Back to Archived Journals » Research and Reports in Neonatology » Volume 5
Current perspectives on neonatal hypoglycemia, its management, and cerebral injury risk
Authors Chandran S, Rajadurai V, Alim A, Hussain K
Received 17 October 2014
Accepted for publication 1 December 2014
Published 2 February 2015 Volume 2015:5 Pages 17—30
DOI https://doi.org/10.2147/RRN.S55353
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Robert Schelonka
Suresh Chandran,1–4 Victor Samuel Rajadurai,1–3 Abdul Alim Abdul Haium,1–3 Khalid Hussain5,6
1Department of Neonatology, KK Women’s and Children’s Hospital, Singapore; 2Duke-NUS Graduate School of Medicine, Singapore; 3Yong Loo Lin School of Medicine, National University of Singapore, Singapore; 4Lee Kong Chian School of Medicine, Nanyang Technological University, Singapore; 5Department of Paediatric Endocrinology, Great Ormond Street Hospital for Children, NHS Foundation Trust, London, 6The Institute of Child Health, University College London, London, UK
Abstract: Glucose is an essential substrate for mammalian cells; in particular, the brain needs glucose continuously as a primary source of energy. Hypoglycemia is the most common biochemical finding in the neonatal period. However, despite the common occurrence, there is still controversy on the definition of hypoglycemia in the newborn period. This has led to the development of guidelines designed to identify infants “at-risk” and the implementation of an “operational threshold” for physicians to consider intervention. In healthy term infants, the optimal hormonal and metabolic adaptations during the immediate neonatal period ensure an adequate energy substrate for the vital organs, whereas the abnormal glucose homeostasis observed in preterm and growth-retarded infants is multifactorial in origin. For these high-risk infants, it is important to identify, screen, and prevent significant hypoglycemia. Detailed investigations are warranted in infants with severe and persistent hypoglycemia. Neonatal hypoglycemia is a major cause of brain injury. The speculated mechanisms of cellular injury include excitatory neurotoxins active at N-methyl-D-aspartate receptors, increased mitochondrial free radical generation with initiation of apoptosis and altered cerebral energetic characteristics. This hypoglycemic brain injury predominantly affects parieto-occipital regions causing cognitive, sensory, psychomotor, and behavioral deficits in children. This state of the art review article covers the fetal–neonatal metabolic adaptation, glucose homeostasis in normal and abnormal conditions, management strategies, and late neurological sequelae of hypoglycemia.
Keywords: hyperinsulinism, hypoglycemic brain injury
Perinatal glucose physiology
Fetal blood glucose regulation
During fetal life, glucose is the principal energy substrate. Throughout pregnancy, the fetus receives a supply of substrates essential for growth and for the deposition of fuel stores required after birth through the placental circulation from the mother. Amino acids and lactate in addition to glucose function as energy stores in the fetus.1 Glucose contributes nearly 65% of the total energy requirement of the fetus and the rest is contributed predominantly by lactate.1 Maternal hepatic glucose production increases by 16%–30% through gestation to supply the fetus with energy.2
By facilitated diffusion, glucose is transported across the placenta, but the fetus is capable of endogenous glucose production in the face of placental insufficiency.1 The placenta transfers to the fetus only 40%–50% of the maternal glucose it receives and converts nearly 60% of the glucose transferred from the maternal circulation to lactate, which is delivered to the fetus and mother in a ratio of 1:3.3 The fetus receives high amounts of amino acids through the placenta leading to higher fetal amino acid concentrations than in maternal blood.4 As pregnancy advances, the amount of glucose transported to the fetus increases, leading to significant deposition of glycogen and fat stores, especially in the third trimester.
The fetal blood glucose level normally corresponds to 60%–80% of maternal levels.5,6 Glycogenic enzymes are present in fetal liver at as early as 8 weeks of gestation and the hepatic glycogen content increases from 3.4 mg/g at 8 weeks of gestation to 50 mg/g at term.5,6 Amino acids are actively transported to the fetus against a transplacental gradient. The fetal uptake of products of β-oxidation of fatty acids is greater than that of glucose, and the fate of these ketone bodies is both their incorporation into brain lipids and also their use as a cerebral energy source.7 Glucose is converted to fats in the third trimester in preparation for changes at birth.6
Fetal metabolism is an anabolic process. The fetus has the ability to regulate its blood glucose concentration independently of maternal glucose levels and placental diffusion.8 In placental insufficiency or maternal starvation, the fetus can activate gluconeogenesis at the expense of growth and storage.8 On the contrary, suboptimal control of diabetes in the mother will lead to increased secretion of insulin resulting in greater than normal fetal growth.6,8
A healthy fetus differs from adults in that there is a blunted insulin response to high glucose concentrations, and that insulin secretion is more sensitive to amino acids than to glucose.9 The fetus is less sensitive than the neonate to the glycogenolytic actions of glucagon but sensitivity increases with advancing gestational age.10,11 In severe and prolonged placental insufficiency, the ensuing failure of glucose homeostasis leads to prolonged fetal hypoglycemia.12
The fetal liver is enriched with stores to the extent that the liver has three times more glycogen than an adult liver,13 which can function as an energy source in the neonatal period. Fetal liver has the enzymes required for gluconeogenesis and glycogenolysis, and in the face of placental insufficiency or maternal starvation these enzymes becomes active. The ketone body production rate is low in fetal life and fetus dependency is more so on amino acid and glucose oxidation for energy rather than on ketones.13
Fetal endocrine milieu
Insulin dominates the fetal endocrine milieu. Development of the pancreas follows a similar sequence in all mammals, but the stage of gestation at which the specific events occur may vary among species.14 Insulin remains the main anabolic hormone in fetal life and islet β-cells can be detected in the pancreas at as early as the 10th to 12th week of gestation.15 In the fetus, the main phase of islet cell development occurs in the second trimester and the remodeling of the islet cell continues through late gestation and early childhood, up to 4 years of life.16,17
The glucagon secreting α-cells predominate for most of the gestation and are the first cell type to be identified clearly in the pancreas.14 Both α- and β-cells are sensitive to a range of stimuli including metabolites, neurotransmitters, and hormones.18,19 The fetal β-cells respond well to changes in glucose and amino acid levels in late gestation.16 Unlike neonatal and adult α-cells, fetal α-cells appear to be relatively unresponsive to changes in the glucose levels but they do respond rapidly to variations in amino acid and catecholamine levels.19
Fetal intestinal transport and enzyme maturation
Epithelial glucose transport has been shown in the jejunum by 20 weeks of gestation in human embryos.20 Lactose, a disaccharide in mammalian milk, is hydrolyzed to the monosaccharide’s glucose and galactose by the lactase present in the brush border of the intestinal villi. Intestinal lactase activity is detectable at as early as 8 weeks of gestation in the fetal gut.20 Lactase activity rises gradually from 8 weeks to 34 weeks, and then rises rapidly from that time to term gestation. With oral feeding in the postnatal period, lactase activity increases in both preterm and term infants.21,22 Intestinal lactase activity in the gut is directly related to the gestational age;20 hence, maintaining normoglycemia while on milk feeds in infants of varying gestational age is critical to the availability of lactase at the intestinal villi.
Metabolic and endocrine adaptation at birth
At birth, the continuous flow of energy substrates from mother to the fetus is curtailed with severance of the umbilical cord. The umbilical cord venous blood glucose concentration is 80%–90% of maternal venous blood.23 A normal term infant accomplishes this transition via a series of metabolic and hormonal adaptations which allow it an independent existence. This fetal–neonatal adaptation fails to a variable extent in both preterm and small-for-gestational-age (SGA) infants.
Endocrine changes occur soon after birth with a decrease in plasma insulin levels and increases of catecholamine and glucagon.10,24 The infant switches on the endogenous production of glucose following birth until exogenous nutritional supply is established. After birth, an endocrine stress response leads insulin and glucagon to play a different role in glucose homeostasis, where hepatic glycogenolysis, lipolysis, and ß-oxidation of fatty acids, which lead to ketone body production and lactate generation due to proteolysis, take place, providing an adequate substrate for gluconeogenesis.10
Liver glycogen stores in a healthy full-term infant last for only 10–12 hours from birth.25 Hepatic glucose production in a full-term infant is 4–6 mg/kg/min, which is proportional to the estimated brain weight in newborn infants.26 The newborn infant has essential enzymes needed for gluconeogenesis from alanine, pyruvate, glycerol, and lactate (Figure 1).27 Medina et al reported that lactate may be a preferred cerebral fuel over glucose and ketone bodies during transition to extrauterine life.28 Alanine and glycerol contributes 5%–10% of the hepatic glucose production, whereas lactate may contribute up to 30% for gluconeogenesis.29 SGA infants have low energy stores but high levels of glucose precursors, suggesting inadequacy of gluconeogenesis. It has been shown that insulin sensitivity is reduced in liver but increased in the periphery in growth-retarded infants.23 Rate of glucose generation and lipolysis are ineffective in SGA infants but gluconeogenesis from glycerol is efficient.30
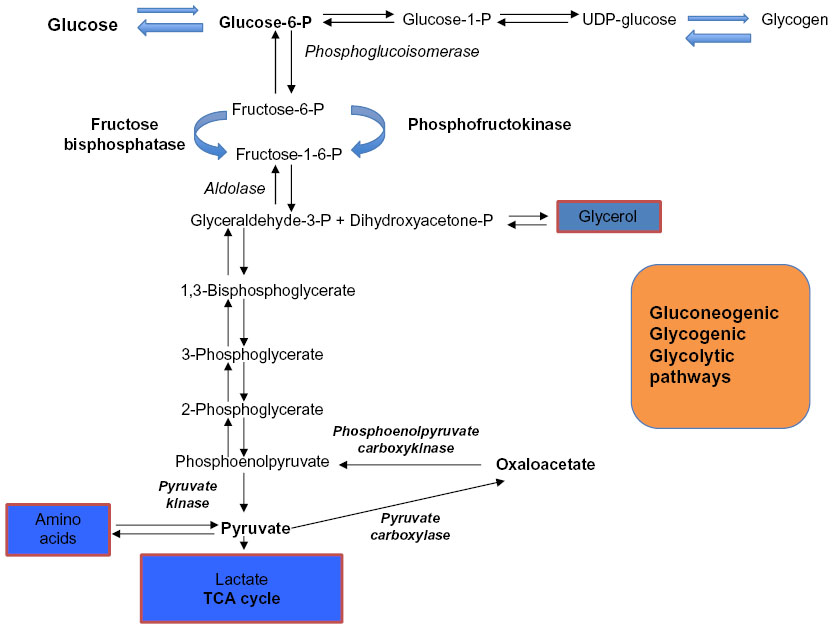 | Figure 1 The gluconeogenic, glycolytic, glycogenolysis, and glycogen synthesis pathways. |
Prior to initiation of breast-feeding, depot fat constituting 15% of birth weight is mobilized for generating energy. A high rate of lipolysis occurs in newborn infants soon after birth and is stimulated by thyroid stimulating hormone (TSH) – a dominating lipolytic hormone – immediately after birth.31 Changes in environmental temperature after birth cause thermal stimulation of the infant’s cutaneous thermoreceptors, leading to activation of the hypothalamic–pituitary–TSH axis, resulting in maximal TSH release, leading to induction of the lipolytic process.32 Triacylglycerol, rich in depot fat, undergoes hydrolysis and releases nonesterified fatty acids. These fatty acids following hepatic β-oxidation become an important energy source. Lipolysis also generates glycerol, which can enter the gluconeogenic pathway (Figure 1).27
The metabolism in the newborn depends greatly on the function of vital enzymes: hepatic glycogen phosphorylase for glycogenolysis, phosphoenolpyruvate carboxykinase for gluconeogenesis, and carnitine acyltransferase for ketogenesis.8,30 Glucagon, as the major glucoregulatory hormone, induces glycogenolysis, gluconeogenesis, and ketogenesis in liver. Catecholamines, cortisol, and growth hormone do play a key role in glucose homeostasis as counter-regulatory hormones. While on breast feeds, when blood glucose concentrations are low, these infants will have high ketone body levels and the ketone bodies function as alternative fuels, preventing neuronal injury.8,33 Formula-fed infants generate low ketone body levels compared to breast-fed infants.34,35
Gastrointestinal adaptation at birth
Gastrointestinal adaptation of the newborn is critical for absorption of milk feeds and, therefore, the substrate availability for energy supply to the vital cells, eg neurons. The introduction of milk feeds trigger the secretion of gastrointestinal peptides and hormones, which in turn induce gut growth, mucosal differentiation, and induction of motor activity, facilitating digestion and absorption. Following establishment of a continuous energy source through regular feeds, the infant successfully adapts to an extrauterine life.34,35
Glucose physiology in the immediate neonatal period
Following the abrupt cessation of glucose supply to the neonate from the mother at birth, neonatal glucose concentration falls rapidly, reaching a nadir at 1 hour of age and then stabilizing by about 3 hours of age spontaneously or in response to milk feeds in healthy full-term infants.36,37 This fall in glucose levels that is noticed after birth appears essential to facilitate the physiological transition for neonatal survival, which includes acceleration of glucose production by gluconeogenesis, stimulation of appetite, adaptation to fast/feed cycles, and promotion of oxidative fat metabolism using lipid from fat stores and ingested milk feeds.37 Cowett and Farrag reported that the range of plasma glucose concentrations in the neonate at delivery depends on the last maternal meal, the duration of labor, the route of delivery, and the type of parenteral fluid administered to the mother.38 During this transient hypoglycemic phase, the plasma insulin levels fall and the catecholamine and glucagon levels rise.39 These vital endocrine changes switch on the essential enzymes for gluconeogenesis, glycogenolysis, lipolysis, and ketogenesis.
The low insulin/glucagon molar ratio is critical in neonatal adaptation at birth for glucose homeostasis, resulting from the mobilization of glycogen stores. These changes in pancreatic hormone secretion could be due to a catecholamine surge following the stress of birth.40 Once the glycogen stores are utilized over the first 10–12 hours of birth in a term infant, further maintenance of plasma glucose levels depend on the milk feeds.25,41
Definition of hypoglycemia
A numerical definition for neonatal hypoglycemia remains controversial even today. Defining hypoglycemia by a single blood glucose value in the immediate neonatal period is questionable as the blood glucose values are a continuum. The critical level below which the incidence of brain injury increases depends on a number of factors including the availability of alternate energy fuels and the baby’s energy demands.
As the newborn infant is transitioning between the complete dependence of the fetus and the complete independence of the adult, the glucose concentration decreases rapidly during the first few hours of life, which is physiological. During pregnancy, the lower limit of fetal glucose level is 3 mmol/L, and the glucose level declines to approximately 2.8 mmol/L by 2 hours and equilibrates at 3.9 mmol/L by 72 hours of life.42,43 Euglycemia following this depends on the availability of substrates (glycogen, amino acids, lactate, and glycerol); intactness of the glycogenolytic, gluconeogenic, and lipolytic pathways; and production of the glucose-regulating hormones insulin, glucagon, adrenaline, growth hormone, and cortisol.43
Hypoglycemia should be considered as a failure of the metabolic adaptation and a more pragmatic approach has been adopted at defining operational thresholds, which might help guide the clinician in terms of practical management.
An “operational threshold” is that concentration of plasma or whole blood glucose at which clinicians should consider intervention, based on the evidence currently available in the literature.44 In an expert recommendation by Cornblath et al, it had been suggested that in any infant who shows neurologic symptoms or signs suggestive of hypoglycemia, intervention should be taken if the blood sugar values are less than 2.5 mmol/L. In asymptomatic “at-risk” infants, urgent interventions should be considered to raise blood sugar levels if they are less than 2.0 mmol/L on two consecutive occasions or if a single blood sugar level is less than 1.0 mmol/L.44 Cornblath and Reisner suggested that a blood glucose concentration less than 2.2 mmol/L or more than 6.9 mmol/L after 3 days of life is abnormal.45
Srinivasan et al published data on blood glucose concentration after evaluating term infants who were appropriate for gestational age weighing between 2,500 and 4,000 g. In this data, nadir of blood glucose was between 1 and 2 hours, and there was a significant rise during the 3rd hour. They suggested “cut off values” for blood glucose concentration of less than 1.9 mmol/L in the first 3 hours, 2.2 mmol/L between 3 and 24 hours, and 2.5 mmol/L after 24 hours of postnatal life to be considered in the hypoglycemic range.37
Heck and Erenberg evaluated the blood glucose concentration in term infants during the first 48 hours of life and concluded that values of 2.2 mmol/L from 3 to 24 hours and 2.5 mmol/L after 24 hours should be considered as hypoglycemia. However, in infants with suspected hyperinsulinemic hypoglycemia (HH) due to lack of alternative energy fuels, the blood glucose levels should be maintained >3.5 mmol/L.46
Definition, method, site of sampling, symptomatology, significance of asymptomatic status, and management and its effect on neurodevelopmental outcome are the areas of disagreement in neonatal hypoglycemia.47,48 Cornblath et al described four possible approaches to define hypoglycemia in the neonate: clinical; statistical; neurophysiological; and neurodevelopmental.44 Clinical manifestations in neonatal hypoglycemia are not unique as similar symptomatology can be seen in a wide variety of pathological conditions. So the authors suggested Whipple’s triad (reliable glucose measurement; signs and symptoms consistent with hypoglycemia; and resolution of the signs and symptoms on restoration of normal blood glucose values) to be fulfilled to attribute the clinical manifestations to hypoglycemia in infants.49,50 The concern with this approach is that several asymptomatic infants have extremely low blood glucose levels and the coexisting conditions often seen in these infants, like hypoxic ischemic encephalopathy, sepsis, and metabolic disorders, limits its use in neonates.
In the statistical approach, a blood glucose concentration of >2 standard deviations below the mean is considered as hypoglycemia based on the normal distribution curve of blood glucose levels for healthy term and preterm infants. Other factors to be considered when using this approach include gestational and postnatal age of the baby, physiological state (pre-feed or post-feed), and the type of milk given (breast milk or formula milk). Above all, feeding practices and unit management policies to low glucose levels are variable, limiting this approach.51
The physiological response approach was based on changes in neurophysiological functions, cerebral blood flow, and hormonal responses relative to different glucose concentrations.52,53 Koh et al evaluated the same using an auditory evoked response waveform on patients in hypoglycemic and euglycemia states.53 Other studies failed to show a similar response in both auditory evoked response and electroencephalogram.54,55 The data on the approach based on neurodevelopmental outcome relative to symptomatic or asymptomatic hypoglycemia is limited. Lucas et al made a landmark observation in this approach.56 Using Bayley motor and mental developmental scores, they evaluated 661 preterm neonates at 18 months of age and scores were assigned blindly. Two-thirds of these infants had hypoglycemia for a period of 3–30 days. These scores were correlated with their glucose levels in the newborn period. They noted the highest regression coefficient at a plasma concentration of less than 2.6 mmol/L, and if these glucose levels remained low for 5 or more consecutive or separate days, the scores were less than 70, indicating higher neurodevelopmental deficits. On the contrary, a 15-year follow-up of a cohort of less-than-32-week gestation preterm infants with frequent hypoglycemic episodes in the first 10 days of life with matched controls by Tin et al did not provide valid data on the neurodevelopmental outcome.57
Causes of neonatal hypoglycemia
Neonatal hypoglycemia can be transient or persistent. The most common cause of transient hypoglycemia is failure of metabolic adaptation in the “at-risk” infant during the period of establishment of feeding (Table 1). Transient forms can follow perinatal stress/asphyxia or sepsis, or be due to transient hyperinsulinism in infants of diabetic mothers (IDM) and SGA (birth weight <10th percentile for the gestational age) infants. In perinatal asphyxia, increasing metabolic and energy demands is achieved transiently by increasing glucose production through glycogenolysis.58 Due to hypoxia, the infant switches to anaerobic metabolism, resulting in rapid exhaustion of glycogen stores, hypoglycemia, and lactic academia.
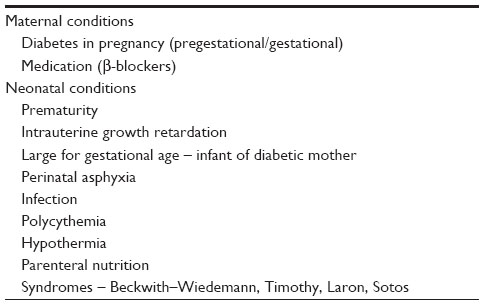 | Table 1 Infants at risk of hypoglycemia |
Hyperinsulinism has also been reported as a cause of hypoglycemia in perinatal asphyxia.59 In sepsis, the hypoglycemia results from an imbalance between the increasing demand and the supply of glucose as the energy substrate. Infants with polycythemia often present with hypoglycemia. It is thought to be due either to increased glucose consumption by the increased red cell mass, or reduced plasma volume and, therefore, decreasing glucose carrying capacity.60 The pathogenesis of hypoglycemia in IDM is multifactorial. It might be due to fetal hyperglycemia due to poor maternal glycemic profiles in pregnancy leading to increased fetal pancreatic β-cell function causing hyperinsulinism.61 Abrupt cessation of maternal glucose supply in the background of hyperinsulinism in IDM leads to hypoglycemia. But the question still remains as to why the IDM who had tightly controlled diabetes, as shown by normal HbA1c levels, have hypoglycemia.62 This is attributed to unrecorded HbA1c levels while having intermittent hyperglycemia in pregnancy.63 Defective counter-regulatory hormonal regulation and lower endogenous glucose production has been implicated in the pathogenesis of hypoglycemia in IDM.64,65 In addition, excessive maternal glucose infusion during the intrapartum period can lead to hypoglycemia in the immediate postnatal period due to increased insulin secretion and blunting of the glycogenolytic and gluconeogenic response of the newborn infant.66 Transient neonatal hypoglycemia often resolves in a few days.58 The metabolic and endocrine causes of neonatal hypoglycemia are summarized in Table 2.27
 | Table 2 Metabolic and endocrine causes of neonatal hypoglycemia |
Prolonged HH is a recognized entity in SGA infants.67 These infants respond well to therapy with diazoxide and a complete resolution occurs over a few months.58 The etiology of prolonged HH in SGA infants could be due to a lack of exogenous substrate supply, depletion of hepatic glycogen stores, defective gluconeogenesis, hyperinsulinism and increased sensitivity to insulin, or adrenocortical insufficiency.67 The possible genetic etiology of prolonged HH due to growth restriction in SGA infants is still unclear.68
Congenital hyperinsulinism (CHI) is the most common cause for persistent or recurrent hypoglycemia.58 CHI is a rare genetic disorder characterized by inappropriate and unregulated insulin secretion in the face of severe hypoglycemia. Mutations in the nine different key genes (ABCC8, KCNJ11, GLUD1, GCK, HADH, SLC16A1, HNF4A, HNF1A, and UCP2) regulating insulin secretion have been identified in patients with CHI. Pancreatic β-cell glucose metabolism raises the cytosolic adenosine triphosphate (ATP):adenosine diphosphate ratio, which closes the ATP sensitive KATP channel (heterooctameric complex composed of sulfonylurea receptor [SUR1] and potassium channel pore-forming [Kir6.2] subunits, encoded by ABCC8 and KCNJ11 genes) leading to cell membrane depolarization and influx of Ca+ into the cell via voltage gated calcium channels. This increase in intracellular calcium triggers insulin release from the β-cells.58,69 Focal and diffuse forms are the two histological types of CHI. Diffuse forms of CHI are most commonly due to recessive mutations in ABCC8/KCNJ11, whereas focal CHI is due to a paternally inherited ABCC8/KCNJ11 mutation and somatic loss of heterozygosity for the 11p allele inside the focal lesion.69
Hyperinsulinism–hyperammonemia syndrome is the second most common form of CHI caused by activating missense mutations in the GLUD1 gene, which encodes glutamate dehydrogenase enzyme.70 Patients usually present with hypoglycemia both while fasting and following protein meals. Mutations in the mitochondrial HADH gene (which encodes the enzyme 3-hydroxyacyl-coenzyme A dehydrogenase (SCHAD)) causes protein induced hyperinsulinaemic hypoglycemia. HADH catalyzes the penultimate reaction of the fatty acid β-oxidation cycle.69 The enzymes glutamate dehydrogenase and SCHAD have a direct protein–protein interaction, which is lost in patients with HADH gene mutation and can lead to severe neonatal or mild late-onset HH.70 HNF4α, HNF1α, and GCK gene mutations cause both CHI and maturity-onset diabetes of the young.58,69
Clinical manifestations
The symptoms of neonatal hypoglycemia are often nonspecific and a high level of suspicion is necessary. Symptoms such as change in neurologic behavior, exaggerated Moro reflex, irritability, jitteriness, tremors, high-pitched cry, seizures, lethargy, or poor feeding should arouse suspicions of hypoglycemia. Other symptoms include cyanosis, hypothermia, apnea, and floppiness.58,71
Diagnosis
A detailed history related to pregnancy (diabetes/diet/insulin), delivery (asphyxia), gestational age (SGA, large for gestational age [LGA]), and birth weight (low birth weight/macrosomia) is essential. Family history of diabetes (gestational/pregestational) may point toward HNF4α mutations and parental consanguinity might point toward a recessively inherited cause of the hypoglycemia. History of siblings having infantile seizures may indicate hypoglycemia as a cause for the disease.72,73
Physical examination of the infant is valuable. Late preterm (34–36+6 weeks), LGA, and SGA infants are at high risk for hypoglycemia. Macrosomia, macroglossia, ear pits, hemihypertrophy, and omphalocele may suggest Beckwith–Wiedemann syndrome, a condition with uniparental disomy of chromosome 11, with a high predisposition for hyperinsulinism. Midline defects, micropenis, and hypoglycemia point to congenital hypopituitarism.74 Hypoglycemia, dehydration, and shock in the presence of ambiguous genitalia point to a diagnosis of congenital adrenal hyperplasia.58 Sudden cardiorespiratory collapse, acidosis, and hypoglycemia following feeds in an otherwise healthy neonate indicate a metabolic disorder.58,73
Investigations
Screening high-risk neonates
Reagent test strips are routinely used in bedside screening for glycemic status. These strips are not reliable at low glucose levels and hence laboratory plasma glucose levels should be obtained.75 Because of the high water content, plasma glucose values are 10%–18% higher than whole blood glucose values.76 A recent systematic review on accuracy of blood glucose measurements using arterial blood gas analyzers observed higher blood sugar values with than with glucose meters using capillary or arterial blood samples.77 In “at-risk” infants, continuous interstitial glucose monitoring was found to be safe and reliable to detect more low glucose levels than blood glucose measurement. Blood glucose concentrations can change rapidly in the immediate neonatal period and, due to delay in glucose diffusion from blood into interstitial space, the glucose concentrations in the latter can lag behind that of blood by as long as 20 minutes. Harris et al showed a good correlation between continuous interstitial and blood glucose measurements and concluded that continuous interstitial glucose monitoring can potentially be advantageous in measuring the duration, severity, and frequency of low glucose concentrations in high-risk infants.78
Screening and management strategies
Evidence for optimal timing and intervals to screen the “at-risk” infants is limited.78 In healthy term infants, neonatal glucose levels fall rapidly after birth, reaching a nadir at 1 hour of age and then stabilizing by about 3 hours of age.37 The value of screening these infants during this period is limited unless they are symptomatic. In a review of neonatal hypoglycemia by the World Health Organization (1997), it was recommended to screen “at-risk” infants at 4–6 hours of life as no study has demonstrated any harm from a few hours of asymptomatic hypoglycemia.47 The Canadian Pediatric Society guidelines (2004) recommended that screening of “at-risk” infants be initiated at 2 hours of age (after an initial feed) and should be continued until the period of risk (36 hours) is considered over (level 5 evidence: expert opinion).79
The frequency and duration of blood glucose monitoring should be decided in relation to the risk factors specific to the individual case. Most often, the “at-risk” groups include IDM, SGA, and late-preterm infants. It remains controversial whether to screen LGA infants born to mothers with no history of diabetes, as it is difficult to rule out diabetes or prediabetic states in pregnancy using the standard glucose tolerance test.80 Any infant having symptoms suggestive of hypoglycemia must be screened and confirmed with plasma glucose as soon as possible. Asymptomatic hypoglycemia may develop in IDM by as early as 1 hour of life, and in LGA and SGA infants by 3 hours of life.76 A clinical report of the American Academy of Pediatrics76 recommended screening of “at-risk” infants within the first hours of birth. Late-preterm and SGA infants should be fed every 2–3 hours with monitoring of pre-feed blood glucose levels for multiple feed–fast cycles for at least 24 hours, and this should be continued only if plasma glucose levels remain below 2.5 mmol/L. In symptomatic infants, a blood glucose value less than 2.2 mmol/L warrants parenteral glucose infusion to maintain the glucose levels above 2.5 mmol/L.76 However, in asymptomatic infants, a practical approach based on mode of feeding, risk factors, and age can be considered when treatment is planned. Glucose monitoring should be continued for 12 hours in macrosomic IDM and for 24 hours in late-preterm infants.76
Transient hypoglycemia in the immediate newborn period is common, which usually resolves with feeding or intravenous dextrose. For infants with intractable hypoglycemia, appropriate samples for the hormones and intermediary metabolites should be collected during an episode of hypoglycemia (Table 3).81
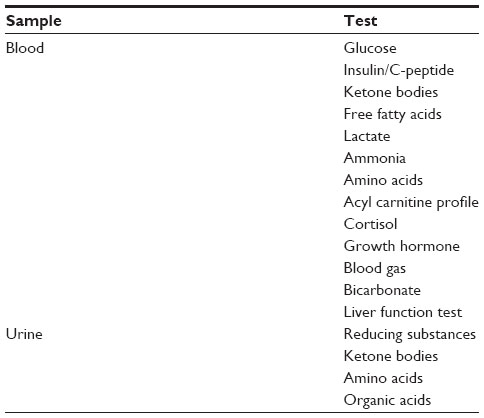 | Table 3 Investigations for cases of severe/persistent hypoglycemia |
Early establishment of feeding and maintaining normothermia is essential. Feed volume and frequency can be increased as tolerated in asymptomatic infants, if they are able to tolerate oral feeds, with monitoring of blood glucose.75 Gavage feeding may be alternatively considered in infants who fail nippling.76 A recent report by Mola-Schenzle et al showed the occurrence of low interstitial blood glucose levels exceeding values lower than 2.5 mmol/L in asymptomatic, stable, healthy, developing, very-low-birth-weight infants.82 Even though the clinical significance of these low glucose values is unclear, these infants should also be considered as high-risk.
Buccal administration of 40% dextrose gel has been practiced in some centers as the initial management of hypoglycemia as it is simple, safe, effective, and allows mother and baby to be together and establish breast-feeding early. Harris et al have recommended oral dextrose gel as initial management in term and late preterm infants in the first 48 hours of life.83 A recent randomized, double-blind, placebo-controlled study by Harris et al found that buccal administration of 40% dextrose gel compared to placebo in term and late-preterm “at-risk” infants with hypoglycemia is effective in reducing the number of treatment failures without any serious adverse events.83
Hypoglycemia is a common neonatal emergency. Treatment of symptomatic infants should not be delayed. In symptomatic infants with severe hypoglycemia, the blood glucose level must be corrected with “minibolus” of intravenous glucose (2 mL/kg of 10% dextrose) followed by gradually increasing the glucose infusion rate, as needed.76 A glucose infusion rate of >8 mg/kg/min warrants further investigations for HH. A central line access should always be secured early, as the concentration of infused glucose might need to be increased.84 If hypopituitarism is suspected due to midline defects or biochemical workup suggests growth hormone deficiency, these infants should be thoroughly investigated for midline defects/hypopituitarism while the low blood glucose level is appropriately corrected. Today, an increasing number of countries screen newborn infants for inborn error of metabolism, the results of which may help to rule out metabolic disorder as a cause of hypoglycemia.73
Once the diagnosis of HH is confirmed (detectable insulin/C-peptide in the face of hypoglycemia, hypoketonemia, hypofattyacidemia, or glycemic response to glucagon in an infant receiving a glucose infusion rate of more than 8 mg/kg/min), diazoxide is the drug of choice (5–20 mg/kg/day given orally).69 A thiazide diuretic (7–10 mg/kg/day) is combined to combat the fluid retention side effect of diazoxide.58 Diazoxide, being a KATP channel agonist, keeps the channel open and prevents depolarization of the β-cell and thereby insulin secretion. In diazoxide unresponsive cases, intravenous glucagon (5–10 μg/kg/hr) and/or octreotide (5–30 μg/kg/day) can be used to stabilize the blood glucose levels in addition to high concentrations of dextrose infusion. Long acting (once-a-month) somatostatin analogue, lanreotide acetate, is a safe alternative to octreotide pump therapy. Nifedipine, being a calcium channel antagonist, has been tried with variable response in the literature.58,69
If the patient is unresponsive to diazoxide, genetic studies should be done to look for the common genetic mutations in ABCC8 and KCNJ11 genes.69 Once the genetic study gives an indication of the type of disease, an [18F] fluoro-L-DOPA positron emission tomography/computed tomography scan can be done to localize the focal lesion, which is curable by lesionectomy.58 In diffuse disease, the treatment option is still a near total pancreatectomy (95%–98%) with the morbidity of diabetes mellitus and exocrine pancreatic insufficiency in future (Figure 2).69 Preliminary reports on the use of sirolimus, a mammalian target of rapamycin inhibitor, in diffuse forms of HH is promising with clear glycemic response in four patients who were unresponsive to high doses of diazoxide and octreotide.85
 | Figure 2 Outline of the management of congenital hyperinsulinism. |
Neuroprotection during hypoglycemia
Hypoglycemia in newborn infants is a surrogate marker of neuronal energy deficiency. During the periods of hypoglycemia, several neuroprotective mechanisms are believed to play a role in guarding against neuronal injury.86 Substitution of alternative cerebral substrates – lactate, ketone bodies, pyruvate, amino acids, free fatty acids, and glycerol – in the face of low blood glucose levels is the first mechanism to prevent neuronal injury. These alternative energy fuels are transported across the blood–brain barrier by monocarboxylate glycoprotein transporters (MCT 1 and 2).86 Ketone bodies are low until milk feeds are established.
Healthy term infants can mount the counter-regulatory ketogenic response whereas preterm and SGA infants fail to trigger an adequate response.52,86 During episodes of HH, the ketone bodies and free fatty acids are low or undetectable, thereby nonavailability of alternative energy fuels leads to brain injury. Secondly, an increase in serum epinephrine concentration and the cerebral blood flow has been reported during hypoglycemic episodes.52,87,88 Thirdly, it has been postulated that an immature brain normally needs decreased cerebral energy fuels in the newborn period compared to a child or adult. As the brain matures, the need for energy fuels increases.86,87 Lastly, when hypoglycemic, the limited glycogen stores in astrocytes provide an immediate supply of glucose to the neurons.52,87 These mechanisms can protect the brain for limited periods but if recurrent and persistent hypoglycemia prevails, these protective mechanisms fail, resulting in permanent neuronal injury. Moreover, recurrent episodes of insulin-induced hypoglycemia have been shown to blunt or completely prevent the secretion of counter-regulatory hormones, termed as “hypoglycaemia-associated autonomic failure”.89,90
What is known about hypoglycemic brain injury?
Large scale prospective randomized control trials have not been performed to establish the safe range of blood glucose levels for newborn infants. Many infants who are hypoglycemic do not exhibit clinical manifestations, while others are symptomatic and at increased risk for the occurrence of permanent brain damage.86 No uniform diagnostic criteria for newborn hypoglycemic brain injury (NHBI) has been available.91 Wang et al has proposed the following diagnostic criteria for NHBI, which includes documented biochemical neonatal hypoglycemia: clinical symptoms such as paroxysmal cyanosis, tremors, convulsions, apnea, and decreased responsiveness; exclusion of other disorders (eg, hypoxic-ischemic encephalopathy and infectious diseases) that may cause brain injury; and confirmatory findings on brain imaging.92
Several cohort studies have reported that persistent or recurrent hypoglycemia may lead to long-term neurodevelopmental sequelae.53,56,93–97 Recurrent hypoglycemia (<2.6 mmol/L) was found to be associated with brain injury in preterm infants. In preterm infants with recurrent hypoglycemia for 5 or more days, Lucas et al found an increased risk of cerebral palsy, developmental delay, and low mental scores at 18 months of age.56 The study by Duvanel et al reported reduced head circumference and lower psychomotor scores at 3.5 and 5 years of age in preterm babies who had recurrent hypoglycemia.94
In term infants, even asymptomatic hypoglycemia (2.6 mmol/L) led to abnormal evoked potentials.53 IDM born at term with severe hypoglycemia (<1.5 mmol/L) had magnetic resonance imaging (MRI) findings of occipital cerebral injury between 4 and 7 days.95 Stenninger et al reported increased incidence of minimal brain dysfunction and deficits in attention, motor control, and perception at 8 years of age in IDM who had asymptomatic hypoglycemia.93 Therefore, aggressive treatment needs to be considered even in infants with asymptomatic hypoglycemia. A 3.7-fold increase in corticospinal tract injury was reported at 1 year of age in term infants at risk of neonatal encephalopathy who had recurrent hypoglycemic episodes (<2.6 mmol/L).97 The risk factors, the threshold value for blood glucose levels at which NHBI occurred, and the neurodevelopmental sequelae are summarized in Table 4.53,56,93–97
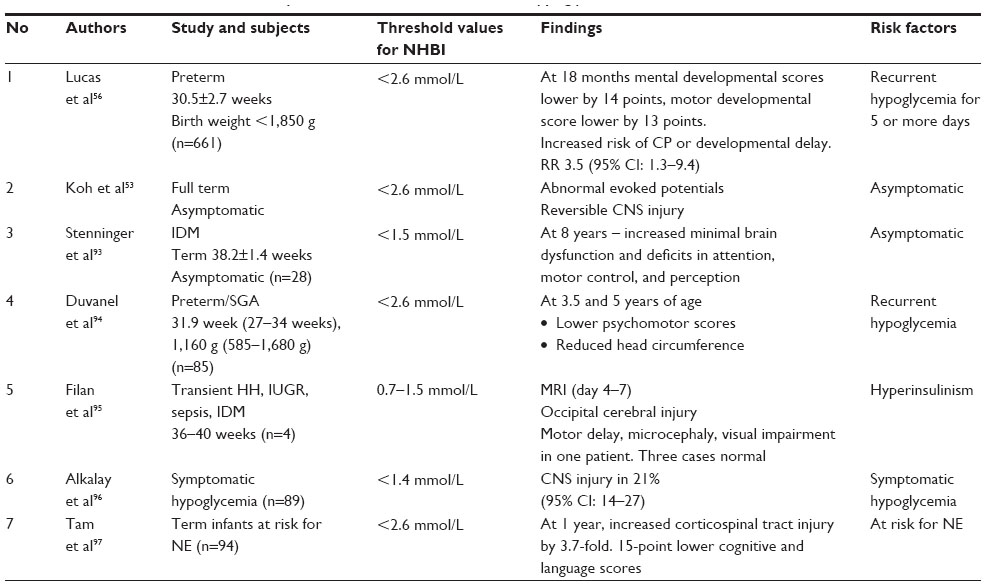 | Table 4 Risk factors and neurodevelopmental outcome of infants with hypoglycemia |
A systemic review of 18 studies by Boluyt et al could not find conclusive evidence on abnormal neurodevelopmental outcome in infants who had hypoglycemia in the 1st week of life. Of the 18 studies on the basis of methodological quality, the authors found only two of them of high quality and hence recommended an optimal future study.98
The NHBI depends on the duration of hypoglycemia rather than its severity.91 A study of neonatal rhesus monkeys by Schrier et al seems to favor this concept. They found that a duration of 6.5 hours of hypoglycemia was not associated with cognitive or behavioral deficits, but 10 hours resulted in motivational and adaptability problems at 8 months of age.99 Recurrent or protracted hypoglycemia seems to be a more sinister risk factor rather than a single low value of blood sugar.56,94 The neuronal injury is also influenced by the existence of other comorbidities such as hypoxic ischemia, prematurity, and SGA.56,94,97 Hypoglycemia combined with hypoxic ischemia has been more deleterious to the immature brain than either condition alone.86 Experimental studies have revealed that hypoglycemia before hypoxic ischemia can further exacerbate brain damage whereas hyperglycemia can significantly reduce it.100,101
Udani et al have reported that neonatal hypoglycemia was the most common etiology for remote symptomatic epilepsy with onset in the first 3 years of life, and infantile spasms was the most common seizure type.102 A study by Kumaran et al revealed that neonatal HH of varying severity is associated with development of late-onset infantile spasms. As the latent periods before the onset of infant spasms is variable, long-term neurodevelopmental follow-up is recommended.103
Pathological changes of NHBI
Swelling of the neuronal and glial cells, necrosis, gyrus atrophy, and white matter demyelination have been observed in association with NHBI.91,104 In some cases, changes due to hypoxic injury may coexist along with the changes that are due to severe hypoglycemia. However, the distribution of pathological changes is distinctly different in both the disorders. NHBI does not follow vascular territory and bilateral posterior parieto-occipital regions are most commonly involved in term as well as preterm infants.91
Diffuse white matter involvement particularly in the watershed areas is more common with hypoxic ischemia, whereas cortical injury is more commonly seen in NHBI. In NHBI, the cerebellum and brainstem are often spared and hemorrhagic lesions are not present. Rarely, abnormal changes have been observed in the deeper gray matter nuclei (basal ganglia, thalami), internal capsule, splenium of the corpus callosum, and the corona radiata.91,104,105
Neuroimaging findings
Cranial ultrasound and computed tomography scans of the brain lack sensitivity and specificity, and have been replaced by MRI for diagnosing NHBI.106 In order to detect acute manifestations, MRI should be performed between 3 and 7 days after the hypoglycemic insult, and the study should include diffusion weighted imaging (DWI).105,107 DWI plays an essential role in the early diagnosis and prognosis of NHBI. Such images done within 7 days revealed marked parieto-occipital hyperintensity signals which were not detected in the T1 and T2 weighted images of conventional MRI scans.91,106 In a study, Tam et al have demonstrated pathological DWI changes that correlated well with abnormal performance of visual evoked potentials, when the investigations were done within 7 days of the initial episode of hypoglycemia.107 Magnetic resonance spectroscopy studies following the acute phase of hypoglycemia have revealed increased lactate and free fatty acid peaks, reduced acetyl aspartic acid peaks, and altered ATP/lactic acid ratio indicating altered brain function.108
How hypoglycemia leads to brain damage
It is unquestionably clear that hypoglycemia causes neonatal encephalopathy and may result in permanent brain injury. However, the specific mechanisms responsible for the brain injury remain poorly defined. The exact reason for the vulnerability of the occipital and parietal lobes to hypoglycemic injury is not clear. Animal work has suggested abnormal anatomy of the circle of Willis and variations in regional blood flow and glucose utilization during hypoglycemia.109 Increased blood flow to the occipital lobes has been demonstrated during hypoglycemia.52,110 There was also a significant reduction of regional cerebral glucose use in the occipital lobes, which may render these regions more vulnerable.90,110
Other causative factors predisposing to occipital injury could be intensive neuronal migration and synaptogenesis occurring in the neonatal period.111,112 As compared against other areas of the cerebral cortex, the fourth lamina of the visual cortex is thicker with more neurons and synapses, thus requiring more energy and also being more susceptible to damage.111 Occipital lobes have well-developed excitatory amino acid receptors, which are excited by high levels of aspartate during hypoglycemia. Thus, excitatory amino acids play a key role in NHBI.113 Increased activation of the N-methyl-D-aspartate receptors due to excitatory amino acids induces excessive influx of sodium and calcium ions into the cells and alters the transmembrane ion gradient.114
Oxygen free radical injury could be another etiopathogenetic mechanism in NHBI. Hypoglycemia results in a lack of energy substrate such as ATP and creatine phosphate, impairing the normal transmembrane concentration of gradient recovery for sodium and calcium ions. This results in excessive calcium influx into the cells and the cellular phosphatase and protease are activated. This alters the mitochondrial metabolism and triggers the formation of free radicals contributing to necrosis of neurons.91,105,108,114 McGowan et al have shown significantly higher superoxide and hydrogen peroxide generation in mitochondria compared against normoglycemic controls in piglets after 2 hours of hypoglycemia.108 The increased mitochondrial reactive oxygen species production could result in alterations in brain structure and function due to oxidant injury to mitochondrial proteins and DNA or changes in oxidant-sensitive signal transduction pathways in the brain.108
Additional contributing factors could be due to a regional developmental deficit in expression or function of the glucose membrane transporter proteins.112
Conclusion
Hypoglycemia remains the most common biochemical disorder in neonates, whilst there is still no consensus on the definition, diagnosis, and treatment. In infants born at term, transient low blood glucose levels are often noted during the metabolic transition to the extra uterine environment. The lower level of blood glucose in a neonate, which would impose the risk of neuronal injury and long-term neurodevelopmental outcome, remains controversial. Screening the “at-risk” infants (such as SGA infants and IDM), early breast feeding, monitoring pre-feed glucose levels while on feeds, treating symptomatic infants with low glucose levels, and identifying and aggressively managing the persistent hyperinsulinemic infants can prevent neuronal injury. Hypoglycemia associated with elevated levels of ketone bodies and lactate is less damaging to the brain when compared to HH, where the infant has hypoketonemia and hypofattyacidemia, the key metabolic milieu for neuroglycopenic brain injury.
Disclosure
The authors report no conflicts of interest in this work.
References
Hay WW Jr, Sparks JW. Placental, fetal, and neonatal carbohydrate metabolism. Clin Obstet Gynecol. 1985;28(3):473–485. | |
Diderholm B, Stridsberg M, Ewald U, Lindeberg-Nordén S, Gustafsson J. Increased lipolysis in non-obese pregnant women studied in the third trimester. BJOG. 2005;112:713–718. | |
Ward Platt M, Deshpande S. Metabolic adaptation at birth. Semin Fetal Neonatal Med. 2005;10(4):341–350. | |
Moe AJ. Placental amino acid transport. Am J Physiol. 1995;268(6 Pt 1):C1321–C1331. | |
Capková A, Jirásek JE. Glycogen reserves in organs of human fetuses in the first half of pregnancy. Biol Neonate. 1968;13:129–142. | |
Shelly HJ. Glycogen reserves and their changes at birth and in anoxia. Br Med Bull. 1961;17:137–143. | |
Adam PA, Räihä N, Rahiala EL, Kekomäki M. Oxidation of glucose and D- B-OH-butyrate by the early human fetal brain. Acta Paediatr Scand. 1975;64(1):17–24. | |
Hawdon J. Glucose homeostasis in the healthy fetus and neonate. In: Rennie JM, editor. Metabolic and Endocrine Disorders. Textbook of Neonatology. London: Elsevier, Churchill Livingstone; 2005:851–852. | |
Obenshain SS, Adam PA, King KC, et al. Human fetal insulin response to sustained maternal hyperglycemia. N Engl J Med. 1970;283(11):566–570. | |
Sperling MA, Grajwer LA, Leake R, Fisher DA. Role of glucagon in perinatal glucose homeostasis. Metabolism. 1976;25(11 Suppl 1):1385–1386. | |
Sperling MA, Ganguli S, Leslie N, Landt K. Fetal-perinatal catecholamine secretion: role in perinatal glucose homeostasis. Am J Physiol. 1984;247:E69–E74. | |
Soothill PW, Nicolaides KH, Campbell S. Perinatal asphyxia, hyperlacticaemia, hypoglycaemia, and erythroblastosis in growth retarded fetuses. Br Med J (Clin Res Ed). 1987;294(6579):1051–1053. | |
Chobot A, Otto-Buczkowska E. Glucose homeostasis from foetus through childhood. Diabetologia Doświadczalna i Kliniczna. 2011; 11(1):29–38. | |
Van Assche FA, Aerts L. The fetal endocrine pancreas. Contrib Gynecol Obstet. 1979;5:44–57. | |
Piper K, Brickwood S, Turnpenny LW, et al. Beta cell differentiation during early human pancreas development. Journal of Endocrinology. 2004;181:11–23. | |
Bonwens L, Lu WG, De Krijgar R. Proliferation and differentiation in the human fetal endocrine pancreas. Diabetologia. 1997;40:398–404. | |
Robb P. The development of the islets of Langerhans in the human foetus. Q J Exp Physiol Cogn Med Sci. 1961;46:335–343. | |
Fowden AL. Pancreatic endocrine function and carbohydrate metabolism in the fetus. In: Abrecht E, Pepe GJ, editors. Research in Perinatal Medicine IV. Ithaca, NY: Perinatology Press; 1985:71–90. | |
Fowden AL. Endocrine regulation of fetal growth. Reprod Fertil Dev. 1995;7:351–363. | |
Weaver LT, Laker MF, Nelson R. Neonatal intestinal lactase activity. Arch Dis Child. 1986;61;896–899. | |
Boellner SW, Beard AG, Panos TC. Impairment of intestinal hydrolysis of lactose in newborn infants. Pediatrics. 1965;36(4):542–550. | |
Mayne A, Hughes CA, Sule D, Brown GA, McNeish AS. Development of intestinal disaccharidases in preterm infants. Lancet. 1983;2(8350):622–623. | |
Mericq V, Ong KK, Bazaes R, et al. Longitudinal changes in insulin sensitivity and secretion from birth to age three years in small- and appropriate-for-gestational-age children. Diabetologia. 2005;48:2609–2614. | |
Hägnevik K, Faxelius G, Irestedt L, Lagercrantz H, Lundell B, Persson B. Catecholamine surge and metabolic adaptation in the newborn after vaginal delivery and caesarean section. Acta Paediatr Scand. 1984;73(5):602–609. | |
Girard J. Gluconeogenesis in late fetal and early neonatal life. Biol Neonate. 1986;50:237–258. | |
Bier DM, Leake RD, Haymond MW, et al. Measurement of “true” glucose production rates in infancy and childhood with 6,6-dideuteroglucose. Diabetes. 1977;26:1016–1023. | |
Chandran S, Yap F, Hussain K. Genetic Disorders Leading to Hypoglycaemia. J Genet Syndr Gene Ther. 2013;4:192. | |
Medina JM, Fernandez E, Bolaros JP, Vicario C, Arizmendi L. Fuel supply to the brain during the early postnatal period. In: Cueza JM, Pasaud-Leone AM, Patel MS, editors. Endocrine development of the Fetus and Neonate. New York: Plenum Press; 1990:175–194. | |
Gustafsson J. Neonatal energy substrate production. Indian J Med Res. 2009;130:618–623. | |
Diderholm B, Ewald U, Ahlsson F, Gustafsson J. Energy substrate production in infants born small for gestational age. Acta Paediatr. 2007;96:29–34. | |
Marcus C, Ehrén H, Bolme P, Arner P. Regulation of lipolysis during the neonatal period. Importance of thyrotropin. J Clin Invest. 1988;82(5):1793–1797. | |
Marchini G, Persson B, Jonsson N, Marcus C. Influence of body temperature on thyrotropic hormone release and lipolysis in the newborn infant. Acta Paediatr. 1995;84(11):1284–1288. | |
Hawdon JM, Ward Platt MP, Aynsley-Green A. Patterns of metabolic adaptation for preterm and term infants in the first neonatal week. Arch Dis Child. 1992;67:357–365. | |
Aynsley-green A. Metabolic and endocrine relationships in the human fetus and neonate: an overview the control of adaptation to postnatal nutrition. In: Linblad BA, editor. Perinatal Nutrition. New York: Academic press; 1988:162–191. | |
Lucas A, Aynsley Green A, Bloom SR. Gut hormones and the first meals. Clin Sci (Lond). 1981;60:349–353. | |
Heck LJ, Erenberg A. Serum glucose levels in term neonates during the first 48 hours of life. J Pediatr. 1987;110(1):119–122. | |
Srinivasan G, Pildes RS, Cattamanchi G, Voora S, Lilien LD. Plasma glucose values in normal neonates: a new look. J Pediatr. 1986; 109(1):114–117. | |
Cowett RM, Farrag HM. Neonatal glucose homeostasis. In: Cowett RM, editor. Principles of Perinatal Neonatal Metabolism. 2nd ed. New York: Springer Verlag; 1998:683–722. | |
Sperling MA, DeLamater PV, Phelps D, Fiser RH, Oh W, Fisher DA. Spontaneous and amino acid-stimulated glucagon secretion in the immediate postnatal period. Relation to glucose and insulin. J Clin Invest. 1974;53(4):1159–1166. | |
Padbury JF, Roberman B, Oddie TH, Hobel CJ, Fisher DA. Fetal catecholamine release in response to labor and delivery. Obstet Gynecol. 1982;60(5):607–611. | |
Lang TF, Hussain K. Pediatric hypoglycemia. Adv Clin Chem. 2014;63:211–245. | |
Kalhan SC. Metabolism of glucose and methods of investigation in the fetus and the newborn. In: Polin RA, Fox WW, Abman SH, editors. Fetal and Neonatal Physiology. Philadelphia: Saunders/Elsevier; 2004:449–464. | |
Cowett RM, Farrag HM. Selected principles of perinatal-neonatal glucose metabolism. Semin Neonatol. 2004;9(1):37–47. | |
Cornblath M, Hawdon JM, Williams AF, et al. Controversies Regarding Definition of Neonatal Hypoglycemia: Suggested Operational Thresholds. Pediatrics. 2000;105(5):1141–1145. | |
Cornblath M, Reisner SH. Blood glucose in the neonate and its clinical significance. N Engl J Med. 1965;273:378–381. | |
Hussain K, Blankenstein O, De Lonlay P, Christesen HT. Hyperinsulinaemic hypoglycaemia: biochemical basis and the importance of maintaining normoglycaemia during management. Arch Dis Child. 2007;92(7):568–570. | |
Williams AF. Hypoglycaemia of the newborn: a review. Bull World Health Organ. 1997;75:261–290. | |
Aynsley-Green A, Hawdon JM. Hypoglycaemia in the neonate: current controversies. Acta Paediatr Jpn. 1997;39 Suppl 1:S12–S16. | |
Whipple AO, Frantz VK. Adenoma of Islet Cells with Hyperinsulinism: A Review. Ann Surg. 1935;101(6):1299–1335. | |
Cornblath M, Ichord R. Hypoglycaemia in the neonate. Semin Perinatol. 2000;24:136–149. | |
Tin W. Defining neonatal hypoglycaemia: a continuing debate. Semin Fetal Neonatal Med. 2014;19(1):27–32. | |
Pyrds O, Christensen NJ, Friis-Hansen B. Increased cerebral blood flow and plasma epinephrine in hypoglycaemic, preterm neonates. Pediatrics. 1990;85:172–176. | |
Koh TH, Aynsley-Green A, Tarbit M, Eyre JA. Neural dysfunction during hypoglycaemia. Arch Dis Child. 1988;63:1353–1358. | |
Greisen G, Pryds O. Neonatal hypoglycaemia. Lancet. 1989;1:1332–1333. | |
Pryds O, Greisen G, Friis-Hansen B. Compensatory increase of CBF in preterm infants during hypoglycaemia. Acta Paediatr Scand. 1988;77:632–637. | |
Lucas A, Morley R, Cole TJ. Adverse neurodevelopmental outcome of moderate neonatal hypoglycaemia. BMJ. 1988;297:1304–1308. | |
Tin W, Brunskill G, Kelly T, Fritz S. 15-year follow up of recurrent “hypoglycemia” in preterm infants. Pediatrics. 2012;130:e1497–e1503. | |
Arya VB, Senniappan S, Guemes M, Hussain K. Neonatal Hypoglycemia. Indian J Pediatr. 2014;81(1):58–65. | |
Collins JE, Leonard JV. Hyperinsulinism in asphyxiated and small-for-dates infants with hypoglycaemia. Lancet. 1984;2(8398):311–313. | |
Rosenkrantz TS, Philipps AF, Skrzypczak PS, Raye JR. Cerebral metabolism in the newborn lamb with polycythemia. Pediatr Res. 1988;23:329–333. | |
Nold JL, Georgieff MK. Infants of diabetic mothers. Pediatr Clin North Am. 2004;51(3):619–637. | |
Taylor R, Lee C, Kyne-grzebalski D, Marshall SM, Davison JM. Clinical outcomes of pregnancy in women with type 1 diabetes(1). Obstet Gynecol. 2002;99:537–541. | |
Kyne-Grzebalski D, Wood L, Marshall SM, Taylor R. Episodic hyperglycaemia in pregnant women with well-controlled Type 1 diabetes mellitus: a major potential factor underlying macrosomia. Diabet Med. 1999;16:702–706. | |
Bloom SR, Johnston DI. Failure of glucagon release in infants of diabetic mothers. Br Med J. 1972;4:453–454. | |
Kalhan SC, Savin SM, Adam PA. Atteunated glucose production rate in newborn infants of insulin dependent diabetic mothers. N Engl J Med. 1977;296:375–376. | |
Sunehag A, Ewald U, Larsson A, Gustafsson J. Attenuated hepatic glucose production but unimpaired lipolysis in newborn infants of mothers with diabetes. Pediatr Res. 1997;42:492–497. | |
Chong JH, Chandran S, Agarwal P, Rajadurai VS. Delayed presentation of prolonged hyperinsulinaemic hypoglycaemia in a preterm small-for-gestational age neonate. BMJ Case Rep. 2013;2013. | |
Arya VB, Flanagan SE, Kumaran A, et al. Clinical and molecular characterisation of hyperinsulinaemic hypoglycaemia in infants born small-for-gestational age. Arch Dis Child Fetal Neonatal Ed. 2013;98(4):F356–F358. | |
Senniappan S, Arya VB, Hussain K. The molecular mechanisms, diagnosis and management of congenital hyperinsulinism. Indian J Endocrinol Metab. 2013;17(1):19–30. | |
Chandran S, Yap F, Hussain K. Molecular mechanisms of protein induced hyperinsulinaemic hypoglycaemia. World J Diabetes. 2014 Oct 15;5(5):666–77. | |
Vanhaltren K, Malhotra A. Characteristics of infants at risk of hypoglycaemia secondary to being ‘infant of a diabetic mother’. J Pediatr Endocrinol Metab. 2013;26(9–10):861–865. | |
Aynsley-Green A, Hussain K, Hall J, et al. Practical management of hyperinsulinism in infancy. Arch Dis Child Fetal Neonatal Ed. 2000;82:F98–F107. | |
Hussain K. Investigations for neonatal hypoglycaemia. Clin Biochem. 2011;44:465–466. | |
Munns CF, Batch JA. Hyperinsulinism and Beckwith-Wiedemann syndrome. Arch Dis Child Fetal Neonatal Ed. 2001;84(1):F67–F69. | |
Deshpande S, Ward Platt M. The investigation and management of neonatal hypoglycaemia. Semin Fetal Neonatal Med. 2005;10:351–361. | |
Committee on Fetus and Newborn, Adamkin DH. Postnatal glucose homeostasis in late-preterm and term infants. Pediatrics. 2011;127(3):575–579. | |
Inoue S, Egi M, Kotani J, Morita K. Accuracy of blood-glucose measurements using glucose meters and arterial blood gas analyzers in critically ill adult patients: systematic review. Crit Care. 2013; 17(2):R48. | |
Harris DL, Battin MR, Weston PJ, Harding JE. Continuous glucose monitoring in newborn babies at risk of hypoglycemia. J Pediatr. 2010;157(2):198–202. | |
No authors listed. Screening guidelines for newborns at risk for low blood glucose. Paediatr Child Health. 2004;9(10):723–740. | |
de Rooy L, Hawdon J. Nutritional factors that affect the postnatal metabolic adaptation of full-term small- and large-for-gestational-age infants. Pediatrics. 2002;109:E42. | |
Cook P, Walker V. Investigation of the child with an acute metabolic disorder. J Clin Pathol. 2011;64:181–191. | |
Mola-Schenzle E, Staffler A, Klemme M, et al. Clinically stable very low birthweight infants are at risk for recurrent tissue glucose fluctuations even after fully established enteral nutrition. Arch Dis Child Fetal Neonatal Ed. Epub November 7, 2014. | |
Harris DL, Weston PJ, Signal M, Chase JG, Harding JE. Dextrose gel for neonatal hypoglycaemia (the Sugar Babies Study):a randomised, double-blind, placebo-controlled trial. Lancet. 2013;382(9910):2077–2083. | |
Peters CJ, Hindmarsh PC. Management of neonatal endocrinopathies – best practice guidelines. Early Hum Dev. 2007;83:553–561. | |
Senniappan S, Alexandrescu S, Tatevian N, et al. Sirolimus therapy in infants with severe hyperinsulinemic hypoglycemia. N Engl J Med. 2014;370(12):1131–1137. | |
Vannucci RC, Vannucci SJ. Hypoglycemic brain injury. Semin Neonatol. 2001;6:147–155. | |
Hawdon JM. Hypoglycemia and the neonatal brain. Eur J Pediatr. 1999;158 Suppl 1:S9–S12. | |
Rozance PJ, Hay WW. Hypoglycemia in newborn infants: Features associated with adverse outcome. Biol Neonate. 2006;90:74–86. | |
Cryer PE. Hypoglycemia-associated autonomic failure in diabetes. Am J Physiol Endocrinol Metab. 2001;281(6):E1115–E1121. | |
Zhou D, Qian J, Liu CX, Chang H, Sun RP. Repetitive and profound insulin-induced hypoglycemia results in brain damage in newborn rats: an approach to establish an animal model of brain injury induced by neonatal hypoglycemia. Eur J Pediatr. 2008;167(10):1169–1174. | |
Su J, Wang L. Research advances in neonatal hypoglycemic brain injury. Translational Pediatrics. 2012;1(2):108–115. | |
Wang L, Fan G, Ji X, Sun B, Guo Q. MRI findings of brain damage due to neonatal hypoglycemia. Zhonghua Fang She Xue Za Zhi. 2009,43:42–45. | |
Stenninger E, Flink R, Eriksson B, SahlÈn C. Long-term neurological dysfunction and neonatal hypoglycaemia after diabetic pregnancy. Arch Dis Child Fetal Neonatal Ed. 1998;79:F174–F179. | |
Duvanel CB, Fawer CL, Cotting J, Hohlfeld P, Matthieu JM. Long-term effects of neonatal hypoglycemia on brain growth and psychomotor development in small-for-gestational-age preterm infants. J Pediatr. 1999;134:492–498. | |
Filan PM, Inder TE, Cameron FJ, Kean MJ, Hunt RW. Neonatal hypoglycemia and occipital cerebral injury. J Pediatr. 2006;148:552–555. | |
Alkalay AL, Flores Sarnat L, Sarnat HB, Farber SJ, Simmons CF. Plasma glucose concentrations in profound neonatal hypoglycemia. Clin Pediatr (Phila). 2006;45:550–558. | |
Tam EW, Haeusslein LA, Bonifacio SL, et al. Hypoglycemia is associated with increased risk for brain injury and adverse neurodevelopmental outcome in neonates at risk for encephalopathy. J Pediatr. 2012;161:88–93. | |
Boluyt N, van Kempen A, Offringa M. Neurodevelopment after neonatal hypoglycemia: a systematic review and design of an optimal future study. Pediatrics. 2006;117(6):2231–2243. | |
Schrier AM, Brady Wilhelm P, Church RM, et al. Neonatal hypoglycemia in the rhesus monkey: Effect on development and behavior. Infant Behav Dev. 1990;13(2):189–207. | |
Yang Y, Chen HJ, Qian LH, et al. Effect of glucose levels on the cerebral synthesis of glucose transporter (GLUT) 3 in neonatal rats with hypoxic ischemia. Lin Chuang Er Ke Za Zhi. 2004;22:42–45. | |
Chen C, Chen HJ, Qian LH, et al. Influence of Different Blood Glucose Levels on Cerebral Pathology under Light Microscopy in Neonatal Rats with Hypoxic-Ischemia. Shi Yong Er Ke Lin Chuang Za Zhi. 2006;21:894–897. | |
Udani V, Munot P, Ursekar M, Gupta S. Neonatal hypoglycemic brain – injury a common cause of infantile onset remote symptomatic epilepsy. Indian Pediatr. 2009;46(2):127–132. | |
Kumaran A, Kar S, Kapoor RR, Hussain K. The clinical problem of hyperinsulinemic hypoglycemia and resultant infantile spasms. Pediatrics. 2010;126(5):e1231–e1236. | |
Burns CM, Rutherford MA, Boardman JP, Cowan FM. Patterns of cerebral injury and neurodevelopmental outcomes after symptomatic neonatal hypoglycemia. Pediatrics. 2008;122:65–74. | |
Kim SY, Goo HW, Lim KH, Kim ST, Kim KS. Neonatal hypoglycaemic encephalopathy: diffusion-weighted imaging and proton MR spectroscopy. Pediatr Radiol. 2006;36:144–148. | |
Yanagawa Y, Isoi N, Tokumaru AM, Sakamoto T, Okada Y. Diffusion-weighted MRI predicts prognosis in severe hypoglycemic encephalopathy. J Clin Neurosci. 2006;13:696–699. | |
Tam EW, Widjaja E, Blaser SI, Macgregor DL, Satodia P, Moore AM. Occipital lobe injury and cortical visual outcomes after neonatal hypoglycemia. Pediatrics. 2008;122(3):507–512. | |
McGowan JE, Chen L, Gao D, Trush M, Wei C. Increased mitochondrial reactive oxygen species production in newborn brain during hypoglycemia. Neurosci Lett. 2006;399:111–114. | |
Alfonso I, Rerecich A. Neonatal hypoglycemia and occipital cerebral injury. J Pediatr. 2007;151:e1–e2. | |
Mujsce DJ, Christensen MA, Vannucci RC. Regional cerebral blood flow and glucose utilization during hypoglycemia in newborn dogs. Am J Physiol. 1989;256:H1659–H1666. | |
Alkalay AL, Sarnat HB, Flores-Sarnat L, Flores-Sarnat L, Simmons CF. Neurologic aspects of neonatal hypoglycemia. Isr Med Assoc J. 2005;7:188–192. | |
Caraballo RH, Sakr D, Mozzi M, et al. Symptomatic occipital lobe epilepsy following neonatal hypoglycemia. Pediatr Neurol. 2004;31:24–29. | |
Barkovich AJ, Ali FA, Rowley HA, Bass N. Imaging patterns of neonatal hypoglycemia. AJNR Am J Neuroradiol. 1998;19:523–528. | |
Straussman S, Levitsky LL. Neonatal hypoglycemia. Curr Opin Endocrinol Diabetes Obes. 2010;17(1):20–24. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.