Back to Journals » Research Reports in Clinical Cardiology » Volume 5
Current treatments for acute heart failure: focus on serelaxin
Authors Bennett R
Received 6 March 2014
Accepted for publication 5 June 2014
Published 16 July 2014 Volume 2014:5 Pages 169—182
DOI https://doi.org/10.2147/RRCC.S44470
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Robert G Bennett
VA Nebraska-Western Iowa Health Care System and Department of Internal Medicine, University of Nebraska Medical Center, Omaha, NE, USA
Abstract: Acute heart failure remains an enormous health concern worldwide, and is a major cause of death and hospitalization. In spite of this, the treatment strategies for acute heart failure have remained largely unchanged for the past 2 decades. Several large randomized, placebo-controlled clinical trials have recently been conducted to attempt to improve the treatment and outcomes of acute decompensated heart failure. Some studies, including the EVEREST (tolvaptan) and ASCEND (nesiritide) showed efficacy at relieving early symptoms, but failed to improve long-term outcomes. Others, including PROTECT (rolofylline) and ASTRONAUT (aliskiren) showed little benefit in the relief of early symptoms or long-term outcomes. The recent RELAX-AHF studies using serelaxin, a recombinant form of relaxin, have shown considerable promise. Importantly, serelaxin improved congestion (dyspnea) and other early targets of acute decompensated heart failure treatment, but also improved mortality at 180 days. The purpose of this review is to provide an overview of current treatment strategies for acute decompensated heart failure, and a discussion of the recent clinical trials, with an emphasis on the serelaxin studies.
Keywords: acute heart failure, dyspnea, relaxin, serelaxin
Introduction
Heart failure is an enormous health concern, with over 5.1 million cases in the United States and over 21 million worldwide.1,2 Acute decompensated heart failure (ADHF) is generally defined as new symptoms or worsening of existing symptoms, often accompanied by dyspnea, venous congestion, edema and fatigue, that results in hospitalization or unplanned emergency care.3,4 It is estimated that 88% of persons who present with ADHF have a history of chronic heart failure.5 ADHF accounts for over 19% of all emergency room visits associated with cardiovascular-related causes, with over 650,000 cases per year in the United States.6 The estimates of mortality in ADHF patients while in the hospital range from 4%–12%, with 60 day mortality of 10%–20%, 1 year mortality at 30%, and readmission within 6 months at 50%.7–14
The characteristics associated with ADHF are variable, but usually include symptoms of systemic and pulmonary congestion, including dyspnea, edema, jugular distension, and fatigue.3,11 The majority of patients are normotensive or hypertensive, and display increased pulmonary capillary wedge pressure (PCWP).8,11,15 Due to congestion and impaired cardiac output, hypoperfusion and ischemia is common, and is thought to contribute to myocardial and renal dysfunction. For these reasons, the early approach to treatment focuses on stabilization of the patient by targeting the hemodynamic impairments and dyspnea. The early course of treatment usually involves loop diuretics and normalization of blood pressure.11,16 Despite frequent positive responses to these approaches, many patients experience worsening cardiac and renal function while hospitalized and after discharge, which contributes to readmission and mortality.17 Therefore, developing new approaches to limiting organ damage in the early treatment of ADHF is a major target of ongoing research.
Recent clinical trials of ADHF
Despite the enormity of the problem of ADHF, the treatment options for ADHF have progressed very little in the past decades.18,19 Several large randomized, placebo-controlled trials have been conducted in attempts to identify new treatments with increased effectiveness and reduced mortality, but most have been unsuccessful. Recent large randomized, placebo-controlled studies are summarized in Table 1 and are described to follow, with an emphasis on clinical studies of relaxin, which is the first pharmacological treatment for ADHF shown to result in improved outcomes from short-term treatment.
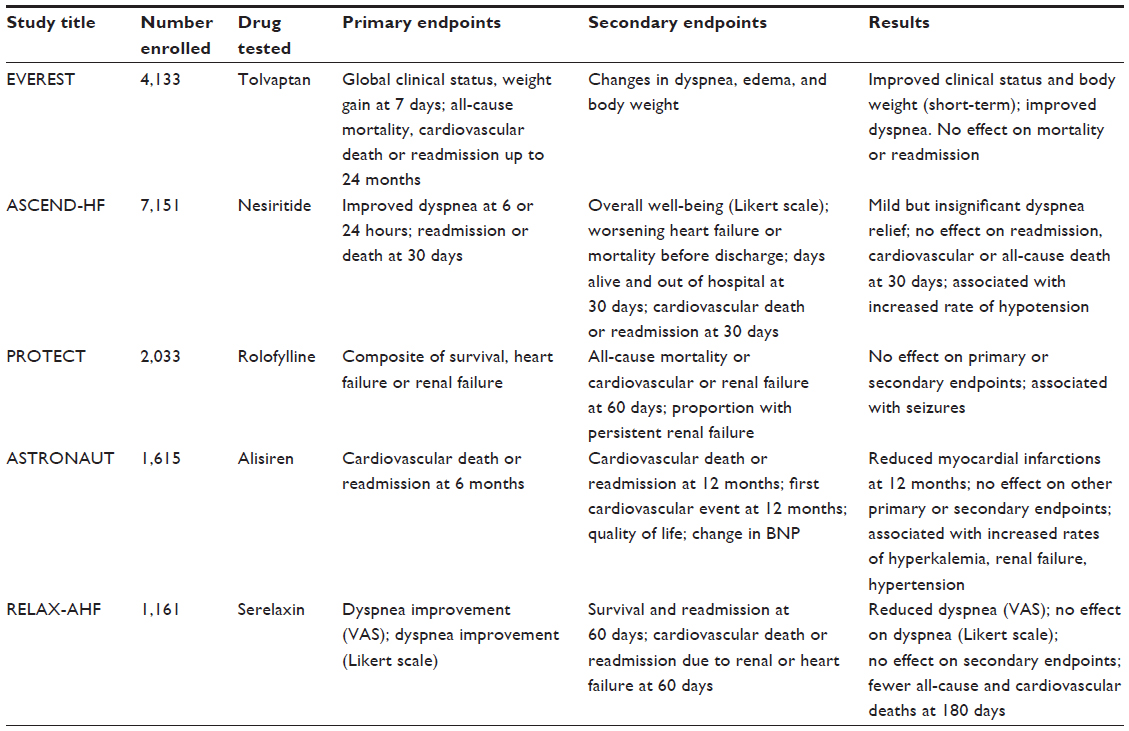 | Table 1 Recent large, randomized placebo-controlled trials of ADHF |
Tolvaptan
Hyponatremia, or low plasma sodium level, is a frequent complication of ADHF, usually in the setting of fluid overload, or hypovolemic hyponatremia. The prevalence of hyponatremia in ADHF has been estimated as 20%–27% when low serum sodium was defined as below 136 mEq/L.20,21 Multiple studies have found an association between hyponatremia in ADHF and adverse effects, including days hospitalized and mortality.20,22 One major factor in the development of hyponatremia in ADHF is the nonosmotic secretion of arginine vasopressin, which in addition to its vasopressive effects, acts as an antidiuretic in the kidney through stimulation of aquaporin-2 in the renal collecting ducts. Arginine vasopressin acts through three different but related receptors known as V1a, V1b and V2. Several nonpeptide antagonists of these receptors have been developed, collectively known as vaptans. The receptor responsible for the antidiuretic effects is the V2 receptor, and specific antagonists targeting this receptor have been developed for the treatment of hyponatremia. One of these antagonists, tolvaptan, is selective for V2 over other receptors. The Acute and Chronic Therapeutic Impact of a Vasopressin Antagonist in Congestive Heart Failure (ACTIV in CHF) study was a phase II trial of tolvaptan in ADHF.23,24 This was a dose-finding study, with patients randomized within 96 hours of presentation. Tolvaptan was administered daily for up to 60 days. The primary endpoints were change in body weight at 24 hours, and worsening heart failure or death at 60 days. There were a number of secondary endpoints, including dyspnea, body weight at discharge, serum electrolytes, and length of hospital stay. The results for the primary endpoints were a significant reduction in body weight at 24 hours, but no difference in worsening heart failure, or death at 60 days. However, post hoc analysis suggested that high risk patients (impaired renal function or severe systemic congestion) had reduced 60 day mortality, and provided the basis for a larger, phase III trial.
Tolvaptan was used in a phase III trial of ADHF patients in the Efficacy of Vasopressin Antagonism in Heart Failure Outcome Study With Tolvaptan (EVEREST) studies, which were actually three related studies. The short-term clinical status trials were two identical studies with enrollments of 2,048 and 2,085 patients with ADHF and congestion.25 The third study was a longer-term outcomes study that included all 4,133 enrolled patients followed for up to 24 months.26 The primary endpoint for the short-term studies was a composite of global clinical status (based on a visual analog scale) and body weight change, at 7 days or discharge. For the outcomes study, the primary endpoints were all-cause mortality (superiority or noninferiority), and cardiovascular death or readmission due to heart failure (superiority only). Secondary endpoints included changes in dyspnea, global clinical status, body weight, and edema. The short-term primary composite endpoint was significantly improved by tolvaptan, largely due to a significant effect on body weight. There were also significant improvements in dyspnea and edema, and other measures of congestion and fluid balance. However, there was no effect on all-cause mortality, cardiovascular death, or readmission.
Why did tolvaptan in the EVEREST study fail to show an improvement in outcomes despite weight loss and dyspnea relief? It has also been suggested that long-term tolvaptan treatment causes a compensatory increase in vasopressin levels, and stimulation of the vasopressin V1a receptor, resulting in increased aldosterone levels.27 One other likely explanation lies in the patient population. In the EVEREST study, most patients were not hyponatremic. A post hoc analysis revealed that only 11.5% of the patients enrolled in the EVEREST trial had hyponatremia as defined by serum sodium below 135 mEq/L.28 However, among the hyponatremic subjects there was a trend toward improved outcomes in response to tolvaptan, and in severe hyponatremia (<130 mEq/L), tolvaptan significantly decreased cardiovascular death or rehospitalization.28 Therefore, future studies that focus on the subjects most likely to respond to tolvaptan, those with hyponatremia, may reveal improved outcomes. Indeed, several trials are currently underway to determine the efficacy of tolvaptan in a number of subgroups of patients with ADHF, including those with renal insufficiency or hyponatremia, or resistance to loop diuretics (NCT01584557), hyponatremia (NCT01635517), high levels of the vasopressin precursor copeptin (NCT01733134), and volume overload with renal insufficiency (NCT01644331).
Nesiritide (recombinant human B-type natriuretic peptide)
Volume overload in heart failure triggers left ventricular release of the natriuretic peptides atrial natriuretic peptide (ANP) and B-type (or brain) natriuretic peptide (BNP).29–31 These peptides serve to promote a number of physiological responses, such as natriuresis, diuresis, vasodilation, and inhibition of the renin–angiotensin–aldosterone system, and therefore are thought to function as a cardioprotective response to injury. With stabilization, the levels of ANP and BNP decline. Therefore, natriuretic peptides, particularly BNP, are often used as surrogate markers for heart failure.32 Due to the physiological effects of ANP and BNP, and the transient nature of their release, it was hypothesized that administration of these peptides in heart failure would lead to improved outcomes. Studies using synthetic BNP in chronic heart failure revealed multiple effects including reduced PCWP, systemic vascular resistance, and increased natriuresis and diuresis.33 A phase II trial of 24 hours infusion of recombinant human BNP (nesiritide) in ADHF found reduced PCWP and vascular resistance, and increased cardiac index, but no significant change in urine output or sodium excretion.34 This was followed by a phase III trial of nesiritide in ADHF with both efficacy and comparative arms.35 The efficacy trial enrolled 127 patients randomized to receive nesiritide at two doses (0.015 or 0.030 μg/kg/min) or placebo for 6 hours. The results were that nesiritide, at either dose, reduced PCWP, reduced dyspnea and fatigue, and improved global clinical status. The comparative trial randomized 305 patients to receive the same nesiritide doses or standard care for 7 days. In this arm, nesiritide produced the same improvements in dyspnea, fatigue, and global clinical status as standard care. Another phase III trial, the VMAC (Vasodilation in the Management of Acute CHF) compared the effect of 3 hours infusion of nesiritide, nitroglycerin or placebo, followed by 48 hours infusion of nesiritide or nitroglycerine in 489 patients hospitalized for ADHF with dyspnea at rest.36 After 3 hours of infusion, nesiritide resulted in significantly lower PCWP than either nitroglycerin or placebo, and improved self-reported dyspnea more than placebo. At 24 hours, the effect on PCWP remained lower with nesiritide, but there was no significant difference in dyspnea or global clinical status, and no effects on mortality (7 days or 6 months) or 30-day rehospitalization due to heart failure. However, due to concerns about the low statistical power of the studies to assess safety, meta-analyses were performed using data compiled from the studies above, other small phase II studies, and data from drug approval records from the US Food and Drug Administration.37,38 The results of these meta-analyses suggested that nesiritide may be associated with an increased risk of worsening renal function and mortality at 30 days, and that larger, appropriately powered studies were needed.
The Acute Study of Clinical Effectiveness of Nesiritide in Decompensated Heart Failure (ASCEND-HF) trial studied the effectiveness of nesiritide in 7,151 subjects with ADHF, with primary endpoints of improved dyspnea at 6 and 24 hours, or a composite of readmission or death at 30 days. Although an improvement in dyspnea was reported, it did not meet the predetermined criteria for significance, and there was no change in readmission or death at 30 days, therefore the study failed to meet either of its primary endpoints.39 A smaller study, Renal Optimization Strategies Evaluation (ROSE) was recently completed, and compared low-dose nesiritide with low-dose dopamine versus (vs) placebo in 360 subjects with ADHF and renal dysfunction.40 This study also failed to show a significant benefit of nesiritide.
There are several possible explanations for the failure of the ASCEND-HF trial to show an improvement in dyspnea or long-term morbidity and mortality. One concern is the use of dyspnea as a primary endpoint, given that this determinant is patient-reported and therefore subjective, and that there is no clear benchmark for “significant” dyspnea relief.41 Furthermore, standard care improves dyspnea in most patients, and by the time of enrollment (within 24 hours of receiving intravenous treatment for ADHF), dyspnea was likely already responding to treatment.41 Another possibility lies in the nature of the major receptor for ANP and BNP, the natriuretic receptor-A (NPR-A). In the presence of excess or prolonged exposure to ligand, the signaling of NPR-A becomes blunted, a phenomenon known as desensitization.42 Therefore, in the setting of heart failure, the response of NPR-A declines despite the elevated levels of ANP and BNP. This may explain why supplementing with additional BNP failed to produce long-term improvement in outcomes.
Rolofylline
Adenosine is a metabolite of adenosine triphosphate, and regulates a number of physiological processes through its four receptors, known as the A1, A2a, A2b, and A3 receptors.43,44 In the kidney, adenosine activates the A1 receptor to cause vasoconstriction of cortical afferent arterioles, reduce glomerular filtration rate (GFR), and increase sodium resorption. In addition, activation of the A1 receptor also stimulates the tubuloglomerular feedback response that normally decreases GFR and diuresis in response to increased tubular sodium intake. This feedback mechanism contributes to resistance to diuretics in some patients. In contrast, activation of the A2 receptors generally promotes vasodilation and cardioprotection. In heart failure, adenosine levels rise dramatically, and therefore it was hypothesized that selective blockage of the A1 receptors could improve renal function, especially those patients resistant to loop diuretics.45,46 Several selective A1 receptor antagonists have been developed, and were found to induce diuresis and inhibit the tubuloglomerular feedback response, while preserving GFR.43,44 Based on these findings, clinical trials were conducted to study the effects of one of these antagonists, rolofylline, in ADHF.
The Placebo-Controlled Randomized Study of the Selective A1 Adenosine Receptor Antagonist Rolofylline for Patients Hospitalized with Acute Decompensated Heart Failure and Volume Overload to Assess Treatment Effect on Congestion and Renal Function (PROTECT) study was based on a pilot study that suggested that rolofylline, an adenosine A1 receptor antagonist, improved dyspnea and reduced worsening renal function in patients with ADHF and renal dysfunction.47 The larger phase III PROTECT study (2,033 subjects) had a primary endpoint of treatment success (defined as improvement in dyspnea at both 24 and 48 hours), failure (defined as death or readmission for heart failure through day 7, worsening heart failure for more than 24 hours after treatment initiation, or persistent worsening renal function), or no change in a composite clinical condition (survival, heart failure or renal failure).48 However, there was no difference between groups in the primary endpoint, nor were there differences detected in the secondary endpoints of persistent renal failure or survival. Furthermore, some patients receiving rolofylline developed seizures, a known adverse effect of adenosine A1 receptor antagonists.
One possibility for the failure of the PROTECT study to meet its primary endpoints may lie in the rather low number of patients (15% rolofylline, 13.7% placebo) enrolled who developed worsening renal function.48 A subgroup analysis showed a trend toward improved response to rolofylline in the subjects with the lowest creatinine clearance.49 Therefore, it is possible that only patients with the highest renal impairment, or those taking high-dose loop diuretics would benefit from rolofylline. A second possibility is the specificity of rolofylline. Among the adenosine A1 receptor blockers developed, rolofylline has the lowest specificity for the A1 receptor, with 32-fold preference for the A1 receptor over the A2 receptor, compared to ~400-fold for tonapofylline (BG9928) and ~900 fold for SLV320, and therefore it was possible that more selective blockage of the A1 receptor would allow the cardioprotective effects through activation of the A2a receptor to remain intact.44 However, in a clinical trial of tonapofylline in heart failure, the outcomes were similar to those in the rolofylline study (worsening of renal failure and increased incidence of seizures), and the study was terminated by the data safety monitoring board.50 A clinical study of SLV320 in ADHF was terminated by the sponsor for strategic reasons unrelated to the results.51 Therefore, there is currently little evidence that this class of drugs is beneficial in the treatment of ADHF, and development of adenosine A1 receptor antagonists for the treatment of ADHF appears to have been abandoned. However, partial agonists of the A1 receptor have been developed, with the idea that partial agonism would result in an overall lower renal effect, while preserving cardioprotective effects mediated through the A1 receptor.52 A phase II trial is currently underway to study the effect of a partial adenosine A1 agonist in chronic heart failure (NCT02040233).
Aliskiren
The renin–angiotensin–aldosterone system is activated in chronic heart failure, and anti-hypertensive drugs targeting this system have been used in the treatment of heart failure for many years.53,54 The drugs currently used for this purpose are either angiotensin converting enzyme (ACE) inhibitors, or angiotensin receptor blockers (ARBs). While these agents have been effective in decreasing morbidity and mortality in heart failure, they do not completely block the renin–angiotensin system, and cause an increase in the levels of renin, and with long-term use angiotensin-II can return to basal levels, in a phenomenon known as angiotensin reactivation and aldosterone escape (or aldosterone breakthrough).55 In contrast, direct renin inhibitors, such as aliskiren, prevent renin from converting angiotensinogen to angiotensin, which is the initial step in activation of the renin–angiotensin system.56 Therefore, it was thought that direct renin inhibitors might block the system more completely, avoid the elevated levels of angiotensin-I and other components of the system, and be less prone to cause aldosterone escape.57 Indeed, clinical studies revealed that aliskiren was at least as effective as other classes of renin–angiotensin system inhibitors in reducing blood pressure.58–60 Due to these properties, it was hypothesized that direct renin inhibitors might be useful in the treatment of heart failure. An early safety and efficacy study, ALOFT (Aliskiren Observation of Heart Failure Treatment) studied the effect of aliskiren (150 mg/day) in 302 patients with stable heart failure, elevated BNP (≥100 pmol/mL), and hypertension despite the use of ACE inhibitors or ARBs and beta-blockers.61 Because of earlier studies associating aliskiren with hypotension, hyperkalemia or renal failure in subjects at high risk, patients with systolic blood pressure below 90 mmHg, serum potassium ≥5.1 mM, or serum creatinine >177 μM were excluded. In addition to safety and tolerability, a reduction in BNP level was a primary endpoint. The treatment was well-tolerated, no significant differences in adverse effects compared to placebo. Aliskiren caused a significant decrease in BNP levels, as well as reduced urine aldosterone, and there was evidence of decreased left ventricular remodeling. On the other hand, another study, the Aliskiren Study in Post-MI Patients to Reduce Remodelling (ASPIRE), examined the effect of aliskiren in patients with recent myocardial infarction (within 2–6 weeks) and left ventricle ejection fraction <45%.62 In this setting, there were no beneficial effects of aliskiren, and there were significantly higher rates of hypotension and hyperkalemia. Therefore, while aliskiren showed evidence of beneficial effects on chronic heart failure, it is not useful for post-myocardial infarction cases. The findings from the ALOFT trial formed the basis for a larger trial of ADHF.
In the Aliskiren Trial on Acute Heart Failure Outcomes (ASTRONAUT) trial, 1,615 patients with impaired left ventricular ejection fraction, fluid overload, and elevated BNP peptides were randomized to placebo or aliskiren, with primary endpoint of cardiovascular death or readmission at 6 months.63 The secondary endpoints were cardiovascular death or readmission at 12 months, first cardiovascular event at 12 months, change in BNP peptides, and quality of life. No difference was found in the primary or secondary endpoints apart from a reduction in myocardial infarctions at 12 months. Furthermore, aliskiren was associated with increased rates of hyperkalemia, renal failure, and hypotension.
There has been much speculation about the reasons for the failure of aliskiren to improve outcomes in ADHF. One possibility could be related to the use of BNP in the ALOFT study as a surrogate marker of heart failure.64 In both the ALOFT and ASTRONAUT studies, aliskiren decreased BNP levels, but this did not correlate to more direct measurements of cardiac function. Another issue relates to the notion that direct renin inhibitors would be less prone to angiotensin reactivation and aldosterone escape than ACE inhibitors or ARBs. However, a recent study showed evidence that aliskiren, either alone or in combination with a ARB, did not reduce the rate of aldosterone escape.65 The patient selection criteria may also have been a major factor in the failure of the study to show improved outcomes. An analysis of the ALOFT study suggested that the effect of aliskiren was greater in patients with elevated blood pressure despite high-dose ACE inhibitors.66 Another study showed that aliskiren was associated with poor outcomes and even death in patients with diabetes who were taking other ACE inhibitors or ARBs.67 In the ASTRONAUT study, subgroup analysis suggested that diabetic patients had poorer outcomes with aliskiren, while nondiabetic subjects had improved outcomes.68 Therefore, future studies may benefit by more selective inclusion and exclusion criteria in the patient population to reflect those who are most likely to respond favorably to aliskiren and less likely to experience adverse outcomes. For example, by including patients with elevated blood pressure despite ACE inhibitor or ARB use, and excluding normotensive subjects, or diabetic patients taking renin–angiotensin–aldosterone system blockers, it is possible that aliskiren may be beneficial for some ADHF patients.
Serelaxin
Relaxin is a polypeptide hormone of the insulin/relaxin superfamily.69,70 There are three relaxin genes in humans, and two in rodents. The product of the relaxin 2 gene is orthologous to the relaxin 1 gene in rodents, and is responsible for the effects germane to this discussion, and therefore endogenous relaxin or purified relaxin used for treatment will be referred to simply as relaxin hereon. The first identified role for relaxin was in pregnancy, as the circulating levels of relaxin, produced by the corpus luteum of the ovary, peak during the first trimester.71 During pregnancy, relaxin is thought to have a role in implantation, as well as other species-dependent functions.69 One other important role attributed to relaxin is in the cardiovascular events that occur in pregnancy.72,73 Concurrent with the peak of circulating relaxin is a large decrease in systemic vascular resistance, and an increase in cardiac output. This is accompanied by vasodilation in the kidney and other organs, and an increase in GFR rate.72,73 Furthermore, there is resistance to the effects of vasodilators such as angiotensin II. Importantly, many of these changes have been detected in response to relaxin in nonpregnant female and male animal models. Chronic relaxin treatment of normal or spontaneously hypertensive rats decreased systemic vascular resistance, and increased cardiac output and global arterial compliance.74,75 Short-term (≤6 hours) relaxin treatment improved cardiac output, arterial compliance, and systemic vascular resistance in angiotensin II-induced hypertensive rats, but not normal or spontaneously hypertensive rats, suggesting that short-term responsiveness may depend on the cause of the hypertension.75
The cardioprotective properties of relaxin have long been known. Relaxin treatment reduced cardiac ischemia-reperfusion tissue injury and increased cardiac blood flow and contractility in rodent and swine models.76–79 In addition to dilation of small vessels in the heart, relaxin decreased inflammation and promoted the formation of new blood vessels at the sites of injury through vascular endothelial growth factor and basic fibroblast growth factor.77,80–82 Relaxin also had a positive inotropic effect on human atria.83 Interestingly, it has been reported that relaxin is released from the failing heart, producing circulating levels that can approach those reached during pregnancy, although the role played by cardiac-produced relaxin is unclear.84–90 However, there are concerns about the lack of validation of the assay used for these studies, and therefore further study is needed to prove that relaxin is produced by the heart.72 In addition to the heart, relaxin protects against ischemia-reperfusion injury in other organs including kidney, lung, liver, brain, and intestine.91–96
A feature that contributes to the morbidity and mortality in heart disease is cardiac fibrosis.97 One of the best characterized functions of relaxin is in remodeling of extracellular matrix proteins and protection from fibrosis.98–100 Targeted disruption of the relaxin gene results in development of age-related fibrosis in many tissues, including heart, lungs, kidney, skin, and reproductive tract.101 Using animal models of fibrosis, treatment with relaxin showed effectiveness in the reduction of fibrosis in experimental models of cardiac disease.79,102–106 In addition, relaxin has shown effectiveness in the treatment of models of pulmonary, renal, and hepatic fibrosis.99,107–112 The ability of relaxin to induce short-term hemodynamic effects and longer-term effects on organ damage made it a prime candidate for the treatment of acute heart failure in clinical trials.
Phase I dose-finding and safety study in chronic heart failure
A phase I clinical study was carried out on patients with stable compensated HF.113,114 The aim of this open-label study was to determine safety, tolerability, and pharmacokinetics, as well as hemodynamic properties of intravenously administered recombinant human relaxin. The subjects were males with New York Heart Association Class II-III, left ventricular ejection fraction <35%, and were on standard chronic heart failure medication (ARBs or ACE inhibitors, beta-blockers, diuretics, and antiplatelet treatment). The exclusion criteria included PCWP <16 mmHg, cardiac index >2.5 L/min/m2, recent myocardial infarction or cardiac surgery, acute coronary syndrome, significant recent arrhythmia, systolic blood pressure under 85 mmHg, or recent stroke. In the initial study (Group A), four patients were infused intravenously with relaxin over 24 hours, with escalating doses of 10, 30, and 100 μg/kg/day (8 hours each). The patients were monitored during the infusion period, and then for 24 hours after withdrawal of relaxin. Patients were also examined on day 9, and contacted by phone after 30 days to evaluate serious adverse events. The drug was well-tolerated, and therefore a second study was performed with six patients as above except with escalating 8-hour infusions of relaxin at 240, 480, and 960 μg/kg/day (Group B). Again, all doses were well-tolerated, and therefore the highest dose was selected for a 24 hour infusion period in six subjects (Group C). For all study groups, the subjects were monitored during the infusion period, and then for 24 hours after withdrawal of relaxin. Patients were also examined on day 9, and contacted by phone after 30 days to evaluate serious adverse events.
A number of beneficial effects were observed, although there was no clear correlation to relaxin dose. For example, PCWP significantly improved in Group A during the 30 and 100 μg/kg/day infusions and in Group C, but not in Group B. Cardiac output significantly improved during the 240 μg/kg/day infusion period in Group B and in Group C, but not Group A. The highest number of beneficial effects was seen after 24 hours in Group C, with significant improvements in cardiac index, cardiac output, and both systemic and pulmonary vascular resistance. Improvement in serum levels of NT-pro-BNP was also detected in Groups A and C, but not Group B. While in general, the effects were maintained for at least 8 hours after the infusion period, most parameters returned to baseline by 24 hours. As expected from previous studies, relaxin infusion caused decreased serum creatinine levels, significantly in Groups A and C, but not in Group B. Interestingly, in Group C, serum creatinine levels were significantly higher than baseline 9 days after infusion, suggesting a possible rebound effect, but creatinine levels had returned to normal by day 30. There were no adverse renal effects reported, and the elevation in creatinine was below the level (0.3–0.5 mg/dL) widely used as indicative of worsening renal function in other clinical studies.37,115
The study established that relaxin was well-tolerated, and produced some beneficial hemodynamic effects in stable compensated HF. One interesting aspect of the results was the lack of a dose effect of relaxin. Significant changes were observed with the lower-dose escalation study (Group A) and in the high dose continuous study (Group C), but only cardiac output was significantly changed in the intermediate group (Group B).
Phase II study: Pre-RELAX-AHF
The Preliminary Study of RELAXin in Acute Heart Failure (Pre-RELAX-AHF) was a randomized, placebo-controlled, double-blind multicenter study conducted in 2007–2008, and was aimed at identifying an optimal dose of relaxin for improving parameters in acute heart failure.116 As this was a Phase IIb dose-finding trial, there was no set endpoint, but a number of potential targets were monitored in aggregate. The subjects enrolled were males presenting with acute heart failure, defined as the presence of dyspnea, pulmonary congestion, and elevated BNP or NT-pro-BNP. Subjects must have been enrolled within 16 hours of presentation, had normal or elevated systolic blood pressure (>125 mmHg), and moderately impaired GFR (30–75 mL/min/1.73 m2). Subjects receiving loop diuretics or nitrates (if systolic blood pressure exceeded 150 mmHg) were included, but were excluded if intravenous vasodilators or inotropic drugs had been administered. Other exclusion criteria included recent acute coronary syndrome, severe pulmonary disease, or stenotic valvular disease. The subjects were randomly assigned to receive placebo (60 subjects), or relaxin at 10, 30, 100, or 250 μg/kg/day (40, 42, 37 or 49 subjects, respectively), infused intravenously for a 48 hour period. Dyspnea was assessed by patient-reported methods, both visual analog scale (VAS) and the Likert scale. Other physician-reported parameters monitored included symptoms of worsening heart or renal failure, jugular distension, and edema. Assessments were made during the infusion period, and periodically for 2 weeks. Follow-up contact was made to determine readmission and vital status for up to 180 days.
Relaxin treatment resulted in significant improvement of dyspnea as estimated by the Likert scale early (6–24 hours) at the 30 μg/kg/day dose, but not at the other doses, and not at the later time points. Using the VAS scale, there was a trend toward improved dyspnea after 5 and 14 days at all relaxin doses, with the greatest effect achieved by the 30 μg/kg/day dose, which approached statistical significance at 14 days. Overall, relaxin treatment resulted in trends toward improvement in worsening heart failure, length of stay in the hospital, 60- or 180-day cardiovascular-related and all-cause readmission or death, and all-cause mortality. The most effective dose was 30 μg/kg/day relaxin, which resulted in decreased cardiovascular death or readmission that approached significance at 60 days, and was statistically significant at 180 days. There was also a trend toward improvements in a number of physician-reported parameters, such as edema, rales, jugular pressure, and use of nitrates and diuretics. Once again, the overall effectiveness was greatest at the 30 μg/kg/day dose. This phenomenon has been reported in other studies using relaxin, in which u-shaped or bell-shaped response curves have been observed in vitro and in in vivo animal studies.70
Treatment with relaxin had a significant effect on blood pressure compared to placebo when all subject data were included. Interestingly, when subjects above and below the median systolic blood pressure (140 mmHg) were analyzed, those above the mean had a significantly larger decrease in blood pressure with relaxin compared to placebo. This is consistent with earlier findings showing that relaxin had vasodilatory effects on angiotensin-II induced hypertension in vivo and preconstricted vessels in vitro.85,117–119 There were few adverse effects that could be attributed to relaxin treatment. There were more subjects in the relaxin-treated groups who experienced hypotension that met the criteria for study discontinuation. There were two subjects that experienced serious adverse events related to decreased blood pressure, both in the highest-dose relaxin group. Unlike the phase I trial of stable compensated heart failure,113,114 there was no significant improvement in renal function in the relaxin group, but there was a trend toward an increase in the percentage of subjects experiencing an increase greater than 26 μmol/L (0.29 mg/dL) at day 14, which was significant at the highest relaxin dose.
Overall, the phase II trial produced promising results which suggested that relaxin improved dyspnea, several cardiovascular markers, and decreased cardiovascular-related death and readmission. The most effective dose for most parameters was 30 μg/kg/day, which also had a similar safety profile to placebo. Therefore, this dose was chosen for a phase III trial.
Phase III: RELAX-AHF
The phase III study began in October 2009, and used the same inclusion and exclusion criteria as the phase II study.120,121 In this randomized, double-blind, placebo controlled study, a total of 1,161 subjects were enrolled, and randomly assigned to receive 30 μg/kg/day recombinant human relaxin (renamed serelaxin by the pharmaceutical manufacturer Novartis International AG, Basel, Switzerland) or placebo (581 or 580 subjects, respectively) by intravenous infusion over 48 hours. There were two primary endpoints for this study, both focused on dyspnea. The first was the cumulative VAS area under the curve from baseline through 5 days. The second was improvement of dyspnea by the Likert scale, defined as reported improvement at all time points (6, 12, and 24 hours). There were also two secondary endpoints. The first was survival and readmission at day 60, and the second was death due to cardiovascular events or readmission due to heart or kidney failure. Because of the higher percentage of subjects experiencing large decreases in blood pressure in the Pre-RELAX-AHF study, a dose-adjustment protocol was included. If there was a decrease of greater than 40 mmHg in systolic blood pressure, but it remained above 100 mmHg, the dose was reduced by half. If systolic blood pressure dropped below 100 mmHg, the treatment was discontinued.
The primary endpoints both involved improvement of dyspnea. Using the VAS scale, the serelaxin group had modest but significantly improved area under the curve compared to placebo from baseline to 5 days, and also from baseline to 14 days. The change from baseline was increased at all time points in the serelaxin group, and was significant from 24 hours and through the 14 day period. For the other primary endpoint, relief of dyspnea by the Likert scale, the serelaxin group had improved scores at each time point, and approached significance at 12 hours (P=0.051), but did not reach statistical significance overall. Nevertheless, the prespecified condition of achieving one primary endpoint was met, due to the VAS scale measurements. The general wellbeing score was significantly improved by serelaxin using the VAS scale, but not the Likert scale. However, serelaxin subjects reported significantly earlier dyspnea improvement than placebo subjects using the Likert scale.
Serelaxin did not significantly improve the secondary endpoints of overall or cardiovascular- or renal-related death or readmission, at day 60. However, there was a significant reduction in the number of cardiovascular deaths at 180 days. Furthermore, serelaxin significantly decreased all-cause mortality at 180 days. Treatment with serelaxin significantly reduced the number of subjects with worsening heart failure, and improved several markers of congestion including edema, rales, jugular pressure, orthopnea, and dyspnea on exertion. The serelaxin subjects also had significantly reduced length of stay in the hospital, and fewer days in the intensive care or cardiac care units.
Decreases in systolic blood pressure were greater in the serelaxin group at the end of the infusion period (48 hours), and at 24 hours after the end of infusion, than in the placebo group. As described above, a dose-reduction protocol was in place in case of a drop in systolic blood pressure greater than 40 mmHg, or withdrawal from the study if blood pressure dropped below 100 mmHg. Significantly more subjects receiving serelaxin required either dose reduction, withdrawal, or both, than placebo (29% versus [vs] 18% respectively). However, adverse events related to hypotension were not different between the two groups, and serelaxin-treated subjects had significantly reduced renal impairment. This was likely due to the dose-reduction protocol that limited adverse effects to the high responders to serelaxin treatment.
The RELAX-AHF studies were the first to show a significant effect of short-term pharmacologic treatment of ADHF on long-term outcomes. There are some important limitations to the study, however. Because of the selective nature of patient selection (such as the requirement for preserved or elevated blood pressure), it is not known if the findings will translate to other subgroups of ADHF. In addition, mortality at 180 days was not a secondary endpoint, and the study was not powered to detect this difference. Another phase III trial is underway to replicate the findings of RELAX-AHF, with an estimated enrollment of over 6,000, and with a primary endpoint of time to cardiovascular-related death up to 180 days (NCT01870778).
Phase II: hemodynamic effects of serelaxin in AHF
The results of the earlier clinical trials suggested that the rapid effects of the relatively short-term serelaxin infusion influenced long-term outcomes. The purpose of this trial was to define the rapid effects of serelaxin on hemodynamic parameters in AHF. The study was conducted in a randomized, double-blind, placebo controlled manner, with an enrollment of 71 participants (34 serelaxin, 37 placebo), and was conducted during 2012.122 For eligibility for enrollment, the time to randomization had to be within 48 hours from presentation, which is a longer window than was used in the phase II/III RELAX-AHF trials (within 16 hours from presentation).116,120 Patients must have had a PCWP equal to or greater than 18 mmHg, and must have been under hemodynamic monitoring for at least 1 hour, with no planned change after stabilization in loop diuretics from 4 hours pretreatment to 1 hour posttreatment. Exclusion criteria included systolic blood pressure lower than 115 mmHg (compared to 125 mmHg in the phase II/III RELAX-AHF studies), or severe renal impairment (GFR less than 30 mL/minute/1.73 m2). Patients enrolled were randomized to receive intravenous infusion of serelaxin (30 μg/kg/day) or placebo continuously for 20 hours, followed by a 4 hour washout period. Similar to the RELAX-AHF study, a dose-reduction or withdrawal protocol was in place. Systolic blood pressure that decreased by greater than 40 mmHg, but remained above 90 mmHg in two consecutive readings 15 minutes apart, mandated the dose to be reduced by half. If systolic blood pressure dropped below 90 mmHg, the treatment was discontinued. The two primary endpoints were change in baseline in PCWP and cardiac index at 8 hours of treatment. Secondary endpoints included blood pressure, pulmonary arterial pressure, cardiac output, right atrial pressure, systemic and pulmonary vascular resistance, renal function, and NT-pro-BNP levels.
Hemodynamic determinations were taken at baseline and during the infusion period at frequent intervals (0.5, 2, 4, 6, 8, and 20 hours), and during the washout period (21, 22, and 24 hours). The peak PCWP decreased rapidly in the serelaxin group, and was significantly lower during the intervals from 0–8 hours. The effect was also significant between 8–20 hours, or during the entire infusion period (0–20 hours). Interestingly, at the 20 hour measurement, the placebo-treated group had dropped to nearly the same level as the serelaxin group. As a result, although the peak PCWP in the serelaxin group remained low during the washout period, it was not significantly different from placebo. No changes in the second primary endpoint, cardiac index, were detected at any time point, which is in contrast to the preclinical data and the phase I relaxin study.74,75,113 However, because of the low number of patients in both clinical trials (16 in the phase I, 63 in the phase II), the large difference in the doses studied, and the difference in the patient population (stable chronic HF in the phase I study vs ADHF in phase II), it is difficult to compare the effect on cardiac index between these trials, and clearly more studies are needed to resolve this question. Serelaxin significantly reduced peak systolic, diastolic and mean pulmonary artery pressure at all time points during infusion. During the washout period, the pressures remained reduced with serelaxin treatment, but significantly only for diastolic pulmonary artery pressure. Significant improvements were also detected in right atrial pressure, and both systemic and pulmonary vascular resistance, especially during the early treatment period (0–8 hours).
Patients on serelaxin rapidly experienced significantly reduced systolic and diastolic blood pressure, which was maintained throughout the washout period. Furthermore, when assessed during follow-up at 44 hours, diastolic blood pressure remained significantly lower than in the placebo group. Hypotension-related adverse effects were higher in the serelaxin group compared to placebo (15% vs 9%, respectively), and three patients in the serelaxin group were removed from the study before completion due to the predefined criteria for blood pressure decrease. There was a significantly higher creatinine clearance rate in the serelaxin group compared to placebo, but most measures of renal function were not different between the groups. The levels of NT-pro-BNP decreased 13% in the serelaxin group during the infusion period, with a 3% increase in the placebo group at the same time. Importantly, the levels of NT-pro-BNP remained significantly reduced in the serelaxin group vs placebo even at the 44 hour point (33% vs 14% decrease, respectively). Adverse effects were similar in each group.
Several important points were revealed by this study. The first is that the hemodynamic effects of serelaxin began quickly after commencing treatment, and were sustained at least as long as the infusion lasted. Secondly, the effects of serelaxin on blood pressure were sustained for as long as 44 hours, even though the infusion period (20 hours) was less than that in the RELAX-AHF study (48 hours). Thirdly, the effects on renal and cardiac function were also sustained for an extended period of time after drug withdrawal. This is consistent with the findings of the Pre-RELAX-AHF and RELAX-AHF studies, and provides additional evidence that early changes in hemodynamics with short-term treatment with serelaxin can result in longer term benefits.
Discussion
The findings of the RELAX-AHF trial represent the first findings of a significant effect on long-term outcomes resulting from a short-term intervention in ADHF. The recent failures of the ASCEND, EVEREST, and other studies to show changes in long-term outcomes raises the question – why did the RELAX-AHF study succeed in this regard? One reason may lie in the mechanisms by which relaxin works. The early effects of relaxin on systemic and pulmonary vascular resistance and blood pressure, serve to relieve congestion, as evidenced by the improvements on dyspnea. This action itself likely contributed to the outcomes, as a retrospective analysis of the data from Pre-RELAX-AHF revealed that failure to relieve persistent dyspnea was an independent risk factor for worse outcomes.123 Secondly, relaxin increases blood flow in organs such as the kidney, reducing the ischemic damage, and acts to limit fibrotic damage in target organs. Indeed, markers of cardiac, renal, and hepatic damage were associated with the 180-day mortality in RELAX-AHF studies, and relaxin improved all of these parameters.124 Finally, the signaling properties of relaxin at the cellular level provide a unique ability to produce rapid and extended effects, and interested readers are directed to a recent excellent review on the topic.125
The design of RELAX-AHF also likely contributed to the success of the study. The time from presentation to treatment was relatively short (within 16 hours), thereby increasing the chances of intervention before under-perfusion had caused extensive organ damage. Most other studies of ADHF had a much broader presentation-to-treatment window. Secondly, the inclusion criteria likely led to increased chances of success. In particular, by limiting inclusion to those with normal to high systolic blood pressure, elevated NT-pro-BNP, and moderate renal impairment, the study population was enriched in subjects more likely to respond to serelaxin. In the event that serelaxin is approved for use in ADHF, one can envision the clinical scenario for the proper use of the drug. If the patient is within 16 hours of presentation, has elevated systolic blood pressure and moderate renal impairment, dyspnea at rest and elevated natriuretic peptides, they might be a good candidate for serelaxin treatment. However, a normotensive patient would be less likely to benefit from serelaxin treatment, and other options should be considered.
The relatively restricted patient inclusion criteria in the RELAX-AHF study is in contrast with most previous interventional studies of the treatment of ADHF, in which all patients were enrolled excluding only those with severe hypotension. In most of the previous studies, the patients most likely to benefit from the pharmaceutical were a minority in the overall population, and this was most likely a major factor in the failure of many of those trials. In various commentaries and reviews of these clinical studies, it is suggested that treatments must be applicable to a broad patient population to be considered therapeutically useful, and this has been considered a major limitation of the RELAX-AHF study. However, given the highly homogeneous characteristics of ADHF patients upon presentation, a “one-size-fits-all” approach is less likely to succeed in either clinical practice or clinical trials. A recent analysis of the RELAX-AHF study revealed that the subjects enrolled represented ~20% of the total patient population admitted for ADHF.126 While only a portion of the total number of patients, this represents over 130,000 persons per year in the United States alone who may be good candidates for serelaxin treatment, which is not an unsubstantial number. In light of the increasing call for a “personalized medicine” approach to patient care, future clinical studies that match the subject selection to the expected physiological and pharmacological properties of the intervention may be more likely to succeed.4,64,127–129 Hopefully, the ongoing studies of targeted subgroups of ADHF patients, such as those discussed above for tolvaptan, will reveal positive outcomes on subgroups of ADHF patients and produce additional treatment options. If so, one can envision a panel of potential treatments that could be tailored to the patient’s individual situation, and lead to more effective treatments for ADHF.
Disclosure
The author reports no conflicts of interest in this work.
References
Go AS, Mozaffarian D, Roger VL, et al. Executive summary: heart disease and stroke statistics – 2014 update: a report from the American Heart Association. Circulation. 2014;129(3):399–410. | |
Bui AL, Horwich TB, Fonarow GC. Epidemiology and risk profile of heart failure. Nat Rev Cardiol. 2011;8(1):30–41. | |
Felker GM, Adams KF Jr, Konstam MA, O’Connor CM, Gheorghiade M. The problem of decompensated heart failure: Nomenclature, classification, and risk stratification. Am Heart J. 2003;145(Suppl 2):S18–S25. | |
Gheorghiade M, Zannad F, Sopko G, et al. Acute heart failure syndromes: current state and framework for future research. Circulation. 2005;112(25):3958–3968. | |
Fonarow GC, Abraham WT, Albert NM, et al. Organized Program to Initiate Lifesaving Treatment in Hospitalized Patients with Heart Failure (OPTIMIZE-HF): rationale and design. Am Heart J. 2004;148(1):43–51. | |
Schappert SM, Rechtsteiner EA. Ambulatory medical care utilization estimates for 2006. Natl Health Stat Reports. 2008;(8):1–29. | |
Adams KF Jr, Fonarow GC, Emerman CL, et al. Characteristics and outcomes of patients hospitalized for heart failure in the United States: Rationale, design, and preliminary observations from the first 100,000 cases in the acute decompensated heart failure national registry (ADHERE). Am Heart J. 2005;149(2):209–216. | |
Nieminen MS, Brutsaert D, Dickstein K, et al. EuroHeart Failure Survey II (EHFS II): a survey on hospitalized acute heart failure patients: description of population. Eur Heart J. 2006;27(22):2725–2736. | |
Follath F, Yilmaz MB, Delgado JF, et al. Clinical presentation, management and outcomes in the Acute Heart Failure Global Survey of Standard Treatment (ALARM-HF). Intensive Care Med. 2011;37(4):619–626. | |
Fonarow GC, Abraham WT, Albert NM, et al. Influence of a performance-improvement initiative on quality of care for patients hospitalized with heart failure: Results of the organized program to initiate lifesaving treatment in hospitalized patients with heart failure (OPTIMIZE-HF). Arch Intern Med. 2007;167(14):1493–1502. | |
Gheorghiade M, Pang PS. Acute heart failure syndromes. J Am Coll Cardiol. 2009;53(7):557–573. | |
Solomon SD, Dobson J, Pocock S, et al. Influence of nonfatal hospitalization for heart failure on subsequent mortality in patients with chronic heart failure. Circulation. 2007;116(13):1482–1487. | |
Kociol RD, Hammill BG, Fonarow GC, et al. Generalizability and longitudinal outcomes of a national heart failure clinical registry: Comparison of Acute Decompensated Heart Failure National Registry (ADHERE) and non-ADHERE medicare beneficiaries. Am Heart J. 2010;160(5):885–892. | |
Giamouzis G, Kalogeropoulos A, Georgiopoulou V, et al. Hospitalization epidemic in patients with heart failure: Risk factors, risk prediction, knowledge gaps, and future directions. J Card Fail. 2011;17(1):54–75. | |
Gheorghiade M, Abraham WT, Albert NM, et al. Systolic blood pressure at admission, clinical characteristics, and outcomes in patients hospitalized with acute heart failure. JAMA. 2006;296(18):2217–2226. | |
Poss J, Link A, Bohm M. Pharmacological treatment of acute heart failure: Current treatment and new targets. Clin Pharmacol Ther. 2013;94(4):499–508. | |
Gheorghiade M, Pang PS, Ambrosy AP, et al. A comprehensive, longitudinal description of the in-hospital and post-discharge clinical, laboratory, and neurohormonal course of patients with heart failure who die or are re-hospitalized within 90 days: Analysis from the EVEREST trial. Heart Fail Rev. 2012;17(3):485–509. | |
Yancy CW, Jessup M, Bozkurt B, et al. 2013 ACCF/AHA guideline for the management of heart failure: A report of the American College of Cardiology Foundation/American Heart Association task force on practice guidelines. J Am Coll Cardiol. 2013;62(16):e147–e239. | |
McMurray JJ, Adamopoulos S, Anker SD, et al. ESC guidelines for the diagnosis and treatment of acute and chronic heart failure 2012: The task force for the diagnosis and treatment of acute and chronic heart failure 2012 of the european society of cardiology developed in collaboration with the heart failure association (HFA) of the ESC. Eur Heart J. 2012;33(14):1787–1847. | |
De Luca L, Klein L, Udelson JE, et al. Hyponatremia in patients with heart failure. Am J Cardiol. 2005;96(12A):19L–23L. | |
Klein L, O’Connor CM, Leimberger JD, et al. Lower serum sodium is associated with increased short-term mortality in hospitalized patients with worsening heart failure: Results from the outcomes of a prospective trial of intravenous milrinone for exacerbations of chronic heart failure (OPTIME-CHF) study. Circulation. 2005;111(19):2454–2460. | |
Schrier RW, Sharma S, Shchekochikhin D. Hyponatraemia: More than just a marker of disease severity? Nat Rev Nephrol. 2013;9(1):37–50. | |
Gheorghiade M, Gattis WA, O’Connor CM, et al. Effects of tolvaptan, a vasopressin antagonist, in patients hospitalized with worsening heart failure: A randomized controlled trial. JAMA. 2004;291(16):1963–1971. | |
Rossi J, Bayram M, Udelson JE, et al. Improvement in hyponatremia during hospitalization for worsening heart failure is associated with improved outcomes: Insights from the acute and chronic therapeutic impact of a vasopressin antagonist in chronic heart failure (ACTIV in CHF) trial. Acute Card Care. 2007;9(2):82–86. | |
Gheorghiade M, Konstam MA, Burnett JC Jr, et al. Short-term clinical effects of tolvaptan, an oral vasopressin antagonist, in patients hospitalized for heart failure: The EVEREST clinical status trials. JAMA. 2007;297(12):1332–1343. | |
Konstam MA, Gheorghiade M, Burnett JC Jr, et al. Effects of oral tolvaptan in patients hospitalized for worsening heart failure: The EVEREST outcome trial. JAMA. 2007;297(12):1319–1331. | |
Pitt B, Gheorghiade M. Vasopressin V1 receptor-mediated aldosterone production as a result of selective V2 receptor antagonism: A potential explanation for the failure of tolvaptan to reduce cardiovascular outcomes in the EVEREST trial. Eur J Heart Fail. 2011;13(12):1261–1263. | |
Hauptman PJ, Burnett J, Gheorghiade M, et al. Clinical course of patients with hyponatremia and decompensated systolic heart failure and the effect of vasopressin receptor antagonism with tolvaptan. J Card Fail. 2013;19(6):390–397. | |
Burnett JC Jr, Kao PC, Hu DC, et al. Atrial natriuretic peptide elevation in congestive heart failure in the human. Science. 1986;231(4742):1145–1147. | |
Mukoyama M, Nakao K, Saito Y, et al. Increased human brain natriuretic peptide in congestive heart failure. N Engl J Med. 1990;323(11):757–758. | |
Mukoyama M, Nakao K, Hosoda K, et al. Brain natriuretic peptide as a novel cardiac hormone in humans evidence for an exquisite dual natriuretic peptide system, atrial natriuretic peptide and brain natriuretic peptide. J Clin Invest. 1991;87(4):1402–1412. | |
Yasue H, Yoshimura M, Sumida H, et al. Localization and mechanism of secretion of B-type natriuretic peptide in comparison with those of A-type natriuretic peptide in normal subjects and patients with heart failure. Circulation. 1994;90(1):195–203. | |
Yoshimura M, Yasue H, Morita E, et al. Hemodynamic, renal, and hormonal responses to brain natriuretic peptide infusion in patients with congestive heart failure. Circulation. 1991;84(4):1581–1588. | |
Mills RM, LeJemtel TH, Horton DP, et al. Sustained hemodynamic effects of an infusion of nesiritide (human b-type natriuretic peptide) in heart failure: A randomized, double-blind, placebo-controlled clinical trial. J Am Coll Cardiol. 1999;34(1):155–162. | |
Colucci WS, Elkayam U, Horton DP, et al. Intravenous nesiritide, a natriuretic peptide, in the treatment of decompensated congestive heart failure. N Engl J Med. 2000;343(4):246–253. | |
Publication Committee for the VMAC Investigators. Intravenous nesiritide vs nitroglycerin for treatment of decompensated congestive heart failure: A randomized controlled trial. JAMA. 2002;287(12):1531–1540. | |
Sackner-Bernstein JD, Skopicki HA, Aaronson KD. Risk of worsening renal function with nesiritide in patients with acutely decompensated heart failure. Circulation. 2005;111(12):1487–1491. | |
Sackner-Bernstein JD, Kowalski M, Fox M, Aaronson K. Short-term risk of death after treatment with nesiritide for decompensated heart failure: a pooled analysis of randomized controlled trials. JAMA. 2005;293(15):1900–1905. | |
O’Connor CM, Starling RC, Hernandez AF, et al. Effect of nesiritide in patients with acute decompensated heart failure. N Engl J Med. 2011;365(1):32–43. | |
Chen HH, Anstrom KJ, Givertz MM, et al. Low-dose dopamine or low-dose nesiritide in acute heart failure with renal dysfunction: The ROSE acute heart failure randomized trial. JAMA. 2013;310(23):2533–2543. | |
Vaduganathan M, Greene SJ, Ambrosy AP, Gheorghiade M, Butler J. The disconnect between phase II and phase III trials of drugs for heart failure. Nat Rev Cardiol. 2013;10(2):85–97. | |
Kuhn M. Structure, regulation, and function of mammalian membrane guanylyl cyclase receptors, with a focus on guanylyl cyclase-A. Circ Res. 2003;93(8):700–709. | |
Vallon V, Miracle C, Thomson S. Adenosine and kidney function: Potential implications in patients with heart failure. Eur J Heart Fail. 2008;10(2):176–187. | |
Hocher B. Adenosine A1 receptor antagonists in clinical research and development. Kidney Int. 2010;78(5):438–445. | |
Funaya H, Kitakaze M, Node K, Minamino T, Komamura K, Hori M. Plasma adenosine levels increase in patients with chronic heart failure. Circulation. 1997;95(6):1363–1365. | |
Gottlieb S. Adenosine A1 antagonists and the cardiorenal syndrome. Curr Heart Fail Rep. 2008;5(2):105–109. | |
Cotter G, Dittrich HC, Weatherley BD, et al. The PROTECT pilot study: A randomized, placebo-controlled, dose-finding study of the adenosine A1 receptor antagonist rolofylline in patients with acute heart failure and renal impairment. J Card Fail. 2008;14(8):631–640. | |
Massie BM, O’Connor CM, Metra M, et al. Rolofylline, an adenosine A1-receptor antagonist, in acute heart failure. N Engl J Med. 2010;363(15):1419–1428. | |
Voors AA, Dittrich HC, Massie BM, et al. Effects of the adenosine A1 receptor antagonist rolofylline on renal function in patients with acute heart failure and renal dysfunction: Results from PROTECT (placebo-controlled randomized study of the selective A1 adenosine receptor antagonist rolofylline for patients hospitalized with acute decompensated heart failure and volume overload to assess treatment effect on congestion and renal function). J Am Coll Cardiol. 2011;57(19):1899–1907. | |
Cleland JGF, Coletta AP, Clark AL. Clinical trials update from the European Society of Cardiology Heart Failure Meeting 2010: TRIDENT 1, BENEFICIAL, CUPID, RFA-HF, MUSIC, DUEL, handheld BNP, phrenic nerve stimulation, CHAMPION and CABG with CRT study. European Journal of Heart Failure. 2010;12(8):883–888. | |
Pang PS, Mehra M, Maggioni AP, et al. Rationale, design, and results from RENO-DEFEND 1: A randomized, dose-finding study of the selective A1 adenosine antagonist SLV320 in patients hospitalized with acute heart failure. Am Heart J. 2011;161(6):1012–1023. e3. | |
Albrecht-Kupper BE, Leineweber K, Nell PG. Partial adenosine A1 receptor agonists for cardiovascular therapies. Purinergic Signal. 2012;8(Suppl 1):91–99. | |
Weber KT. Aldosterone in congestive heart failure. N Engl J Med. 2001;345(23):1689–1697. | |
Shearer F, Lang CC, Struthers AD. Renin-angiotensin-aldosterone system inhibitors in heart failure. Clin Pharmacol Ther. 2013;94(4):459–467. | |
Bomback AS, Klemmer PJ. The incidence and implications of aldosterone breakthrough. Nat Clin Pract Nephrol. 2007;3(9):486–492. | |
Wood JM, Maibaum J, Rahuel J, et al. Structure-based design of aliskiren, a novel orally effective renin inhibitor. Biochem Biophys Res Commun. 2003;308(4):698–705. | |
Hollenberg NK, Fisher ND, Price DA. Pathways for angiotensin II generation in intact human tissue: Evidence from comparative pharmacological interruption of the renin system. Hypertension. 1998;32(3):387–392. | |
Gradman AH, Schmieder RE, Lins RL, Nussberger J, Chiang Y, Bedigian MP. Aliskiren, a novel orally effective renin inhibitor, provides dose-dependent antihypertensive efficacy and placebo-like tolerability in hypertensive patients. Circulation. 2005;111(8):1012–1018. | |
Pool JL, Schmieder RE, Azizi M, et al. Aliskiren, an orally effective renin inhibitor, provides antihypertensive efficacy alone and in combination with valsartan. Am J Hypertens. 2007;20(1):11–20. | |
Andersen K, Weinberger MH, Egan B, et al. Comparative efficacy and safety of aliskiren, an oral direct renin inhibitor, and ramipril in hypertension: A 6-month, randomized, double-blind trial. J Hypertens. 2008;26(3):589–599. | |
McMurray JJ, Pitt B, Latini R, et al. Effects of the oral direct renin inhibitor aliskiren in patients with symptomatic heart failure. Circ Heart Fail. 2008;1(1):17–24. | |
Solomon SD, Shin SH, Shah A, et al. Effect of the direct renin inhibitor aliskiren on left ventricular remodelling following myocardial infarction with systolic dysfunction. Eur Heart J. 2011;32(10):1227–1234. | |
Gheorghiade M, Bohm M, Greene SJ, et al. Effect of aliskiren on postdischarge mortality and heart failure readmissions among patients hospitalized for heart failure: The ASTRONAUT randomized trial. JAMA. 2013;309(11):1125–1135. | |
Mentz RJ, Felker GM, Ahmad T, et al. Learning from recent trials and shaping the future of acute heart failure trials. Am Heart J. 2013;166(4):629–635. | |
Bomback AS, Rekhtman Y, Klemmer PJ, Canetta PA, Radhakrishnan J, Appel GB. Aldosterone breakthrough during aliskiren, valsartan, and combination (aliskiren + valsartan) therapy. J Am Soc Hypertens. 2012;6(5):338–345. | |
Sidik NP, Solomon SD, Latini R, et al. Effect of aliskiren in patients with heart failure according to background dose of ACE inhibitor: A retrospective analysis of the aliskiren observation of heart failure treatment (ALOFT) trial. Cardiovasc Drugs Ther. 2011;25(4):315–321. | |
Parving HH, Brenner BM, McMurray JJ, et al. Cardiorenal end points in a trial of aliskiren for type 2 diabetes. N Engl J Med. 2012;367(23):2204–2213. | |
Maggioni AP, Greene SJ, Fonarow GC, et al. Effect of aliskiren on post-discharge outcomes among diabetic and non-diabetic patients hospitalized for heart failure: Insights from the ASTRONAUT trial. Eur Heart J. 2013;34(40):3117–3127. | |
Sherwood OD. Relaxin’s physiological roles and other diverse actions. Endocr Rev. 2004;25(2):205–234. | |
Bathgate RA, Halls ML, van der Westhuizen ET, Callander GE, Kocan M, Summers RJ. Relaxin family peptides and their receptors. Physiol Rev. 2013;93(1):405–480. | |
Eddie LW, Bell RJ, Lester A, et al. Radioimmunoassay of relaxin in pregnancy with an analogue of human relaxin. Lancet. 1986;1(8494):1344–1346. | |
Du X, Bathgate RAD, Samuel CS, Dart AM, Summers RJ. Cardiovascular effects of relaxin: From basic science to clinical therapy. Nat Rev Cardiol. 2010;7(1):48–58. | |
Conrad KP. Maternal vasodilation in pregnancy: The emerging role of relaxin. Am J Physiol Regul Integr Comp Physiol. 2011; 301(2):R267–R275. | |
Conrad KP, Debrah DO, Novak J, Danielson LA, Shroff SG. Relaxin modifies systemic arterial resistance and compliance in conscious, nonpregnant rats. Endocrinology. 2004;145(7):3289–3296. | |
Debrah DO, Conrad KP, Danielson LA, Shroff SG. Effects of relaxin on systemic arterial hemodynamics and mechanical properties in conscious rats: Sex dependency and dose response. J Appl Physiol (1985). 2005;98(3):1013–1020. | |
Masini E, Bani D, Bello MG, Bigazzi M, Mannaioni PF, Sacchi TB. Relaxin counteracts myocardial damage induced by ischemia-reperfusion in isolated guinea pig hearts: Evidence for an involvement of nitric oxide. Endocrinology. 1997;138(11):4713–4720. | |
Bani D, Masini E, Bello MG, Bigazzi M, Sacchi TB. Relaxin protects against myocardial injury caused by ischemia and reperfusion in rat heart. Am J Pathol. 1998;152(5):1367–1376. | |
Perna AM, Masini E, Nistri S, et al. Novel drug development opportunity for relaxin in acute myocardial infarction: Evidences from a swine model. FASEB J. 2005;19(11):1525–1527. | |
Zhang J, Qi Y, Geng B, et al. Effect of relaxin on myocardial ischemia injury induced by isoproterenol. Peptides. 2005;26(9):1632–1639. | |
Lewis M, Deshpande U, Guzman L, et al. Systemic relaxin administration stimulates angiogenic cytokine expression and vessel formation in a rat myocardial infarct model. In: Tregear G, Ivell R, Bathgate R, Wade J, editors. Relaxin 2000. Springer Netherlands; 2001:159–167. | |
Nistri S, Chiappini L, Sassoli C, Bani D. Relaxin inhibits lipopolysaccharide-induced adhesion of neutrophils to coronary endothelial cells by a nitric oxide-mediated mechanism. FASEB J. 2003;17(14):2109–2111. | |
Masini E, Nistri S, Vannacci A, Bani Sacchi T, Novelli A, Bani D. Relaxin inhibits the activation of human neutrophils: Involvement of the nitric oxide pathway. Endocrinology. 2004;145(3):1106–1112. | |
Dschietzig T, Alexiou K, Kinkel HT, Baumann G, Matschke K, Stangl K. The positive inotropic effect of relaxin-2 in human atrial myocardium is preserved in end-stage heart failure: Role of G(i)-phosphoinositide-3 kinase signaling. J Card Fail. 2011;17(2):158–166. | |
Dschietzig T, Richter C, Bartsch C, et al. The pregnancy hormone relaxin is a player in human heart failure. FASEB J. 2001;15(12):2187–2195. | |
Fisher C, MacLean M, Morecroft I, et al. Is the pregnancy hormone relaxin also a vasodilator peptide secreted by the heart? Circulation. 2002;106(3):292–295. | |
Fisher C, Berry C, Blue L, Morton JJ, McMurray J. N-terminal pro B type natriuretic peptide, but not the new putative cardiac hormone relaxin, predicts prognosis in patients with chronic heart failure. Heart. 2003;89(8):879–881. | |
Fisher C, Al-Benna S, Kirk A, Morton JJ, McMurray JJ. Transcardiac and transpulmonary gradients in the putative new cardiovascular hormone relaxin. Heart. 2003;89(7):789–790. | |
Hocher B, Ziebig R, Krause R, et al. Relaxin is an independent risk factor predicting death in male patients with end-stage kidney disease. Circulation. 2004;109(19):2266–2268. | |
Kupari M, Mikkola TS, Turto H, Lommi J. Is the pregnancy hormone relaxin an important player in human heart failure? Eur J Heart Fail. 2005;7(2):195–198. | |
Xu Q, Lekgabe ED, Gao X, et al. Endogenous relaxin does not affect chronic pressure overload-induced cardiac hypertrophy and fibrosis. Endocrinology. 2008;149(2):476–482. | |
Masini E, Cuzzocrea S, Mazzon E, et al. Protective effects of relaxin in ischemia/reperfusion-induced intestinal injury due to splanchnic artery occlusion. Br J Pharmacol. 2006;148(8):1124–1132. | |
Boehnert MU, Armbruster FP, Hilbig H. Relaxin as a protective substance in preservation solutions for organ transplantation, as shown in an isolated perfused rat liver model. Transplant Proc. 2008;40(4):978–980. | |
Wilson BC, Rappaport R. An in vitro study of the protective effect of relaxin on brain tissue under ischemic stress. Ann N Y Acad Sci. 2009;1160:265–268. | |
Alexiou K, Matschke K, Westphal A, Stangl K, Dschietzig T. Relaxin is a candidate drug for lung preservation: Relaxin-induced protection of rat lungs from ischemia-reperfusion injury. J Heart Lung Transplant. 2010;29(4):454–460. | |
Alexiou K, Wilbring M, Matschke K, Dschietzig T. Relaxin protects rat lungs from ischemia-reperfusion injury via inducible NO synthase: Role of ERK-1/2, PI3K, and forkhead transcription factor FKHRL1. PLoS One. 2013;8(9):e75592. | |
Yoshida T, Kumagai H, Kohsaka T, Ikegaya N. Relaxin protects against renal ischemia-reperfusion injury. Am J Physiol Renal Physiol. 2013;305(8):F1169–F1176. | |
Burchfield JS, Xie M, Hill JA. Pathological ventricular remodeling: Mechanisms: Part 1 of 2. Circulation. 2013;128(4):388–400. | |
Samuel CS. Relaxin: Antifibrotic properties and effects in models of disease. Clin Med Res. 2005;3(4):241–249. | |
Samuel CS, Lekgabe ED, Mookerjee I. The effects of relaxin on extracellular matrix remodeling in health and fibrotic disease. Adv Exp Med Biol. 2007;612:88–103. | |
Bennett RG. Relaxin and its role in the development and treatment of fibrosis. Transl Res. 2009;154(1):1–6. | |
Samuel CS, Zhao C, Bathgate RA, et al. The relaxin gene-knockout mouse: A model of progressive fibrosis. Ann N Y Acad Sci. 2005;1041:173–181. | |
Samuel CS, Unemori EN, Mookerjee I, et al. Relaxin modulates cardiac fibroblast proliferation, differentiation, and collagen production and reverses cardiac fibrosis in vivo. Endocrinology. 2004;145(9):4125–4133. | |
Samuel CS, Hewitson TD, Zhang Y, Kelly DJ. Relaxin ameliorates fibrosis in experimental diabetic cardiomyopathy. Endocrinology. 2008;149(7):3286–3293. | |
Lekgabe ED, Kiriazis H, Zhao C, et al. Relaxin reverses cardiac and renal fibrosis in spontaneously hypertensive rats. Hypertension. 2005;46(2):412–418. | |
Samuel CS, Cendrawan S, Gao X, et al. Relaxin remodels fibrotic healing following myocardial infarction. Lab Invest. 2011;91(5):675–690. | |
Parikh A, Patel D, McTiernan CF, et al. Relaxin suppresses atrial fibrillation by reversing fibrosis and myocyte hypertrophy and increasing conduction velocity and sodium current in spontaneously hypertensive rat hearts. Circ Res. 2013;113(3):313–321. | |
Williams EJ, Benyon RC, Trim N, et al. Relaxin inhibits effective collagen deposition by cultured hepatic stellate cells and decreases rat liver fibrosis in vivo. Gut. 2001;49(4):577–583. | |
Hewitson TD, Ho WY, Samuel CS. Antifibrotic properties of relaxin: In vivo mechanism of action in experimental renal tubulointerstitial fibrosis. Endocrinology. 2010;151(10):4938–4948. | |
Mookerjee I, Solly NR, Royce SG, Tregear GW, Samuel CS, Tang ML. Endogenous relaxin regulates collagen deposition in an animal model of allergic airway disease. Endocrinology. 2006;147(2):754–761. | |
Mookerjee I, Tang ML, Solly N, Tregear GW, Samuel CS. Investigating the role of relaxin in the regulation of airway fibrosis in animal models of acute and chronic allergic airway disease. Ann N Y Acad Sci. 2005;1041:194–196. | |
Samuel CS, Mookerjee I, Masterson R, Tregear GW, Hewitson TD. Relaxin regulates collagen overproduction associated with experimental progressive renal fibrosis. Ann N Y Acad Sci. 2005;1041:182–184. | |
Royce SG, Cheng V, Samuel CS, Tang MLK. The regulation of fibrosis in airway remodeling in asthma. Mol Cell Endocrinol. 2012;351(2):167–175. | |
Dschietzig T, Teichman S, Unemori E, et al. Intravenous recombinant human relaxin in compensated heart failure: A safety, tolerability, and pharmacodynamic trial. J Card Fail. 2009;15(3):182–190. | |
Dschietzig T, Teichman S, Unemori E, et al. First clinical experience with intravenous recombinant human relaxin in compensated heart failure. Ann N Y Acad Sci. 2009;1160:387–392. | |
Voors AA, Davison BA, Felker GM, et al. Early drop in systolic blood pressure and worsening renal function in acute heart failure: Renal results of pre-RELAX-AHF. Eur J Heart Fail. 2011;13(9):961–967. | |
Teerlink JR, Metra M, Felker GM, et al. Relaxin for the treatment of patients with acute heart failure (pre-RELAX-AHF): A multicentre, randomised, placebo-controlled, parallel-group, dose-finding phase IIb study. Lancet. 2009;373(9673):1429–1439. | |
Debrah DO, Conrad KP, Jeyabalan A, Danielson LA, Shroff SG. Relaxin increases cardiac output and reduces systemic arterial load in hypertensive rats. Hypertension. 2005;46(4):745–750. | |
McGuane JT, Debrah JE, Sautina L, et al. Relaxin induces rapid dilation of rodent small renal and human subcutaneous arteries via PI3 kinase and nitric oxide. Endocrinology. 2011;152(7):2786–2796. | |
Li Y, Brookes ZLS, Kaufman S. Acute and chronic effects of relaxin on vasoactivity, myogenic reactivity and compliance of the rat mesenteric arterial and venous vasculature. Regul Pept. 2005;132(1–3):41–46. | |
Teerlink JR, Cotter G, Davison BA, et al. Serelaxin, recombinant human relaxin-2, for treatment of acute heart failure (RELAX-AHF): A randomised, placebo-controlled trial. Lancet. 2013;381(9860):29–39. | |
Ponikowski P, Metra M, Teerlink JR, et al. Design of the RELAXin in acute heart failure study. Am Heart J. 2012;163(2):149–155. e1. | |
Ponikowski P, Mitrovic V, Ruda M, et al. A randomized, double-blind, placebo-controlled, multicentre study to assess haemodynamic effects of serelaxin in patients with acute heart failure. Eur Heart J. 2014;35(7):431–441. | |
Metra M, Teerlink JR, Felker GM, et al. Dyspnoea and worsening heart failure in patients with acute heart failure: Results from the pre-RELAX-AHF study. Eur J Heart Fail. 2010;12(10):1130–1139. | |
Metra M, Cotter G, Davison BA, et al. Effect of serelaxin on cardiac, renal, and hepatic biomarkers in the relaxin in acute heart failure (RELAX-AHF) development program: Correlation with outcomes. J Am Coll Cardiol. 2013;61(2):196–206. | |
Du XJ, Hewitson TD, Nguyen MN, Samuel CS. Therapeutic effects of serelaxin in acute heart failure. Circ J. 2014;78:542–552. | |
Wang TS, Hellkamp AS, Patel CB, Ezekowitz JA, Fonarow GC, Hernandez AF. Representativeness of RELAX-AHF clinical trial population in acute heart failure. Circulation: Cardiovascular Quality and Outcomes. 2014;7(2):259–268. | |
Givertz MM, Teerlink JR, Albert NM, et al. Acute decompensated heart failure: Update on new and emerging evidence and directions for future research. J Card Fail. 2013;19(6):371–389. | |
Konstam MA, Pang PS, Gheorghiade M. Seeking new heights in acute heart failure syndromes: Lessons from ASCEND and EVEREST. Eur Heart J. 2013;34(18):1345–1349. | |
O’Connor CM, Fiuzat M. Lessons learned from clinical trials in acute heart failure: Phase 3 drug trials. Heart Fail Clin. 2011;7(4):451–456. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.