Back to Journals » International Journal of Nanomedicine » Volume 13
Cyclic RGD-conjugated Pluronic® blending system for active, targeted drug delivery
Authors Lim C, Moon J, Sim T, Hoang NH ![]() , Won WR, Lee ES, Youn YS
, Won WR, Lee ES, Youn YS ![]() , Choi HG, Oh K
, Choi HG, Oh K ![]() , Oh KT
, Oh KT ![]()
Received 20 April 2018
Accepted for publication 8 June 2018
Published 10 August 2018 Volume 2018:13 Pages 4627—4639
DOI https://doi.org/10.2147/IJN.S171794
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Thomas Webster
Chaemin Lim,1,* Junseong Moon,1,* Taehoon Sim,1 Ngoc Ha Hoang,1 Woong Roeck Won,1 Eun Seong Lee,2 Yu Seok Youn,3 Han-Gon Choi,4 Kyungsoo Oh,1 Kyung Taek Oh1
1College of Pharmacy, Chung-Ang University, Seoul, Republic of Korea; 2Division of Biotechnology, The Catholic University of Korea, Bucheon, Republic of Korea; 3School of Pharmacy, SungKyunKwan University, Suwon, Republic of Korea; 4College of Pharmacy, Institute of Pharmaceutical Science and Technology, Hanyang University, Ansan, Republic of Korea
*These authors contributed equally to this work
Background: Blending micellar systems of different types of polymers has been proposed as an efficient approach for tailor-made drug formulations. The lamellar structure of hydrophobic polymers may provide a high drug loading capacity, and hydrophilic polymers may provide good colloidal stability.
Methods: In this study, the anticancer model drug docetaxel was loaded onto a nanosized blending micellar system with two pluronics (L121/F127). To achieve increased antitumor activity, the cyclic arginine-glycine-aspartic acid tripeptide (cRGD) as an active tumor targeting ligand was conjugated to the blending system.
Results: The docetaxel-loaded Pluronic blending system exhibited a higher drug loading capacity than that of F127 and showed high colloidal stability with a spherical structure. cRGD conjugates demonstrated enhanced drug cellular uptake and anticancer activity against αvβ3 integrin-overexpressing U87MG cancer cells. In vivo animal imaging also revealed that the prepared cRGD-conjugated nanoparticles effectively accumulated at the targeted tumor site through an active and passive targeting strategy.
Conclusion: Accordingly, the prepared nanosized system shows potential as a tailor-made, active targeting, nanomedicinal platform for anticancer therapy. We believe that this novel nanoplatform will provide insights for advancement of tumor therapy.
Keywords: blending micellar system, docetaxel, cyclic RGD, Pluronic L121/F127, active targeting, nanomedicine
Introduction
For decades, nanomedicines have attracted considerable attention in pharmaceutical science for drug delivery and, more specifically, for cancer treatment. Many researchers have used nanoparticles to improve the bioavailability of drugs as well as to reduce toxicity and side effects.1–3 In particular, nanoparticles can provide controlled and targeted drug release in tumor tissue that maximizes drug efficacy, and so various types of drug delivery systems, including polymeric micelles, liposomes, polyionic complexes, carbon nanotubes, nanogels, and solid lipid nanoparticles, have been extensively developed for cancer treatment.4–7 Among the various nanoparticles, commercialized Pluronic® block copolymers, which have an A-B-A type of triblock structure (EOx-POy-EOx, EO: ethylene oxide, PO: propylene oxide), have been intensively investigated in preclinical and pharmaceutical science applications.8,9 These copolymers have amphiphilic properties characterized by their hydrophilic–lipophilic balance (HLB) values, which mainly depend on the length of EO and PO in the triblock copolymer and can be self-assembled into core–shell micelles, with a PO core capable of loading insoluble drugs and a hydrophilic EO shell that provides colloidal stability. EO and PO length in relation to the HLB values can significantly affect particle size, drug loading capacity, and stability, as well as the drug release profile.10,11 When the length of the hydrophobic part increases, the loading capacity of the micelles may also be enhanced, but the colloidal stability of nanoparticles with low HLB values could decrease, falling outside of the preferred size range for drug delivery. On the other hand, when the length of the hydrophilic part increases, the loading capacity of the micelles could decrease, but the colloidal stability of nanoparticles could be improved above the critical micelle concentration range.12–14
To compensate for the drawbacks of each polymeric nanoparticle and optimize the physicochemical properties of nanoparticles for drug delivery, polymeric blending has been widely investigated. For example, a blend of hydrophobic L61 and hydrophilic F127 has been developed for doxorubicin delivery and is currently being evaluated in clinical trials.15 In addition, a mixture of P105 and L101 was developed as a paclitaxel delivery system and demonstrates outstanding cell killing effects against multidrug-resistant cells.16 A blending system with L121 and P123 was also developed as a nanosized drug carrier and yields improved colloidal stability and higher drug loading capacity than the monosystems of L121 or P123.17 In our previous work, we developed a Pluronic blending system composed of L121 and F127 to take advantage of the blending systems that allow tailored mixing of block copolymers with different physicochemical properties. The lamellar structure of L121 provides hydrophobic cargo for enhanced drug loading capacity and the F127 with long chains of EO provides improved colloidal stability and a spherical structure for L121.18 In the present study, to demonstrate the application of this L121/F127 blending system in cancer treatment, the anticancer hydrophobic model drug, docetaxel (DTX) was incorporated into the L121/F127 blending system. In addition, to improve the tumor targeting ability of the nanosized blending system, cyclic arginine-glycine-aspartic acid (cRGD) was conjugated to the nanoparticles (Figure 1). The αvβ3 integrins that are specifically overexpressed on tumor endothelium and tissues are reported to have a high binding affinity with various cRGD sequences.19,20 Thus, conjugation of the cRGD moiety to nanoparticles could enhance the cellular uptake efficiency of nanoparticles via integrin αvβ3 receptor-mediated endocytosis, resulting in improved anticancer activity. We prepared and evaluated the cRGD-conjugated DTX-loading L121/F127 blending system for drug loading capacity, stability, targeting ability, and therapeutic efficacy as a promising tailor-made drug delivery system.
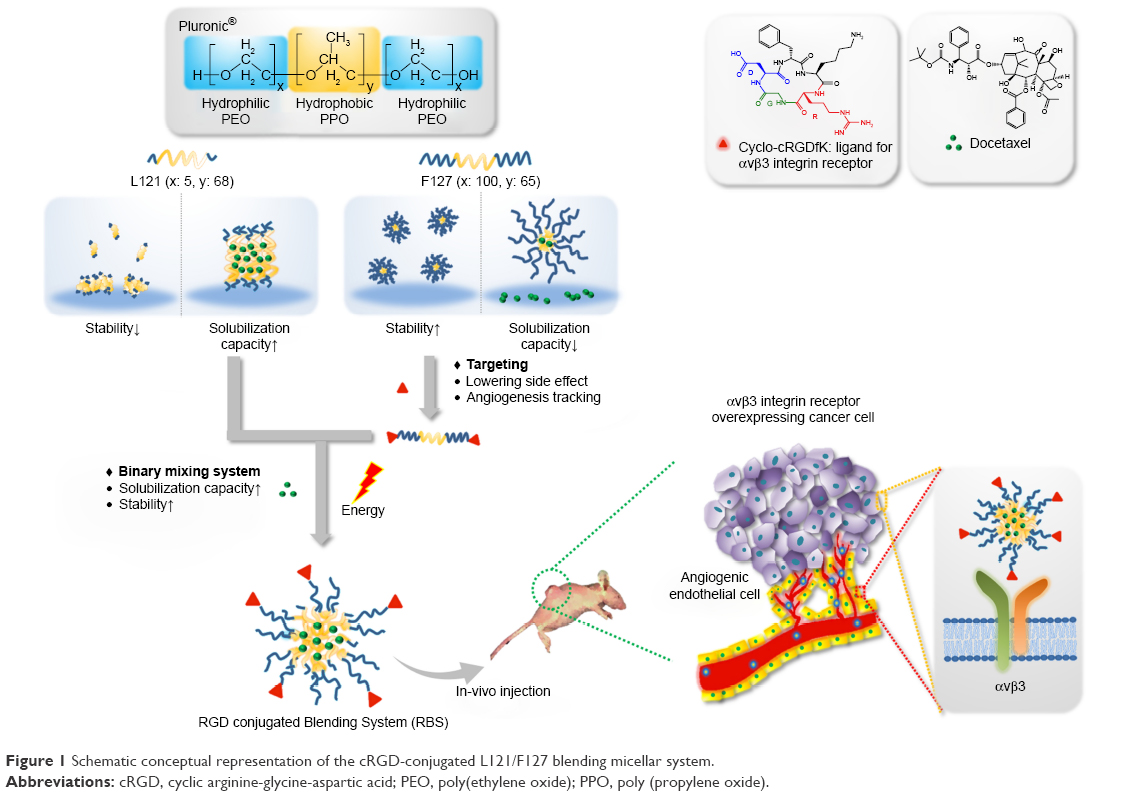 | Figure 1 Schematic conceptual representation of the cRGD-conjugated L121/F127 blending micellar system. |
Materials and methods
Materials
Pluronic F127 (F127), Pluronic L121 (L121), N, N-dicyclohexylcarbodiimide (DCC), ethylene diamine, potassium tert-butoxide, and ethyl bromoacetate were obtained from Sigma-Aldrich (St Louis, MO, USA). Tetrahydrofuran, dimethylsulfoxide (DMSO), and dichloromethane were purchased from Honeywell Burdick & Jackson® (Muskegon, MI, USA). cRGD was obtained from ApexBio® (Houston, TX, USA). DTX was purchased from Samyang Bio Pharmaceutical® (Deajeon, South Korea). 1-Ethyl-3-(3-dimethyl aminopropyl) carbodiimide hydrochloride (EDC), N-hydroxysulfosuccinimide (sulfo-NHS), and fluorescein-5-isothiocyanate (FITC) were obtained from Thermo Fisher Scientific (Waltham, MA, USA). Diethyl ether and hexane were purchased from Samchun Chemical (Seoul, South Korea). U87 malignant glioma cells (U87MG cells, KCLB No 30014) and MCF-7 breast cancer cells (MCF-7 cells, KCLB No 30022) were obtained from the Korean Cell Line Bank (Seoul, South Korea). RPMI medium, DPBS, penicillin–streptomycin solution, trypsin-EDTA solution, and fetal bovine serum were purchased from Welgene (Seoul, South Korea). Cell Counting Kit-8 (CCK-8) was obtained from Dojindo Molecular Technologies (Tokyo, Japan).
Synthesis of carboxylated F127 and cRGD-conjugated F127
F127 was carboxylated as described in the literature.21 Briefly, 10 mg of F127 was dissolved in toluene at 60°C and activated with an excess amount of potassium tert-butoxide for 3 hours. Ethyl bromoacetate was then added to the reactant solution and stirred for 12 hours at 45°C. Modified F127 was obtained by precipitating the reaction mixture in an excess amount of diethyl ether, and the dried polymer was hydrolyzed in 1 N NaOH solution for 30 minutes followed by polymer extraction using an equal volume of methylene chloride. Finally, carboxylated F127 was obtained by precipitating the polymer solution in an excess amount of diethyl ether, filtering, and drying under vacuum. cRGD-conjugated F127 was synthesized by coupling the carboxylated F127 with cRGD using EDC and sulfo-NHS chemistry. Carboxylated F127 was dissolved in 5 mL of phosphate buffer (pH 7.0, 0.1 M) and activated with EDC and sulfo-NHS for 30 minutes at room temperature. Then, 2-mercaptoethanol and cRGD were added and the coupling reaction was carried out for 3 hours at room temperature. Salts and free reactants were removed by dialysis (molecular weight cutoff [MWCO] 10 kDa; Spectrum Laboratories, Rancho Domingues, CA, USA) against distilled water, and cRGD-conjugated F127 was finally obtained by freeze-drying. The cRGD conjugation was confirmed by the presence of an 1H nuclear magnetic resonance peak at δ 2.1 and comparison of the molecular weight (MW) of cRGD-F127 with that of F127 using gel permeation chromatography (PLgel 10 μm MIXED-B column, mobile phase: tetrahydrofuran, flow rate: 1.0 mL/min, temperature: 30°C; Agilent Technologies, Santa Clara, CA, USA). FITC-conjugated F127 was synthesized by coupling the aminated F127 with FITC (Figure S1).
Preparation of the micelle blending system and DTX-loaded micelle blending system
Micelles based on a Pluronic blending system were prepared by solvent evaporation method.22 A mixture of F127 and L121 was dissolved in 5 mL of dichloromethane and stirred for 1 hour at room temperature. Dichloromethane was then completely evaporated by magnetic stirring for 12 hours at 45°C, forming a film layer at the bottom of the vial. Micelles were formed by adding 5 mL of distilled water into the vial with stirring for 3 hours and subsequently sonicating for 2 minutes using a probe sonicator at 500 W (Vibra cell, Sonics & Material, Inc. Newtown, CT, USA). For DTX-loaded micelles, DTX (1 mg) and polymer (9 mg) were dissolved in 5 mL of dichloromethane and prepared in the same manner. The micelle solution was filtered through a 0.2 μm filter membrane to eliminate unloaded drug. Drug-loading capacity and efficiency were calculated using the following equations:
|

|

The concentration of DTX was determined using high-performance liquid chromatography (Agilent 1200 series, Agilent Technologies Santa Clara, CA, USA). A reverse phase C18 column (150×4.6 mm id., pore size 5 μm; Microsolv Tech. Corp., Eatontown, NJ, USA) was used, and the mobile phase, consisting of a mixture of acetonitrile and DW (55:45, v/v%), was delivered at a flow rate of 1 mL/min with a pump. The column effluent was detected at 230 nm, and the concentration of DTX was calculated using a linear calibration curve of standard DTX solutions (R2=0.9999; Figure S2).
Particle size and zeta potential measurements using dynamic light scattering
The effective hydrodynamic diameters (Deff) and zeta potentials of the micelles were measured by photon correlation spectroscopy using a Zetasizer Nano-ZS (Malvern Instruments, Malvern, UK) equipped with the Multi-angle Sizing Option (BI-MAS). The average particle size and zeta potentials of the micelles were calculated from three measurements performed on each sample (n=3).
Morphology of micelles using field emission-scanning electron microscopy (FE-SEM)
To observe the morphologies of micelles, the dilute micelle solution was placed onto a glass slide, dried in vacuo and coated with platinum using ion sputtering. The morphology of the blending system was obtained using FE-SEM (Carl Zeiss AG, Jena, Germany).
In vitro drug release
To test drug release, free DTX or DTX-loaded micelle solutions (2 mL) were placed in triplicate into spectra/Por dialysis membrane bags (MWCO 6K–8K Da), respectively, and incubated in PBS (pH 7.4, containing 0.5% Tween 80 [w/v] to maintain a sink condition), under agitation at 100 rpm (shaking water bath; Nexus Tech., Seoul, South Korea) at 37°C. The outer phase of the dialysis membrane bag was withdrawn for analysis of drug concentration and replaced with fresh PBS at predetermined time intervals to maintain a sink condition. The concentration of DTX was measured by high-performance liquid chromatography.
In vitro cellular uptake study
The cellular uptake of micelles into MCF-7 and U87MG cells was studied using confocal microscopy and flow cytometry. For confocal images, each cell was seeded at a density of 1.5×105 cells/well onto six-well plates with a cover glass in each well. The next day, each cell was exposed to FITC-conjugated micelles or cRGD and FITC-conjugated micelles. After 4 hours of incubation, the cells on the cover glass were washed three times with cold PBS and fixed with paraformaldehyde solution (4%) in PBS for 15 minutes. The fixed cells were stained with 4′,6-diamidino-2-phenylindole (Thermo Fisher Scientific) and the cell-plated cover glasses were mounted on glass slides using Permount® mounting medium (Thermo Fisher Scientific). The prepared cells were observed using a confocal microscope (Carl Zeiss AG Jena, Germany). For flow cytometry, MCF-7 and U87MG cells were seeded at a density of 4×105 cells/well onto six-well plates. The next day, each cell was exposed to FITC-conjugated micelles or cRGD and FITC-conjugated micelles. After 4 hours of incubation, the cells were washed three times with cold PBS, harvested, and analyzed with a FACSCalibur flow cytometer using the Cell Quest Pro software (BD Biosciences, San Diego, CA, USA).
In vitro anticancer effects
MCF-7 and U87MG cells in growth medium were seeded at a density of 5×103 cells/well in a 96-well plate 24 hours prior to the cytotoxicity test. Free DTX and DTX-loaded micelles in RPMI 1640 medium were prepared immediately before use. The medium was removed from the 96-well plate and DTX at different concentrations was added and incubated for 48 hours. Cell viability was assessed using the CCK-8 assay. CCK-8 assay is based on colorimetric assays with the highly water-soluble tetrazolium salt and it produces a water-soluble formazan dye upon reduction in the presence of an electron mediator in viable cells. Briefly, fresh medium (90 μL) with 10 μL of CCK solution was added to each well and the plate was incubated for an additional 3 hours. The absorbance of each well was read on a Flexstation three microplate reader (Molecular Devices, Sunnyvale, CA, USA) at a wavelength of 450 nm.
In vivo fluorescence imaging
In vivo experiments were performed using 3-week-old female nude mice (BALB/c, nu/nu mice; Nara Biotech, Seoul, South Korea). The mice were maintained under the guidelines of a protocol approved by the Institutional Animal Care and Use Committee of Chung-Ang University of Korea, and all experiments were performed in compliance with the relevant laws and institutional guidelines (National Institute of Health guidelines “Principles of laboratory animal care”, approval number: 2017–00036). For real-time tumor imaging animal experiments, carboxylated F127 was reacted with the amine of Cy5.5 (1:1 molar ratio) using DCC and NHS chemistry. Carboxylated F127 was dissolved in 5 mL of DMSO and activated with DCC and NHS for 30 minutes at room temperature. Cy5.5-NH2 dissolved in DMSO (1 mL) was added to that solution and the coupling reaction was carried out for 24 hours at room temperature. The free reactants were removed by dialysis (MWCO 3.5 kDa; Spectrapore) against DMSO for 2 days and Cy5.5-conjugated F127 was finally obtained by freeze-drying.23 αvβ3-overexpressing U87MG cells were introduced into 4-week-old female nude mice by subcutaneous injection of 5×105 cells and when the tumor volume reached 100 mm3, Cy5.5-labeled micelles were injected intravenously through the tail vein. A fluorescence labeled organism bioimaging instrument (FOBI) fluorescence live imaging system (IFLIS; NeoScience, Suwon, South Korea) was used to obtain live fluorescence images of the mice. At 24 hours postinjection, the nude mice were sacrificed and the excised organs were analyzed.
Results and discussion
RGD-F127 synthesis
In this study, cRGD was conjugated to carboxylated F127 in the presence of sulfo-NHS and EDC (Figure 2A). The conjugation of cRGD to F127 was confirmed by the presence of an 1H nuclear magnetic resonance peak at δ 2.6 (Figure 2B), change of MW in the gel permeation chromatography (GPC) analysis (Figure 2C) and change of spectrum peak in Fourier-transform infrared analysis (Figure S3). One molecule of cRGD was conjugated to F127 and the MW of the cRGD-F127 was ~17,000 Da.
 | Figure 2 (A) Overall scheme for the synthesis of cRGD-conjugated F127. (B) 1H-NMR spectra of carboxylated F127 and cRGD-conjugated F127. (C) Characteristics of block copolymer by 1H NMR and GPC. aNumber average molecular weights are determined from the integration ratio of PPO segments at 1.1 ppm and to that of PEO segments at 3.6 ppm and RGD segments at 2.1 ppm in the 1H-NMR. b,cWeight average molecular weight and PDI were measured by GPC. |
Pluronic blending system preparation and characterization
It is well known that blending systems composed of different types of polymers may compensate for the drawbacks of each individual polymeric system and optimize the physicochemical properties of nanoparticles in drug delivery applications.15–17 In our previous study, we demonstrated that a Pluronic-based blending system with L121/F127 (1:1) had a higher drug loading capacity than a monosystem of F127 and it showed high thermodynamic stability in aqueous solution.18 Thus, to take the advantage of a micellar blending system, L121/F127 (1:1) was prepared as a drug carrier and the cRGD peptide as an αvβ3 integrin receptor-specific targeting moiety was conjugated to F127 to enhance drug delivery efficacy into targeted tumor sites. In particular, to optimize the anchoring ratio of cRGD ligand to nanoparticle, L121 was fixed at 50 wt% and different ratios of F127 and cRGD-F127 were prepared and characterized (Figure 3A and B). As the amount of cRGD conjugated to F127 increased in the mixed micellar system, the particle size did not change significantly. However, an increase in cRGD-F127 ratio led to drastic decreases in the zeta potentials of the RGD-conjugated blending system (RBS), which may be due to the presence of a carboxyl group in cRGD-F127. The surface charge of nanoparticles plays an important role in endocytic pathways, and the cellular uptake efficiency of highly negatively charged nanoparticles could decrease due to electrostatic repulsion between nanoparticles and the negatively charged cell membrane. In addition, when nanoparticles are intravenously administered, the highly negatively charged nanoparticles tend to be arrested easily by the reticuloendothelial system and removed by the liver and spleen.24–26 Thus, to improve the circulatory half-life and carrier transport capability while preserving the target affinity of the surface-anchored cRGD peptide, we selected RBS30, which has a small particle size with moderate zeta potential value, as a drug carrier for further experiments.25 The average particle size of RBS30 was ~125 nm with a narrow size distribution and a zeta potential of −17 mV in distilled water. Based on FE-SEM, the morphology of RBS30 consisted of regular spherical particles with smooth surfaces (Figure 3C).
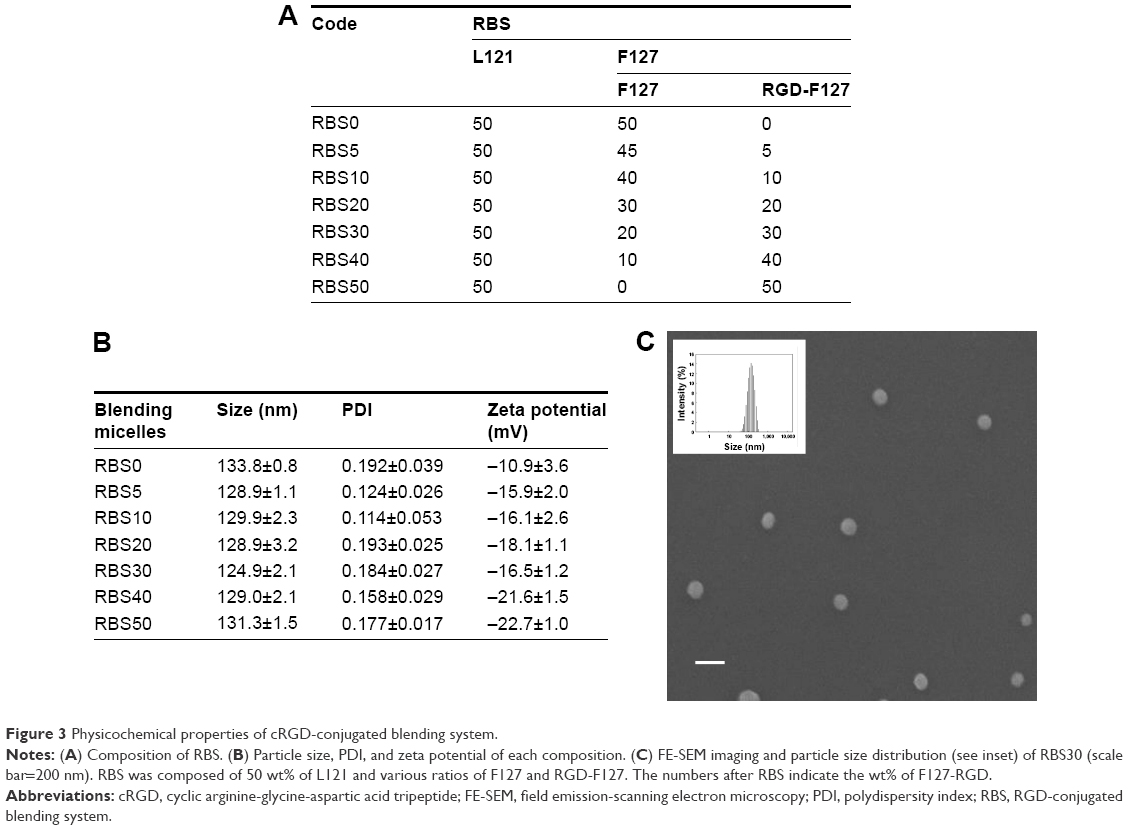 | Figure 3 Physicochemical properties of cRGD-conjugated blending system. |
Drug formulation and characterization
One of the most efficient anticancer drugs, DTX, has been widely used to treat cancer in multiple locations, including breast, prostate, lung, and stomach.27 We chose DTX as a model drug to validate the enhanced delivery and activity for cancer treatment of the cRGD-conjugated Pluronic blending system. DTX-loaded micelles prepared by a solvent evaporation method are characterized and shown in Figure 4A. As the target loading amount of drug increased from 5% to 15%, the loading content of RBS30 also increased without much change in particle size. The maximum drug loading content was 11.38% and was highly stable in aqueous conditions without precipitation for >1 week. When the target loading amount of DTX for RBS30 increased to 20%, the loading content and loading efficiency dropped drastically due to increased hydrophobic interactions between the drug molecule and hydrophobic core of the micelles, resulting in particle aggregation and precipitation. Based on these results, 10% targeted RBS30 was selected as an anticancer nanomedicine due to its small size with high loading efficiency. Compared to blank micelles, there were no big differences in particle size, zeta potential, or PDI values of DTX-loaded RBS30. The DTX release profiles from micelles in PBS (pH 7.4, containing 0.5% Tween 80 [w/v] to maintain a sink condition) are illustrated in Figure 4B. Free DTX was rapidly released within 12 hours, but DTX in RBS30 or RBS0 exhibited an initial burst of release followed by a sustained release. Sustained and controlled release of the developed formulation could be advantageous in clinical therapy because a continuous low dose of DTX could be an effective treatment for cancer with fewer adverse events for patients.28 There was no significant difference in the drug release profile between RBS30 and RBS0, indicating that cRGD conjugation to nanoparticles did not affect the crystallinity or fluidity of the micelle core.29 The prepared DTX-loaded RBS30 showed good stability in particle size without any precipitation for >1 week (Figure 4C). The freeze-drying and reconstitution process with DTX-loaded RBS30 is in progress to make it more favorable for clinical applications.30
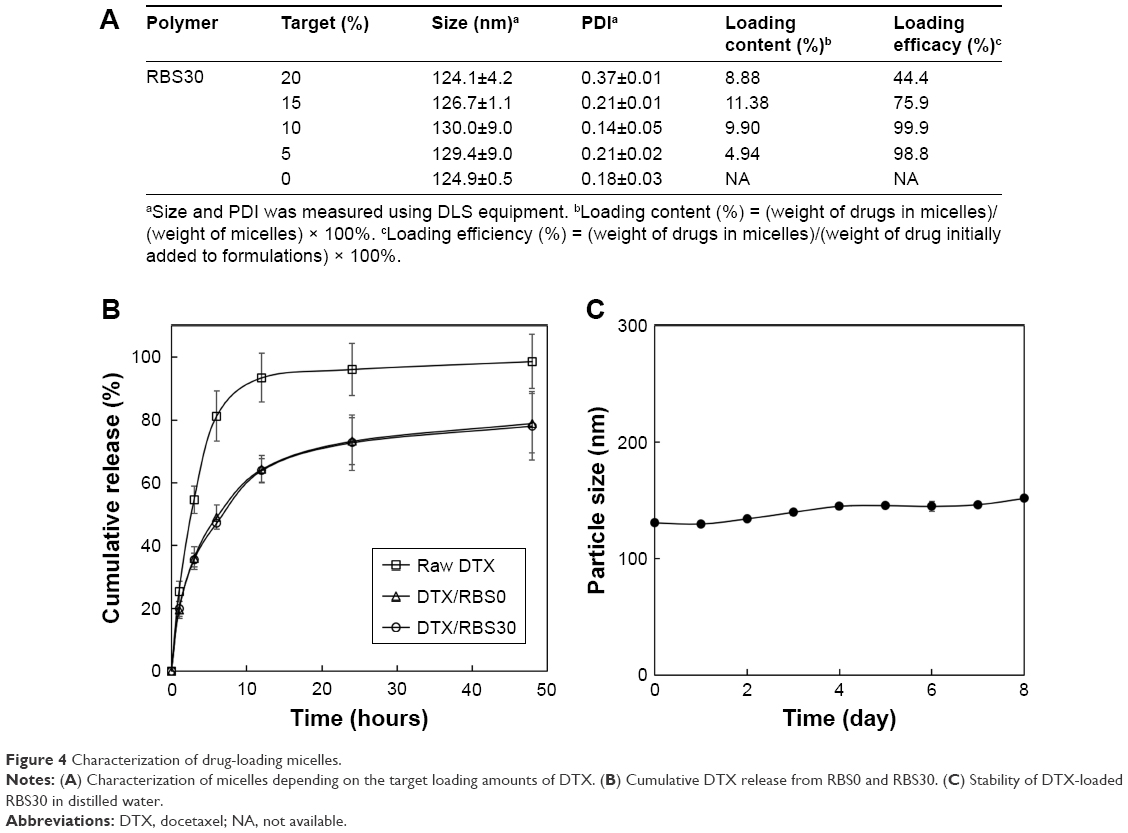 | Figure 4 Characterization of drug-loading micelles. |
Cellular uptake study
αvβ3 reportedly plays an important role in angiogenesis for tumor growth and is overexpressed on tumoral endothelial cells as well as in U87MG cells. The triple peptide arginine-glycine-aspartic acid (RGD) is known to bind preferentially to the integrin αvβ3 receptor.19,20 In terms of intracellular absorption of nanoparticles, the receptor-mediated endocytosis pathway is known to be more efficient than the general endocytosis pathway.31 Thus, we attached cRGD to the micelle surface via sulfo-NHS chemistry to enhance drug delivery efficacy. Confocal microscopy and flow cytometry were used to characterize the effect of cRGD conjugation on the cellular uptake of the nanoparticles in U87MG and MCF-7 cells. There were no significant differences in FITC fluorescence intensity from MCF-7 cells treated with RBS0 or RBS30, as seen under confocal fluorescence microscopy (Figure 5A). However, the FITC fluorescence intensity from αvβ3 integrin-overexpressing U87MG cells incubated with RBS30 was higher than that of cells incubated with RBS0 (Figure 5B), demonstrating that the cRGD moiety-conjugated delivery platform promoted cellular uptake via the ligand–receptor interaction in an αvβ3 integrin-overexpressing cancer cell line. The enhanced uptake of FITC-labeled RBS30 was confirmed by flow cytometry (Figure 5). In the flow cytometry analysis, there was no big difference in the FITC intensity in MCF-7 cells incubated with RBS0 or RBS30, but the FITC intensity in U87MG cells incubated with RBS30 was ~3.3 times higher than that of cells incubated with RBS0.
 | Figure 5 Confocal microscopy images and FACS analysis of (A) MCF-7 and (B) U87MG cells treated with FITC-labeled RBS0 or RBS30 for 4 hours. |
In vitro anticancer effects
Targeting the tumor cells by cRGD moiety conjugation strategy is a promising approach for delivering drugs with improved anticancer activity. cRGD-conjugated nanoparticles could be more preferentially taken up by the tumor cells via integrin αvβ3 receptor-mediated endocytosis, enhancing the anticancer activity.32,33 To examine cRGD ligand conjugation on nanoparticles, the in vitro cytotoxicity of DTX-loaded RBS0 and RBS30 against U87MG cells and MCF-7 cells was evaluated by a cell viability test. Cells were exposed to a series of equivalent concentrations of DTX for 48 hours and the viability of the cells was quantified using a CCK assay. As shown in Figure 6A, there were no significant differences between DTX-loaded RBS0 and DTX-loaded RBS30 in anticancer activity against MCF-7 cells. However, when the samples were added to αvβ3-overexpressing U87MG cells, DTX-loaded RBS30 achieved better anticancer activity than that in cells treated with free DTX or DTX-loaded RBS0 (Figure 6B), demonstrating that the addition of cRGD moieties in αvβ3-overexpressing cells leads to receptor-mediated endocytosis. Since αvβ3 integrin is typically overexpressed on various malignant tumors as well as tumoral endothelial cells, cRGD-based active tumor targeting could be a great strategy in developing anticancer nanomedicines. In addition, the particle size of drug-loaded RBS30 was about 130 nm with a narrow size distribution, taking advantage of the enhanced permeability and retention effect.34,35 Thus, in vivo, the combination of the active and passive targeting ability of RBS30 could lead to the penetration of nanoparticles into the tumor tissue more efficiently, followed by enhanced cellular uptake and improved anticancer activity.
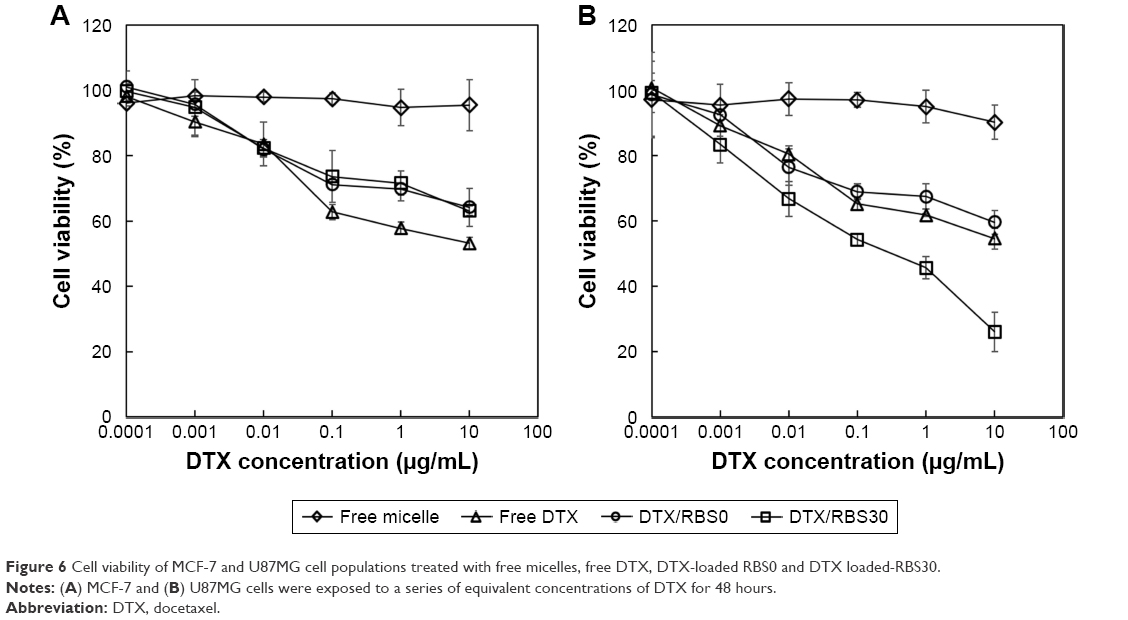 | Figure 6 Cell viability of MCF-7 and U87MG cell populations treated with free micelles, free DTX, DTX-loaded RBS0 and DTX loaded-RBS30. |
In vivo imaging of cRGD-conjugated nanoparticles composed of F127/L121 (1:1) was designed with the assumption that the cRGD peptide could be used as an active targeting ligand both in vivo and in vitro. Thus, to evaluate the active targeting ability of RBS30 in vivo, free Cy5.5 and Cy5.5-labeled RBS0 and RBS30 were intravenously injected into αvβ3-overexpressing tumor-bearing nude mice. As shown in Figure 7A, only RBS0 and RBS30 facilitated high-resolution in vivo Fluorescence intensity (FI) at the tumor site, demonstrating the Enhanced permeability and retention (EPR) effect of nanoparticles in blood stream circulation. To investigate the cRGD-based active targeting ability more precisely, nude mice were sacrificed and the main organs were excised at 24 hours postinjection (Figure 7B). The fluorescence intensity of tumors treated with RBS30 was 1.4 times higher than that of those treated with RBS0, indicating that RBS30 was more efficiently delivered to the tumor site via active and passive targeting (Figure 7C). The different targeting efficacy of RBS30 between in vitro and in vivo conditions is related to the EPR effect and heterogeneity and complexity of the in vivo environment.36,37
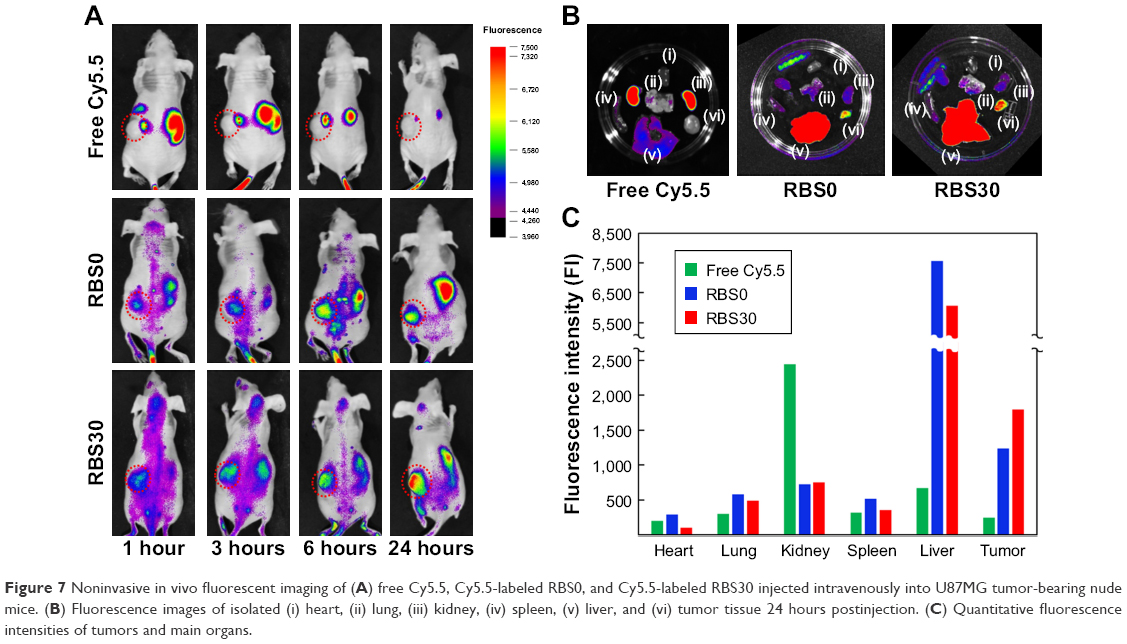 | Figure 7 Noninvasive in vivo fluorescent imaging of (A) free Cy5.5, Cy5.5-labeled RBS0, and Cy5.5-labeled RBS30 injected intravenously into U87MG tumor-bearing nude mice. (B) Fluorescence images of isolated (i) heart, (ii) lung, (iii) kidney, (iv) spleen, (v) liver, and (vi) tumor tissue 24 hours postinjection. (C) Quantitative fluorescence intensities of tumors and main organs. |
Conclusion
In this research, cRGD-conjugated blending systems of L121/F127 were prepared and evaluated for potential use in drug delivery as an active targeting delivery platform. As proposed in Figure 1, L121 and F127 were blended to improve DTX-loading capacity as well as particle stability. In addition, the cRGD moiety was conjugated to the micellar system to enhance drug delivery efficacy. The hydrophobic DTX was successfully loaded into the core of micelles with an average size of 100 nm with demonstrated stability of more than a week. The cRGD moiety-conjugated nanoparticle showed improved anticancer activity and tumor targeting ability against αvβ3 integrin-overexpressing U87MG cancer cells both in vitro and in vivo. Based on these findings, the cRGD moiety-conjugated blending system shows considerable potential for antitumor activity and as a tailor-made drug delivery platform to encapsulate various drugs by changing the ratio of blended polymers. We believe that this novel nanoplatform will provide insights for advancement of tumor therapy.
Acknowledgments
This research was supported by a Chung-Ang University Graduate Research Scholarship in 2017 and supported by National Research Foundation of Korea (NRF) grants funded by the Korean government (MSIP) (NRF-2015R1A5A1008958).
Disclosure
The authors report no conflicts of interest in this work.
References
Doane TL, Burda C. The unique role of nanoparticles in nanomedicine: imaging, drug delivery and therapy. Chem Soc Rev. 2012;41(7):2885–2911. | ||
Wagner V, Dullaart A, Bock AK, Zweck A. The emerging nanomedicine landscape. Nat Biotechnol. 2006;24(10):1211–1217. | ||
Ma X, Williams RO. Polymeric nanomedicines for poorly soluble drugs in oral delivery systems: an update. Int J Pharm Investig. 2018;48(1):61–75. | ||
Hoang NH, Lim C, Sim T, Oh KT, Kt O. Triblock copolymers for nano-sized drug delivery systems. J Pharm Investig. 2017;47(1):27–35. | ||
Moghimi SM, Hunter AC, Murray JC. Long-circulating and target-specific nanoparticles: theory to practice. Pharmacol Rev. 2001;53(2):283–318. | ||
Son G-H, Lee B-J, Cho C-W. Mechanisms of drug release from advanced drug formulations such as polymeric-based drug-delivery systems and lipid nanoparticles. J Pharm Investig. 2017;47(4):287–296. | ||
Uhrich KE, Cannizzaro SM, Langer RS, Shakesheff KM. Polymeric systems for controlled drug release. Chem Rev. 1999;99(11):3181–3198. | ||
Dumortier G, Grossiord JL, Agnely F, Chaumeil JC. A review of poloxamer 407 pharmaceutical and pharmacological characteristics. Pharm Res. 2006;23(12):2709–2728. | ||
Danson S, Ferry D, Alakhov V, et al. Phase I dose escalation and pharmacokinetic study of pluronic polymer-bound doxorubicin (SP1049C) in patients with advanced cancer. British Journal of Cancer. 2004;90(11):2085–2091. | ||
Xiong XY, Tam KC, Gan LH. Polymeric nanostructures for drug delivery applications based on Pluronic copolymer systems. J Nanosci Nanotechnol. 2006;6(9–10):2638–2650. | ||
Han LM, Guo J, Zhang LJ, Wang QS, Fang XL. Pharmacokinetics and biodistribution of polymeric micelles of paclitaxel with Pluronic P123. Acta Pharmacol Sin. 2006;27(6):747–753. | ||
Torchilin VP. Structure and design of polymeric surfactant-based drug delivery systems. J Control Release. 2001;73(2–3):137–172. | ||
Yun JM, Park S-Young, Lee ES, et al. Physicochemical characterizations of amphiphilic block copolymers with different MWs and micelles for development of anticancer drug nanocarriers. Macromol Res. 2012;20(9):944–953. | ||
Allen C, Maysinger D, Eisenberg A. Nano-engineering block copolymer aggregates for drug delivery. Colloids Surf B Biointerfaces. 1999;16(1–4):3–27. | ||
Alakhov V, Klinski E, Li S, et al. Block copolymer-based formulation of doxorubicin. From cell screen to clinical trials. Colloids Surf B Biointerfaces. 1999;16(1–4):113–134. | ||
Wang Y, Yu L, Han L, Sha X, Fang X. Difunctional Pluronic copolymer micelles for paclitaxel delivery: synergistic effect of folate-mediated targeting and Pluronic-mediated overcoming multidrug resistance in tumor cell lines. Int J Pharm. 2007;337(1–2):63–73. | ||
Lee ES, Oh YT, Youn YS, et al. Binary mixing of micelles using Pluronics for a nano-sized drug delivery system. Colloids Surf B Biointerfaces. 2011;82(1):190–195. | ||
Oh KT, Bronich TK, Kabanov AV. Micellar formulations for drug delivery based on mixtures of hydrophobic and hydrophilic Pluronic block copolymers. J Control Release. 2004;94(2–3):411–422. | ||
Danhier F, Vroman B, Lecouturier N, et al. Targeting of tumor endothelium by RGD-grafted PLGA-nanoparticles loaded with paclitaxel. J Control Release. 2009;140(2):166–173. | ||
Danhier F, Le Breton A, Préat V. RGD-based strategies to target alpha(v) beta(3) integrin in cancer therapy and diagnosis. Mol Pharm. 2012;9(11):2961–2973. | ||
Zalipsky S, Barany G. Facile synthesis of α-hydroxy-ω-carboxymethylpolyethylene oxide. J Bioact Compat Polym. 1990;5(2):227–231. | ||
Lee ES, Lim C, Song HT, et al. A nanosized delivery system of superparamagnetic iron oxide for tumor MR imaging. Int J Pharm. 2012;439(1–2):342–348. | ||
Jeong JY, Hong EH, Lee SY, et al. Boronic acid-tethered amphiphilic hyaluronic acid derivative-based nanoassemblies for tumor targeting and penetration. Acta Biomater. 2017;53:414–426. | ||
Fröhlich E. The role of surface charge in cellular uptake and cytotoxicity of medical nanoparticles. Int J Nanomedicine. 2012;7:5577–5591. | ||
He C, Hu Y, Yin L, Tang C, Yin C. Effects of particle size and surface charge on cellular uptake and biodistribution of polymeric nanoparticles. Biomaterials. 2010;31(13):3657–3666. | ||
Lim C, Sim T, Hoang NH, et al. A charge-reversible nanocarrier using PEG-PLL (-g-Ce6, DMA)-PLA for photodynamic therapy. Int J Nanomedicine. 2017;12:6185–6196. | ||
Choi YH, Han H-K. Nanomedicines: current status and future perspectives in aspect of drug delivery and pharmacokinetics. J Pharm Investig. 2018;48(1):43–60. | ||
Kao C-L, Cha T-L, Kao C-C, et al. Weekly low-dose docetaxel is an effective treatment with fewer adverse events for metastatic castration-resistant prostate cancer in Taiwanese patients. Urol Sci. 2015;26(4):267–270. | ||
Hoang NH, Lim C, Sim T, et al. Characterization of a triblock copolymer, poly(ethylene glycol)-polylactide-poly(ethylene glycol), with different structures for anticancer drug delivery applications. Polym Bull. 2017;74(5):1595–1609. | ||
di Tommaso C, Como C, Gurny R, Möller M. Investigations on the lyophilisation of MPEG-hexPLA micelle based pharmaceutical formulations. Eur J Pharm Sci. 2010;40(1):38–47. | ||
Xu S, Olenyuk BZ, Okamoto CT, Hamm-Alvarez SF. Targeting receptor-mediated endocytotic pathways with nanoparticles: rationale and advances. Adv Drug Deliv Rev. 2013;65(1):121–138. | ||
Li Z, Huang P, Zhang X, et al. RGD-conjugated dendrimer-modified gold nanorods for in vivo tumor targeting and photothermal therapy. Mol Pharm. 2010;7(1):94–104. | ||
Yonenaga N, Kenjo E, Asai T, et al. RGD-based active targeting of novel polycation liposomes bearing siRNA for cancer treatment. J Control Release. 2012;160(2):177–181. | ||
Maeda H, Bharate GY, Daruwalla J. Polymeric drugs for efficient tumor-targeted drug delivery based on EPR-effect. Eur J Pharm Biopharm. 2009;71(3):409–419. | ||
Fang J, Nakamura H, Maeda H. The EPR effect: unique features of tumor blood vessels for drug delivery, factors involved, and limitations and augmentation of the effect. Adv Drug Deliv Rev. 2011;63(3):136–151. | ||
Choi J, Rustique E, Henry M, et al. Targeting tumors with cyclic RGD-conjugated lipid nanoparticles loaded with an IR780 NIR dye: in vitro and in vivo evaluation. Int J Pharm. 2017;532(2):677–685. | ||
Bae YH, Park K. Targeted drug delivery to tumors: myths, reality and possibility. J Control Release. 2011;153(3):198–205. |
Supplementary materials
Synthesis of fluorescein isothiocyanate (FITC)-conjugated F127
Aminated F127 was prepared by the coupling reaction between carboxylated F127 and ethylene diamine. Carboxylated F127 was activated with N-hydroxysulfosuccinimide (0.2 mmol) and N, N-dicyclohexylcarbodiimide (0.2 mmol) in dichloromethane for 24 hours at room temperature, followed by the addition of ethylene diamine. The coupling reaction was carried out under a nitrogen environment at room temperature for 3 hours and precipitated in diethyl ether. FITC-F127 was synthesized by the coupling reaction of aminated F127 with FITC. Briefly, aminated F127 (0.2 mmol) and FITC (0.4 mmol) were dissolved in 5 mL of dimethyl sulfoxide and the coupling reaction was carried out for 3 hours at room temperature. The reactant solutions were dialyzed against distilled water and collected by freeze-drying.
FITC was conjugated to amine residues in the modified F127 and the conjugation was confirmed by the presence of an 1H nuclear magnetic resonance peak at δ 6.4 and change of molecular weight in gel permeation chromatography analysis (Figure S1). Two molecules of FITC were conjugated to each F127 molecule.
Attenuated total reflectance Fourier transform infrared analysis
The infrared spectra of F127, carboxylated F127, and cyclic arginine-glycine-aspartic acid tripeptide (cRGD)-conjugated F127 were analyzed by using a Perkin-Elmer attenuated total reflectance Fourier transform infrared analysis spectrum at room temperature. The spectral region of 600–4,000 cm−1 was obtained from four scans with a resolution of 2 cm−1 and peak fitting was done by using OMNIC software (Thermo Nicolet Corporation, Madison, WI, USA).
When F127 was carboxylated, the intensities of infrared (IR) peak centered at ≈1,730 cm−1 appeared, which are ascribed to the saturated carboxyl group stretching. When F127-COOH was reacted with cRGD, the intensities of IR peak centered at ≈1,730 cm−1 decreased and a new IR peak at ≈1,650 cm−1 (amide C=O) appeared. These results distinctly indicated that F127 was successfully conjugated to cRGD, as we confirmed by 1H nuclear magnetic resonance and gel permeation chromatography analysis.
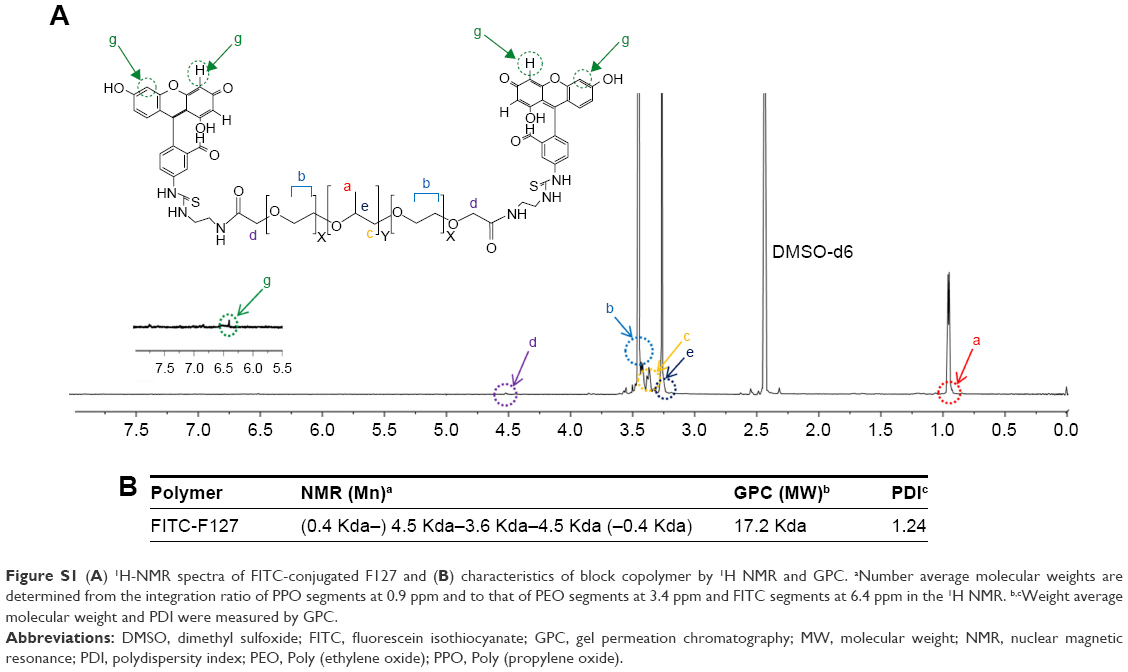 | Figure S1 (A) 1H-NMR spectra of FITC-conjugated F127 and (B) characteristics of block copolymer by 1H NMR and GPC. aNumber average molecular weights are determined from the integration ratio of PPO segments at 0.9 ppm and to that of PEO segments at 3.4 ppm and FITC segments at 6.4 ppm in the 1H NMR. b,cWeight average molecular weight and PDI were measured by GPC. |
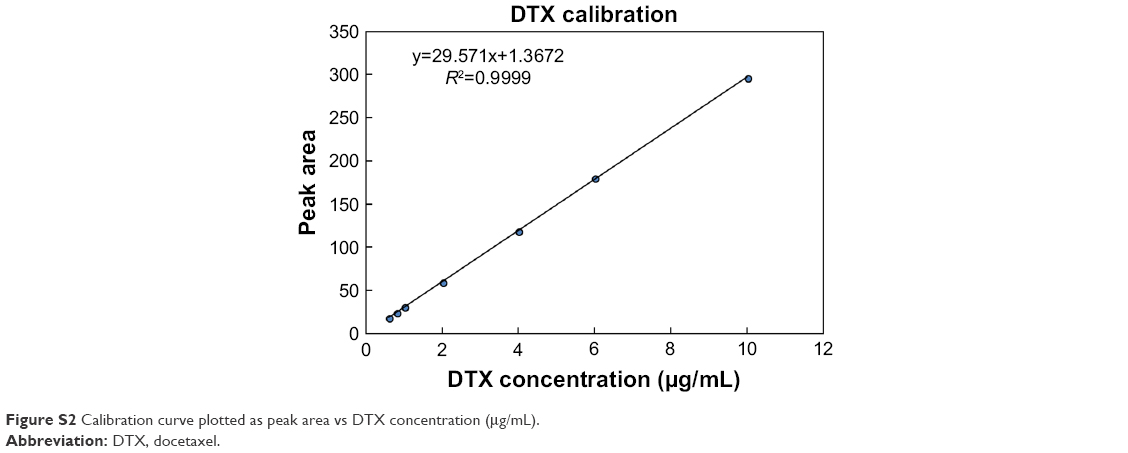 | Figure S2 Calibration curve plotted as peak area vs DTX concentration (μg/mL). |
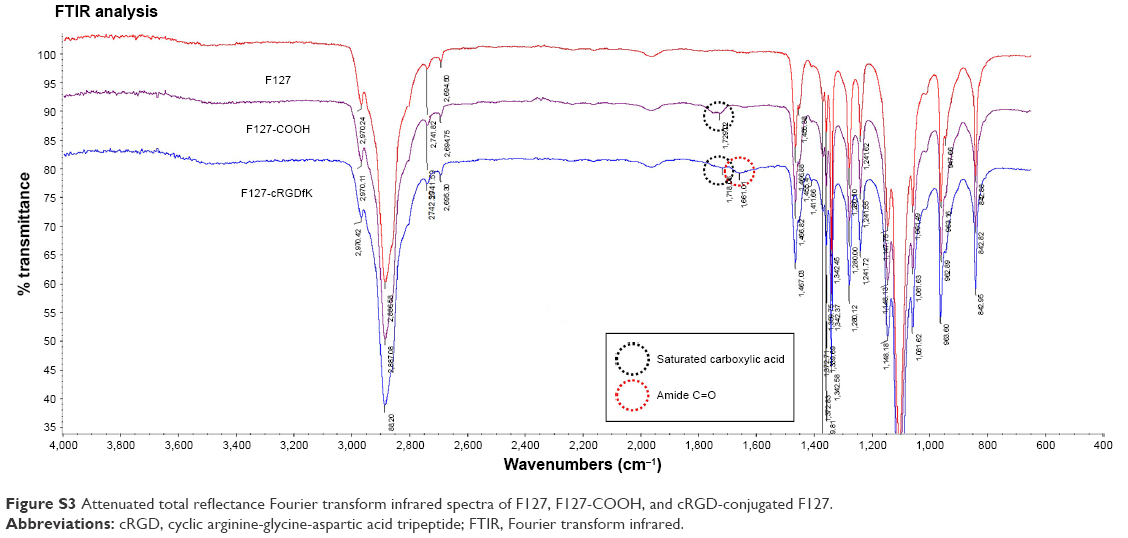 | Figure S3 Attenuated total reflectance Fourier transform infrared spectra of F127, F127-COOH, and cRGD-conjugated F127. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.