brazilian population.43 These mutations are less frequent in Asians and African-Americans.
Table 1 summarizes the frequency of CYP2C9 *2 and CYP2C9 *3 alleles in various ethnic groups around the world.
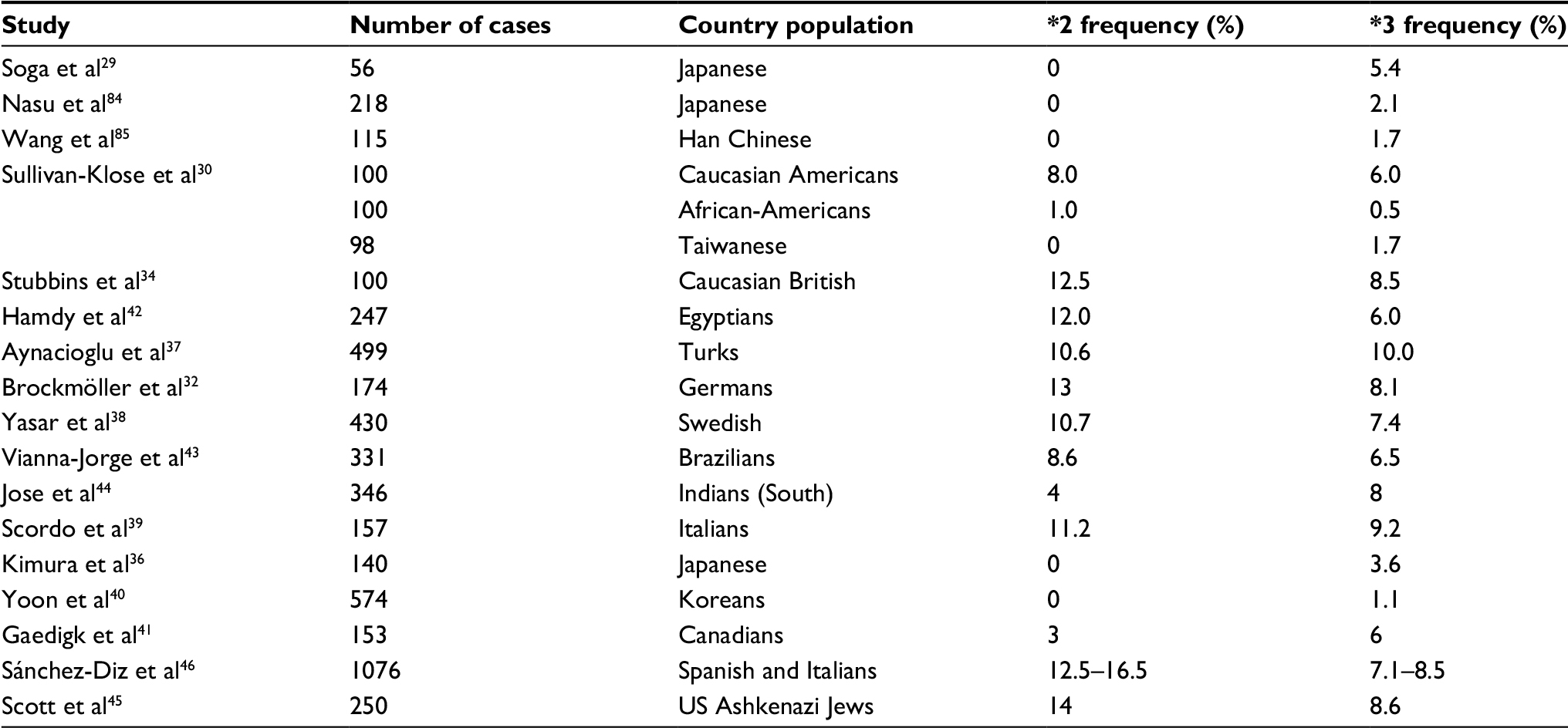 | Table 1 Allele frequency of CYP2C9 *2 and CYP2C9 *3 in normal volunteers |
Subsequent studies on individuals identified with variants CYP2C9 *2 and CYP2C9 *3 showed significantly reduced enzymatic activity,35,8,46,9 as individuals who presented these variants had a reduction of 27–54% of the PHT metabolism47 exhibiting a higher predisposition to PHT toxicity at the usual dosages.8,28,48 Therefore, it has been proposed that PHT dosing in these patients should be individualized according to their pharmacogenetic characteristics as determined by genotyping profile.28,49 While there is some difference in the frequency distribution of *2 and *3 alleles in patients with epilepsy (ranging from 4.5 to 13.6%), this appears to be very similar to the difference due to the ethnic population to which these patients belong.28
Table 2 summarizes the allele frequency of CYP2C9 determined in studies on patients with epilepsy, and Table 3 shows the frequency of mutant alleles.
 | Table 2 Allele frequency of CYP2C9 *2 and CYP2C9 *3 in patients with epilepsy |
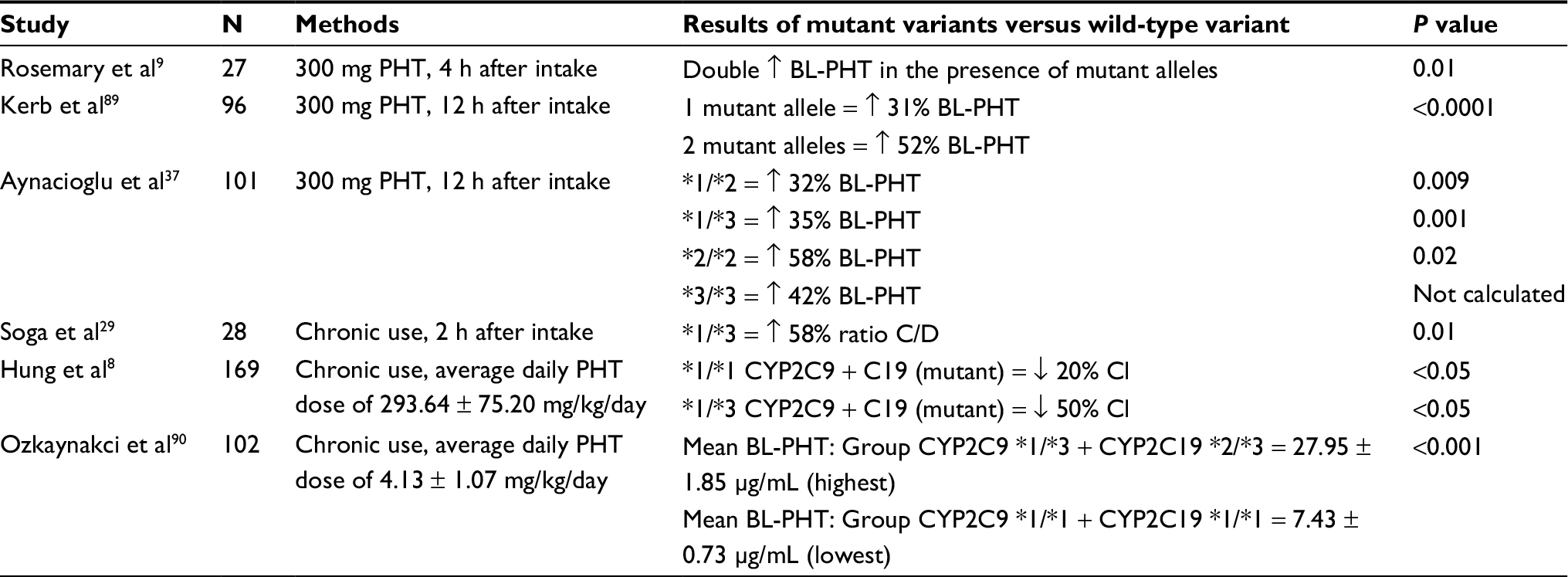 | Table 3 Phenytoin metabolic changes comparing both CYP2C9 wild-type and mutant variants Abbreviations: N, number of patients; PHT, phenytoin; BL-PHT, blood level of phenytoin; C/D, serum phenytoin concentration/dose of phenytoin; Cl, clearance of phenytoin. |
Other variants are rare and have been reported in different ethnic groups; the CYP2C9 *4 is an extremely rare variant identified in Japanese epileptic patients,52 the CYP2C9 *5 variant has been found in African-Americans53–55 and the variant CYP2C9 *6 has been identified in African-Americans and is a result of a null allele due to deletion of A nucleotide at position 818 in exon 5, which leads to almost 90–95% reduction in PHT clearance as compared to that of the wild-type enzyme.10,56
Some studies showed a statistically significant increase in blood level of PHT in patients with epilepsy and alleles CYP2C9 *2 and CYP2C9 *3, compared to patients with the wild-type genotype (Table 3). The presence of only one mutant allele was associated with an increase of 31–58% of PHT blood level. In the presence of two mutant alleles, the PHT blood level increase was from 42 to 58%.
The CYP2C19 gene is also responsible for the metabolization of PHT to a lesser extent (10%). Genetic polymorphism of the CYP2C19 gene has been reported to cause point mutations in exon 5 (variant CYP2C19 *2) and exon 4 (variant CYP2C19 *3) on chromosome 10, and these variants also explain the low metabolism of PHT in Japanese9 and Turkish epileptic patients.37
Adverse effects of PHT
PHT could cause a variety of idiosyncratic or dose-related effects, which can be significantly more common in patients who have the CYP2C9 *2 and CYP2C9 *3 variants.19,29,57–59 Conversely, a recent study of epileptic children in Northern India showed that patients with alleles *2 and *3 had a significant increase in serum PHT, but without a higher incidence of side effects.60
Reversible cosmetic changes, such as hirsutism and coarse facial features, could be problematic, especially in female adolescents.61 Gingival hypertrophy is another frequent problem, although the mechanisms underlying its development are still debatable. Inflammation resulting from plaque, increase in sulfated glycosaminoglycans, population differences in gingival immunoglobulin content and fibroblast count, epithelial growth factors and pharmacokinetics of tissue-binding factors have all been reported as possible causative agents.5,61 Individuals carrying alleles *3 and *2 showed no higher gingival hyperplasia than those carrying the wild-type alleles,8,30 suggesting that this symptom should be due to other factors.
The risk of craniofacial abnormalities in children after fetal exposure to PHT had been reported; however, this was not associated with the maternal CYP2C9 genotype, but rather with the presence of polymorphisms of the rare maternal alleles EPHX1, Y113H and H139R, which compromises the metabolization of arene oxide intermediate to inactive compounds. Variation in EPHX1 activity could affect fetal exposure to the reactive oxide intermediate.62
PHT could induce acute cutaneous reactions such as maculopapular exanthema or severe manifestations such as Stevens–Johnson syndrome (SJS), toxic epidermal necrolysis (TEN) or drug reactions with eosinophilia and systemic symptoms, which occur primarily in the first eight weeks of treatment.59,63–65 A study showed that Korean epileptic patients with genotype CYP2C9 *1/*3 developed more maculopapular rash associated with PHT when compared to controls with or without exposure to PHT (an increase in relative risk of about 170-fold).59 Similarly, Thai epileptic patients with the CYP2C9 *3 genotype had significantly more SJS and TEN due to PHT than patients with the wild-type allele (23% versus 6.5%), but without significant differences in the HLA-B*15:02.10 It is not clear if either the role of CYP2C9 alleles in non-Asian ethnic groups or slow dose titration of PHT itself is responsible for reducing the risk of cutaneous acute reactions as compared to those occurring with lamotrigine.6
The neurotoxic side effects of PHT (drowsiness, dysarthria, tremor, ataxia, and cognitive impairment) are very likely to occur when plasma levels exceed 20 mg/mL.18,50 The presence of polymorphism of CYP2C9 could increase the incidence and severity of neurotoxic side effects. The presence of a null allele for the CYP2C9 enzyme can result in the most severe PHT intoxication.56
Studies show that prolonged use of PHT correlates directly with the development of cerebellar atrophy (CA).66,67 It is suspected that acute exposure to high doses of PHT may also be associated with the development of this complication,68–70 and CA can be induced experimentally in animals by the administration of PHT leading to loss of Purkinje cells and glial damage to the granular layer.71
The incidence of CA in patients using PHT, either acutely or chronically,70,72 can be as high as 50% of patients.73 Older patients in particular are at a higher risk, as CA was observed in this group more frequently, indicating that either advanced age itself or a longer duration of the epilepsy could also be an important factor in the pathogenesis of cerebellar degeneration.67
Administration of PHT in newborn mice can result in a reduction in both cerebellar weight and size.74 Also, changes in white matter distribution and neuronal cell counts in both the Purkinje cells and granular layers have been reported in rats after exposure to this drug.75 Twardowschy et al have recently demonstrated that the presence of variant alleles *2 and *3 may be associated with a smaller cerebellar white matter and volume in patients with epilepsy, treated with PHT for at least 1 year.76
Clinical implications of use of PHT in patients with CYP2C9 polymorphisms
PHT is a highly protein-bound drug (90%), which can cause significant dose-dependent toxicity to the central nervous system. Because of its narrow therapeutic range and nonlinear pharmacokinetic profile, the therapeutic and toxic effects of PHT show a relatively good relationship with its serum concentration.77
Table 3 shows that the frequency of mutant alleles *2 and *3 in patients with epilepsy is 4.5–13.6%. These patients have reduced PHT metabolization, thus being at an increased risk of severe or frequent side effects during the use of PHT at a dosage of 5–10 mg/kg PO. Risks are even higher and more relevant in the emergency management of status epilepticus, when the recommended dose of PHT is a IV bolus of 20 mg/kg, especially if patients have hypoalbuminemia or if patients are in intensive care unit.76 In patients with CYP2C9 *1/*2 or *1/*3 alleles (intermediate metabolizers), the PHT maintenance dose must be reduced by 25%, and in those with CYP2C9 *2/*2, *2/*3 or *3/*3 alleles (poor metabolizers), the dose must be reduced by 50%.6,8,79,80
Patients positive for HLA-B*15:02 could also be at an increased risk of PHT-induced SJS/TEN, particularly if they have CYP2C9 *2 or *3 genotypes. The Clinical Pharmacogenetics Implementation Consortium Guidelines for CYP2C9 and HLA-B Genotypes and Phenytoin Dosing and the US Food and Drug Administration proposed that neither PHT nor fosphenytoin should be used in these patients.81
It is also very important to consider the interactions of PHT with other drugs.
PHT meets all the pharmacogenomic prerequisites necessary for change in drug effect to be considered clinically important, as proposed by MacKenzie and Hall:82
- The effect of the polymorphism on the total active drug moiety should be considerable.
- There should be a clear concentration-versus-response relation.
- There should be severe concentration-dependent adverse effects.
- There should be a narrow therapeutic index.
Future use of pharmacogenomics, particularly for the intensive care unit patients, will be more helpful, given our current inability to predict either the efficacy or the occurrence of unwanted adverse effects of drugs in a population at high risk of being administered large doses of drugs, usually as an IV bolus, more prone to severe adverse effects and with little physiological reserve to compensate them.82
Conclusion
PHT is a very effective drug to treat epilepsy and one of the best options for treating status epilepticus, but its dosages should be individualized to reduce the risk of side effects that could justify its withdrawal, even when effective. The presence of CYP2C9 polymorphism reduces 25–50% of the metabolization of PHT, increasing the risk of side effects with the use of usual (5–10 mg/kg) PHT daily dose.
It is still controversial whether CYP2C9 genotyping should be performed on a regular basis before starting PHT treatment. We need prospective well-controlled studies to prove that individualizing antiepileptic doses in any given patient, either avoiding or reducing the risk of serious side effects, is superior to clinical observation and blood level assessments. There are many other confounding factors that could influence the metabolization and incidence of side effects associated with PHT use, such as age, body weight, pregnancy, drug interactions and so on, in addition to CYP2C9 polymorphisms.6,80,83
As soon as genetic testing for detection of polymorphisms becomes more readily available and cost-effective, we will be able to estimate the metabolization rates and drug interactions, before the prescription and administration of a drug, thus providing a real personalized medicine.
Disclosure
The authors report no conflicts of interest in this work.
References
Merritt HH, Houston Merritt H. Sodium diphenyl hydantoinate in the treatment of convulsive disorders. JAMA. 1938;111(12):1068–1073. | ||
Perucca E. NICE guidance on newer drugs for epilepsy in adults. BMJ. 2004;328(7451):1273–1274. | ||
Yaari Y, Selzer ME, Pincus JH. Phenytoin: mechanisms of its anticonvulsant action. Ann Neurol. 1986;20(2):171–184. | ||
Thorn CF, Whirl-Carrillo M, Leeder JS, Klein TE, Altman RB. PharmGKB summary: phenytoin pathway. Pharmacogenet Genomics. 2012;22(6):466–470. | ||
Ieiri I, Goto W, Hirata K, et al. Effect of 5-(p-hydroxyphenyl)-5-phenylhydantoin (p-HPPH) enantiomers, major metabolites of phenytoin, on the occurrence of chronic-gingival hyperplasia: in vivo and in vitro study. Eur J Clin Pharmacol. 1995;49(1–2):51–56. | ||
Franco V, Perucca E. CYP2C9 polymorphisms and phenytoin metabolism: implications for adverse effects. Expert Opin Drug Metab Toxicol. 2015;11(8):1269–1279. | ||
Yukawa E, Mamiya K. Effect of CYP2C19 genetic polymorphism on pharmacokinetics of phenytoin and phenobarbital in Japanese epileptic patients using Non-linear Mixed Effects Model approach. J Clin Pharm Ther. 2006;31(3):275–282. | ||
Hung CC, Lin CJ, Chen CC, Chang CJ, Liou HH. Dosage recommendation of phenytoin for patients with epilepsy with different CYP2C9/CYP2C19 polymorphisms. Ther Drug Monit. 2004;26(5):534–540. | ||
Rosemary J, Surendiran A, Rajan S, Shashindran CH, Adithan C. Influence of the CYP2C9 AND CYP2C19 polymorphisms on phenytoin hydroxylation in healthy individuals from south India. Indian J Med Res. 2006;123(5):665–670. | ||
Tassaneeyakul W, Prabmeechai N, Sukasem C, et al. Associations between HLA class I and cytochrome P450 2C9 genetic polymorphisms and phenytoin-related severe cutaneous adverse reactions in a Thai population. Pharmacogenet Genomics. 2016;26(5):225–234. | ||
Wang B, Wang J, Huang SQ, Su HH, Zhou SF. Genetic polymorphism of the human cytochrome P450 2C9 gene and its clinical significance. Curr Drug Metab. 2009;10(7):781–834. | ||
Cavalleri GL, McCormack M, Alhusaini S, Chaila E, Delanty N. Pharmacogenomics and epilepsy: the road ahead. Pharmacogenomics. 2011;12(10):1429–1447. | ||
Roden DM, Altman RB, Benowitz NL, et al; Pharmacogenetics Research Network. Pharmacogenomics: challenges and opportunities. Ann Intern Med. 2006;145(10):749–757. | ||
Seven M, Batar B, Unal S, Yesil G, Yuksel A, Guven M. The effect of genetic polymorphisms of cytochrome P450 CYP2C9, CYP2C19, and CYP2D6 on drug-resistant epilepsy in Turkish children. Mol Diagn Ther. 2014;18(2):229–236. | ||
Lakhan R, Kumari R, Singh K, Kalita J, Misra UK, Mittal B. Possible role of CYP2C9 & CYP2C19 single nucleotide polymorphisms in drug refractory epilepsy. Indian J Med Res. 2011;134:295–301. | ||
Lopez-Garcia MA, Feria-Romero IA, Fernando-Serrano H, Escalante-Santiago D, Grijalva I, Orozco-Suarez S. Genetic polymorphisms associated with antiepileptic metabolism. Front Biosci (Elite Ed). 2014;6:377–386. | ||
Franco V, Perucca E. The pharmacogenomics of epilepsy. Expert Rev Neurother. 2015;15(10):1161–1170. | ||
Perucca E. Clinically relevant drug interactions with antiepileptic drugs. Br J Clin Pharmacol. 2006;61(3):246–255. | ||
Spina E, Pisani F, de Leon J. Clinically significant pharmacokinetic drug interactions of antiepileptic drugs with new antidepressants and new antipsychotics. Pharmacol Res. 2016;106:72–86. | ||
Herranz JL. Farmacogenética, farmacogenómica y terapia antiepiléptica individualizada [Pharmacogenetics, pharmacogenomics and individualised antiepileptic therapy]. Rev Neurol. 2006;43 Suppl 1:S43–S49. Spanish [with English abstract]. | ||
Anderson GD. Pharmacogenetics and enzyme induction/inhibition properties of antiepileptic drugs. Neurology. 2004;63(10 Suppl 4):S3–S8. | ||
Klotz U. The role of pharmacogenetics in the metabolism of antiepileptic drugs: pharmacokinetic and therapeutic implications. Clin Pharmacokinet. 2007;46(4):271–279. | ||
Wilkinson GR. Drug metabolism and variability among patients in drug response. N Engl J Med. 2005;352(21):2211–2221. | ||
Ingelman-Sundberg M. Pharmacogenetics of cytochrome P450 and its applications in drug therapy: the past, present and future. Trends Pharmacol Sci. 2004;25(4):193–200. | ||
Wilkinson GR. Drug metabolism and variability among patients in drug response. N Engl J Med. 2005;352(21):2211–2221. | ||
Ethell BT, Anderson GD, Burchell B. The effect of valproic acid on drug and steroid glucuronidation by expressed human UDP-glucuronosyltransferases. Biochem Pharmacol. 2003;65(9):1441–1449. | ||
Zhou SF, Zhou ZW, Huang M. Polymorphisms of human cytochrome P450 2C9 and the functional relevance. Toxicology. 2010;278(2):165–188. | ||
Dai DP, Xu RA, Hu LM, et al. CYP2C9 polymorphism analysis in Han Chinese populations: building the largest allele frequency database. Pharmacogenomics J. 2013;14(1):85–92. | ||
Soga Y, Nishimura F, Ohtsuka Y, et al. CYP2C polymorphisms, phenytoin metabolism and gingival overgrowth in epileptic subjects. Life Sci. 2004;74(7):827–834. | ||
Sullivan-Klose TH, Ghanayem BI, Bell DA, et al. The role of the CYP2C9-Leu359 allelic variant in the tolbutamide polymorphism. Pharmacogenetics. 1996;6(4):341–349. | ||
Wang SL, Huang J, Lai MD, Tsai JJ. Detection of CYP2C9 polymorphism based on the polymerase chain reaction in Chinese. Pharmacogenetics. 1995;5(1):37–42. | ||
Brockmöller J, Rost KL, Gross D, Schenkel A, Roots I. Phenotyping of CYP2C19 with enantiospecific HPLC-quantification of R- and S-mephenytoin and comparison with the intron4/exon5 G-->A-splice site mutation. Pharmacogenetics. 1995;5(2):80–88. | ||
Stubbins MJ, Harries LW, Smith G, Tarbit MH, Wolf CR. Genetic analysis of the human cytochrome P450 CYP2C9 locus. Pharmacogenetics. 1996;6(5):429–439. | ||
Stubbins MJ, Harries LW, Smith G, Tarbit MH, Wolf CR. Genetic analysis of the human cytochrome P450 CYP2C9 locus. Pharmacogenetics. 1996;6(5):429–439. | ||
Nasu K, Kubot T, Ishizaki T. Genetic analysis of CYP2C9 polymorphism in a Japanese population. Pharmacogenetics. 1997;7(5):405–409. | ||
Kimura M, Ieiri I, Mamiya K, Urae A, Higuchi S. Genetic polymorphism of cytochrome P450s, CYP2C19, and CYP2C9 in a Japanese population. Ther Drug Monit. 1998;20(3):243–247. | ||
Aynacioglu AS, Brockmöller J, Bauer S, et al. Frequency of cytochrome P450 CYP2C9 variants in a Turkish population and functional relevance for phenytoin. Br J Clin Pharmacol. 2001;48(3):409–415. | ||
Yasar Ü, Eliasson E, Dahl M-L, Johansson I, Ingelman-Sundberg M, Sjöqvist F. Validation of methods for CYP2C9 genotyping: frequencies of mutant alleles in a Swedish population. Biochem Biophys Res Commun. 1999;254(3):628–631. Erratum in Biochem Biophys Res Commun. 1999;258(1):227. | ||
Scordo MG, Aklillu E, Yasar U, Dahl M-L, Spina E, Ingelman-Sundberg M. Genetic polymorphism of cytochrome P450 2C9 in a Caucasian and a black African population. Br J Clin Pharmacol. 2001;52(4):447–450. | ||
Yoon YR, Shon JH, Kim MK, et al. Frequency of cytochrome P450 2C9 mutant alleles in a Korean population. Br J Clin Pharmacol. 2001;51(3):277–280. | ||
Gaedigk A, Casley WL, Tyndale RF, Sellers EM, Jurima-Romet M, Leeder JS. Cytochrome P4502C9 (CYP2C9) allele frequencies in Canadian Native Indian and Inuit populations. Can J Physiol Pharmacol. 2001;79(10):841–847. | ||
Hamdy SI, Hiratsuka M, Narahara K, et al. Allele and genotype frequencies of polymorphic cytochromes P450 (CYP2C9, CYP2C19, CYP2E1) and dihydropyrimidine dehydrogenase (DPYD) in the Egyptian population. Br J Clin Pharmacol. 2002;53(6):596–603. | ||
Vianna-Jorge R, Perini JA, Rondinelli E, Suarez-Kurtz G. CYP2C9 genotypes and the pharmacokinetics of tenoxicam in Brazilians. Clin Pharmacol Ther. 2004;76(1):18–26. | ||
Jose R, Chandrasekaran A, Sam SS, et al. CYP2C9 and CYP2C19 genetic polymorphisms: frequencies in the south Indian population. Fundam Clin Pharmacol. 2005;19(1):101–105. | ||
Scott SA, Edelmann L, Kornreich R, Erazo M, Desnick RJ. CYP2C9, CYP2C19 and CYP2D6 allele frequencies in the Ashkenazi Jewish population. Pharmacogenomics. 2007;8(7):721–730. | ||
Sánchez-Diz P, Estany-Gestal A, Aguirre C, et al. Prevalence of CYP2C9 polymorphisms in the south of Europe. Pharmacogenomics J. 2009;9(5):306–310. | ||
Dagenais R, Wilby KJ, Elewa H, Ensom MHH. Impact of genetic polymorphisms on phenytoin pharmacokinetics and clinical outcomes in the Middle East and North Africa Region. Drugs R D. 2017;17(3):341–361. | ||
Tate SK, Depondt C, Sisodiya SM, et al. Genetic predictors of the maximum doses patients receive during clinical use of the anti-epileptic drugs carbamazepine and phenytoin. Proc Natl Acad Sci U S A. 2005;102(15):5507–5512. | ||
Adithan C, Gerard N, Vasu S, Balakrishnan R, Shashindran CH, Krishnamoorthy R. Allele and genotype frequency of CYP2C9 in Tamilnadu population. Eur J Clin Pharmacol. 2003;59(8–9):707–709. | ||
Thakkar AN, Bendkhale SR, Taur SR, Gogtay NJ, Thatte UM. Association of CYP2C9 polymorphisms with phenytoin toxicity in Indian patients. Neurol India. 2012;60(6):577–580. | ||
López M, Dorado P, Monroy N, et al. Pharmacogenetics of the antiepileptic drugs phenytoin and lamotrigine. Drug Metabol Drug Interact. 2011;26(1):5–12. | ||
Imai J, Ieiri I, Mamiya K, et al. Polymorphism of the cytochrome P450 (CYP) 2C9 gene in Japanese epileptic patients: genetic analysis of the CYP2C9 locus. Pharmacogenetics. 2000;10(1):85–89. | ||
Dickmann LJ, Rettie AE, Kneller MB, et al. Identification and functional characterization of a new CYP2C9 variant (CYP2C9*5) expressed among African Americans. Mol Pharmacol. 2001;60(2):382–387. | ||
Allabi AC, Gala JL, Horsmans Y, et al. Functional impact of CYP2C95, CYP2C96, CYP2C98, and CYP2C911 in vivo among black Africans. Clin Pharmacol Ther. 2004;76(2):113–118. | ||
Allabi AC, Gala JL, Horsmans Y. CYP2C9, CYP2C19, ABCB1 (MDR1) genetic polymorphisms and phenytoin metabolism in a Black Beninese population. Pharmacogenet Genomics. 2005;15(11):779–786. | ||
Kidd RS, Curry TB, Gallagher S, Edeki T, Blaisdell J, Goldstein JA. Identification of a null allele of CYP2C9 in an African–American exhibiting toxicity to phenytoin. Pharmacogenetics. 2001;11(9):803–808. | ||
Depondt C, Godard P, Espel RS, Da Cruz AL, Lienard P, Pandolfo M. A candidate gene study of antiepileptic drug tolerability and efficacy identifies an association of CYP2C9 variants with phenytoin toxicity. Eur J Neurol. 2011;18(9):1159–1164. | ||
Kesavan R, Narayan SK, Adithan C. Influence of CYP2C9 and CYP2C19 genetic polymorphisms on phenytoin-induced neurological toxicity in Indian epileptic patients. Eur J Clin Pharmacol. 2010;66(7):689–696. | ||
Lee AY, Kim MJ, Chey WY, Choi J, Kim BG. Genetic polymorphism of cytochrome P450 2C9 in diphenylhydantoin-induced cutaneous adverse drug reactions. Eur J Clin Pharmacol. 2004;60(3):155–159. | ||
Chaudhary N, Kabra M, Gulati S, Gupta YK, Pandey RM, Bhatia BD. Frequencies of CYP2C9 polymorphisms in North Indian population and their association with drug levels in children on phenytoin monotherapy. BMC Pediatr. 2016;16:66. | ||
Brodie MJ, Dichter MA. Antiepileptic drugs. N Engl J Med. 1996; 334(3):168–175. | ||
Azzato EM, Chen RA, Wacholder S, Chanock SJ, Klebanoff MA, Caporaso NE. Maternal EPHX1 polymorphisms and risk of phenytoin-induced congenital malformations. Pharmacogenet Genomics. 2010;20(1):58–63. | ||
Arif H, Buchsbaum R, Weintraub D, et al. Comparison and predictors of rash associated with 15 antiepileptic drugs. Neurology. 2007;68(20):1701–1709. | ||
Chung W-H, Chang W-C, Lee Y-S, et al. Genetic variants associated with phenytoin-related severe cutaneous adverse reactions. JAMA. 2014;312:525–534. | ||
Chang C-C, Ng C-C, Too C-L, et al. Association of HLA-B*15:13 and HLA-B*15:02 with phenytoin-induced severe cutaneous adverse reactions in a Malay population. Pharmacogenomics J. 2017;17:170–173. | ||
Ney GC, Lantos G, Barr WB, Schaul N. Cerebellar atrophy in patients with long-term phenytoin exposure and epilepsy. Arch Neurol. 1994;51(8):767–771. | ||
De Marcos FA, Ghizoni E, Kobayashi E, Li LM, Cendes F. Cerebellar volume and long-term use of phenytoin. Seizure. 2003;12(5):312–315. | ||
Kuruvilla T, Bharucha NE. Cerebellar atrophy after acute phenytoin intoxication. Epilepsia. 1997;38(4):500–502. | ||
Alioğlu Z, Sari A, Velioğlu SK, Ouml;zmenoğlu M. Cerebellar atrophy following acute phenytoin intoxication. J Neuroradiol. 2000;27(1):52–55. | ||
Zuin DR, Neme R, Porta L, Vera J, Lopez OL. Atrofia cerebelosa aguda por intoxicación con difenilhidantoína producto de interacción medicamentosa [Acute cerebellar atrophy caused by diphenylhydantoin intoxication resulting from drug interaction]. Rev Neurol. 2003;36(2):195–196. Spanish [with English abstract]. | ||
Ohmori H, Ogura H, Yasuda M, et al. Developmental neurotoxicity of phenytoin on granule cells and Purkinje cells in mouse cerebellum. J Neurochem. 1999;72(4):1497–1506. | ||
Tan EK, Chan LL, Auchus AP. Phenytoin cerebellopathy without epilepsy. Acta Neurol Scand. 2001;104(1):61–62. | ||
Del Negro A, Dantas CD, Zanardi V, Montenegro MA, Cendes F. Relação dose-dependente do uso crônico de fenitoína e atrofia cerebelar em pacientes com epilepsia [Dose-dependent relationship of chronic use of phenytoin and cerebellar atrophy in patients with epilepsy]. Arq Neuropsiquiatr. 2000;58:276–281. Portuguese [with English abstract]. | ||
Ohmori H, Kobayashi T, Yasuda M. Neurotoxicity of phenytoin administered to newborn mice on developing cerebellum. Neurotoxicol Teratol. 1992;14(3):159–165. | ||
Tauer U, Knoth R, Volk B. Phenytoin alters Purkinje cell axon morphology and targeting in vitro. Acta Neuropathol. 1998;95(6):583–591. | ||
Twardowschy CA, Werneck LC, Scola RH, Borgio JG, De Paola L, Silvado C. The role of CYP2C9 polymorphisms in phenytoin-related cerebellar atrophy. Seizure. 2013;22(3):194–197. | ||
Patsalos PN, Berry DJ, Bourgeois BF, et al. Antiepileptic drugs--best practice guidelines for therapeutic drug monitoring: a position paper by the subcommission on therapeutic drug monitoring, ILAE Commission on Therapeutic Strategies. Epilepsia. 2008;49(7):1239–1276. | ||
Soriano VV, Tesoro EP, Kane SP. Characterization of free phenytoin concentrations in end-stage renal disease using the Winter-Tozer equation. Ann Pharmacother. 2017;51(8):669–674. | ||
van der Weide J, Steijns LS, van Weelden MJ, de Haan K. The effect of genetic polymorphism of cytochrome P450 CYP2C9 on phenytoin dose requirement. Pharmacogenetics. 2001;11(4):287–291. | ||
Samer CF, Lorenzini KI, Rollason V, Daali Y, Desmeules JA. Applications of CYP450 testing in the clinical setting. Mol Diagn Ther. 2013;17(3):165–184. | ||
Caudle KE, Rettie AE, Whirl-Carrillo M, et al. Clinical pharmacogenetics implementation consortium guidelines for CYP2C9 and HLA-B genotypes and phenytoin dosing. Clin Pharmacol Ther. 2014;96(5):542–548. | ||
MacKenzie M, Hall R. Pharmacogenomics and pharmacogenetics for the intensive care unit: a narrative review. Can J Anaesth. 2017;64(1):45–64. | ||
Zaccara G, Perucca E. Interactions between antiepileptic drugs, and between antiepileptic drugs and other drugs. Epileptic Disord. 2014;16(4):409–431. | ||
Nasu K, Kubot T, Ishizaki T. Genetic analysis of CYP2C9 polymorphism in a Japanese population. Pharmacogenetics. 1997;7(5):405–409. | ||
Wang SL, Huang J, Lai MD, Tsai JJ. Detection of CYP2C9 polymorphism based on the polymerase chain reaction in Chinese. Pharmacogenetics. 1995;5(1):37–42. | ||
Hashimoto Y, Otsuki Y, Odani A, et al. Effect of CYP2C polymorphisms on the pharmacokinetics of phenytoin in Japanese patients with epilepsy. Biol Pharm Bull. 1996;19(8):1103–1105. | ||
Odani A, Hashimoto Y, Otsuki Y, et al. Genetic polymorphism of the CYP2C subfamily and its effect on the pharmacokinetics of phenytoin in Japanese patients with epilepsy. Clin Pharmacol Ther. 1997;62(3):287–292. | ||
Twardowschy CA, Werneck LC, Scola RH, De Paola L, Silvado CE. CYP2C9 polymorphism in patients with epilepsy: genotypic frequency analyzes and phenytoin adverse reactions correlation. Arq Neuropsiquiatr. 2011;69(2A):153–158. | ||
Kerb R, Aynacioglu AS, Brockmöller J, et al. The predictive value of MDR1, CYP2C9, and CYP2C19 polymorphisms for phenytoin plasma levels. Pharmacogenomics J. 2001;1(3):204–210. | ||
Ozkaynakci A, Gulcebi MI, Ergeç D, et al. The effect of polymorphic metabolism enzymes on serum phenytoin level. Neurol Sci. 2015;36(3):397–401. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.