Back to Archived Journals » Cell Health and Cytoskeleton » Volume 7
Current perspectives on cadherin-cytoskeleton interactions and dynamics
Authors Liang X, Gomez G, Yap A
Received 20 October 2014
Accepted for publication 2 December 2014
Published 5 January 2015 Volume 2015:7 Pages 11—24
DOI https://doi.org/10.2147/CHC.S76107
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Denis Wirtz
Xuan Liang, Guillermo A Gomez, Alpha S Yap
Division of Cell Biology and Molecular Medicine, Institute for Molecular Bioscience, The University of Queensland, Brisbane, Australia
Abstract: Cells are linked together dynamically by adhesion molecules, such as the classical cadherins. E-cadherin, which mediates epithelial cell–cell interactions, plays fundamental roles in tissue organization and is often perturbed in diseases such as cancer. It has long been recognized that the biology of E-cadherin arises from cooperation between adhesion and the actin cytoskeleton.
A major feature is the generation of contractile forces at junctions, yielding patterns of tension that contribute to tissue integrity and patterning. Here we discuss recent developments in understanding how cadherin junctions integrate signaling and cytoskeletal dynamics to sense and generate force.
Keywords: E-cadherin, actomyosin, morphogenesis, cell extrusion, wound closure, mechanobiology
Introduction
Epithelia are fundamental tissues of the body. In post-developmental life, epithelia form the principal barriers between the external environment and the bodies of metazoan organisms. During development, many precursor tissues constitute epithelia. One defining feature of an epithelium is the physical linkage of its constituent cells together to form a mechanically integrated, multicellular structure. To paraphrase John Donne, no cell is an island, as least as far as epithelia are concerned.
The physical coupling of epithelial cells is achieved by cell–cell adhesion. Many different molecular adhesion systems are now known. Of these, the classical cadherin family plays a central role, both during morphogenesis and in post-developmental tissue homeostasis. This subfamily of the cadherin superfamily comprises approximately 20 members, including E-cadherin, N-cadherin, and VE-cadherin, which are predominantly expressed in epithelial, neuronal, and endothelial tissues, respectively.1 Their biological impact arises from their ability to coordinate cell adhesion with cell signaling and the actin cytoskeleton. Indeed, it has long been appreciated that biochemical and functional links with the actin cytoskeleton play a vital role in cadherin biology.2 This was often conceived as reflecting an impact of the cytoskeleton upon cell adhesion.2 Increasingly, we are coming to realize that the impact of cadherin-actin cooperation also reflects how adhesion couples the contractile actin cytoskeletons of cells together.
Rather than being solely about how the cytoskeleton supports adhesion, the terms of the discussion can also be turned around. Of course, this dualistic discussion is too simplistic, as might be expected of any cooperative system. Accordingly, it becomes important to understand how functional cooperation between adhesion and the cytoskeleton is generated and how this translates into biological outcomes. Here an important advance has been the realization that cadherin junctions and their cytoskeletons provide an apparatus for force to be generated and resisted at cell–cell junctions.36 Junctions can be considered to represent active mechanical agents that mediate cell–cell interactions and can potentially scale-up to the tissue level. Here we discuss some advances in our understanding of the functional architecture of E-cadherin-cytoskeletal cooperation, considering their dynamic properties together with their role in the mechanobiology of junctions. Finally, we present some examples of how coupling adhesion to contractility can influence epithelial biology.
E-cadherin receptors and adhesive junctions
In polarized epithelia, E-cadherin is often found throughout cell–cell contacts. It commonly concentrates in the subapical region to form a belt-like structure known as the zonula adherens (ZA), (Figure 1A).1,2 E-cadherin also distributes along the lateral junctions below the ZA, where it forms discrete mobile adhesive clusters (Figure 1C),6–8 which can interchange with the cadherin pool of the ZA.8 Crystallographic analysis of several classical cadherins suggests that their ectodomains can engage in two types of interactions: trans-interactions between cadherins presented on neighboring cells and cis-interactions between the ectodomains presented on the same cell surface. Trans-interactions are mediated by strand-swapping at the first extracellular repeat, while cis-interactions are mediated by the second extracellular repeat.9,10 The combination of trans-interactions and cis-interactions has been proposed to contribute to the cadherin clustering that is commonly observed at adhesive contacts. However, this may not be sufficient, because clustering also requires the cytoplasmic tail and can be influenced by intracellular elements of the cytoskeleton.11–13
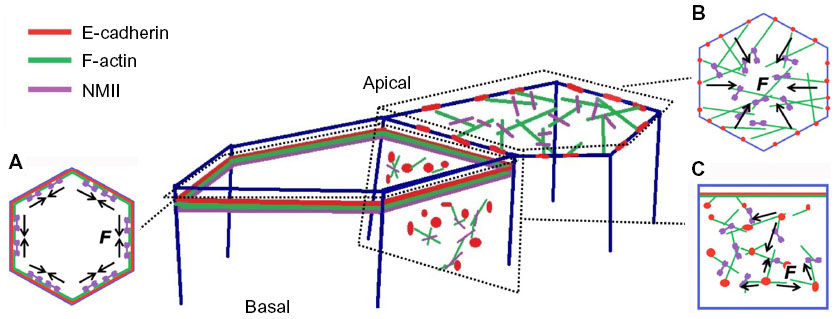 | Figure 1 E-cadherin, F-actin, and NMII form cortical cytoskeleton systems in the epithelium. (A) Circumferential actomyosin cytoskeleton decorating E-cadherin at the zonula adherens. (B) Apical actomyosin meshwork. (C) Cortical F-actin and NMII interaction with E-cadherin through the basolateral membrane. Mechanical forces (F in the figure) are generated by coupling of E-cadherin to the actomyosin cytoskeleton in these different cortical contexts. |
Classical cadherins exist as membrane-spanning macromolecular complexes. Cadherins can interact with a variety of other proteins, especially through their conserved cytoplasmic tails (Figure 2). In particular, three main proteins, the so-called catenins (α-catenin, β-catenin, and p120-catenin), are best understood to associate with cadherin cytoplasmic tails.1 β-catenin binds directly to the C-terminal in a region of approximately 96 amino acids, to then serve as a scaffold for α-catenin, whereas p120-catenin associates directly with the membrane-proximal region of the cytoplasmic tail.14 This said, a host of other proteins are known to interact directly or indirectly with classical cadherins. Many of these interactions are likely to be substoichiometric and/or dynamic. However, this does not mean that they are functionally insignificant. Instead, cadherins may be better understood as anchoring diverse molecular complexes, the composition of which may depend on the cellular context.
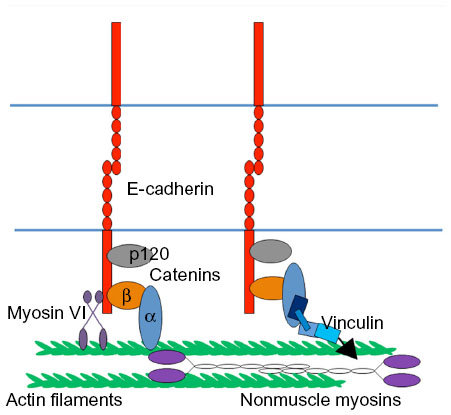 | Figure 2 Force apparatus at the zonula adherens. The junctional cytoskeleton composed of F-actin and nonmuscle myosin II. E-cadherin cytoplasmic binding proteins (eg, α-catenin, myosin VI, and vinculin) link the E-cadherin receptors to the junctional cytoskeleton system. |
Cadherin and the contractile cytoskeleton
E-cadherin junctions are distinguished by the cortical accumulation of an actomyosin cytoskeleton (Figure 1). At the ZA, this commonly takes the form of a prominent circumferential actin ring (Figure 1A),15 which is decorated with non-muscle myosin II.13,16,17 At lateral junctions, the actomyosin network can be found as a two-dimensional network of shorter actin strands that are decorated with and linked by myosin (Figure 1C).8 Actin integrity is essential for cadherin function. Treatment with actin-depolymerizing drugs, such as cytochalasin or latrunculin,18,19 or depletion of actin regulatory proteins,20,21 can disrupt both the junctional cytoskeleton and adhesive interactions.13,16,22–25 In addition, maneuvers that inhibit myosin II also disrupt adhesion, junctional integrity, and tissue organization.21 These findings, taken with increasing evidence for actin regulators at cadherin junctions, have formed the basis for the concept that cadherin function arises from close cooperation between its adhesion system and the actomyosin cytoskeleton. This cooperation reflects three broad processes: the capacity for the cadherin molecular complex to physically associate with F-actin; the ability of cadherin adhesions to regulate the dynamic cytoskeleton; and the ability to recruit and activate myosin II coupled to junctions.
Physical association of cadherin complexes and actin filaments
A long-prevailing model proposed that β-catenin functionally linked the cortical F-actin filament network to cadherin through α-catenin (Figure 2).2 This was founded on early observations that α-catenin can bind directly to F-actin26–28 and indirect evidence from assays such as detergent solubility, which were interpreted as evidence that the cadherin complex, including the E-cadherin cytoplasmic tail, might associate with the actin cytoskeleton.29,30 Confidence in this model was shaken when it proved impossible to reconstitute the quaternary complex (cadherin tail, β-catenin, α-catenin, F-actin) from purified proteins in solution.31 Further, α-catenin homodimers appeared to have greater affinity for F-actin than did the monodimers.32 Recently, using Drosophila embryos engineered to express fusion proteins, it has been reported that E-cadherin-associated monomeric α-catenin interacts with F-actin directly at the junctions and the α-catenin–cadherin complex is essential for maintenance of the adherens junction during embryonic development.25 Similarly, α-catenin in Caenorhabditis elegans requires both an ability to bind β-catenin and its F-actin-binding site to support development.33 This conundrum has recently been resolved by the observation that a more stable interaction between the cadherin-catenin complex and F-actin occurs when these elements are engaged under force.34 This tension-sensitive process is attributable to the formation of catch-bonds between the cadherin-catenin complex and actin filaments.
However, it is important to note that there are many other ways in which cadherins can associate with cortical actin filaments (Figure 2). α-catenin is reported to interact with a host of other F-actin-binding proteins, including EPLIN35 and vinculin;36,37 the cadherin may associate directly with proteins such as myosin VI;38,39 and it may recruit actin regulators with the capacity to bind F-actin. Indeed, perturbation of EPLIN, vinculin, or myosin VI was shown to disrupt junctional actin.35,37,38,40,41 It is therefore unlikely that a single molecular mechanism is responsible for coupling cadherins to the actin cytoskeleton. Instead, different mechanisms may be used in different functional contexts. Such contexts may be regulated by processes such as cell signaling and also the mechanical forces that act upon cell–cell junctions. Indeed, the recruitment of vinculin to cadherin junctions is sensitive to tension, being reduced when contractility is decreased.37,40
Junctional F-actin cytoskeleton and dynamics
Actin filaments are intrinsically dynamic. Within cells, actin assembly and filament elongation preferentially occurs at the barbed ends of filaments, while filament turnover can occur via depolymerization at the pointed ends as well as filament severing. This applies to the junctional cytoskeleton itself: fluorescence recovery after photobleaching analysis of fluorescently-tagged actin revealed that F-actin at established cell–cell junctions turns over with half-times of tens of seconds.13,15,32 Thus, even at a steady-state, the junctional cytoskeleton undergoes continuous turnover, suggesting that filaments are being continually disassembled and replenished.
Actin assembly in the junctional cytoskeleton
The rate-limiting step in actin filament assembly is the process of nucleation, ie, the growth of a polymer from the initial monomer to a trimer (Figure 3).42,43 Subsequent filament elongation can be driven by self-assembly, although it is also regulated by proteins such as those of the Ena/VASP family, which promote barbed end elongation.44–46 Because it is energetically unfavorable, F-actin nucleation is catalyzed by several families of proteins, some of which are found at E-cadherin junctions. These include the formins47,48 and the Arp2/3 complex.20,21
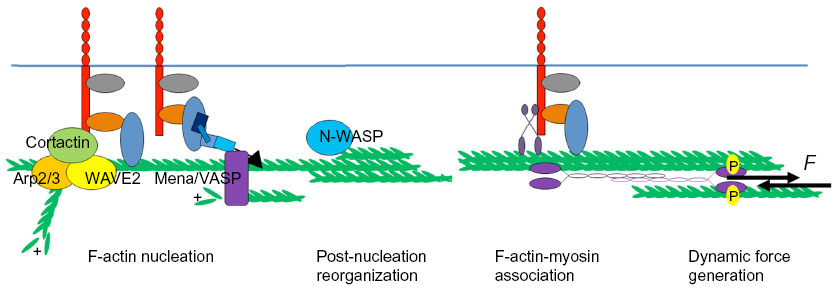 | Figure 3 Dynamic assembly of the junctional contractile apparatus capable of generating tensile forces (F in the figure) at the zonula adherens. Generation of the junctional cytoskeleton involves multiple steps, including F-actin nucleation, post-nucleation filament growth and reorganization, and association of myosin II to generate force. |
The formins are a large family of proteins47,48 that typically nucleate actin assembly at the barbed ends of unbranched filaments.49 In keratinocytes, formin-1 was identified as an α-catenin-binding protein that supported the junctional cytoskeleton, while mDia1 has also been found at the junctions of simple epithelia.47,48 In Drosophila mesoderm cells Diaphanous also promoted stable connections between the contractile actin networks of the apical poles (often called the medial–apical network) and cell–cell junctions (Figure 1B).50 Indeed, given that prominent actin bundles are often found adjacent to the ZA in simple epithelia, it has been attractive to postulate that proteins which nucleate assembly in unbranched filaments might contribute to the junctional cytoskeleton.
Interestingly, however, a major site of actin nucleation can also be found at the junctional membrane, apparently located between the actomyosin bundles at the ZA (Figure 3).15,37,51 Thus, after photobleaching, green fluorescent protein-actin fluorescence recovered first at the membrane and then extended outwards to encompass the apical actin bundles.15 Similarly, labeling of free barbed ends by incorporation of fluorescently labeled G-actin occurred most prominently at this location, rather than in the bundles.15,37 The Arp2/3 complex is found there, can coimmunoprecipitate with E-cadherin,19,20,52 and contributes to nucleation at this site. Chemical inhibition of Arp2/3 complex function or depletion by RNA interference decreased barbed end labeling at the plasma membrane, leading to disruption of both junctional F-actin and E-cadherin contact integrity.19–21 Surprisingly, under the same circumstances, inhibition of formin activity using a broad-spectrum inhibitor, SMIFH2, did not affect the reassembly of junctional F-actin after latrunculin treatment.19
The Arp2/3 complex is a seven-subunit protein complex consisting of two actin-related proteins (Arp), ie, Arp2 and Arp3, and five other ARP complex proteins (ARPC1–5).53–55 Arp2 and Arp3 resemble monomeric actin and bind to the sides of a pre-existing (“mother”) filament, promoting the growth of a new (“daughter”) filament.56 The daughter filament is oriented at a 70 degree angle to the mother filament, with the pointed ends directed towards the mother filament.56 This orientation means that Arp2/3 tends to generate branched actin networks.55,57 Isolated Arp2/3 has a relatively poor intrinsic nucleating activity, which therefore requires that its activity in cells be stimulated by accessory proteins.54 Two classes of F-actin nucleation promoting factors (NPFs) have been identified to activate Arp2/3.58 Class I NPFs possess a characteristic VCA domain, which is composed of the verprolin-homology domain for actin binding, the cofilin-homology (C) domain, and the acidic (A) domain for Arp2/3 binding.57 Class 1 NPFs include Wiskott–Aldrich syndrome protein (WASP) and neuronal-WASP (N-WASP, which is actually ubiquitous), and the WASP-family verprolin-homologous protein (WAVE) family of proteins.57 Of these, WAVE2 contributes to Arp2/3-based actin assembly at the epithelial ZA in mammalian cells.21,57,59 Class II NPFs lack VCA domains but, instead, possess acidic domains that bind the Arp2/3 complex and tandem repeat domains that can bind to filamentous actin.57,58 Cortactin is a Class II NPF that is found at cell–cell junctions in association with E-cadherin and contributes to junctional actin assembly.60,61 This entails its ability to recruit both Arp2/3 and WAVE2 to junctions.21,60,61
Together, these observations suggest that junctional filaments at the ZA may be nucleated by Arp2/3 at the membrane as branched networks and then incorporated into perijunctional bundles. This implies that reorganization of the actin network may occur as a post-nucleation step to promote the formation of perijunctional bundles (Figure 3). One protein involved in this step is the actin regulator, N-WASP.15 Although N-WASP is best understood as a catalytic activator of Arp2/3, its depletion in epithelial cells was shown to compromise actin bundles and the integrity of the ZA, without disrupting actin nucleation.8,15 Instead, N-WASP could stabilize actin filaments at the ZA.15 This suggested that N-WASP might act at a post-nucleation step. Consistent with this, the effects of N-WASP depletion could be restored by expression of an N-WASP mutant lacking the VCA domain that is necessary to activate Arp2/3.15 Instead, its biological activity at the ZA mapped to its N-terminus, the region that binds the WASP-interacting family proteins.15 Exactly how N-WASP stabilizes filaments to promote network reorganization remains to be elucidated.
F-actin disassembly at cadherin junctions
Less is known about how actin filaments undergo turnover and disassembly at cell–cell junctions. A variety of proteins can induce filament turnover within cells, notably members of the ADF/cofilin family that induce filament severing and pointed-end disassembly.62,63 What role they may play in junctional actin dynamics has yet to be characterized. Gelsolin, another filament severing protein, was identified at N-cadherin contacts between fibroblasts where it contributed to actin dynamics.64 Finally, nonmuscle myosin II can induce turnover of F-actin at the cell–cell surface8 through its ability to generate stresses that can buckle and sever filaments.65–67
Nonmuscle myosins at cadherin junctions
Nonmuscle myosin II (NMII) is the principal contractile force-generator in eukaryotic cells. NMII forms a heterohexamer composed of two 230 kDa heavy chains, two 20 kDa essential light chains that stabilize the heavy chain, and two regulatory light chains (RLC) that regulate myosin activity.68,69 The NMII heavy chain consists of an N-terminal head domain that bears the F-actin-binding and ATP-binding sites responsible for motor activity, a neck-region, and a coiled-coil rod domain.69,70 Single myosins are poorly processive motors and are therefore unable to effectively generate contractile force.71,72 Instead, within cells, the rod domains of NMII allow it to assemble into bipolar minifilaments consisting of 16–30 myosins that support productive contractility.71,72 As defined by the heavy chain, a single NMII gene is found in Drosophila, whereas three NMII paralogs, ie, NMII A, B, and C, are found in mammalian cells and are encoded by MYH9, MYH10, and MYH14, respectively.68 Despite broad structural similarities, these mammalian paralogs have different properties and functions. NMIIA has higher ATPase activity, while NMIIB possesses a higher duty ratio and ADP removal kinetics.73,74 Drosophila myosin and the mammalian NMII paralogs have all been identified at E-cadherin junctions and their inhibition perturbs junctional integrity.13,16,22,23,50,75
A variety of signaling pathways influence the recruitment and/or motor activation of myosins at E-cadherin junctions. Activation of NMII motor function is best understood to occur through phosphorylation of its RLC. S19 of the RLC constitutes the primary phosphorylation site and T18 the secondary phosphorylation site. Their phosphorylation can be induced by a variety of protein kinases, including myosin light chain kinase, myotonic dystrophy kinase-related CDC42-binding kinase alpha, and Rho kinase.2 Although Rho kinase can phosphorylate RLC directly in vitro, its ability to promote RLC phosphorylation within cells may be principally due to inhibition of myosin phosphatase.76,77 Phosphorylation of S19 increases F-actin binding and myosin ATPase activity, and also promotes the formation of myosin minifilaments, all of which are molecular features that contribute to contractility. These parameters are further increased when T18 is also phosphorylated.78 Activated NMII, marked by the presence of phosphorylated RLC, is found at adherens junctions and can be induced by homophilic ligation of E-cadherin itself.16,24 Both Rho kinase and myosin light chain kinase promote the recruitment of NMIIA to epithelial cell–cell junctions, suggesting that recruitment may be closely linked to motor activation. On the other hand, myosin IIB junctional localization is dominantly influenced by Src and Rap1 kinases.13,79 Further, the myosin heavy chain can be phosphorylated by kinases, such as protein kinase C, often leading to inhibition of minifilament formation.78 However, whether these events influence actomyosin at cell–cell junctions is not yet known.
The recruitment of myosin II to junctions is also influenced by the integrity and dynamics of F-actin. Both NMIIA and NMIIB failed to concentrate at junctions when actin assembly by the WAVE2/Arp2/3 mechanism was inhibited.21 This likely reflects the central role that F-actin binding plays in recruiting NMII to E-cadherin adhesions. Indeed, myosin can be found in a number of patterns that coincide with junctional F-actin. In the apical rings adjacent to the ZA, NMII is often found to overlay with the F-actin bundles in punctae8,13,80 and sometimes in sarcomere-like patterns.81 At the lateral junctions, myosin punctae more commonly appear to bridge between short F-actin cables or strands.8 Other myosin-binding proteins, such as anillin,82 are found at epithelial cell–cell junctions and influence junctional integrity, but whether these may couple NMII to cadherins remains to be evaluated.
Junctional tensile apparatus and forces
Generating force at junctions
The coupling of an actomyosin contractile apparatus to E-cadherin adhesions can ultimately generate contractile stress at junctions. Such stresses may serve many biological functions (to be discussed further in the section on cellular applications of junctional contractility). However, these stresses can vary, both in their direction and magnitude.
A variety of spatial patterns of stress have been observed to be exerted at cell–cell junctions (Figure 1A–C). These include stresses that are oriented perpendicular or orthogonal to the junctions.83 For example, in embryonic cells that undergo apical constriction, contractile pulses in the medial–apical actomyosin networks cause pulsatile orthogonal movements of their associated cell–cell junctions (Figure 1B).84 Similarly, as endothelial cells form junctions with one another, it is common to observe bundles of F-actin that are oriented approximately perpendicular to the junctions, inserting into VE-cadherin punctae at those contacts.85,86 These appear to generate orthogonal pulling forces on the punctae.
In other contexts, dominant patterns of stress are found to be oriented parallel to the junctions (Figure 1A). This was revealed when those junctions were focally cut with lasers: the residual portions of the junctions retracted, suggesting that tension was oriented principally along the junctions.8,21,80,87 This apparent line tension within junctions has been observed in Drosophila and mammalian epithelial cells. One potential mechanism for contractility to generate an apparent line tension is the organization of actomyosin into cables aligned with the junctions.8,21,79 Thus, the mechanisms that determine the orientation of actomyosin, vis-a-vis the junctions, may critically determine the dominant vectors of stress that are generated when contractility is coupled to adhesion.
The spatial and temporal dynamics of actomyosin networks can also collaborate with this subcellular distribution to influence the overall patterns of contractility that are generated within cells. In Drosophila embryos, this is exemplified by the flow of NMII that occurs from the medial–apical regions of cells (their apical areas) to the ZA as the epithelia remodel.84 This polarized flow of actomyosin towards epithelial junctions is proposed as a critical mechanism for the shrinkage of dorsal–ventral-oriented junctions during embryonic development.84 The flow was influenced by the polarized distribution of E-cadherin and depletion of E-cadherin compromised the flow.84 NMII flow requires myosin activation by RLC phosphorylation, and dynamic cycles of phosphorylation and dephosphorylation are necessary for morphogenetic cell shape changes to occur.88
In addition to differences in the orientation of contractile stress, cell–cell junctions can also display differences in the detectable magnitude of stress that is exerted upon them. This can even occur within individual cell–cell junctions. For example, although the lateral junctions of polarized epithelial cells possess a contractile actomyosin network that exerts force upon cadherins (Figure 1C), the stresses in this region can be substantially lower than those found at the apical ZA.8 Stresses in these lateral junctions are limited by myosin-induced actin filament turnover.8 Effectively, myosin-induced contractility caused actin networks to condense and then be turned over, thereby allowing stress to be dissipated.8 One reason for stress-induced filament turnover may be that, at the lateral cortex, myosin often bridges between short actin cables that are not aligned with one another, which would tend to cause filament buckling.8
A further implication is that mechanisms must exist within the cytoskeleton of the ZA that antagonize or compensate for potential stress-induced turnover. Indeed, this appears to be the case. One such mechanism is filament stabilization by N-WASP, as mentioned earlier. Consistent with this, depletion of N-WASP reduces tension at the ZA to levels similar to those found at the lateral junctions.8 Another mechanism is tension-sensitive actin assembly.37,89 We recently observed that actin assembly at the ZA is tuned in response to contractility, ie, decreasing when contractility is inhibited and increasing when contractility is stimulated.37 This reflected the tension-sensitive recruitment of vinculin to E-cadherin junctions.37,90,91 Vinculin associated with members of the Ena/VASP family, which can promote actin assembly by functioning as an actin polymerase and/or protecting barbed ends from premature capping.92 Disabling tension-sensitive actin assembly by inhibiting either vinculin or its ability to recruit Ena/VASP proteins decreased ZA tension, while ectopically targeting Ena/VASP to junctions could restore tension even to vinculin-deficient cells.37
Mechanosensing and signal transduction at cadherin junctions
The ability of E-cadherin adhesion to promote actomyosin assembly indicates that the adhesion system can serve as an active agent in the generation of force at junctions (Figure 4). Increasingly, it is also apparent that cell–cell junctions are sites where forces are sensed to elicit proportional cellular responses.83,93 These responses often entail roles for the cytoskeleton, suggesting that mechanotransduction ultimately elicits signals that regulate cytoskeletal function. This is supported by evidence that junctional homeostasis can be influenced by myosins.16 Similarly, it has been observed in endothelial cells that tugging on junctions increases the length of those junctions, suggesting a role for mechanoresponsiveness at VE-cadherin adhesions.85 Indeed, the use of cadherin fusion proteins bearing FRET-based tension sensors demonstrated that both E-cadherin and VE-cadherin at cell–cell junctions are themselves under molecular-level tension; in the case of VE-cadherin, this was also altered in response to shear flow.94–96 Further, the direct application of force to E-cadherin molecules can elicit cellular responses. This was demonstrated when magnetic beads coated with E-cadherin antibodies were allowed to engage cadherins on the surfaces of epithelial cells.97 Application of an oscillating magnetic field to twist the beads, and thereby exert force on the attached cellular cadherin, caused the cells to stiffen, a process that required an intact actomyosin cytoskeleton.97 In another example, pulling forces exerted on C-cadherin in Xenopus blastomeres induced protrusive activity at the pole opposite to the pulling force.98 Recently, it has been shown that applying anisotropic stresses to the junctions between pairs of S180 cells deformed the contacts between the cells, leading to asymmetric distribution of E-cadherin, actin, and myosin.99 Together, these observations suggest that cell–cell junctions possess mechanosensors.
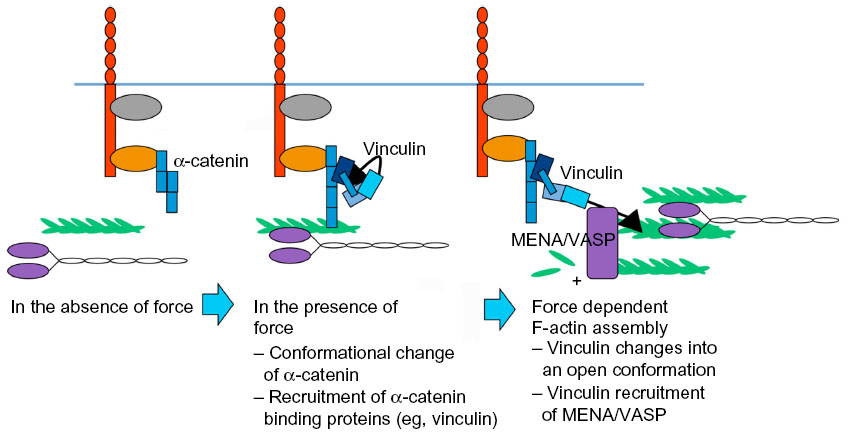 | Figure 4 Force sensing and responding at E-cadherin receptors through vinculin recruitment and tension-sensitive actin assembly. In the absence of force, α-catenin is in a close conformation; when the cadherin receptors are under force, α-catenin opens its VH2 domain for vinculin binding; vinculin transits into a open conformation under force, and recruits Mena/VASP, assembling F-actin in a force-dependent manner. |
How, then, might forces be sensed at junctions? One paradigm involves tension-sensitive proteins in the cytoskeletal network that is connected to cadherins. This is exemplified by vinculin, which, as discussed earlier, is recruited to α-catenin in response to force (Figure 4).86,97,100 Vinculin was necessary for the cellular stiffening that occurred when E-cadherin adhesions were subjected to force by twisting cadherin-bound magnetic beads.97 Although the precise mechanism was not identified in those original studies, it may involve tension-sensitive actin assembly (Figure 4). There are also multiple ways in which vinculin may participate in force sensing. One mechanism involves its recruitment to junctions by α-catenin. Vinculin can directly bind α-catenin36,37,101 in a region bearing cryptic epitopes that can be revealed in cells in response to contractility.100 As crystallographic analysis indicated that α-catenin could adopt an autoinhibited conformation,90,102 molecular tension might unfold α-catenin to promote the binding of vinculin. It was recently demonstrated that, when isolated α-catenin molecules were stretched using magnetic tweezers, the vinculin-binding domain was unfolded to activate vinculin binding, which then stabilized α-catenin in its open conformation.91
This example conforms to a broader model whereby molecular function is directly modulated by force-induced changes in protein conformation, a model that appears increasingly prevalent in many cellular contexts.3,103 Indeed, other cytoskeletal components at cadherin junctions have the potential to be mechanosensitive. These include myosin VI,38 which is sensitive to mechanical load,39,104 and EPLIN, the localization of which to junctions was modulated by contractility.35 Myosin VI can bind directly to E-cadherin,41 whereas EPLIN is recruited by α-catenin.35 Further, junctional tension can promote the recruitment and stabilization of NMII at the junctions,17,105 perhaps reflecting the preference of myosin to interact with prestressed actin filaments.17,105 Thus, mechanosensitivity may reside at multiple loci within cadherin junctions.
Another paradigm for mechanosensing involves activation of cell signaling pathways by force-sensitive molecules. Here, attractive candidates might be the cadherins themselves, especially given their capacity to experience molecular force at cell–cell junctions.95,96 Classical cadherins can participate in cell signaling in many ways. These include modulation of signaling by Rho and Rap family GTPases,106–108 lipid kinases such as phosphatidylinositol-3 kinase,109 and protein kinases such as the Src tyrosine kinase.110,111 The extent to which such pathways are activated by force at junctions remains to be thoroughly characterized. It was recently demonstrated that vinculin at cell–cell junctions is tyrosine-phosphorylated in a fashion that depends on cellular contractility, suggesting that it is the target for a mechanosensitive signaling pathway.112 Vinculin phosphorylation required the Abelson tyrosine kinase and E-cadherin adhesion,112 but how these elements are linked remains to be determined. One possibility is that force experienced by the cadherin when it is engaged in adhesion might elicit cell signaling. However, it is also possible that cadherins can participate as scaffolds in mechanosensing. This is illustrated by the role of VE-cadherin in sensing fluid shear stress upon endothelia.96 Here, VE-cadherin served as an adaptor molecule linking platelet endothelial cell adhesion molecule to vascular endothelial growth factor receptor 2 signaling.113 Adhesive ligation of VE-cadherin was not necessary for its function in this signaling pathway, implying that it was not the direct mechanoreceptor, although it is possible that it received mechanical inputs from other receptors. Indeed, the initial application of fluid shear stress reduced mechanical tension on VE-cadherin.96 Clearly, much remains to be learnt about how cadherins participate in mechanotransduction at cell–cell junctions.
Examples of cellular applications of junctional contractility
Morphogenesis during development
The cadherin-actomyosin system allows cell–cell adhesion to transmit forces between cells. During development, this is used in a number of ways to achieve morphogenetic outcomes. One example occurs when epithelial sheets undergo folding events, such as occurs during ventral furrow formation in the Drosophila embryo. In this process, individual cells undergo constriction of their apical poles which, through adhesive coupling, leads to folding of the whole tissue itself. Here the principal force-generator appears to reside in the aforementioned medial-apical actomyosin network of cells.114–117 This displays a pulsatile pattern of contractility that is transmitted to the E-cadherin junctions to induce apical constriction.114 The molecular mechanisms responsible for coupling this contractile network to cadherin adhesion remain to be fully elucidated, but includes the scaffolding protein, Canoe (afadin in vertebrates), loss of which caused the contractile network to uncouple from junctions.116
Pulsatile contractility in a medial–apical network is also implicated in another morphogenetic process, ie, neighbor-exchange, where cells rearrange relative to one another. This is commonly seen when cells intercalate during the process of convergent-extension.17,75 The precise mode of contractility that contributes to neighbor-exchange may be developmentally regulated (Figure 5A). For example, in Drosophila, convergence-extension characterizes the process of germband extension. Medial–apical pulsed contractility appears to be a dominant mode of contractility during early germband extension,75,84 but at later stages tonic contractility at junctions becomes prominent, associated with the appearance of prominent myosin accumulation at the junctions themselves.17,118 It is not known how these patterns of actomyosin change during development. However, myosin has been observed to flow from the medial–apical network into the junctions,84 suggesting that changes in local concentrations of actomyosin may be relevant.
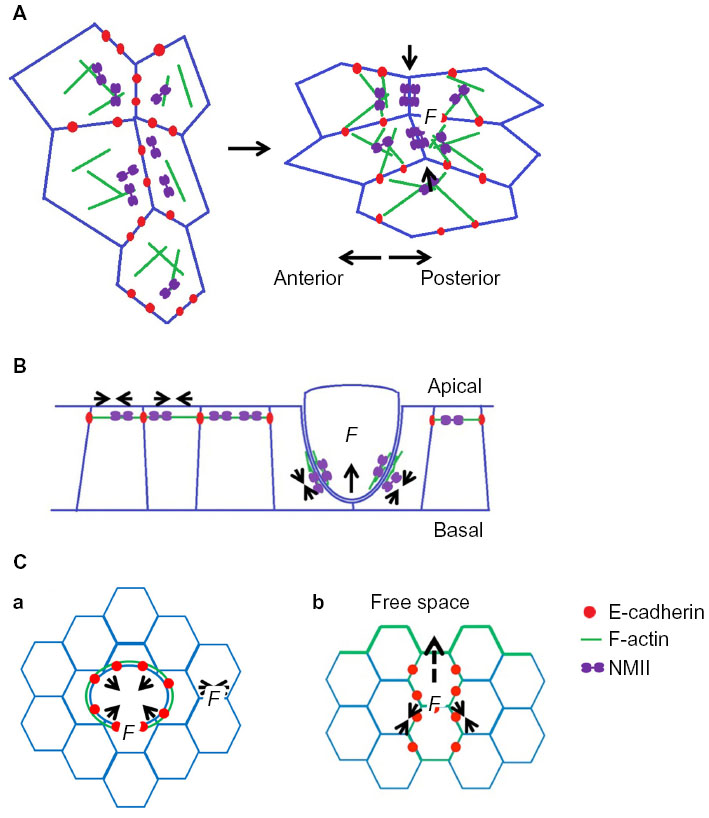 | Figure 5 Examples of cellular applications of junctional contractility mechanical forces (F in the figure). (A) Junctional contractility that drives germ band extension during Drosophila embryonic development. (B) Mechanical forces at cell–cell contacts that contribute to apical epithelial extrusion. (C) Actomyosin dependent tensile forces facilitate the cells moving into free space during (a) wound healing and (b) collective migration. |
This emphasizes that actomyosin and adhesion interact cooperatively during morphogenesis. For example, pulsed contractility induced the redistribution of E-cadherin from the apical to the subapical region,114 indicating that patterns of contractility influenced junctional organization. Conversely, junctional dynamics can influence patterns of contractility. This is exemplified by the observation that medial–apical actomyosin flowed towards junctions in a planar-polarized fashion.84 This actomyosin flow was, in turn, attributable to polarized fluctuations in the levels of E-cadherin at junctions due to polarized endocytosis of the adhesion molecule itself.87,119
Morphogenetic junctional contractility is subject to several layers of cellular regulation. These include local signals that affect the contractile apparatus. Cycles of myosin phosphorylation and dephosphorylation, regulated by Rho kinase and myosin phosphatase, contribute to medial–apical network contractility.88 The junctional contractility is also subject to developmental regulation. For example, during apical constriction in the gastrulating Drosophila embryo, the transcription factor Snail appeared to initiate pulsed contractility in the medial–apical networks, whereas another transcription factor, Twist, stabilized the constricted apical poles.115 Presumably these transcription factors exert their effects indirectly through cascades of downstream effectors that ultimately regulate contractility and its coupling to the cadherin system. Indeed, the coupling process itself may be developmentally regulated. This is suggested by studies in C. elegans embryos, which displayed pulsatile apical actomyosin networks that at first were not mechanically transmitted to the junctions.120 Morphogenetic movements did not occur in this situation, but were only initiated when the junctions became mechanically coupled to actomyosin.120
Epithelial extrusion
Extrusion is a process whereby minorities of cells become physically expelled from epithelia.8 These minorities may result from apoptosis, which was the context where extrusion was first identified, or from other processes, such as bacterial infection or the expression of oncogenes. In these circumstances, extrusion may represent a mechanism to eliminate abnormal cells and restore homeostasis.121 In the case of oncogenic extrusion, however, extrusion may promote the proliferation of transformed cells by removing them from the inhibitory environment of a normal epithelium.122,123 Many instances of extrusion are cell nonautonomous phenomena, which only occur when minorities of abnormal cells are surrounded by normal cells.122,123
This is clearly seen with oncogenic extrusion, which does not occur when a large number of cells express oncogenes, but only when the oncogene-expressing cells are surrounded by nonexpressing cells.122,123 This further implies that the intercellular interface associated with extrusion may bear distinctive properties. Interestingly, extrusion can also affect apparently normal cells when epithelia become overcrowded; here expulsion of the unlucky minority seems to provide a mechanism to restore population homeostasis.124,125
The notion that extrusion might be an active mechanical process was first suggested by the observation that during apoptotic extrusion the neighboring cells assemble an actomyosin ring at their interface with the cell to be extruded.122,126,127 This suggested that contractility might be a key to extrusion. Recently, the role of junctional mechanics has become apparent in the context of oncogenic extrusion. Here we observed that patterns of contractile tension become reorganized specifically at the junction between transformed and nontransformed cells, ie, where extrusion will occur.8 Junctions between nontransformed cells displayed high tension at their apical ZA but lower tension at the lateral junctions located below the ZA (Figure 5B).8 However, this was altered specifically at the extrusion interface, which displayed reduced apical junction tension and increased lateral tension.8 This was attributable, at least in part, to the redistribution of N-WASP from the apical to the lateral junctions.8 Therefore, local changes in junctional F-actin stability could modulate patterns of junctional tension to facilitate extrusion. It is likely that other changes in the junctional cytoskeleton collaborate to induce extrusion. E-cadherin can promote the junctional recruitment of N-WASP, as it regulates other aspects of the junctional actin cytoskeleton.15 Whether these regulatory effects of E-cadherin contribute to extrusion remains to be elucidated. E-cadherin was necessary in the neighboring cells for extrusion to occur.122,123 However, it was recently demonstrated that cleavage of E-cadherin in single cells can be sufficient to promote extrusion,127 suggesting that adhesion alone may be less important than how the patterns of mechanical tension are altered at cell–cell interfaces.
Wound healing
Finally, cadherin-cytoskeletal cooperation may contribute to the process(es) of wound healing (Figure 5C). Its precise role may depend on the size of the wounds that are involved. For example, the closure of small wounds can be critically influenced by the cortical forces of the actomyosin cytoskeleton, as shown in Figure 5C(a).128 As studied in Drosophila, several stages in the process have been described, which begin with the retraction of the wound margins. This is thought to reflect the release of tension that was previously existing in the epithelium (which can be considered as a prestressed material).129 Subsequently, an apparent wave of myosin and F-actin flows in the epithelium towards the cells at the immediate wound margin, likely under the control of Ca2+ signaling.130,131 Lastly, at the very edge of the wounded area, myosin and actin are organized into a contractile ring, which is anchored to junctional E-cadherin.132 This ring generates contractile forces that shrink the area of the open wounds.130,133,134 Here cadherin-actin cooperation may be essential for contractility to be coordinated at the wound margin.
The closure of large wounds also involves many cellular processes, including lamellipodia that drive migration of the cells at the wound front.128 This process can also be considered as a form of collective cell migration, as is readily illustrated in artificial wounds where cells show coordinated directionality of migration, as shown in Figure 5C(b).121,135–137 However, this coordination is disrupted when E-cadherin or elements of the cadherin molecular complex (eg, α-catenin) are depleted. Under these circumstances, cell movements become chaotic within the migrating population.138–140 The precise mechanisms that allow cadherins to support collective migration are not well understood. Cadherin adhesion might be required to maintain the integrity of the migrating population;121,140 it may also transmit forces generated by cells at the margins of the wounds to other cells in the epithelial population.140 This is likely to be a fruitful field for future research.
Conclusion
Epithelia establish and maintain robust tissue architectures out of intrinsically dynamic components. We have highlighted the capacity for cooperation between adhesion and the dynamic actomyosin cytoskeleton to contribute to a range of biological outcomes.57,69 Ultimately, the molecular and cellular mechanisms that integrate adhesion with the cytoskeleton are subject to regulation by external cues, both chemical and mechanical, that impinge upon cells from their local environments.4,5,141 It will be an exciting challenge for the future to characterize how such signals influence tissue homeostasis in the healthy organism, and how they might be perturbed to disturb cell–cell interactions in disease.
Acknowledgments
This work was supported by the National Health and Medical Research Council of Australia (1044041, 1037320,1067405), the China Science Council (to XL), and The Kids Cancer Project of the Oncology Research Foundation.
Disclosure
The authors report no conflicts of interest in this work.
References
Niessen CM, Leckband D, Yap AS. Tissue organization by cadherin adhesion molecules: dynamic molecular and cellular mechanisms of morphogenetic regulation. Physiol Rev. 2011;91(2):691–731. | |
Yonemura S. Cadherin-actin interactions at adherens junctions. Curr Opin Cell Biol. 2011;23(5):515–522. | |
Hoffman BD, Grashoff C, Schwartz MA. Dynamic molecular processes mediate cellular mechanotransduction. Nature. 2011;475(7356):316–323. | |
Huveneers S, de Rooij J. Mechanosensitive systems at the cadherin-F-actin interface. J Cell Sci. 2013;126 Pt 2:403–413. | |
Munjal A, Lecuit T. Actomyosin networks and tissue morphogenesis. Development. 2014;141(9):1789–1793. | |
Vaezi A, Bauer C, Vasioukhin V, et al. Actin cable dynamics and Rho/Rock orchestrate a polarized cytoskeletal architecture in the early steps of assembling a stratified epithelium. Dev Cell. 2002;3(3):367–381. | |
Hong S, Troyanovsky RB, Troyanovsky SM. Spontaneous assembly and active disassembly balance adherens junction homeostasis. Proc Natl Acad Sci U S A. 2010;107(8):3528–3533. | |
Wu SK, Gomez GA, Michael M, et al. Cortical F-actin stabilization generates apical-lateral patterns of junctional contractility that integrate cells into epithelia. Nat Cell Biol. 2014;16(2):167–178. | |
Harrison OJ, Jin X, Hong S, et al. The extracellular architecture of adherens junctions revealed by crystal structures of type I cadherins. Structure. 2011;19(2):244–256. | |
Brasch J, Harrison OJ, Ahlsen G, et al. Structure and binding mechanism of vascular endothelial cadherin: a divergent classical cadherin. J Mol Biol. 2011;408(1):57–73. | |
Yap AS, Brieher WM, Pruschy M, et al. Lateral clustering of the adhesive ectodomain: a fundamental determinant of cadherin function. Curr Biol. 1997;7(5):308–315. | |
Yap AS, Niessen CM, Gumbiner BM. The juxtamembrane region of the cadherin cytoplasmic tail supports lateral clustering, adhesive strengthening, and interaction with p120ctn. J Cell Biol. 1998;141(3):779–789. | |
Smutny M, Cox HL, Leerberg JM, et al. Myosin II isoforms identify distinct functional modules that support integrity of the epithelial zonula adherens. Nat Cell Biol. 2010;12(7):696–702. | |
McCrea PD, Gu D. The catenin family at a glance. J Cell Sci. 2010;123 Pt 5:637–642. | |
Kovacs EM, Verma S, Ali RG, et al. N-WASP regulates the epithelial junctional actin cytoskeleton through a non-canonical post-nucleation pathway. Nat Cell Biol. 2011;13(8):934–943. | |
Shewan AM, Maddugoda M, Kraemer A, et al. Myosin 2 is a key Rho kinase target necessary for the local concentration of E-cadherin at cell-cell contacts. Mol Biol Cell. 2005;16(10):4531–4542. | |
Fernandez-Gonzalez R, Simoes Sde M, Roper JC, et al. Myosin II dynamics are regulated by tension in intercalating cells. Dev Cell. 2009;17(5):736–743. | |
Yamazaki D, Oikawa T, Takenawa T. Rac-WAVE-mediated actin reorganization is required for organization and maintenance of cell-cell adhesion. J Cell Sci. 2007;120(1):86–100. | |
Tang VW, Brieher WM. Alpha-actinin-4/FSGS1 is required for Arp2/3-dependent actin assembly at the adherens junction. J Cell Biol. 2012;196(1):115–130. | |
Verma S, Shewan AM, Scott JA, et al. Arp2/3 activity is necessary for efficient formation of E-cadherin adhesive contacts. J Biol Chem. 2004;279(32):34062–34070. | |
Verma S, Han SP, Michael M, et al. A WAVE2-Arp2/3 actin nucleator apparatus supports junctional tension at the epithelial zonula adherens. Mol Biol Cell. 2012;23(23):4601–4610. | |
Krendel M, Gloushankova NA, Bonder EM, et al. Myosin-dependent contractile activity of the actin cytoskeleton modulates the spatial organization of cell-cell contacts in cultured epitheliocytes. Proc Natl Acad Sci U S A. 1999;96(17):9666–9670. | |
Miyake Y, Inoue N, Nishimura K, et al. Actomyosin tension is required for correct recruitment of adherens junction components and zonula occludens formation. Exp Cell Res. 2006;312(9):1637–1650. | |
Watanabe T, Hosoya H, Yonemura S. Regulation of myosin II dynamics by phosphorylation and dephosphorylation of its light chain in epithelial cells. Mol Biol Cell. 2007;18(2):605–616. | |
Desai R, Sarpal R, Ishiyama N, et al. Monomeric alpha-catenin links cadherin to the actin cytoskeleton. Nat Cell Biol. 2013;15(3):261–273. | |
Rimm DL, Koslov ER, Kebriaei P, et al. Alpha 1(E)-catenin is an actin-binding and -bundling protein mediating the attachment of F-actin to the membrane adhesion complex. Proc Natl Acad Sci U S A. 1995;92(19):8813–8817. | |
Ozawa M. Identification of the region of alpha-catenin that plays an essential role in cadherin-mediated cell adhesion. J Biol Chem. 1998;273(45):29524–29529. | |
Ozawa M, Kemler R. Altered cell adhesion activity by pervanadate due to the dissociation of alpha-catenin from the E-cadherin.catenin complex. J Biol Chem. 1998;273(11):6166–6170. | |
Adams CL, Nelson WJ, Smith SJ. Quantitative analysis of cadherin-catenin-actin reorganization during development of cell-cell adhesion. J Cell Biol. 1996;135(6 Pt 2):1899–1911. | |
Butz S, Kemler R. Distinct cadherin-catenin complexes in Ca(2+)-dependent cell-cell adhesion. FEBS Lett. 1994;355(2):195–200. | |
Yamada S, Pokutta S, Drees F, et al. Deconstructing the cadherin-catenin-actin complex. Cell. 2005;123(5):889–901. | |
Drees F, Pokutta S, Yamada S, et al. Alpha-catenin is a molecular switch that binds E-cadherin-beta-catenin and regulates actin-filament assembly. Cell. 2005;123(5):903–915. | |
Kwiatkowski AV, Maiden SL, Pokutta S, et al. In vitro and in vivo reconstitution of the cadherin-catenin-actin complex from Caenorhabditis elegans. Proc Natl Acad Sci U S A. 2010;107(33):14591–14596. | |
Buckley CD, Tan J, Anderson KL et al. The minimal cadherin-catenin complex binds to actin filaments under force. Science. 2014;346(6209):12542111–125421118. | |
Taguchi K, Ishiuchi T, Takeichi M. Mechanosensitive EPLIN-dependent remodeling of adherens junctions regulates epithelial reshaping. J Cell Biol. 2011;194(4):643–656. | |
Watabe-Uchida M, Uchida N, Imamura Y, et al. Alpha-catenin-vinculin interaction functions to organize the apical junctional complex in epithelial cells. J Cell Biol. 1998;142(3):847–857. | |
Leerberg JM, Gomez GA, Verma S, et al. Tension-sensitive actin assembly supports contractility at the epithelial zonula adherens. Curr Biol. 2014;24(15):1689–1699. | |
Maddugoda MP, Crampton MS, Shewan AM, et al. Myosin VI and vinculin cooperate during the morphogenesis of cadherin cell cell contacts in mammalian epithelial cells. J Cell Biol. 2007;178(3):529–540. | |
Dunn AR, Chuan P, Bryant Z, et al. Contribution of the myosin VI tail domain to processive stepping and intramolecular tension sensing. Proc Natl Acad Sci U S A. 2010;107(17):7746–7750. | |
Peng X, Cuff LE, Lawton CD, et al. Vinculin regulates cell-surface E-cadherin expression by binding to beta-catenin. J Cell Sci. 2010;123 Pt 4:567–577. | |
Mangold S, Wu SK, Norwood SJ, et al. Hepatocyte growth factor acutely perturbs actin filament anchorage at the epithelial zonula adherens. Curr Biol. 2011;21(6):503–507. | |
Drenckhahn D, Pollard TD. Elongation of actin filaments is a diffusion-limited reaction at the barbed end and is accelerated by inert macromolecules. J Biol Chem. 1986;261(27):12754–12758. | |
Pollard TD, Blanchoin L, Mullins RD. Molecular mechanisms controlling actin filament dynamics in nonmuscle cells. Annu Rev Biophys Biomol Struct. 2000;29:545–576. | |
Krause M, Dent EW, Bear JE, et al. Ena/VASP proteins: regulators of the actin cytoskeleton and cell migration. Annu Rev Cell Dev Biol. 2003;19:541–564. | |
Pasic L, Kotova T, Schafer DA. Ena/VASP proteins capture actin filament barbed ends. J Biol Chem. 2008;283(15):9814–9819. | |
Winkelman JD, Bilancia CG, Peifer M, et al. Ena/VASP Enabled is a highly processive actin polymerase tailored to self-assemble parallel-bundled F-actin networks with Fascin. Proc Natl Acad Sci U S A. 2014;111(11):4121–4126. | |
Kobielak A, Pasolli HA, Fuchs E. Mammalian formin-1 participates in adherens junctions and polymerization of linear actin cables. Nat Cell Biol. 2004;6(1):21–30. | |
Carramusa L, Ballestrem C, Zilberman Y, et al. Mammalian diaphanous-related formin Dia1 controls the organization of E-cadherin-mediated cell-cell junctions. J Cell Sci. 2007;120 Pt 21:3870–3882. | |
Chesarone MA, DuPage AG, Goode BL. Unleashing formins to remodel the actin and microtubule cytoskeletons. Nat Rev Mol Cell Biol. 2010;11(1):62–74. | |
Mason FM, Tworoger M, Martin AC. Apical domain polarization localizes actin-myosin activity to drive ratchet-like apical constriction. Nat Cell Biol. 2013;15(8):926–936. | |
Han SP, Gambin Y, Gomez GA, et al. Cortactin scaffolds Arp2/3 and WAVE2 at the epithelial zonula adherens. J Biol Chem. 2014; 289(11):7764–7775. | |
Kovacs EM, Goodwin M, Ali RG, et al. Cadherin-directed actin assembly: E-cadherin physically associates with the Arp2/3 complex to direct actin assembly in nascent adhesive contacts. Curr Biol. 2002;12(5):379–382. | |
Mullins RD, Stafford WF, Pollard TD. Structure, subunit topology, and actin-binding activity of the Arp2/3 complex from Acanthamoeba. J Cell Biol. 1997;136(2):331–343. | |
Goley ED, Ohkawa T, Mancuso J, et al. Dynamic nuclear actin assembly by Arp2/3 complex and a baculovirus WASP-like protein. Science. 2006;314(5798):464–467. | |
Goley ED, Welch MD. The ARP2/3 complex: an actin nucleator comes of age. Nat Rev Mol Cell Biol. 2006;7(10):713–726. | |
Mullins RD, Heuser JA, Pollard TD. The interaction of Arp2/3 complex with actin: nucleation, high affinity pointed end capping, and formation of branching networks of filaments. Proc Natl Acad Sci U S A. 1998;95(11):6181–6186. | |
Rotty JD, Wu C, Bear JE. New insights into the regulation and cellular functions of the ARP2/3 complex. Nat Rev Mol Cell Biol. 2013;14(1):7–12. | |
Suetsugu S. Activation of nucleation promoting factors for directional actin filament elongation: allosteric regulation and multimerization on the membrane. Semin Cell Dev Biol. 2013;24(4):267–271. | |
Takenawa T, Suetsugu S. The WASP-WAVE protein network: connecting the membrane to the cytoskeleton. Nat Rev Mol Cell Biol. 2007;8(1):37–48. | |
Helwani FM, Kovacs EM, Paterson AD, et al. Cortactin is necessary for E-cadherin-mediated contact formation and actin reorganization. J Cell Biol. 2004;164(6):899–910. | |
Shimamura S, Sasaki K, Tanaka M. The Src substrate SKAP2 regulates actin assembly by interacting with WAVE2 and cortactin proteins. J Biol Chem. 2013;288(2):1171–1183. | |
Bravo-Cordero JJ, Magalhaes MA, Eddy RJ, et al. Functions of cofilin in cell locomotion and invasion. Nat Rev Mol Cell Biol. 2013;14(7):405–415. | |
Elam WA, Kang H, De la Cruz EM. Biophysics of actin filament severing by cofilin. FEBS Lett. 2013;587(8):1215–1219. | |
El Sayegh TY, Arora PD, Ling K, et al. Phosphatidylinositol-4,5 bisphosphate produced by PIP5KIgamma regulates gelsolin, actin assembly, and adhesion strength of N-cadherin junctions. Mol Biol Cell. 2007;18(8):3026–3038. | |
Haviv L, Gillo D, Backouche F, et al. A cytoskeletal demolition worker: myosin II acts as an actin depolymerization agent. J Mol Biol. 2008;375(2):325–330. | |
Murrell MP, Gardel ML. F-actin buckling coordinates contractility and severing in a biomimetic actomyosin cortex. Proc Natl Acad Sci U S A. 2012;109(51):20820–20825. | |
Vogel SK, Petrasek Z, Heinemann F, et al. Myosin motors fragment and compact membrane-bound actin filaments. Elife. 2013;2:e00116. | |
Vicente-Manzanares M, Ma X, Adelstein RS, et al. Non-muscle myosin II takes centre stage in cell adhesion and migration. Nat Rev Mol Cell Biol. 2009;10(11):778–790. | |
Preller M, Manstein DJ. Myosin structure, allostery, and mechano-chemistry. Structure. 2013;21(11):1911–1922. | |
Levayer R, Lecuit T. Biomechanical regulation of contractility: spatial control and dynamics. Trends Cell Biol. 2012;22(2):61–81. | |
Finer JT, Simmons RM, Spudich JA. Single myosin molecule mechanics: piconewton forces and nanometre steps. Nature. 1994; 368(6467):113–119. | |
Finer JT, Mehta AD, Spudich JA. Characterization of single actin-myosin interactions. Biophys J. 1995;68(4 Suppl):291S–297S. | |
Sellers JR, Wang F, Chantler PD. Trifluoperazine inhibits the MgATPase activity and in vitro motility of conventional and unconventional myosins. J Muscle Res Cell Motil. 2003;24(8):579–585. | |
Kovacs M, Toth J, Nyitray L, et al. Two-headed binding of the unphosphorylated nonmuscle heavy meromyosin. ADP complex to actin. Biochemistry. 2004;43(14):4219–4226. | |
Bertet C, Sulak L, Lecuit T. Myosin-dependent junction remodelling controls planar cell intercalation and axis elongation. Nature. 2004; 429(6992):667–671. | |
Riddick N, Ohtani K, Surks HK. Targeting by myosin phosphatase-RhoA interacting protein mediates RhoA/ROCK regulation of myosin phosphatase. J Cell Biochem. 2008;103(4):1158–1170. | |
Khasnis M, Nakatomi A, Gumpper K, et al. Reconstituted human myosin light chain phosphatase reveals distinct roles of two inhibitory phosphorylation sites of the regulatory subunit, MYPT1. Biochemistry. 2014;53(16):2701–2709. | |
Heissler SM, Manstein DJ. Nonmuscle myosin-2: mix and match. Cell Mol Life Sci. 2013;70(1):1–21. | |
McLachlan RW, Yap AS. Protein tyrosine phosphatase activity is necessary for E-cadherin-activated Src signaling. Cytoskeleton (Hoboken). 2011;68(1):32–43. | |
Ratheesh A, Gomez GA, Priya R, et al. Central spindlin and alpha-catenin regulate Rho signalling at the epithelial zonula adherens. Nat Cell Biol. 2012;14(8):818–828. | |
Ebrahim S, Fujita T, Millis BA, et al. NMII forms a contractile transcellular sarcomeric network to regulate apical cell junctions and tissue geometry. Curr Biol. 2013;23(8):731–736. | |
Reyes CC, Jin M, Breznau EB, et al. Anillin regulates cell-cell junction integrity by organizing junctional accumulation of Rho-GTP and actomyosin. Curr Biol. 2014;24(11):1263–1270. | |
Gomez GA, McLachlan RW, Yap AS. Productive tension: force-sensing and homeostasis of cell-cell junctions. Trends Cell Biol. 2011;21(9):499–505. | |
Rauzi M, Lenne PF, Lecuit T. Planar polarized actomyosin contractile flows control epithelial junction remodelling. Nature. 2010;468(7327):1110–1114. | |
Liu Z, Tan JL, Cohen DM, et al. Mechanical tugging force regulates the size of cell-cell junctions. Proc Natl Acad Sci U S A. 2010;107(22):9944–9949. | |
Huveneers S, Oldenburg J, Spanjaard E, et al. Vinculin associates with endothelial VE-cadherin junctions to control force-dependent remodeling. J Cell Biol. 2012;196(5):641–652. | |
Levayer R, Lecuit T. Oscillation and polarity of E-cadherin asymmetries control actomyosin flow patterns during morphogenesis. Dev Cell. 2013;26(2):162–175. | |
Vasquez CG, Tworoger M, Martin AC. Dynamic myosin phosphorylation regulates contractile pulses and tissue integrity during epithelial morphogenesis. J Cell Biol. 2014;206(3):435–450. | |
Courtemanche N, Lee JY, Pollard TD, et al. Tension modulates actin filament polymerization mediated by formin and profilin. Proc Natl Acad Sci U S A. 2013;110(24):9752–9757. | |
Ishiyama N, Tanaka N, Abe K, et al. An autoinhibited structure of alpha-catenin and its implications for vinculin recruitment to adherens junctions. J Biol Chem. 2013;288(22):15913–15925. | |
Yao M, Qiu W, Liu R, et al. Force-dependent conformational switch of alpha-catenin controls vinculin binding. Nat Commun. 2014;5:4525. | |
Hansen SD, Mullins RD. VASP is a processive actin polymerase that requires monomeric actin for barbed end association. J Cell Biol. 2010;191(3):571–584. | |
Leckband DE, de Rooij J. Cadherin adhesion and mechanotransduction. Annu Rev Cell Dev Biol. 2014;30:291–315. | |
Grashoff C, Hoffman BD, Brenner MD, et al. Measuring mechanical tension across vinculin reveals regulation of focal adhesion dynamics. Nature. 2010;466(7303):263–266. | |
Borghi N, Sorokina M, Shcherbakova OG, et al. E-cadherin is under constitutive actomyosin-generated tension that is increased at cell-cell contacts upon externally applied stretch. Proc Natl Acad Sci U S A. 2012;109(31):12568–12573. | |
Conway DE, Breckenridge MT, Hinde E, et al. Fluid shear stress on endothelial cells modulates mechanical tension across VE-cadherin and PECAM-1. Curr Biol. 2013;23(11):1024–1030. | |
Le Duc Q, Shi Q, Blonk I, et al. Vinculin potentiates E-cadherin mechanosensing and is recruited to actin-anchored sites within adherens junctions in a myosin II-dependent manner. J Cell Biol. 2010;189(7):1107–1115. | |
Weber GF, Bjerke MA, DeSimone DW. A mechanoresponsive cadherin-keratin complex directs polarized protrusive behavior and collective cell migration. Dev Cell. 2012;22(1):104–115. | |
Engl W, Arasi B, Yap LL, et al. Actin dynamics modulate mechanosensitive immobilization of E-cadherin at adherens junctions. Nat Cell Biol. 2014;16(6):587–594. | |
Yonemura S, Wada Y, Watanabe T, et al. Alpha-catenin as a tension transducer that induces adherens junction development. Nat Cell Biol. 2010;12(6):533–542. | |
Choi HJ, Pokutta S, Cadwell GW, et al. AlphaE-catenin is an autoinhibited molecule that coactivates vinculin. Proc Natl Acad Sci U S A. 2012;109(22):8576–8581. | |
Rangarajan ES, Izard T. Dimer asymmetry defines alpha-catenin interactions. Nat Struct Mol Biol. 2013;20(2):188–193. | |
del Rio A, Perez-Jimenez R, Liu R, et al. Stretching single talin rod molecules activates vinculin binding. Science. 2009;323(5914):638–641. | |
Chuan P, Spudich JA, Dunn AR. Robust mechanosensing and tension generation by myosin VI. J Mol Biol. 2011;405(1):105–112. | |
Uyeda TQ, Iwadate Y, Umeki N, et al. Stretching actin filaments within cells enhances their affinity for the myosin II motor domain. PLoS One. 2011;6(10):e26200. | |
Hogan C, Serpente N, Cogram P, et al. Rap1 regulates the formation of E-cadherin-based cell-cell contacts. Mol Cell Biol. 2004;24(15):6690–6700. | |
Price LS, Hajdo-Milasinovic A, Zhao J, et al. Rap1 regulates E-cadherin-mediated cell-cell adhesion. J Biol Chem. 2004;279(34):35127–35132. | |
Priya R, Yap AS, Gomez GA. E-cadherin supports steady-state Rho signaling at the epithelial zonula adherens. Differentiation. 2013;86(3):133–140. | |
Laprise P, Chailler P, Houde M, et al. Phosphatidylinositol 3-kinase controls human intestinal epithelial cell differentiation by promoting adherens junction assembly and p38 MAPK activation. J Biol Chem. 2002;277(10):8226–8234. | |
Truffi M, Dubreuil V, Liang X, et al. RPTPalpha controls epithelial adherens junctions, linking E-cadherin engagement to c-Src-mediated phosphorylation of cortactin. J Cell Sci. 2014;127 Pt 11:2420–2432. | |
McLachlan RW, Kraemer A, Helwani FM, et al. E-cadherin adhesion activates c-Src signaling at cell-cell contacts. Mol Biol Cell. 2007; 18(8):3214–3223. | |
Bays JL, Peng X, Tolbert CE, et al. Vinculin phosphorylation differentially regulates mechanotransduction at cell-cell and cell-matrix adhesions. J Cell Biol. 2014;205(2):251–263. | |
Tzima E, Irani-Tehrani M, Kiosses WB, et al. A mechanosensory complex that mediates the endothelial cell response to fluid shear stress. Nature. 2005;437(7057):426–431. | |
Dawes-Hoang RE, Parmar KM, Christiansen AE, et al. folded gastrulation, cell shape change and the control of myosin localization. Development. 2005;132(18):4165–4178. | |
Martin AC, Kaschube M, Wieschaus EF. Pulsed contractions of an actin-myosin network drive apical constriction. Nature. 2009; 457(7228):495–499. | |
Sawyer JK, Harris NJ, Slep KC, et al. The Drosophila afadin homologue Canoe regulates linkage of the actin cytoskeleton to adherens junctions during apical constriction. J Cell Biol. 2009;186(1):57–73. | |
Sawyer JK, Choi W, Jung KC, et al. A contractile actomyosin network linked to adherens junctions by Canoe/afadin helps drive convergent extension. Mol Biol Cell. 2011;22(14):2491–2508. | |
Zallen JA, Blankenship JT. Multicellular dynamics during epithelial elongation. Semin Cell Dev Biol. 2008;19(3):263–270. | |
Levayer R, Pelissier-Monier A, Lecuit T. Spatial regulation of Dia and Myosin-II by RhoGEF2 controls initiation of E-cadherin endocytosis during epithelial morphogenesis. Nat Cell Biol. 2011;13(5):529–540. | |
Roh-Johnson M, Shemer G, Higgins CD, et al. Triggering a cell shape change by exploiting preexisting actomyosin contractions. Science. 2012;335(6073):1232–1235. | |
Menko AS, Bleaken BM, Walker JL. Regional-specific alterations in cell-cell junctions, cytoskeletal networks and myosin-mediated mechanical cues coordinate collectivity of movement of epithelial cells in response to injury. Exp Cell Res. 2014;322(1):133–148. | |
Hogan C, Dupre-Crochet S, Norman M, et al. Characterization of the interface between normal and transformed epithelial cells. Nat Cell Biol. 2009;11(4):460–467. | |
Leung CT, Brugge JS. Outgrowth of single oncogene-expressing cells from suppressive epithelial environments. Nature. 2012;482(7385):410–413. | |
Eisenhoffer GT, Loftus PD, Yoshigi M, et al. Crowding induces live cell extrusion to maintain homeostatic cell numbers in epithelia. Nature. 2012;484(7395):546–549. | |
Marinari E, Mehonic A, Curran S, et al. Live-cell delamination counterbalances epithelial growth to limit tissue overcrowding. Nature. 2012;484(7395):542–545. | |
Slattum G, McGee KM, Rosenblatt J. P115 RhoGEF and microtubules decide the direction apoptotic cells extrude from an epithelium. J Cell Biol. 2009;186(5):693–702. | |
Grieve AG, Rabouille C. Extracellular cleavage of E-cadherin promotes epithelial cell extrusion. J Cell Sci. 2014;127 Pt 15:3331–3346. | |
Abreu-Blanco MT, Verboon JM, Liu R, et al. Drosophila embryos close epithelial wounds using a combination of cellular protrusions and an actomyosin purse string. J Cell Sci. 2012;125 Pt 24:5984–5997. | |
Ma X, Lynch HE, Scully PC, et al. Probing embryonic tissue mechanics with laser hole drilling. Phys Biol. 2009;6(3):036004. | |
Antunes M, Pereira T, Cordeiro JV, et al. Coordinated waves of actomyosin flow and apical cell constriction immediately after wounding. J Cell Biol. 2013;202(2):365–379. | |
Takada H, Furuya K, Sokabe M. Mechanosensitive ATP release from hemichannels and Ca2+ influx through TRPC6 accelerate wound closure in keratinocytes. J Cell Sci. 2014;127(19):4159–4171. | |
Abreu-Blanco MT, Verboon JM, Parkhurst SM. Cell wound repair in Drosophila occurs through three distinct phases of membrane and cytoskeletal remodeling. J Cell Biol. 2011;193(3):455–464. | |
Danjo Y, Gipson IK. Actin ‘purse string’ filaments are anchored by E-cadherin-mediated adherens junctions at the leading edge of the epithelial wound, providing coordinated cell movement. J Cell Sci. 1998;111 Pt 22:3323–3332. | |
Abreu-Blanco MT, Watts JJ, Verboon JM, et al. Cytoskeleton responses in wound repair. Cell Mol Life Sci. 2012;69(15):2469–2483. | |
Poujade M, Grasland-Mongrain E, Hertzog A, et al. Collective migration of an epithelial monolayer in response to a model wound. Proc Natl Acad Sci U S A. 2007;104(41):15988–15993. | |
Anon E, Serra-Picamal X, Hersen P, et al. Cell crawling mediates collective cell migration to close undamaged epithelial gaps. Proc Natl Acad Sci U S A. 2012;109(27):10891–10896. | |
Chapnick DA, Liu X. Leader cell positioning drives wound-directed collective migration in TGFbeta-stimulated epithelial sheets. Mol Biol Cell. 2014;25(10):1586–1593. | |
Hwang S, Zimmerman NP, Agle KA, et al. E-cadherin is critical for collective sheet migration and is regulated by the chemokine CXCL12 protein during restitution. J Biol Chem. 2012;287(26):22227–22240. | |
Li L, Hartley R, Reiss B, et al. E-cadherin plays an essential role in collective directional migration of large epithelial sheets. Cell Mol Life Sci. 2012;69(16):2779–2789. | |
Vedula SR, Hirata H, Nai MH, et al. Epithelial bridges maintain tissue integrity during collective cell migration. Nat Mater. 2014;13(1):87–96. | |
Heisenberg CP, Bellaiche Y. Forces in tissue morphogenesis and patterning. Cell. 2013;153(5):948–962. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.