Back to Journals » International Journal of Nanomedicine » Volume 15
CXCL12/CXCR4 Axis-Targeted Dual-Functional Nano-Drug Delivery System Against Ovarian Cancer
Authors Xue J, Li R ![]() , Gao D, Chen F, Xie H
, Gao D, Chen F, Xie H
Received 15 April 2020
Accepted for publication 13 July 2020
Published 7 August 2020 Volume 2020:15 Pages 5701—5718
DOI https://doi.org/10.2147/IJN.S257527
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Mian Wang
Jiyang Xue1 ,* Ruixiang Li2 ,* Dingding Gao,2 Fenghua Chen,3 Hongjuan Xie1
1Department of Pharmacy, Shanghai First Maternity and Infant Hospital, Tongji University School of Medicine, Shanghai 201204, People’s Republic of China; 2Innovation Research Institute of Traditional Chinese Medicine, Shanghai University of Traditional Chinese Medicine, Shanghai 201203, People’s Republic of China; 3Department of Ultrasonography, Shanghai First Maternity and Infant Hospital, Tongji University School of Medicine, Shanghai 201204, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Jiyang Xue; Hongjuan Xie
Department of Pharmacy, Shanghai First Maternity and Infant Hospital, Tongji University School of Medicine, Shanghai 201204, People’s Republic of China
Tel +86-21-20261152
Email [email protected]; [email protected]
Introduction: Traditional chemotherapy for ovarian cancer is limited due to drug resistance and systemic side effects. Although various targeted drug delivery strategies have been designed to enhance drug accumulation at the tumor site, simply improvement of targeting capability has not consistently led to satisfactory outcomes. Herein, AMD3100 was selected as the targeting ligand because of its high affinity to chemokine receptor 4 (CXCR4), which was highly expressed on ovarian cancer cells. Moreover, the AMD3100 has been proved having blockage capability of stromal cell-derived factor 1 (SDF-1 or CXCL12)/CXCR4 axis and to be a sensitizer of chemotherapeutic therapy. We designed a dual-functional targeting delivery system by modifying paclitaxel (PTX)-loaded PEGylation bovine serum albumin (BSA) nanoparticles (NPs) with AMD3100 (AMD-NP-PTX), which can not only achieve specific tumor-targeting efficiency but also enhance the therapeutic outcomes.
Methods: AMD3100 was chemically modified to Mal-PEG-NHS followed by reacting with BSA, then AMD-NP-PTX was synthesized and characterized. The targeting efficiency of AMD-NP was evaluated both in vitro and in vivo. The anticancer effect of AMD-NP-PTX was determined on Caov3 cells and ovarian cancer-bearing nude mice. Finally, the potential therapeutic mechanism was studied.
Results: AMD-NP-PTX was synthesized successfully and well characterized. Cellular uptake assay and in vivo imaging experiments demonstrated that NPs could be internalized by Caov3 cells more efficiently after modification of AMD3100. Furthermore, the AMD-NP-PTX exhibited significantly enhanced inhibition effect on tumor growth and metastasis compared with PTX, NP-PTX and free AMD3100 plus NP-PTX both in vitro and in vivo, and demonstrated improved safety profile. We also confirmed that AMD-NP-PTX worked through targeting CXCL12/CXCR4 axis, thereby disturbing its downstream signaling pathways including epithelial–mesenchymal transition (EMT) processes and nuclear factor κB (NF-κB) pathway.
Conclusion: The AMD-NP-PTX we designed would open a new avenue for dual-functional NPs in ovarian cancer therapy.
Keywords: AMD3100, paclitaxel, nanoparticle, CXCL12/CXCR4 axis, ovarian cancer
Introduction
Ovarian cancer is one of the most common gynecological malignancies, of which the 5-year survival rate is only approximately 30%. Even after standard treatment with surgical resection combined with first-line chemotherapy, more than 70% of patients will eventually die due to tumor recurrence and metastasis.1 In addition, traditional chemotherapy drugs have several side effects, including lack of tumor selectivity and proneness to drug resistance, which are not ideal for improving patients’ survival rate and prognosis.2 Therefore, exploration of new treatment routes in ovarian cancer is urgently needed.
Targeted therapy refers to the design of a corresponding therapeutic drug at the cellular molecular level for a well-defined tumor-related site. Thus, when the drug enters the body, it selectively binds to specific sites of the tumor, causing specific death of the tumor cells without damaging the normal tissue cells.3–5 Several studies have confirmed that nanocarriers could be regarded as the most prospective drug delivery system owing to their extraordinary ability to become enriched in tumor sites through the enhanced permeability and retention effect (EPR), combined with their advantages such as slow-release of drugs, high tumor selectivity and reducing of systemic adverse reactions.6,7 However, the passive targeting of NPs alone is insufficient. An active targeted drug delivery system designed to target specific sites in the tumor has the ability to actively seek its “target” and could greatly improve the targeting efficiency.8,9 Recent years, various targeted drug delivery strategies have been designed to enhance drug accumulation at the tumor site, nevertheless, simply improvement of targeting capability has not consistently led to satisfactory outcomes. Targeting ligands that possess superior binding capacity and simultaneously have strong influence on intracellular signaling cascades may be expected to synergistically enhance the antitumor therapeutic efficacy. To this end, we aim to develop a dual-functional nano-drug delivery system for synergistic targeted therapy of ovarian cancer.
Chemokine receptor 4 (CXCR4) is a member of the chemokine superfamily and is a specific receptor for stromal cell derived factor-1 (SDF-1, CXCL12), which is a highly conserved G protein-coupled 7-transmembrane receptor. Scotton et al found that chemokine CXCL12 is present in 95% of ovarian cancer ascites, while CXCR4 is the only chemokine receptor expressed in ovarian cancer cells and expressed in approximately 80% of ovarian cancer tissues, but not in the normal ovarian epithelium, making it an promising target for targeted therapy of ovarian cancer.10,11 Moreover, ovarian cancer cells expressing CXCR4 could be transported and homing under the control of CXCL12/CXCR4 axis, and the metastatic cancer cells are closely associated with stromal cells under the action of CXCL12 chemokines and thus acquire drug resistance.12,13 Several studies have revealed that the progression and metastasis of ovarian cancer could be inhibited by blockage of the CXCL12/CXCR4 axis.14,15 To this regard, the CXCL12/CXCR4 axis is a potential therapeutic target. Overall, designing a formulation targeting ovarian cancer through CXCR4 receptor, together suppressing interaction between CXCL12/CXCR4 and blocking downstream signaling pathway can provide two-pronged attack strategy and enhance the therapeutic outcomes.
AMD3100, a specific non-peptide blocker of CXCR4, has been proved to effectively inhibit the cell proliferation, migration, and invasion of ovarian cancer by blocking the CXCL12/CXCR4 axis. Righi et al16 showed that AMD3100 promoted the apoptosis of tumor cells and the local T-cell mediated immune responses of ovarian cancer. Kajiyama et al17 demonstrated that AMD3100 was capable of inhibiting metastasis of epithelial ovarian cancer in xenograft mouse models. However, all of these studies exhibit only modest anti-cancer effects, probably because the antagonists only block the activity of the CXCL12/CXCR4 axis without eliminating the cancer cells. Thus, the combination of AMD3100 and antineoplastic drugs is particularly promising. Recently, studies have shown that the CXCR4 antagonist in combination with low-dose chemotherapy drug PTX have a synergistic effect on ovarian cancer.18,19 The effect may be through interrupting the interaction between cancer cells and stromal cells, then mobilizing cancer cells to leave the protective microenvironment and entering the blood circulation system, thereby increasing the sensitivity of cancer cells to chemotherapeutic drugs.20 Whereas although the synergistic antitumor effects have been confirmed, the insufficient pharmacokinetics as well as target-free toxicity make it difficult to apply this combination in patients.
Based on these findings, in this study, AMD3100, the specific non-peptide blocker and high-affinity antagonist of CXCR4, was first selected as a specific ligand to be modified on the surface of the PEGylation albumin nano-delivery system loading with chemotherapy drug PTX in the treatment of ovarian cancer. We suppose the AMD-NP-PTX can target ovarian cancer, but also enhance the therapeutic effect synergistically through the combination of low-dose PTX sustained-released from NP and AMD3100. The AMD-NP-PTX was well characterized by NMR and TEM. The targeting efficiency was evaluated both in vitro and in vivo. The anticancer effect of AMD-NP-PTX was determined on Caov3 cells and ovarian cancer-bearing nude mice, the in vivo toxicity of the drug delivery system was evaluated. Finally, the potential therapeutic mechanism was also studied.
Materials and Methods
Materials, Cells and Animals
AMD3100·8HCl was purchased from Selleckchem, bovine serum albumin (BSA) was purchased from BioFroxx (German). GdCl3·6H2O, PTX were obtained from Meilunbio Co. Ltd. (Dalian, China). Mal-PEG-NHS (MW 2 kDa) was obtained from JenKem Technology Co., Ltd. (Beijing, China). Dulbecco’s modified Eagle’s medium (DMEM), fetal bovine serum (FBS), trypsin, phosphate buffered saline (PBS, 10 mM, pH 7.4), 3-(4,5-dimethlthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT), and Annexin V-FITC Apoptosis Detection Kit were purchased from Gibco Invitrogen Corp. Transwell chamber (24-well, 8.0-μm pore membranes) was purchased from Corning (USA). BD MatrigelTM Basement Membrane Matrix was purchased from BD Biosciences. Bodipy 650/665-X NHS ester was obtained from Life Technologies. 4.6-Diamidino-2-phenylindole (DAPI) was purchased from Beyotime Biotech. Co. Ltd. Primary antibodies of CXCR4, E-cadherin, N-cadherin, vimentin, NF-κB and GADPH were purchased from Abcam. SDF-1 protein was obtained from Sigma.
Ovarian cancer cell line Caov3 cells and the normal ovarian cells (Chinese hamster ovary cell, CHO) were purchased from the Shanghai Institutes for Biological Sciences. The cells were maintained in DMEM supplemented with 10% FBS, 100 U/mL penicillin and streptomycin at 37°C in a humidified atmosphere containing 5% CO2.
The female BALB/c nude mice (5–6 weeks old, 18–20 g) were obtained from the Shanghai SLAC Laboratory Animal Co., Ltd (Shanghai, China). All animal experiments were approved by the Institutional Use and Care of Animals Committee and conducted according to the approved animal protocol of the Animal Centre of Tongji University.
Synthesis and Characterization of AMD-PEG-BSA
AMD-PEG-NHS was synthesized by Michael addition between the secondary amines of AMD3100 and maleimide (Mal) group of Mal-PEG-NHS at an equimolar ratio of AMD3100 and Mal-PEG-NHS, referring to a previously published protocol with minor modification.21 Briefly, Mal-PEG-NHS (50 mg, 0.025 mmol) and AMD3100·8HCl (20 mg, 0.025 mmol) were added into a glass vial containing MeOH/water mixture (2 mL, 7/3, v/v). The pH of the reaction mixture was adjusted to 7.4 by adding of NaOH, then the polymerization was carried out in the dark at 37°C (400 rpm) for 48 h. The reaction was then added dropwise of NaOH so that kept the pH of the mixture around 7.4, and stirred at 50°C (400 rpm) for another 24 h. Then, the resulting solution was cooled to room temperature, freeze-drying after dialysis. The prepared AMD-PEG-NHS was characterized by NMR H spectrum. After that, AMD-PEG-BSA was synthesized based on the reaction mechanism that the amino group on BSA surface reacts with the NHS ester of AMD-PEG-NHS could form amide bond when stirring at room temperature, according to procedures reported previously with some modifications.22 In brief, AMD-PEG-NHS was reacted with BSA at a molar ratio of amino group on BSA and NHS on AMD-PEG-NHS at 1:1.5, the pH of BSA solution was adjusted by NaHCO3 to around 7.4, followed by adding AMD-PEG-NHS to react for the whole night at room temperature. The obtained product was purified using ultracentrifugal filter tube (MWCO: 5KDa), and the filtrate was lyophilized to calculate the yield.
Preparation of NPs
The preparation of NPs was according to a biomineralization approach previously published with minor modifications.23 Briefly, GdCl3.6H2O (5 mL, 0.4 mg/mL) was slowly added to PEG-BSA solution (or AMD-PEG-BSA) (10 mL, 1 mg/mL) under stirring (500 rpm/min). Three minutes later, sodium hydroxide solution (5 mL, 0.2 mg/mL) was added and stirred for 60 min. Then PTX solution (100 ul, 20 mg/mL) dissolved with dimethyl sulfoxide (DMSO) was added, and the mixture was left to interact under vigorous stirring in the dark at room temperature for 30 min. The resulting solution was subjected to exhausted dialysis with deionized water to eliminate excessive PTX, Gd3+ and a small amount of organic solvents, then lyophilized to collect the final product. The schematic diagram of the AMD-PEG-BSA-NP-PTX (AMD-NP-PTX) preparation process was shown in Figure 1.
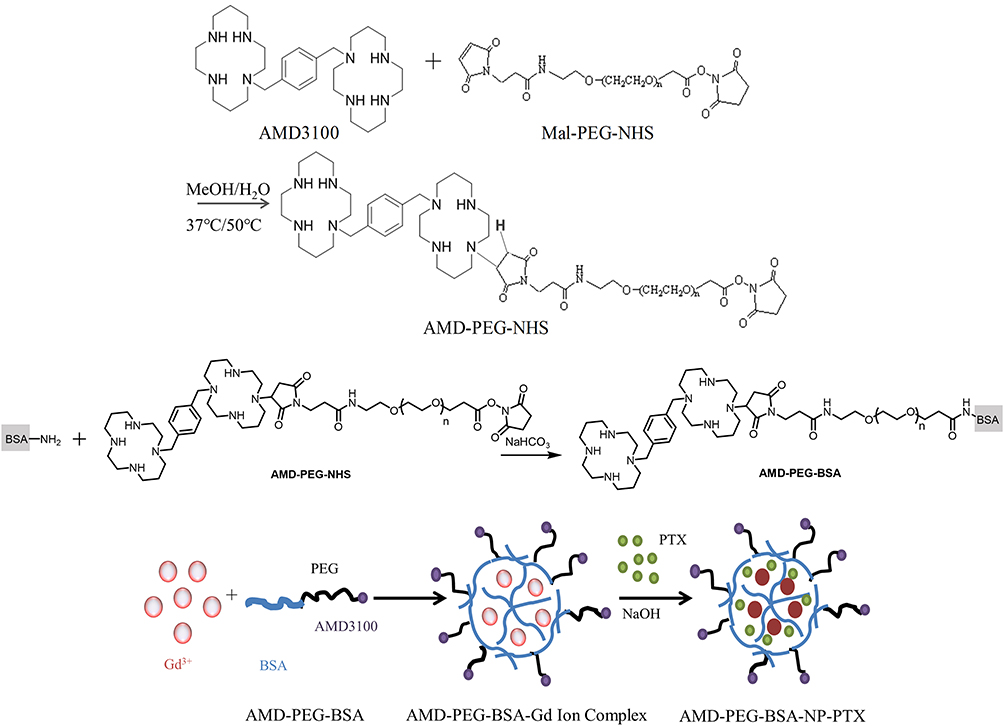 |
Figure 1 Schematic diagram of AMD-NP-PTX preparation process. |
Characterization of NPs
The particle size, polydispersity index (PDI) and zeta potential of NPs were measured by a laser diffraction instrument (Zetasizer Nano ZS, ZEN 3690, Malvern). The morphology of NPs was observed using transmission electron microscopy (JEM-1230, Joel). The drug loading (DL) of PTX in NPs was examined after ultrafiltration and calculated according to the equation: DL (%) = WE/WN × 100%
(WE represents as the encapsulated amount of PTX, and WN represents as the mass of AMD-NP-PTX).
To investigate the in vitro drug release profile of PTX from PTX-solution and AMD-NP-PTX, 3 mL of AMD-NP-PTX and PTX-solution was sealed in a dialysis bag (MWCO: 8000–14,000), respectively. The dialysis bags were then introduced into 100 mL of PBS buffer with 0.5% (v/v) Tween-80 (pH=7.4 or 6.8), stirring at 37 °C at 200 rpm for 96 h. At a predetermined time point, 0.5 mL aliquots were withdrawn and analyzed by high-performance liquid chromatography (HPLC), and the fresh release medium of the same volume was added. The PTX release behavior was then obtained with time versus cumulative PTX release.
Cellular Uptake Assay and AMD3100 Competitive Cellular Uptake Study
To detect the cellular internalization of NP and AMD-NP, Caov3 cells were seeded in a 6-well plate at a density of 1.5 × 105 cells per well for 24 h and then incubated with Bodipy-labeled NP and AMD-NP at 37°C for 4 h. Cells were washed 3 times with PBS, fixed with 4% POM for 10 min, stained with 4ʹ,6-diamidino-2-phenylindole (DAPI) for 3 min, and then imaged with a confocal laser scanning microscope (Nikon, A1R). To testify the selective character of AMD-NP in vitro, the normal ovarian cells (Chinese hamster ovary cell, CHO) without CXCR4 expression were also subjected to cell uptake experiments according to the above procedure.
For the AMD3100 competitive cell uptake study, free AMD3100 was added to the Caov3 cells with the final concentration of 0, 0.2, 2 and 20 μmol/L for 10 min, then the cells were treated with Bodipy-labeled AMD-NP for 4 h. The cellular uptake of Bodipy-labeled AMD-NP was evaluated and calculated using flow cytometry.
In vivo Real-Time Imaging
Bodipy was selected as a fluorescent probe to track the real-time distribution of NPs in ovarian cancer nude mice. At first, the model of ovarian cancer-bearing nude mice was established. Six nude mice were injected with Caov3 cells (4×106 cells) and then divided into 2 groups randomly. Two weeks later, 2 groups of nude mice were intravenously administered with 200 µL of Bodipy-labeled NP and AMD-NP, respectively. At the predetermined time points (1 h, 3 h, 6 h, 24 h), fluorescence images were obtained via an in-vivo imaging system (CRI, Ma, USA). After 24 h of administration, the nude mice were sacrificed, and the organs along with tumor tissues were taken for fluorescence imaging.
Cell Viability Assay
MTT assay was used to evaluate the cell viability of different formulations. Caov3 cells (2000 cells per well) were cultured in 96-well plates for 24 h and then exposed to free PTX, NP-PTX, free AMD3100 plus NP-PTX, AMD-NP-PTX and blank AMD-NP for further incubation (at a PTX concentration of 0.5 μmol/L). After 48 h, MTT solution was added and incubated at 37°C for 3 h, and then 150 μL of DMSO was added. The absorbance was measured at 570 nm using a microplate reader.
Flow Cytometric Analysis of Cell Apoptosis
Flow cytometry was used to study the apoptosis of Caov3 cells. The cells were plated in 6-well plates followed by treatment with free PTX, NP-PTX, free AMD3100 plus NP-PTX and AMD-NP-PTX (at a PTX concentration of 0.5 μmol/L) for 48 h, then digested with trypsin, stained with annexin V-FITC and propidium iodide (PI) using the Annexin V-FITC Apoptosis Detection Kit I (BD Biosciences). After 5–10 min of reaction in the dark, the apoptotic cell was measured and calculated using a flow cytometer.
Cell Migration Experiment
Caov3 cell suspension was inoculated in the upper chamber of Transwell inserts in a 24-well plate, and serum-free medium was placed in the lower chamber. To detect the effects of different formulations on the migration of Caov3 cells, PTX solution, NP-PTX, free AMD3100 plus NP-PTX and AMD-NP-PTX (at a PTX concentration of 0.5 μmol/L) were added to the upper chamber and co-culture with Caov3 cells, serum-free medium and chemokine SDF-1 (CXCL12) were added to the lower chamber, and the cells were cultured in an incubator (37°C, 5% CO2) for 24 h. Under 400 × high magnification, the total number of cells migrated from 5 fields was recorded.
Tumor Growth and Metastasis in Caov3 Ovarian Cancer Xenograft
4×106 Caov3 cells were injected subcutaneously into female BALB/C nude mice. When the tumor volume reached approximately 100 mm3, the nude mice were randomly divided into 5 groups (n = 6). The control group was administered intravenously with normal saline, the treatment group was injected with PTX solution, NP-PTX, free AMD3100 plus NP-PTX and AMD-NP-PTX (at an equal dosage of 10 mg/kg PTX) every other day for 7 times, respectively. Tumor length and width were measured and recorded with Vernier caliper every 2 days after administration, the growth of the transplanted tumor was observed, and the body weight of each mouse was recorded. Nude mice were sacrificed after the experiment was completed, and the tumor tissues were collected and weighed. Then, the morphological changes and apoptosis of tumor tissues in nude mice were evaluated by terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) staining. A commercially available TUNEL kit (R&D Systems, Minneapolis, MN) was used to detect the apoptotic cells in frozen sections of tumor tissues according to the manufacture’s recommendations. Subsequently, the major organs (heart, liver, spleen, lung and kidney) of the mice were separated, fixed and embedded into paraffin. The fixed organs were then sliced, subjected to the hematoxylin and eosin (H&E) staining and optical microscopy study.
In order to investigate the effect of AMD-NP-PTX on tumor metastasis in vivo, we established an intraperitoneal ovarian cancer model in BALB/C nude mice using Caov3 cells. Thirty-six BALB/C nude mice were randomly divided into six groups (n = 6), treatment began at 2 weeks after intraperitoneal injection of tumor cells every other day for 7 times. Each group of nude mice received one of the following administrations: (I) normal saline; (II) CXCL12 (100 ng/mL) plus normal saline; (III) CXCL12 (100 ng/mL) plus PTX; (IV) CXCL12 (100 ng/mL) plus NP-PTX; (V) CXCL12 (100 ng/mL) plus AMD and NP-PTX (VI) CXCL12 (100 ng/mL) plus AMD-NP-PTX (at an equal dosage of 10 mg/kg PTX). CXCL12 was injected intraperitoneally, and other drugs were all administered intravenously. At the end of the observation, the mice were sacrificed and the tumor metastasis in mesentery, abdominal wall, liver, spleen, uterine appendage, heart, kidney, lung and brain were examined.
Western Blotting Analysis
The tissues of different treatment groups were cut into small pieces, and the lysate was added. After splitting, the samples were centrifuged at 12,000g for 15 minutes, the supernatant was taken and quantified with BCA protein Kit (thermo). After protein sample preparation, 50 μg protein of each sample was loaded on 10% sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to polyvinylidene difluoride (PVDF) membranes. Five percent skimmed milk powder was sealed overnight at 4°C, E-cadherin (1:1000; Abcam), N-cadherin (1:1000; Abcam), vimentin (1:1000; Abcam) and NF-κB (1:1000; Abcam) primary antibodies were added, respectively, incubated at room temperature for 2 h, then horseradish peroxidase (HRP)-labeled secondary antibody was added and incubated with membrane at 37°C for 1 h. After washing, ECL chemiluminescence was detected and put into the imaging system for scanning. Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was used as an internal reference.
Statistical Analysis
The results were presented as mean ± standard deviation (SD). The statistical significance was evaluated by Student’s t-test.
Results
Synthesis and Characterization of AMD-PEG-BSA
AMD-PEG-NHS was synthesized from Mal-PEG-NHS and AMD3100 by Michael addition reaction. The material was purified by dialysis with a yield of 76.67%. The material was characterized by NMR H spectrum. As shown in Figure 2, the absorption peak of the Mal group in Mal-PEG-NHS appeared at 6.728 ppm (c) and the aromatic proton peak in AMD-PEG-NHS shown at 7.26 ppm (a). The disappearance of the Mal group and the appearance of aromatic proton peaks proved that Michael addition made AMD3100 successfully connect to PEG-NHS through the reaction of secondary amines with the Mal group. The content of PEG in the copolymers was determined from 1H-NMR integral intensity of the aromatic protons of AMD3100 at 7.26 ppm (a) and PEG methylene protons at 3.58 ppm (b). As we can see in Figure 2, the integral intensity ratio at those two chemical shifts was 4:176, which means a 1:1 ratio between AMD3100 and PEG in the final product of AMD-PEG-NHS. Next, AMD-PEG-NHS was further reacted with BSA at a molar ratio of amino group on BSA and NHS on AMD-PEG-NHS at 1:1.5 to synthesize AMD-PEG-BSA. The material was purified using ultracentrifugal filter tube (MW 5KDa), and the filtrate was lyophilized, the yield was calculated to be around 85.14%. After final calculation, the mass ratio of AMD3100 to AMD-PEG-BSA was about 0.1425/1 (mg/mg).
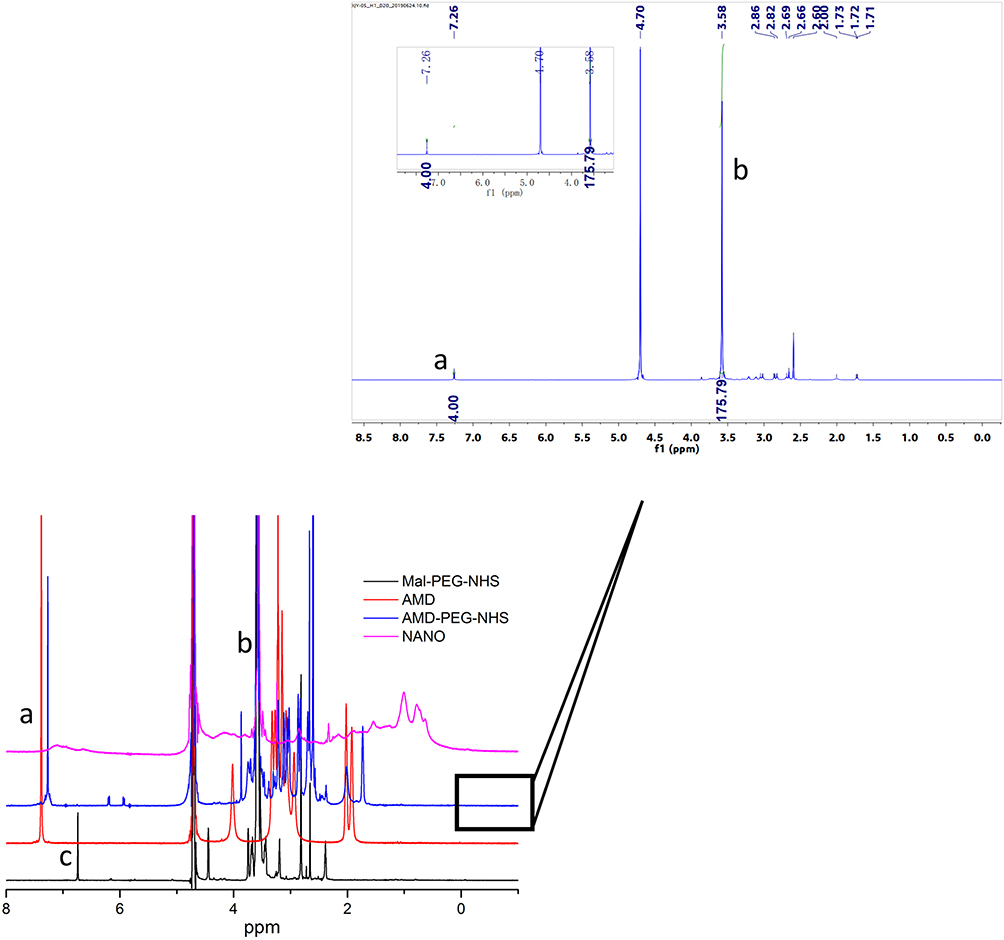 |
Figure 2 NMR H spectra of Mal-PEG-NHS, AMD-PEG-NHS and NANO (AMD-NP-PTX). (a) – aromatic phenylene protons of AMD3100. (b) – methylene protons of PEG. (c) – Maleimide group of Mal-PEG-NHS. |
Preparation and Characterization of NPs
AMD-NP-PTX (or NP-PTX) was synthesized by biomineralization based on AMD-PEG-BSA (or PEG-BSA) and Gd3+. The hydrophobicity of the anticancer drug PTX was utilized to incorporate the drug into the NPs by non-covalent binding. AMD-PEG-BSA-NP-PTX (AMD-NP-PTX) were successfully constructed based on NMR H spectrum characterization (Figure 2). The DL of PTX in AMD-NP-PTX was 13.35%. The particle size of AMD-NP-PTX were 115.8 nm with PDI of 0.126 (Figure 3A), the zeta potential was −10.4 mv. The NPs were smooth and uniform under TEM (Figure 3B). Release experiments in vitro (Figure 3C and D) showed that the AMD-NP-PTX had an obvious sustained release profile, and the drug release rate of the delivery system was significantly faster at faintly acidic pH (6.8) than at neutral pH (7.4).
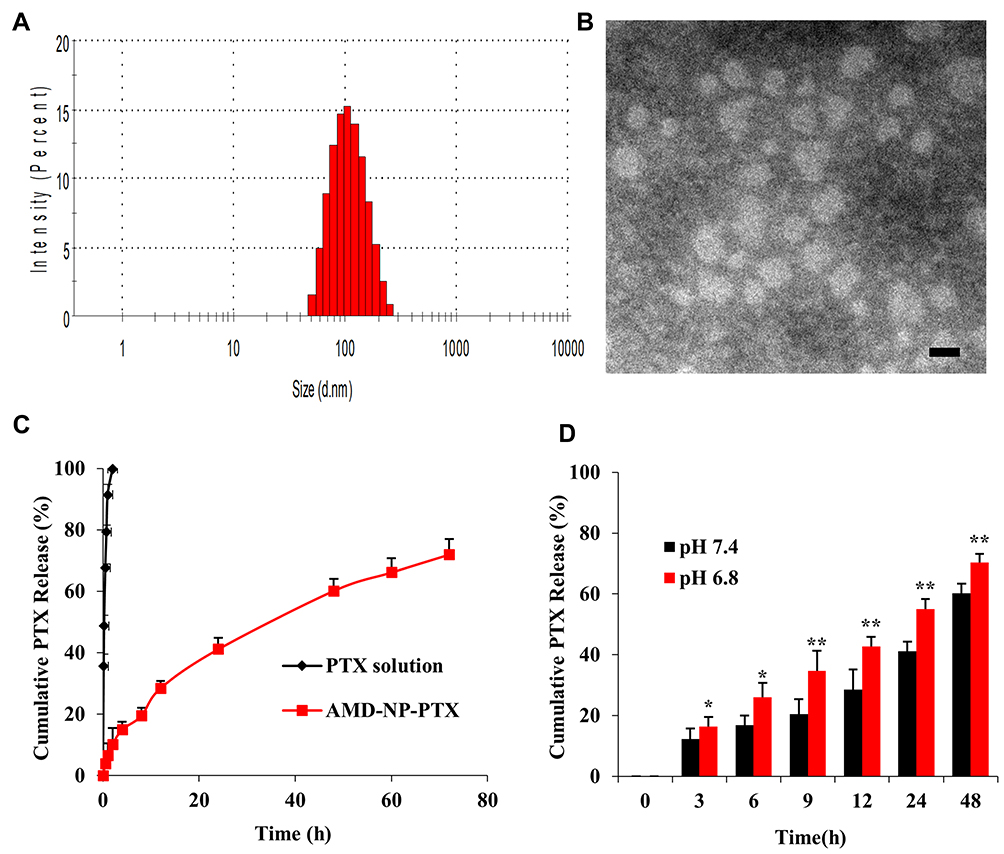 |
Figure 3 Characterization of AMD-NP-PTX (A) Size distributions and Zeta potential of AMD-NP-PTX. (B) TEM characterization of AMD-NP-PTX. Scale bar=100 nm. (C) In vitro drug release curve of PTX solution and AMD-NP-PTX (pH 7.4). Data are presented as the mean±SD (n=3). (D) In vitro drug release profile of AMD-NP-PTX under different pH conditions (pH 6.8 and 7.4). Data are presented as the mean±SD (n=3). *p<0.05, **p<0.01 (significant difference between the two groups). |
Cellular Uptake Assay and AMD3100 Competitive Cellular Uptake Study
The uptake efficiency of AMD3100-modified NPs by Caov3 ovarian cancer cells with high expression of CXCR4 receptor and CHO cells without CXCR4 receptor expression were evaluated by CLSM using Bodipy as tracing fluorescence. The results showed that compared with unmodified NPs, the uptake of the AMD3100-modified drug delivery system by Caov3 cells increased significantly (Figure 4A). The relative uptake ratios were 78.0±6.4%, 18.5±4.2% and 2.6±0.73% for the AMD-NP, NP and free Bodipy, respectively. However, there was no effect on the uptake of CHO cells. These results suggested that the AMD3100-modified NPs we constructed could selectively and efficiently target ovarian cancer cells with high expression of CXCR4, but not normal ovarian cells. For the AMD3100 competitive cell uptake results evaluated and calculated using flow cytometry, the uptake of Bodipy-labeled AMD-NP was competitively inhibited by free AMD3100, and the inhibitory effect was dose-dependent (Figure 4B and C).
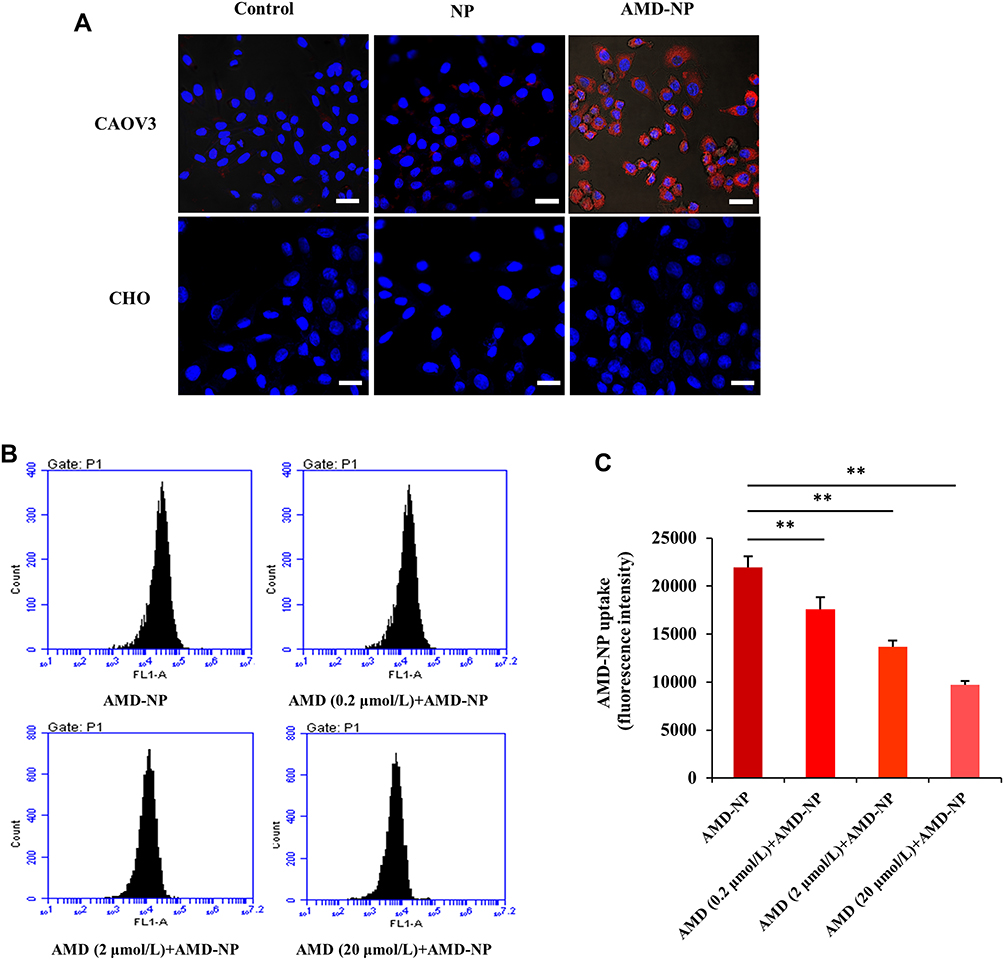 |
Figure 4 (A) Laser confocal assay was used to evaluate the uptake of Bodipy-labeled NP and AMD-NP by Caov3 cells and CHO cells in vitro. Blue represents the nucleus (DAPI) and red represents the fluorescent (Bodipy) labeled NPs. Scale bar=25 μm. (B) Competitive cell uptake study was performed by pre-addition of free AMD3100 with different concentrations using flow cytometry. (C) Quantitative analysis of flow cytometry. Data are presented as the mean±SD (n=3). **p<0.01. |
In vivo Real-Time Imaging
The targeting effect and bio-distribution profile of NP and AMD-NP in tumor-bearing nude mice were observed by infrared fluorescence. As it can be seen in Figure 5A, Bodipy-labeled AMD-NP maintained a strong fluorescence signal at the tumor site until 24 h we observed, while the signal of the NP group at the tumor site was very weak. In vitro fluorescence images of excised tumors and organs removed at 24 h post-administration also confirmed the strong tumor-targeting effect of AMD-NP (Figure 5B). The results of quantitative analysis exhibited that the fluorescence intensity of AMD-NP in tumor tissue was approximately 9.375 times that of the NP group (p<0.01) (Figure 5C).
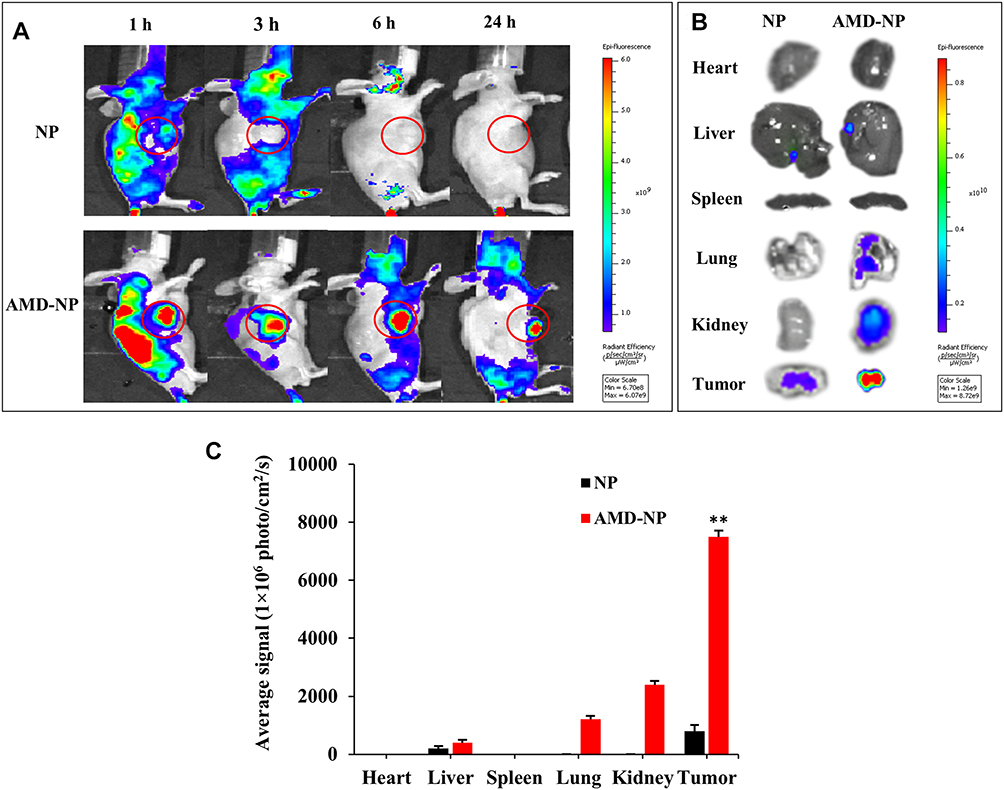 |
Figure 5 The tumor accumulation study in vivo. (A) The whole-body imaging of Caov3 tumor-bearing mice at 1 h, 3 h, 6 h and 24 h after intravenous injection of Bodipy-labeled NP, AMD-NP. (B) Tissue distribution of Bodipy-labeled NP, AMD-NP imaged 24 h post-injection. (C) Quantitative analysis of Bodipy signal from NP and AMD-NP in different organs. Data are presented as the mean±SD (n=3). **p<0.01 compared with NP-PTX group. |
Cell Viability Assay
As seen in Figure 6A, the results of MTT experiment showed that the inhibition of AMD-NP-PTX on the proliferation of Caov3 cells was significantly greater than that of the control group (p<0.01), PTX solution group (p<0.01) and NP-PTX group (p<0.05). Free AMD3100 only slightly sensitized Caov3 cells to NP-PTX, While AMD-NP-PTX displayed more cell killing compared with free AMD3100 plus NP-PTX (p<0.05). These results suggested that the chemical conjugation of AMD3100 with NP-PTX could obtain synergistic inhibition to Caov3 ovarian cancer cells. We also examined the effect of AMD3100 modified blank NPs and they were found to have no significant effect on Caov3 cells.
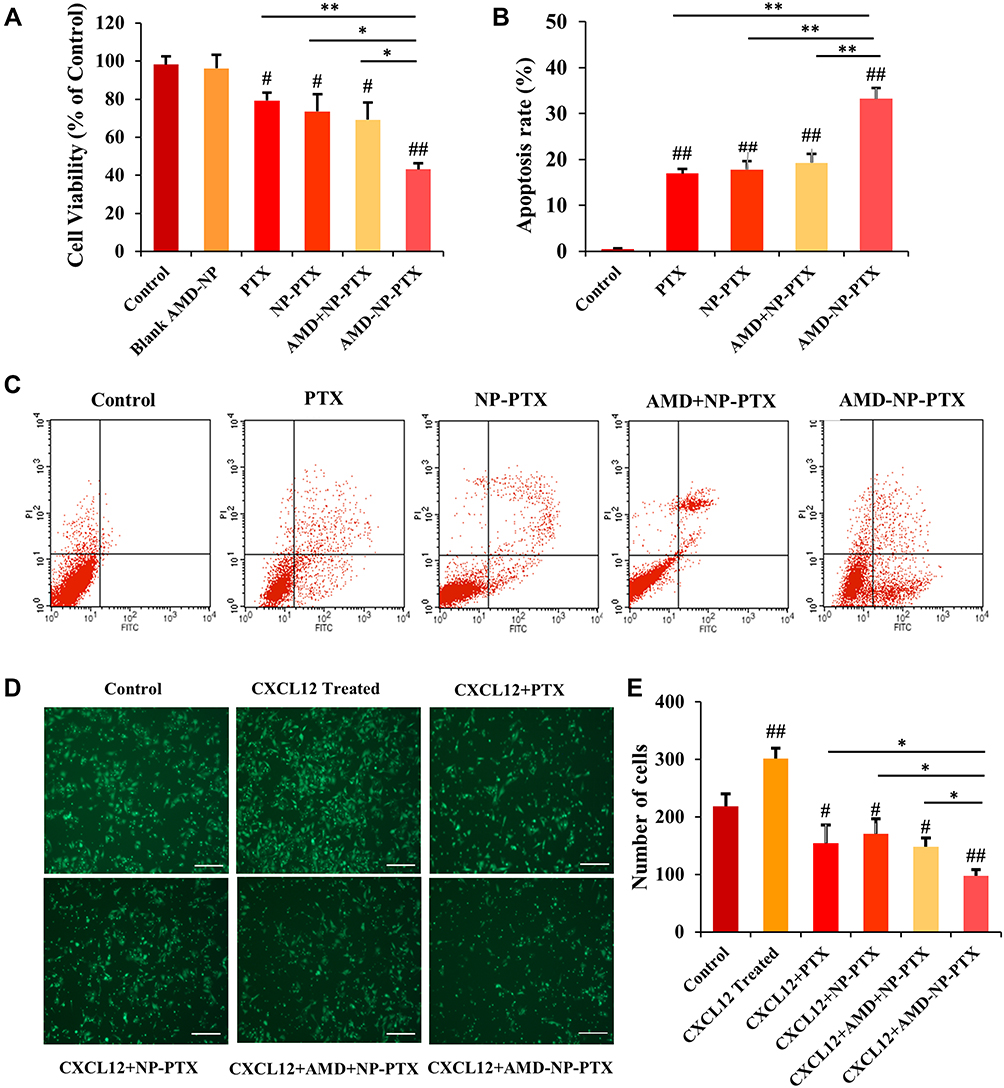 |
Figure 6 Effects of AMD-NP-PTX on proliferation, apoptosis, and migration of Caov3 ovarian cancer cells (A) Cell viability. Data are presented as the mean±SD (n=3). (B) The histogram comparing the cell apoptosis in 5 groups. Data are presented as the mean±SD (n=3). (C) Cell apoptosis mapping measured by flow cytometry. (D) Images of Transwell migration results. Scale bar=200 μm. (E) Number of migrated cells among 6 groups. Data are presented as the mean±SD (n=3). #p<0.05, ##p<0.01 (significant difference compared with the control group); *p<0.05, **p<0.01 (significant difference between different treatment groups). |
Flow Cytometric Analysis of Cell Apoptosis
The effect of different treatment group on the apoptosis of Caov3 cells were examined by AnnexinV-FITC/PI staining method. The results were shown in Figure 6B and C. Compared with the control group, PTX solution group, NP-PTX group and free AMD3100 plus NP-PTX group, the apoptosis rate of Caov3 cells induced by AMD-NP-PTX was significantly increased (p<0.01), which suggested that the connection of AMD3100 onto NP-PTX increased the apoptosis of Caov3 ovarian cancer cells.
Cell Migration Experiment
As shown in Figure 6D and E, the migration number of Caov3 cells increased significantly after the addition of chemokine SDF-1 (CXCL12), while it was inhibited after treatment with different formulations. The number of migrated cells in the AMD-NP-PTX groups was significantly lower than that in the PTX, NP-PTX and non-active targeted free AMD3100 plus NP-PTX group (p<0.05), suggesting that the addition of SDF-1 (CXCL12) chemokine can promote the migration of Caov3 cells, while NP-PTX modified with CXCR4 antagonist AMD3100 blocked the biological axis of CXCL12/CXCR4, thus enhancing the migration inhibition of Caov3 cells.
Tumor Growth and Metastasis in Caov3 Ovarian Cancer Xenograft
We further evaluated the effects of PTX, NP-PTX, free AMD3100 plus NP-PTX and AMD-NP-PTX on tumor progression in Caov3 transplanted ovarian cancer xenograft models, the treatment regimen was presented in Figure 7A. As shown in Figure 7B–D, treatment with PTX or NP-PTX alone only exhibited a moderate tumor growth inhibition effect. Due to the poor pharmacokinetics of free AMD3100, the systemic administration of free AMD3100 did not significantly sensitize ovarian cancer cells to NP-PTX treatment in vivo (p>0.05). However, the systemic injection of AMD-NP-PTX obtained a remarkable synergistic tumor growth inhibition in Caov3 transplanted nude mice compared to treatment with PTX solution, NP-PTX and free AMD3100 plus NP-PTX (p<0.01). The results of TUNEL staining (Figure 7E and F) illustrated that the cancer cells undergoing apoptosis could be obviously seen in the AMD-NP-PTX group, and the number of apoptotic cells was increased significantly compared with the control group and other treatment groups.
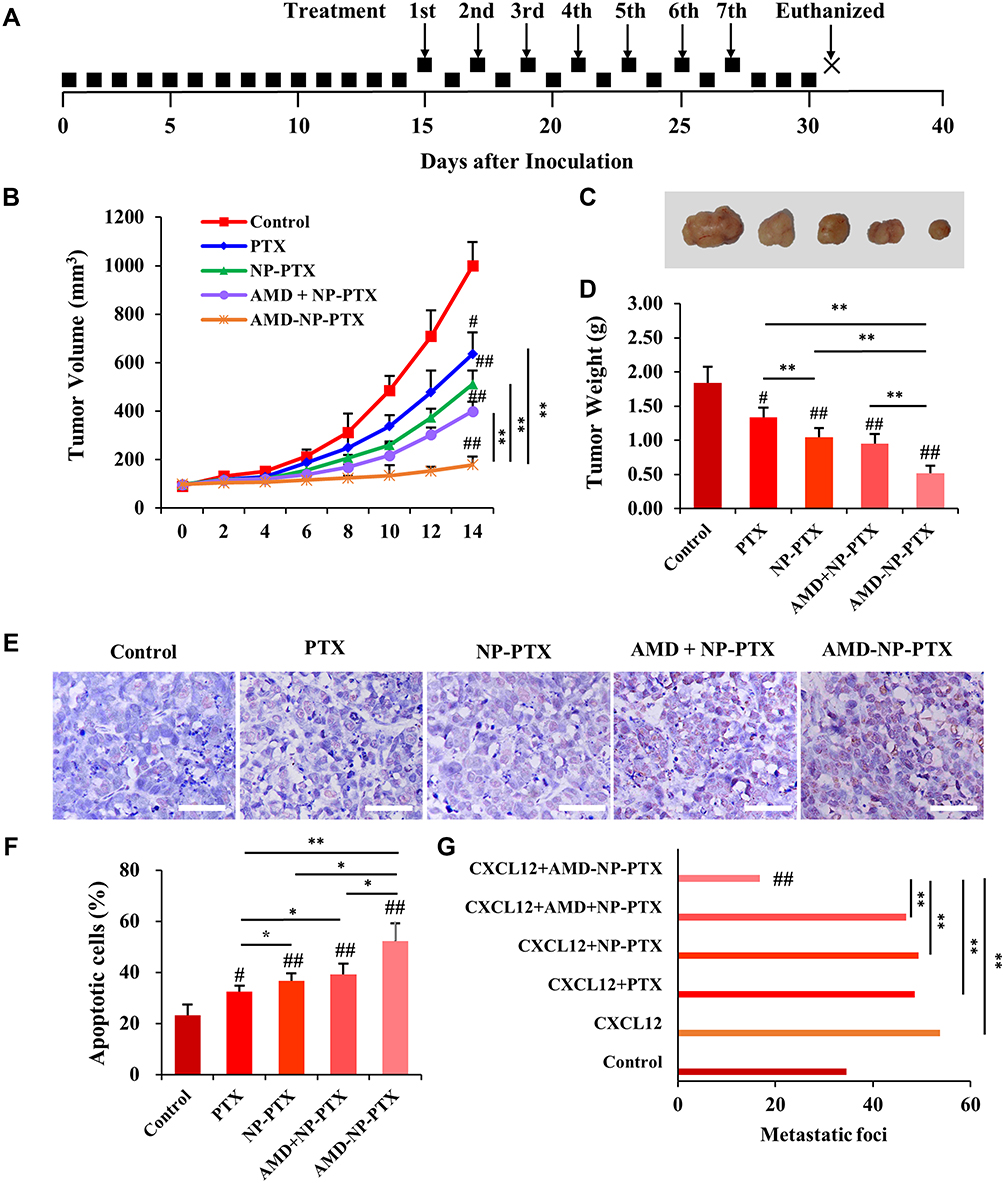 |
Figure 7 In vivo anticancer effect of AMD-NP-PTX on ovarian cancer-bearing nude mice. (A) The treatment regimen. (B) The curve of tumor volume change in each group. (C) The tumor nodules were dissected and photographed in each group after treatment. (D) The average tumor weight in each group. (E) TUNEL staining was used to detect the apoptosis of tumor tissue in nude mice. Scale bar=50 μm. (F) Tumor apoptosis cells assessed by counting the rate of TUNEL-positive cells. (G) Number of metastatic foci in different groups. Data are presented as the mean±SD (n=6). #p<0.05, ##p<0.01 (significant difference compared with the control group); *p<0.05, **p<0.01 (significant difference between different treatment groups). |
For in vivo tumor metastasis assay, 2 weeks after administration, mice were sacrificed and dissected. The results showed that tumor nodules were found in the mesentery, peritoneum, liver and uterine appendages of the mice, but no metastasis was observed in the heart, kidney, lung and brain. Under the CXCL12 stimulation, larger and more metastatic nodules formed (Figure 7G). Compared with control-treated mice, neither treatment with PTX, NP-PTX alone nor NP-PTX in combination with free AMD3100 showed a reduction in tumor metastasis formation (Figure 7G). In contrast, compared with the control group and other treatment groups, nude mice receiving AMD-NP-PTX displayed significantly fewer metastatic lesions (p<0.01).
In vivo Toxicity Evaluation
We recorded the body weight changes of nude mice during the treatment, and assessed the important organs with hematoxylin and eosin (H&E) staining. As indicated in Figure 8A, compared with the initial weight of tumor-bearing mice, body weight of the mice in PTX solution group decreased significantly (p<0.01), mice in NP-PTX treatment group also showed weight loss (p<0.05), but higher than that in PTX solution group, while there was no significant weight loss in the AMD-NP-PTX group during the treatment. In addition, the results of H&E staining analysis (Figure 8B) showed that compared with the saline control group, the PTX solution group had obvious pathological changes. Inflammatory cell infiltration and hepatomegaly can be seen in many liver cells. The lung tissue suffered a series of pathological changes, such as ecchymosis, inflammatory cell infiltration. Blood vessel bleeding point could be found in the heart, incomplete structure of spleen corpuscle could be seen in the spleen, and the kidney morphology also showed obvious changes. The NP-PTX group also suffered some pathological changes but were significantly better than that of the PTX group. In contrast, AMD-NP-PTX treatment group had no obvious pathological symptoms compared to the saline control group, which further confirmed the safety of AMD-NP-PTX in vivo.
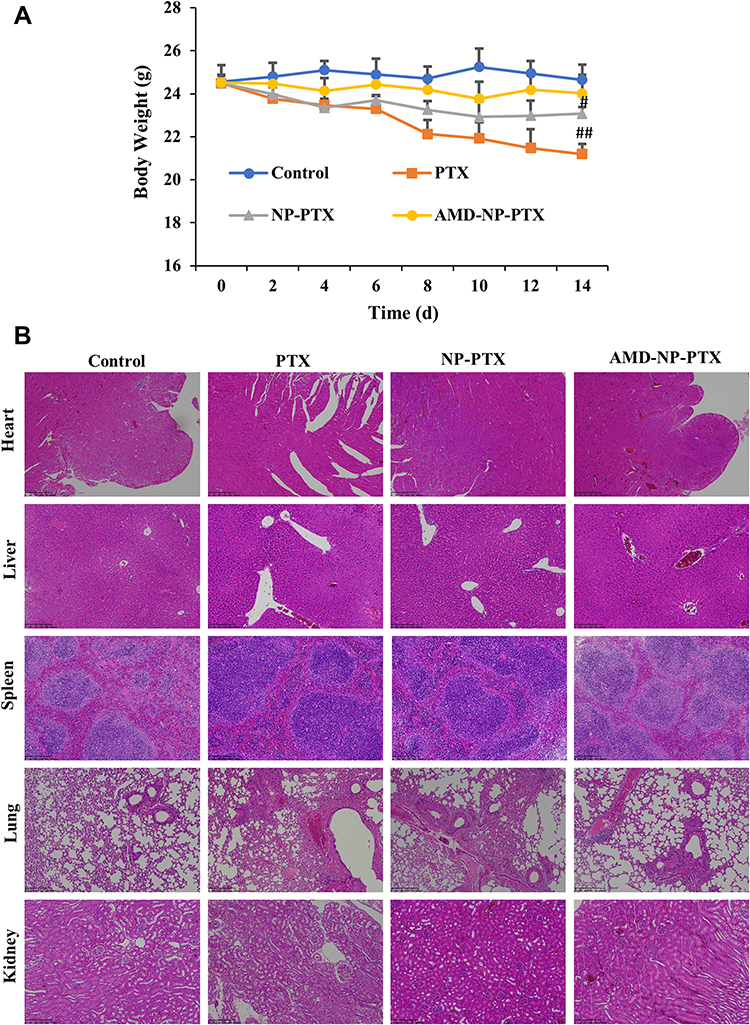 |
Figure 8 In vivo toxicity evaluation. (A) Changes in body weight of nude mice during the treatment. (B) H&E stained images of major organs from mice treated with different formulations. Data are presented as the mean±SD (n=6). Scale bar=200 μm. #p<0.05, ##p<0.01 (significant difference compared to the initial body weight of tumor-bearing nude mice). |
Western Blotting Analysis
To study the therapeutic mechanism of AMD-NP-PTX, we examined the protein expression of CXCL12/CXCR4 related pathways. The expression of E-cadherin, N-cadherin and vimentin (related to EMT process) as well as NF-κB was detected by Western blotting. The results (Figure 9A–E) showed that compared with the control group, the expression of NF-κB, N-cadherin and vimentin in the tumor tissue of nude mice treated with AMD-NP-PTX was significantly inhibited (p<0.01), while E-cadherin increased significantly (p<0.01), and there are also significant differences compared with the PTX group and the NP-PTX group, indicating that chemical bonding of AMD3100 to the PTX-loaded NPs may disturb CXCL12/CXCR4 axis, thus regulate the EMT process and NF-κB signaling pathway of ovarian cancer, thereby exerting synergistic inhibiting effect on tumor growth and metastasis.
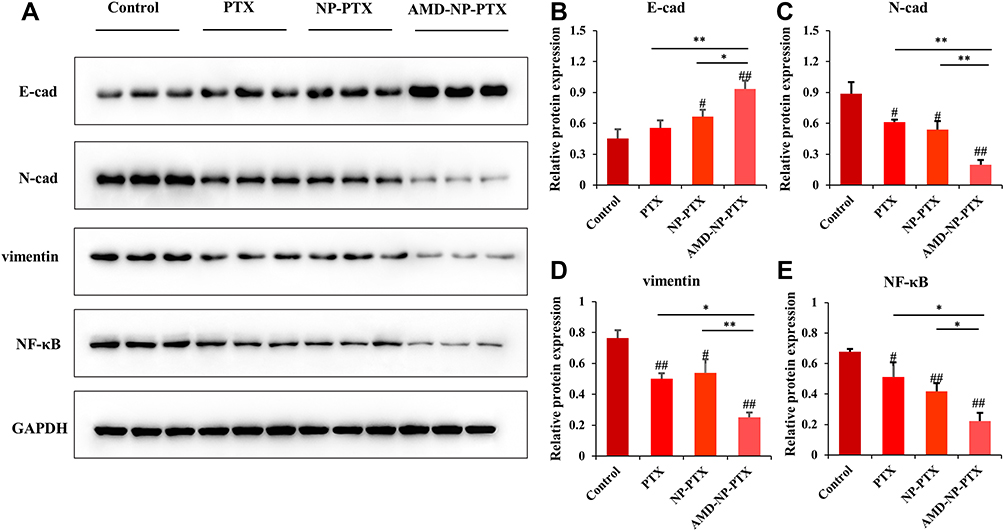 |
Figure 9 Study on the mechanism of AMD-NP-PTX against the growth and metastasis of ovarian cancer through Western blotting. (A) The relative expression of E-cadherin, N-cadherin, vimentin and NF-κB in tumor tissues of different groups measured by Western blotting. The quantification analysis of the expression of (B) E-cadherin, (C) N-cadherin, (D) vimentin and (E) NF-κB in tumor tissues measured by Western blotting. Data are presented as the mean±SD (n=3). #p<0.05, ##p<0.01 (significant difference compared with the control group); *p<0.05, **p<0.01 (significant difference between different treatment groups). |
Discussion
Tumor recurrence and metastasis are major causes of the high mortality of ovarian cancer. Traditional anticancer drug therapy is limited due to drug resistance and systemic side effects produced during conventional chemotherapy. Although various targeted drug delivery strategies have been designed to enhance drug accumulation at the tumor site, simply conversion of active targeting approaches has not consistently shown satisfactory outcomes.24,25 Therefore, in addition to binding capability, targeting groups that could simultaneously disturb intracellular signaling cascades may be expected to synergistically enhance the antitumor therapeutic efficacy.
AMD3100, an antagonist with high affinity to CXCR4 receptor which highly expressed on ovarian cancer cells, has been determined to inhibit progression and metastasis of ovarian cancer by blockage of the CXCL12/CXCR4 axis.26,27 Meanwhile, a previous study had shown that AMD3100 combined with low-dose PTX could increase anti-ovarian cancer efficacy.19 On the basis of these findings, we designed and developed a novel active targeting delivery system by modifying PTX-loaded PEGylation BSA-NPs with the dual-functional ligand AMD3100 (AMD-NP-PTX). It is well known that albumin-based NPs have been widely used for delivery of drugs with low solubility,28,29 and PEGylation strategy for NPs has become necessary to avoid their hydrophobic nature, slow degradation and rapid uptake by Reticuloendothelial System (RES).30,31 Herein, we chemically modified AMD3100 to Mal-PEG-NHS followed by reacting with BSA, synthesized PTX-loaded AMD-PEG-BSA NPs by biomineralization. The mechanism of biomineralization method for the preparation of NPs is similar to the biomineralization of organisms in nature: isolation and interaction with inorganic ions, and then provide a scaffold for the formed minerals. BSA molecules separate Gd ions and entrap them when they are added to the BSA aqueous solution. The scaffolding ability of the BSA molecule is activated by adjusting the pH to 12 and the entrapped ions in a progressive reaction to form gadolinium-based hybrid nanoparticles in situ. We chose this method because of its simple preparation, good repeatability, safety and environmental protection. Moreover, due to the presence of Gd ions, it is expected that the role of this system in disease diagnosis will be tapped in further research to realize the integration of diagnosis and treatment. Then we characterized the NPs with a series of methods. The diameter of the AMD-NP-PTX we prepared was 115.8 nm, and the potential was −10.4 mv. Studies have shown that NPs with a particle size of 100–200 nm have a favorable EPR effect on tumor blood vessels.32 Negatively charged NPs exhibited long-circulation characteristics in the body33 and were more conducive to avoiding non-specific uptake.34 Release experiments in vitro showed that the AMD-NP-PTX had an obvious sustained release profile, and the drug release rate was significantly faster at faintly acidic pH (6.8) than at neutral pH (7.4). This may be because the formation of NPs was based on coordination, which was easier to dissociate in acidic environments. According to a previous study, cancer cells are more acidic than blood or normal tissues,35 so the release characteristics made them more stable in the blood circulation while more released at the tumor site, which was more conducive to controlling drug release sites, thereby enhancing targeted treatment effect and reducing damage to normal tissues.
To explore whether AMD-NP could be internalized by tumor cells more efficiently, cellular uptake assay and in vivo imaging experiments were performed. In this study, Caov3 cells were selected because it has been demonstrated to express higher levels of CXCR4 compared with other ovarian cancer cell lines.13,26 The results of cellular uptake assay suggested that the relative uptake ratios were 78.0±6.4%, 18.5±4.2% and 2.6±0.73% for the AMD-NP, NP and free Bodipy with no effect on the uptake of normal ovarian cells, and the uptake of AMD-NP could be competitively inhibited by free AMD3100 in a dose-dependent manner, which confirmed that AMD-NP significantly increased targeting efficiency of ovarian cancer cells through the binding of AMD3100 and CXCR4. In vivo imaging experiments showed that the AMD-NP group exhibited a much stronger and more extensive distribution in tumor compared with the NP group and other organs within 24 h we observed, which indicated that the modification of AMD3100 on the surface of NPs could significantly improve drug targeting and release at the tumor sites, thereby beneficial to enhance the anti-tumor effect.
Based on the profound tumor targeting and accumulation characteristics, we further examined the antitumor effect of AMD-NP-PTX in vitro and in vivo. As a result, the chemical conjugation of AMD3100 with NP-PTX obtained synergistic inhibition of proliferation and migration to Caov3 ovarian cancer cells, while increased their apoptosis. Similar results were obtained from in vivo anti-cancer evaluation. The tumor growth of the mice was significantly suppressed in the AMD-NP-PTX group than those in the control group and other treatment groups, demonstrating the enhanced sensitivity of cancer cells to chemotherapeutic drugs by AMD3100. With regard to in vivo tumor metastasis assay, the results exhibited that under the CXCL12 stimulation, larger and more metastatic nodules formed, the nude mice receiving AMD-NP-PTX displayed significantly inhibited tumor metastasis while none of the other groups, which indicated that the CXCL12/CXCR4 pathway might serve as a direct regulator of tumor metastasis or indirectly mediate tumor metastasis through stromal cells as reported before,12,13 and the CXCR4-targeted AMD-NP-PTX we developed inhibited the downstream pathway of CXCL12/CXCR4 axis, thereby effectively reducing metastatic burden. Most noteworthy, due to the poor pharmacokinetics of free AMD3100, the systemic administration of free AMD3100 did not significantly sensitize ovarian cancer cells to NP-PTX treatment both in vitro and in vivo, validating the dual-functional effect of AMD-NP-PTX. Previous studies have reported the limitation of simple combination of AMD3100 and low-dose PTX due to the lack of pharmacokinetics together with the targetless toxicity.19 Herein, the active targeted nano-drug delivery system we designed solved these problems simultaneously, which can not only increase tumor targeting, but also achieve more excellent synergistic anti-cancer effect of AMD3100 and NP-released low-dose PTX. Moreover, there is a broad consensus that in vivo toxicity evaluation is essential for systemic drug delivery systems. During the study of antitumor experiment in vivo, we have monitored the body weight of nude mice and assessed the important organs with hematoxylin and eosin (H&E) staining. The results indicated the AMD-NP-PTX had good biocompatibility and safety in vivo.
To investigate the mechanism of tumor growth and metastasis inhibition of AMD-NP-PTX on ovarian cancer, we examined the protein expression of CXCL12/CXCR4 related pathways. The binding of CXCL12 to CXCR4 induces downstream signaling through multiple pathways. It first activates the CXCR4 receptor-coupled G protein, then activates phosphatidyl alcohol-3 kinase (PI3), mitogen-bound protease (MAPK), and the transcription factor NF-κB. Through the cascade reaction, it transmits chemotaxis, growth, proliferation or differentiation signals to the nucleus, exerting its biological effects.36,37 In addition, the CXCL12/CXCR4 axis has been shown to induce EMT processes in various cancers, such as hepatocellular carcinoma38 and pancreatic cancer.39 EMT is a biological process in which epithelial cells lose their polarity and are transformed into cells with interstitial phenotype, and acquire the ability to invade and migrate. Reactivation of EMT can promote the development of tumor diseases such as cancer cell migration, invasion and metastasis phenotype, resistance to anoikis, and drug resistance.40 It can also make tumor cells have the characteristics of stem cells and promote tumor drug resistance and recurrence.41 E-cadherin, N-cadherin and vimentin are considered to be key regulatory transcription factors for EMT, of which E-cadherin controls epithelial genes, while N-cadherin and vimentin activate mesenchymal genes.42,43 Previous studies have shown that silencing the expression of CXCR4 can inhibit the protein expression of EMT-related markers N-cadherin and MMP-2/9, and increase the expression of E-cadherin, thereby inducing apoptosis and inhibiting the migration of oral squamous cell carcinoma.44 Other studies have determined that the CXCL12/CXCR4 axis may induce the migration, invasion and EMT of papillary thyroid carcinoma (PTC) cells by activating the NF-κB signaling pathway.45 In this study, after being treatment with AMD-NP-PTX, the expression of E-cadherin in the tumor tissue increased, while the expression of N-cadherin, vimentin and NF-κB decreased, which suggested that the AMD-NP-PTX we developed may interrupt CXCL12/CXCR4 axis through binding with CXCR4 protein, thus inhibit tumor growth and metastasis through suppressing its downstream signaling pathways including the EMT process and NF-κB pathway.
Conclusion
In summary, we chemically modified AMD3100 to Mal-PEG-NHS followed by reacting with BSA, synthesized PTX-loaded AMD-PEG-BSA NPs by biomineralization. The AMD-PEG-BSA-NP-PTX (AMD-NP-PTX) was synthesized successfully and well characterized by NMR and TEM. Compared with PTX, NP-PTX and free AMD3100 plus NP-PTX, the targeted AMD-NP-PTX exhibited excellent cellular drug uptake, controlled drug release, remarkable anticancer efficacy and biodistribution, good biocompatibility and safety, working both as tumor-targeting drug carriers for the delivery of PTX into ovarian cancer cells and as inhibitors of CXCL12/CXCR4 axis to provide synergistic anticancer effects, which make it a promising targeting strategy for ovarian cancer therapy.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Siegel R, Naishadham D, Jemal A. Cancer statistics. CA Cancer J Clin. 2012;62(1):10–29. doi:10.3322/caac.20138
2. Zhang F, Li M, Wang J, Liang X, Su Y, Wang W. Finding new tricks for old drugs: tumoricidal activity of non-traditional antitumor drugs. AAPS Pharm Sci Tech. 2016;17(3):539–552. doi:10.1208/s12249-016-0518-y
3. Chae YK, Pan AP, Davis AA, et al. Path toward precision oncology: review of targeted therapy studies and tools to aid in defining “actionability” of a molecular lesion and patient management support. Mol Cancer Ther. 2017;16(12):2645–2655. doi:10.1158/1535-7163.mct-17-0597
4. Hong EJ, Choi DG, Shim MS. Targeted and effective photodynamic therapy for cancer using functionalized nanomaterials. Acta Pharm Sin B. 2016;6(4):297–307. doi:10.1016/j.apsb.2016.01.007
5. Aggarwal S, Singh P, Topaloglu O, Isaacs JT, Denmeade SR. A dimeric peptide that binds selectively to prostate-specific membrane antigen and inhibits its enzymatic activity. Cancer Res. 2006;66(18):9171–9177. doi:10.1158/0008-5472.can-06-1520
6. Overchuk M, Zheng G. Overcoming obstacles in the tumor microenvironment: recent advancements in nanoparticle delivery for cancer theranostics. Biomaterials. 2017;156:217–237. doi:10.1016/j.biomaterials.2017.10.024
7. Nel A, Ruoslahti E, Meng H. New insights into “permeability” as in the enhanced permeability and retention effect of cancer nanotherapeutics. ACS Nano. 2017;11(10):9567–9569. doi:10.1021/acsnano.7b07214
8. Zhong L, Xu L, Liu Y, et al. Transformative hyaluronic acid-based active targeting supramolecular nanoplatform improves long circulation and enhances cellular uptake in cancer therapy. Acta Pharm Sin B. 2019;9(2):207–219. doi:10.1016/j.apsb.2018.11.006
9. Dhas NL, Ige PP, Kudarha RR. Design, optimization and in-vitro study of folic acid conjugated-chitosan functionalized PLGA nanoparticle for delivery of bicalutamide in prostate cancer. Powder Technol. 2015;283:234–245. doi:10.1016/j.powtec.2015.04.053
10. Popple A, Durrant LG, Spendlove I, et al. The chemokine, CXCL12, is an independent predictor of poor survival in ovarian cancer. Br J Cancer. 2012;106(7):1306–1313. doi:10.10.38/bjc.2012.49
11. Scotton CJ, Wilson JL, Milliken D, Stamp G, Balkwill FR. Epithelial cancer cell migration: a role for chemokine receptors? Cancer Res. 2001;61(13):4961–4965.
12. Guo Q, Gao BL, Zhang XJ, et al. CXCL12-CXCR4 axis promotes proliferation, migration, invasion, and metastasis of ovarian cancer. Oncol Res. 2015;22(5):247–258. doi:10.3727/096504015x14343704124430
13. Mao TL, Fan KF, Liu CL. Targeting the CXCR4/CXCL12 axis in treating epithelial ovarian cancer. Gene Ther. 2017;24(10):621–629. doi:10.1038/gt.2017.69
14. Gil M, Komorowski MP, Seshadri M, et al. CXCL12/CXCR4 blockade by oncolytic virotherapy inhibits ovarian cancer growth by decreasing immunosuppression and targeting cancer-initiating cells. J Immunol. 2014;193(10):5327–5337. doi:10.4049/jimmunol.1400201
15. Zheng N, Chen J, Li T, et al. Abortifacient metapristone (RU486 derivative) interrupts CXCL12/CXCR4 axis for ovarian metastatic chemoprevention. Mol Carcinog. 2017;56(8):1896–1908. doi:10.1002/mc.22645
16. Righi E, Kashiwagi S, Yuan J, et al. CXCL12/CXCR4 blockade induces multimodal antitumor effects that prolong survival in an immunocompetent mouse model of ovarian cancer. Cancer Res. 2011;71(16):5522–5534. doi:10.1158/0008-5472.can-10-3143
17. Kajiyama H, Shibata K, Terauchi M, Ino K, Nawa A, Kikkawa F. Involvement of SDF-1α/CXCR4 axis in the enhanced peritoneal metastasis of epithelial ovarian carcinoma. Int J Cancer. 2008;122(1):91–99. doi:10.1002/ijc.23083
18. Lee HH, Bellat V, Law B. Chemotherapy induces adaptive drug resistance and metastatic potentials via phenotypic CXCR4-expressing cell state transition in ovarian cancer. PLoS One. 2017;12(2):e0171044. doi:10.1371/journal.pone.0171044
19. Reeves PM, Abbaslou MA, Kools FRW, Poznansky MC. CXCR4 blockade with AMD3100 enhances Taxol chemotherapy to limit ovarian cancer cell growth. Anticancer Drugs. 2017;28(9):935–942. doi:10.1097/cad.0000000000000518
20. Domanska UM, Boer JC, Timmer-Bosscha H, et al. CXCR4 inhibition enhances radiosensitivity, while inducing cancer cell mobilization in a prostate cancer mouse model. Clin Exp Metastasis. 2014;31(7):829–839. doi:10.1007/s10585-014-9673-2
21. Wang Y, Li J, Oupický D. Polymeric Plerixafor: effect of PEGylation on CXCR4 antagonism, cancer cell invasion, and DNA transfection. Pharm Res. 2014;31(12):3538–3548. doi:10.1007/s11095-014-1440-1
22. Li F, Zhao Y, Mao CQ, Kong Y, Ming X. RGD-modified albumin nanoconjugates for targeted delivery of a porphyrin photosensitizer. Mol Pharmaceutics. 2017;14(8):2793–2804. doi:10.1021/acs.molpharmaceut.7b00321
23. Wen Y, Dong HQ, Li Y, Shen AJ, Li YY. Nano-assembly of bovine serum albumin driven by rare-earth-ion (Gd) biomineralization for highly efficient photodynamic therapy and tumor imaging. J Mater Chem B. 2016;4(4):743–751. doi:10.1039/c5tb01962a
24. Sinha R, Kim GJ, Nie S, Shin DM. Nanotechnology in cancer therapeutics: bioconjugated nanoparticles for drug delivery. Mol Cancer Ther. 2006;5(8):1909–1917. doi:10.1158/1535-7163.mct-06-0141
25. Zhang WQ, Yu KF, Zhong T, et al. Does ligand-receptor mediated competitive effect or penetrating effect of iRGD peptide when co-administration with iRGD-modified SSL? J Drug Targeting. 2015;23(10):897–909. doi:10.3109/1061186x.2015.1034279
26. Liu Y, Ren CC, Yang L, Xu YM, Chen YN. Role of CXCL12-CXCR4 axis in ovarian cancer metastasis and CXCL12-CXCR4 blockade with AMD3100 suppresses tumor cell migration and invasion in vitro. J Cell Physiol. 2019;234(4):3897–3909. doi:10.1002/jcp.27163
27. Ray P, Lewin SA, Mihalko LA, Schmidt BT, Luker KE, Luker GD. Noninvasive imaging reveals inhibition of ovarian cancer by targeting CXCL12-CXCR4. Neoplasia. 2011;13(12):1152–1161. doi:10.1593/neo.111076.
28. Takashima T, Kawajiri H, Nishimori T, et al. Safety and efficacy of low-dose nanoparticle albumin-bound paclitaxel for HER2-negative metastatic breast cancer. Anticancer Res. 2018;38(1):379–383. doi:10.21873/anticanres.12233
29. Kinoshita R, Ishima Y, Chuang VTG, et al. Improved anticancer effects of albumin-bound paclitaxel nanoparticle via augmentation of EPR effect and albumin-protein interactions using S-nitrosated human serum albumin dimer. Biomaterials. 2017;140:162–169. doi:10.1016/j.biomaterials.2017.06.021
30. Gong YH, Shu M, Xie JH, et al. Enzymatic synthesis of PEG-poly (amine-co-thioether esters) as highly efficient pH and ROS dual-responsive nanocarriers for anticancer drug delivery. J Mater Chem B. 2019;7(4):651–664. doi:10.1039/c8tb02882f
31. Kong L, Campbell F, Kros A. DePEGylation strategies to increase cancer nanomedicine efficacy. Nanoscale Horiz. 2019;4(2):378–387. doi:10.1039/c8nh00417j
32. Perrault SD, Walkey C, Jennings T, Fischer HC, Chan WC. Mediating tumor targeting efficiency of nanoparticles through design. Nano Lett. 2009;9(5):1909–1915. doi:10.1021/nl.900031y
33. Cho EC, Xie J, Wurm PA, Xia Y. Understanding the role of surface charges in cellular adsorption versus internalization by selectively removing gold nanoparticles on the cell surface with a I2/KI etchant. Nano Lett. 2009;9(3):1080–1084. doi:10.1021/nl803487r
34. Xiao W, Chen WH, Xu XD, et al. Design of a cellular uptake-shielding “plug and play” template for photo controllable drug release. Adv Mater. 2011;23(31):3526–3530. doi:10.1002/adma.201101806
35. Tannock IF, Rotin DV. Acid pH in tumors and its potential for therapeutic exploitation. Cancer Res. 1989;49(16):4373–4384.
36. Song ZY, Gao ZH, Qu XJ. A review of CXCR4/CXCL12 axis in colorectal cancer. Biomed Aging Pathol. 2014;4(3):285–290. doi:10.1016/j.biomag.2014.06.001
37. Zhao Z, Ma X, Ma J, Sun X, Li F, Lv J. Naringin enhances endothelial progenitor cell (EPC) proliferation and tube formation capacity through the CXCL12/CXCR4/PI3K/Akt signaling pathway. Chem-Biol Interact. 2018;286:45–51. doi:10.1016/j.cbi.2018.03.002
38. Li X, Li P, Chang Y, et al. The SDF-1/CXCR4 axis induces epithelial-mesenchymal transition in hepatocellular carcinoma. Mol Cell Biochem. 2014;392(12):77–84. doi:10.1007/s11010-014-2020-8
39. Li X, Ma Q, Xu Q, et al. SDF-1/CXCR4 signaling induces pancreatic cancer cell invasion and epithelial-mesenchymal transition in vitro through non-canonical activation of Hedgehog pathway. Cancer Lett. 2012;322(2):169–176. doi:10.1016/j.canlet.2012.02.035
40. Mallini P, Lennard T, Kirby J, Meeson A. Epithelial-to-mesenchymal transition: what is the impact on breast cancer stem cells and drug resistance. Cancer Treat Rev. 2014;40(3):341–348. doi:10.1016/j.ctrv.2013.09.008
41. Hong D, Fritz AJ, Zaidi SK, et al. Epithelial-to-mesenchymal transition and cancer stem cells contribute to breast cancer heterogeneity. J Cell Physiol. 2018;233(12):9136–9144. doi:10.1002/jcp.26847
42. Peinado H, Olmeda D, Cano A. Snail, Zeb and bHLH factors in tumour progression: an alliance against the epithelial phenotype? Nat Rev Cancer. 2007;7(6):415–428. doi:10.1038/nrc2131
43. Lamouille S, Xu J, Derynck R. Molecular mechanisms of epithelial-mesenchymal transition. Nat Rev Mol Cell Biol. 2014;15(3):178–196. doi:10.1038/nrm3758
44. Duan Y, Zhang S, Wang L, et al. Targeted silencing of CXCR4 inhibits epithelial-mesenchymal transition in oral squamous cell carcinoma. Oncol Lett. 2016;12(3):2055–2061. doi:10.3892/ol.2016.4838
45. Lin Y, Ma Q, Li L, Wang H. The CXCL12-CXCR4 axis promotes migration, invasiveness, and EMT in human papillary thyroid carcinoma B-CPAP cells via NF-κB signaling. Biochem Cell Biol. 2018;96(5):619–626. doi:10.1139/bcb-2017-0074
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.