Back to Archived Journals » Research and Reports in Medicinal Chemistry » Volume 4
Current perspectives on applications of shikimic and aminoshikimic acids in pharmaceutical chemistry
Authors Díaz Quiroz DC, Carmona SB, Bolívar F, Escalante A
Received 13 March 2014
Accepted for publication 7 May 2014
Published 22 July 2014 Volume 2014:4 Pages 35—46
DOI https://doi.org/10.2147/RRMC.S46560
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Dulce Catalina Díaz Quiroz, Susy Beatriz Carmona, Francisco Bolívar, Adelfo Escalante
Department of Cellular Engineering and Biocatalysis, Institute of Biotechnology, Universidad Nacional Autónoma de México, Cuernavaca, Morelos, México
Abstract: Aromatic metabolism comprises the shikimic acid (SA) and the aminoshikimic acid (ASA) pathways. The SA pathway is the common route for the biosynthesis of aromatic amino acids and other metabolites in bacteria, higher plants, fungi, and Apicomplexa parasites, but this pathway is absent in mammals. A variant of the SA pathway known as the ASA pathway branches off from the normal pathway in some bacteria, and its final product, 3-amino-5-hydroxybenzoic acid, is the precursor for many aminoglycoside antibiotics such as kanamycin, neomycin, butirosin, and spectinomycin. The SA pathway includes the key intermediate SA, which is the precursor for the chemical synthesis of the drug oseltamivir phosphate, known commercially as Tamiflu®, an efficient inhibitor of the neuraminidase enzyme of the seasonal influenza viruses types A and B, avian influenza virus H5N1, and human influenza virus H1N1. Meanwhile, the intermediate of the ASA pathway, ASA, is an attractive candidate for use as the core scaffold for the synthesis of combinatorial libraries and is a potential alternative to SA as a precursor for oseltamivir phosphate synthesis. In this review, we discuss the relevance of the key intermediates SA and ASA as scaffold molecules for the synthesis of diverse chemicals. We highlight the current and potential pharmaceutical applications of these molecules and discuss the main strategies for the production of these aromatic compounds from natural sources and the application of metabolic engineering strategies in diverse bacterial strains for production through biotechnological processes.
Keywords: aromatic metabolism, shikimic acid, aminoshikimic acid, oseltamivir phosphate, medical applications, metabolic engineering
Introduction
Biosynthetic aromatic metabolism is an essential pathway in microorganisms, plants, and some eukaryotic organisms. These pathways comprise a great diversity of compounds involved in several essential biological processes, such as the synthesis of the structural blocks for protein synthesis, siderophores, electron-carrier compounds, secondary metabolites such as pigments, and antimicrobial substances. Aromatic metabolism occurs through the shikimic acid (SA) and the aminoshikimic acid (ASA) pathways. The SA pathway is the common route for the biosynthesis of aromatic amino acids and metabolites such as folate cofactors, ubiquinone, and the vitamins E and K. This biosynthetic pathway is present in bacteria, fungi, higher plants, and certain Apicomplexa parasites, specifically Plasmodium falciparum, Toxoplasma gondii, and Cryptosporidium parvum, but it is absent in mammals.1–3 The SA pathway comprises seven enzymatic reactions, beginning with the condensation of the central carbon metabolism (CCM) intermediates phosphoenolpyruvate (PEP) and erythrose-4-phosphate (E4P) to generate 3-deoxy-D-arabino-heptulosonate 7-phosphate (DAHP). The final product of the pathway is chorismic acid (CHA), which is the common metabolic intermediate for the biosynthesis of diverse aromatic compounds including aromatic amino acids (tryptophan, tyrosine, and phenylalanine), pigments, electron-carrier molecules, and some secondary metabolites that are synthesized through specific pathways.1,2,4 Diverse microorganisms belonging to the phyla Actinobacteria and Proteobacteria, including bacteria of the genera Streptomyces, Burkholderia, Pseudomonas, Pectobacterium, and Brevibacterium, possess a metabolic pathway related to the SA pathway involved in the microbial production of a family of more than 100 compounds known as phenazines, which are colored secondary metabolites. Although the biological role of these compounds is not well understood, they exhibit activity as siderophores, electron-carriers, broad-spectrum antimicrobial agents and cytotoxic effects against eukaryotic cells. Additionally, other electron-carrier compounds known as methanophenazines are produced by Methanosarcina mazei, the unique Archaea producing phenazines.5–8
A variant of the SA pathway known as the ASA pathway was proposed to branch off from the normal pathway at the stage of 4-amino-3,4-dideoxyheptulosonic acid 7-phosphate (aminoDAHP).9 This pathway is present in microorganisms such as Amycolatopsis mediterranei, Actinosynnema pretiosum, and Streptomyces collinus, and it includes a 5-deoxy-5-amino-3-dehydroquinic acid (aminoDHQ) synthase that cyclizes aminoDAHP to aminoDHQ, which is in turn dehydrated to generate 5-amino-5-deoxy-3-dehydroshikimic acid (aminoDHS). The final step of this pathway is the biosynthesis of 3-amino-5-hydroxybenzoic acid (AHBA) by AHBA synthase, which aromatizes aminoDHS. AHBA is the precursor for many aminoglycoside antibiotics, such as kanamycin, neomycin, butirosin, and spectinomycin.7,9
SA is a key intermediate of the SA pathway, and it has gained relevance as the substrate for the chemical synthesis of the drug oseltamivir phosphate (OSP), known commercially as Tamiflu® (Genentech, Inc., South San Francisco, CA, USA); this drug is an efficient inhibitor of the surface protein neuraminidase (NA) enzyme of the seasonal influenza virus types A and B, avian influenza virus H5N1, and human influenza virus H1N1.4,10 Because of this important application, the production of SA from different sources has gained great attention and has been extensively reviewed.4,10–13 Scientific and industrial interest in the ASA pathway is mainly focused on the production of AHBA as the precursor for the synthesis of aminoglycoside antibiotics;5,7,9,14 however, another intermediate of this pathway, ASA, is an attractive candidate for use as the core scaffold for the synthesis of combinatorial libraries alternative to SA as a precursor for the synthesis of OSP.14
In this review, we discuss the relevance of the key intermediates SA and ASA as scaffold molecules for the synthesis of diverse chemicals; their current and potential pharmaceutical applications; current strategies for the production of these aromatic compounds from natural sources; and the application of metabolic engineering strategies in diverse bacterial strains for production through biotechnological processes.
Biosynthetic pathways for SA and ASA production
SA is an intermediate of the common biosynthetic pathway for aromatic compounds, the SA pathway, which links CCM to the biosynthesis of aromatic compounds. This pathway begins with the condensation of PEP and E4P to generate DAHP. The Gram-negative bacterium Escherichia coli has three different DAHP synthases (DAHPS), isoenzymes AroF, AroG, and AroH (encoded by the aroF, aroG, and aroH genes, respectively). These three enzymes are subject to feedback inhibition by the aromatic amino acids tyrosine, phenylalanine, and tryptophan, respectively, because each isoenzyme possesses a distinct regulatory domain inserted into the enzyme core.15 DAHP is transformed to generate 3-dehydroquinic acid (DHQ); this reaction is catalyzed by DHQ synthase (encoded by aroB). In the next step, DHQ loses a water molecule and is transformed into 3-dehydroshikimic acid (DHS) by DHQ dehydratase. DHS is converted into SA by SA dehydrogenase. The next step in the pathway is the formation of shikimic acid-3-phosphate; in E. coli, this step is catalyzed by the SA kinase isoenzymes I and II. Subsequently, 3-enolpyruvyl-shikimate-5-P (EPSP) synthase catalyzes the conversion of shikimic acid-3-phosphate into EPSP; finally, CHA synthase converts EPSP to CHA (Figure 1), the common precursor for the biosynthesis of aromatic amino acids and other derivatives (Figure 2). In E. coli, the enzymes DHQ synthase, DHQ dehydratase, and SA dehydrogenase are expressed constitutively, unlike DAHPS and SA kinase II, which are transcriptionally regulated. Furthermore, in E. coli, DHQ and DHS are transformed enzymatically to quinic acid (QA) and gallic acid (GA), other aromatic byproducts of the pathway.1,4,10,16–18
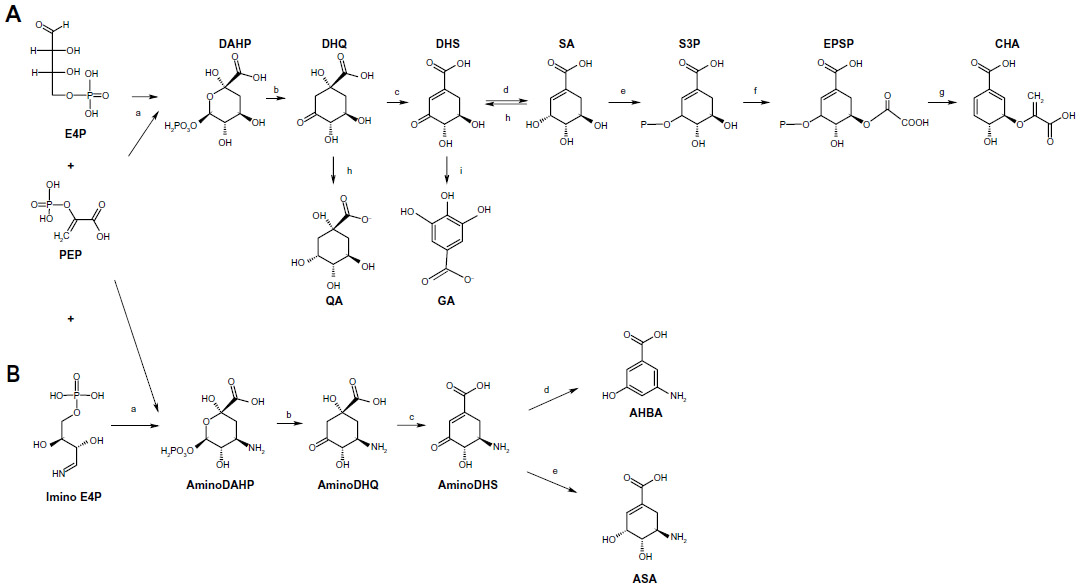 | Figure 1 Microbial SA and ASA biosynthetic pathways. |
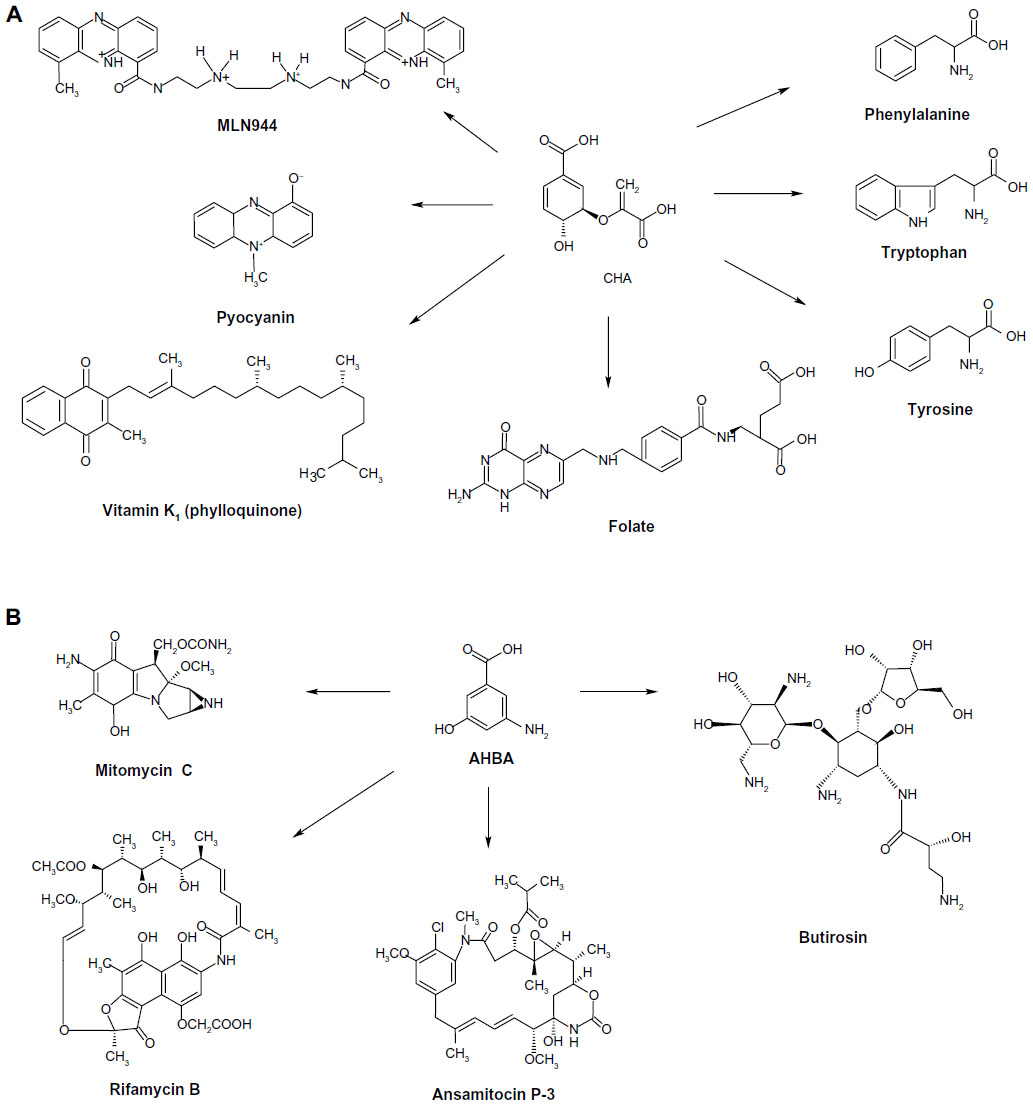 | Figure 2 Diverse pharmaceutical products derived from CHA and AHBA. |
The ASA pathway was proposed to be a variant of the SA pathway because SA was considered the source for the synthesis of C7N units consisting of a six-membered carbocycle with an extra carbon and a nitrogen in a meta arrangement, which is a characteristic structural component of all ansamycin, geldanamycin, and maytansinoid antibiotics; in these antibiotics, the C7N compounds are the starter units from which the chain is assembled by polyketide-type biosynthesis (Figure 2). Furthermore, C7N units are combined with a carbohydrate-derived moiety in the antibiotics of the mitomycin family. However, the SA pathway intermediates SA, QA, and DHQ cannot be incorporated into the C7N units of ansamycins and mitomycins, suggesting that biosynthesis of the C7N unit diverges from the main SA pathway before DHQ.5
Comparison of biosynthetic mechanisms of C7N unit-containing antibiotics enabled the identification of the naturally occurring amino acid AHBA as the possible precursor for ansamycin structures; this amino acid was chemically synthesized in labeled form and efficiently incorporated into naphthalenic and benzenic ansamycins and into mitomycin.5 This conclusion is consistent with physiological complementation experiments using a mutant of A. mediterranei blocked in the early steps of rifamycin biosynthesis;7 the biosynthetic pathway in this mutant was complemented by supplementation of AHBA as a unique substrate. Degradation experiments using the antibiotic mitomycin suggested that aminoDAHP generated from DAHP is the specific precursor of the C7N units.5,7 In the proposed ASA pathway, aminoDAHP is formed by the reaction of E4P with ammonia, generated in situ by hydrolysis of glutamine, to form an imine, which is then condensed with PEP. AminoDAHP fuels a pathway composed of reactions paralleling the normal SA pathway: aminoDAHP is cyclized to aminoDHQ, dehydrated to aminoDHS, and then aromatized to AHBA by a postulated AHBA synthase in a reaction that has no analogous step in the normal SA pathway.7
The first complete microbial AHBA biosynthetic pathway was established in A. mediterranei and involves the intersection of kanosamine biosynthesis, iminoE4P biosynthesis, and a modification of the above-described ASA pathway; this modification involves a reduction of the aminoDHS intermediate by ASA dehydrogenase, which is encoded by the rifI gene (Figure 1).14,19
SA derivatives in the pharmaceutical industry
SA is an industrially important chiral compound that is a precursor for the synthesis of many chemical substances used in the pharmaceutical, chemical, and cosmetic industries.4,10 The pharmaceutical applications of SA include its use as an antipyretic, antioxidant, anticoagulant, antithrombotic, anti-inflammatory, and analgesic agent. Furthermore, SA has a key role in the synthesis of important pharmacological compounds such as anticancer agents, antibacterial agents, and hormones;3,4,20,21 however, the most important application of SA is its use as the substrate for the chemical synthesis of OSP.4,11,20,22 Because the chemical structure of ASA is similar to that of SA, it has similar applications, including its potential use as a substrate for OSP synthesis, which might significantly improve the speed and cost of the production of OSP or other oseltamivir carboxylates (Figure 3).14,19 However, no other applications for the synthesis of pharmaceutically valuable compounds have been observed.
 | Figure 3 Diverse pharmaceutical products derived from SA and ASA. |
Antitumor activity
The polyoxygenated cyclohexane (–)-zeylenone exhibits anticancer, antiviral, and antibiotic activities; it can inhibit nucleoside transport in Ehrlich carcinoma cells, and it is cytotoxic to several human tumor cell lines. An improved route for the enantioselective synthesis of (–)-zeylenone from SA has been observed.23 The 4-epi-shikimic acid skeleton is a constituent of several antitumor products, such as dioxolamycin, the cythiaformines B, C, D, and pericosine A.24 Platinum (II) anticancer drugs are water-soluble complexes that retain antitumor activity and are effectively absorbed after oral administration; SA complexes of platinum (II) are active against L1210 and P388 leukemia and B16 melanoma in vivo.25 Because the use of amines as carrier ligands may provide broad-spectrum antitumor activity, diamine-coupled SA complexes of platinum have been studied.26
Antithrombotic activity
SA can inhibit platelet aggregation induced by ADP (adenosine 5’-diphosphate) and collagen in vitro, as well as platelet aggregation induced by ADP in rats subjected to middle cerebral artery thrombosis.27 SA can elevate the blood coagulation time of mice, reduce the volume of cerebral infarction, decrease neurological deficit scores, abate brain edema, and increase cerebral blood flow in ischemic areas.28 To increase its liposolubility and bioavailability, the triacetyl, monopalmityloxy, and isopropylidene derivatives of SA (among others) have been synthetized and evaluated for their physiological effects as antithrombotic agents and protective agents in brain damage induced by focal cerebral ischemia. Triacetyl SA exhibits anticoagulant and antithrombotic activities in rats.29 In addition to its antithrombotic effects, monopalmityloxy SA can lengthen coagulation time.30 The 3,4-oxo-isopropylidene SA has anticoagulant and antithrombotic activities,31 and it inhibits adhesion of polymorphonuclear leukocytes to tumor necrosis factor alpha (TNF-α)-induced endothelial cells in vitro.32 This derivative has been used to generate drugs for treating ulcerative colitis.33
Miscellaneous anti-biological activities
Because the SA pathway is absent in mammals, the key intermediate SA is considered a potential molecule for the design and synthesis of enzyme inhibitors used as antibacterial, fungicidal, and herbicidal agents, which may selectively inhibit enzyme-catalyzed reactions in microbes and weeds without negative effects in mammals.3,20,34 Notably, this pathway exists in Apicomplexa parasites (causing malaria, pneumonia, and tuberculosis), paving the way for the design of new anti-parasite compounds.3,35 Synthesis of SA and its analogs is an area of active research focused on enzymatic mechanisms and design of inhibitors, as exemplified by the development of the influenza virus NA inhibitor (NAI) OSP, the glyoxalase I inhibitor COCT (2-crotonyloxymethyl-(4R,5R,6R)-4,5,6-trihydroxycyclohex-2-enone),34 and the α-glycosidase inhibitor valiolamine.36
SA and ASA as precursors for OSP synthesis
Influenza is a disease of the respiratory system that affects several million people each winter. Seasonal influenza and subtypes, such as the avian virus H5N1 and the pandemic strain of influenza A virus H1N1 of swine origin, emerged in March–April of 2009, necessitating the development of potent antiviral drugs for the prophylaxis and treatment of the influenza virus infections. SA has attracted worldwide attention because it is the substrate for industrial synthesis of the phosphate salt of the tri-substituted cyclohexene ethyl carboxylate OSP.10
The influenza virus is a single-stranded RNA virus that belongs to the family Orthomyxoviridae with a lipid-containing envelope and an eight-segmented genome encoding eleven or 12 proteins.37 Among the known influenza viruses, types A, B, and C, only type A has caused pandemic illness.38 The virions have three surface proteins: the hemagglutinin (HA), NA, and M2 proteins. Influenza A is further subtyped by the glycoproteins H (16 different molecules) and N (nine molecules).39 The influenza A virus replication cycle has been successfully mapped.40 The virus initiates its life cycle when HA binds to terminal sialic acids on cellular receptors, after which the virus is endocytosed. The low pH of the endosome activates the M2 proton channel in the influenza A viral membrane, which allows protons to enter to the viral core, triggering the fusion of the viral and endosomal membranes with subsequent release of the viral ribonucleoprotein (RNP) to the cytoplasm and its subsequent transport to the nucleus. The nuclear viral RNP then serves as the template for the production of messenger RNA. Beneath the cytoplasmic membrane, the RNP assembles under patches of HA and NA to form virions. The interaction of the viral HA with host sialic acid must be disrupted to release the infectious virion progeny; this is accomplished by the sialidase activity of NA.41
There are two major classes of antivirals licensed for the treatment and prevention of influenza: the M2 inhibitors (adamantanes) and the NAI.42 By blocking the M2 proton channel, the M2 inhibitors prevent release of the viral RNP for migration to the nucleus of the infected cell. The NAI prevent release of newly formed virions from the cell surface. The first class of influenza antivirals, the adamantanes, were developed nearly 5 decades ago, in the 1960s, but several viruses have developed resistance to these drugs, and treatment of influenza infections by this class of drugs is currently not recommended by the World Health Organization (WHO). The NAI were developed in the 1990s. Several specific NAI have been developed; however, only four are currently licensed for the treatment and prophylaxis of influenza infection: zanamivir (RELENZA®, GlaxoSmithKline, London, UK), OSP (Tamiflu®), and the Japan- and South Korea-licensed laninamivir and peramivir. OSP is administered orally and zanamivir and laninamivir by oral inhalation, whereas peramivir is administered by injection.43
Zanamivir and OSP constitute current therapy for the treatment of influenza.20 For the H1N1 virus, OSP therapy is generally associated with faster resolution of symptoms and more rapid clearance of viral shedding in hospitalized patients of all ages, organ transplant recipients, and pregnant women.44,45 Tamiflu® has a serum half-life of approximately 3 hours, whereas RELENZA® has a short half-life, low oral bioavailability, and requires topical application using disk inhaler technology.46
OSP is administered as the prodrug OSP and is converted by hepatic esterases to the active form oseltamivir carboxylate. High-affinity binding of OSP to NA requires reorientation of the residue Glu276 to create a pocket to accommodate the bulky hydrophobic side chain. Many mutations conferring reduced sensitivity to OSP (His274Tyr, Glu119Val, and Arg292Lys) cluster around the hydrophobic pocket, impairing Glu276 rotation.47 Resistance to the NAI can be both drug and virus type or subtype specific. Most mutations of the influenza A/H1 strains that result in high-level resistance to OSP still confer adequate susceptibility to zanamivir. The increased number of NAI-resistant mutations might be overcome by other newer antivirals such as conjugated sialidases (DAS181), inhibitors of the influenza RNA polymerase (T-705), antibodies against HA, and antisense strategies, which require further study.48
Tamiflu® is the only approved orally available drug for treatment and prevention of influenza infections.10,47 Therefore, the high demand for this drug is not adequately met by industrial production using SA, which was developed by Gilead (Gilead Sciences Inc., Foster City, CA, USA) and Roche (Hoffman-La Roche Ltd., Basel, Switzerland) chemists; notably, the current supply of Tamiflu® covers just 2% of the world population.49 To protect people from pandemic human or H5N1 avian influenza, OSP should be manufactured and stocked in every country worldwide.50 Topics of current research are the relatively new techniques for SA isolation from known and novel sources. According to the WHO Pandemic influenza preparedness and response guidance, threats of influenza pandemics will continue to emerge.51 Therefore, maintaining stockpiles of containment drugs such as Tamiflu® is necessary for managing a major outbreak.
SA production
Extraction from plant sources
Because SA is the substrate for the chemical synthesis of OSP, diverse methods have been developed to produce large-scale quantities including its chemical synthesis, plant source extraction, and biotechnological approaches using different engineered microorganisms or enzymes.4,13,52 Although several routes have been described for chemical synthesis, production of SA acid by these methods is too expensive and/or generates waste containing environmental pollutants; therefore, these are not commercially viable.4,10,53–59
Because plants synthesize aromatic amino acids and most polycyclic compounds by their SA pathway, the yield of SA from this source can vary; the factors that influence the yield include the time of harvest, handling, storage, plant tissue, and other factors that regulate plant metabolism resulting in the accumulation of SA in tissues where metabolic processes have slowed or stopped, such as seeds and fruits.60,61 Plants of the Illicium spp. are a good source of SA; the highest content has been reported to range from 4.99% (Illicium anisatum) to 24.42% (Illicium religiosum) (on a dry basis) (Table 1), but care must be taken to avoid the toxic species (>50) that contain the toxin anisatin, which is associated with neurotoxicity in infants.62,63 The fruit of the Chinese star anise (Illicium verum) tree yields, according to diverse sources, up to 17% of SA (dry basis content),62 one of the highest concentrations found in plants and with less anisatin compared to other Illicium spp.63 Studies have examined other plant sources for SA, but dry basis content ranged from 0.25% to 15.64% in 50 plant tissues out of 173 samples tested.62 In addition, efforts have been made to develop efficient methods of SA extraction from plant sources,11,20 including extraction with hot water from grounded seed pods of the Chinese star anise at temperatures ranging from 20°C to 200°C or higher to obtain recoveries of 100%,64 extraction with alcohols (eg, ethanol or methanol),65,66 acids or ionic liquid extraction followed by liquid–liquid separations, precipitation, filtration, and chromatographic purification.67,68
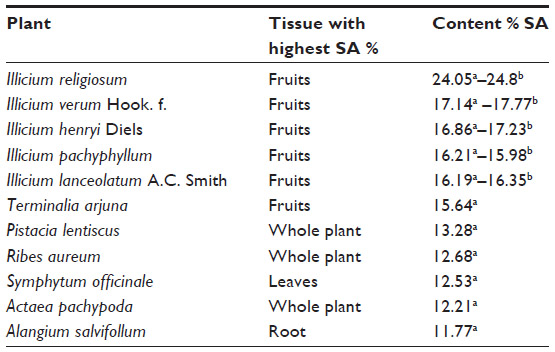 | Table 1 Highest content of SA (<10% weight/weight) in diverse plant sources |
China accounts for 80%–90% of the global star anise production.69 SA is extracted from the pods of the Chinese star anise, which is mainly grown in four mountain provinces in southwest China: Guanxi, Sichuan, Yunnan, and Guizhou. The tree bears fruit approximately 6 years after planting, and the SA manufacturing process is complex, proprietary, and requires considerable time even when the raw materials are available.70 OSP production by Roche starts with dried star anise, which is harvested between March and May. SA is extracted at the start of a ten-stage manufacturing process that takes 1 year, and 90% of the production is already used by Roche for the preparation of Tamiflu®.20 Approximately 30 kg of star anise is required to generate 1 kg of SA,22 and the supply of this source is susceptible to weather changes. The demand for Tamiflu® has increased recently, and Roche cannot meet this demand due to shortage of plant-derived SA.69 Therefore, alternative biotechnological strategies to produce SA, such as those using metabolically engineered bacterial strains of E. coli, Citrobacter freundii, and Bacillus subtilis, are important (Table 2).
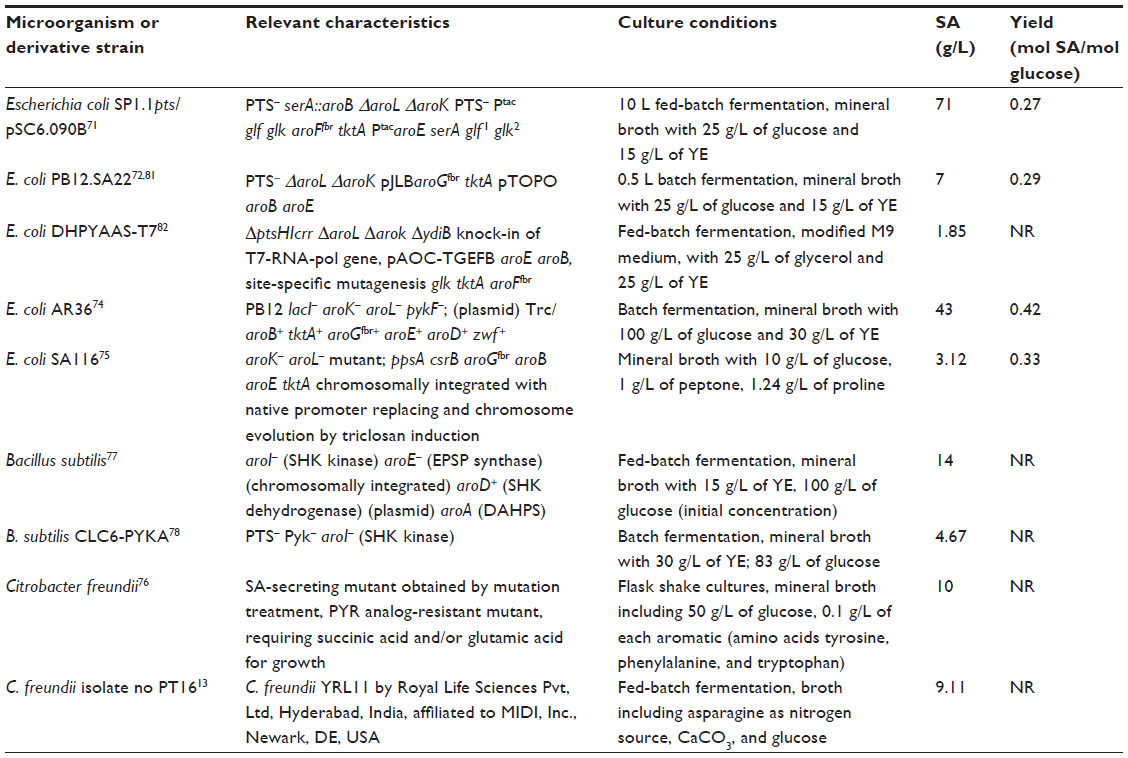 | Table 2 Microbial engineered/mutagenized overproducing strains for SA production |
Microbial production of SA
Most metabolic engineering efforts for SA production have focused on E. coli. Successful metabolically engineered strategies resulting in high SA production in E. coli include the use of PEP:glucose phosphotransferase system (PTS)-deficient mutants (PTS–) strains in which PTS, the main glucose transport system, is inactivated, resulting in high PEP availability;52,71–73 introduction of glucose transport and phosphorylation capabilities (additional copies of the glf and glk genes from Zymomonas mobilis encoding the glucose facilitator system [Glf] and glucokinase [Glk] enzymes, respectively,71 or the use of adaptive evolution to select E. coli derivatives that grow on glucose74); modification of CCM and pentose phosphate pathway including the introduction of an additional plasmid-encoded-copy of the zwf and tktA genes encoding the glucose 6-phosphate-1-dehydrogenase (Zwf) and transketolase I (TktA I) enzymes, respectively, resulting in the increased availability of E4P;71–73 modulation of the carbon flux from PEP to the tricarboxylic acid cycle by inactivation of the pyruvate kinase I (Pyk I) enzyme72,73 or conversion of pyruvate to PEP by a plasmid-encoded copy of ppsA coding for PEP synthase (PpsA) enzyme;71 modification of the SA pathway genes including the introduction of an additional plasmid-encoded copy DAHPS AroFfbr71 encoded by aroFfbr or DAHPS AroGfbr encoded by aroGfbr,72,73 or genes encoding enzymes from the SA pathway including the single or dual inactivation of the genes aroK and aroL encoding the SA kinase I and II enzymes, respectively; and introduction of additional plasmid-borne copies of genes encoding limiting enzymes of the pathway such as aroB, aroD, and aroE (Figure 1).71–73 A promising strategy by which to avoid the disadvantages associated with plasmid in engineered strains involves chromosomal integration of the ppsA, csrB, aroGfbr, aroB, aroE, and tktA genes encoding for PpsA, small regulatory RNA (CsrB), DAHPS AroGfbr, 3-dehydroquinic acid synthase (AroB), shikimic acid dehydrogenase (AroE), and TktA I, respectively, in an aroK– aroL– mutant strain, replacing their native promoters and subsequent chromosome evolution by triclosan induction.75 These genetically modified E. coli strains with specific genetic backgrounds are grown using diverse culture conditions, which has led to the successful overproduction of SA, with yields ranging from 0.08 to 0.42 mol SA/mol glucose (Table 2).
Other microorganisms, such as C. freundii and B. subtilis, have been explored for the production of SA. A C. freundii mutant strain that is resistant to pyruvate, vitamin B1, high SA concentrations, requiring succininc acid or glutamic acid for its growth, which requires succinic acid or glutamic acid for its growth, exhibited improved SA production efficiency and increased capacity to secrete extracellular SA.76 A B. subtilis strain carrying a defective shikimate kinase enzyme, deletion of the aroE gene encoding for EPSP synthase with enhanced shikimate dehydrogenase, and DAHPS activities successfully accumulated SA, while a Pyk−, PTS−, and shikimate kinase-deficient B. subtilis strain successfully accumulated DHS and SA.77,78
In addition to microbial fermentative processes, SA has been produced from glucose in a two-step procedure involving the cell-dried or membrane fractions from Gluconobacter oxydans with the initial conversion of QA → DHQ → DHS catalyzed by quinate dehydrogenase and DHQ dehydratase enzymes. This reaction is then coupled to a second step catalyzed by cytoplasmic enzymes: SA dehydrogenase (NADPH [dihydronicotinamide-adenine dinucleotide phosphate] + DHS → SA + NADP [nicotinamide adenine dinucleotide phosphate]) and the NADPH regenerating glutamate dehydrogenase (glucose + NADP → NADPH + glucono-δ-lactone), with a conversion efficiency of 57%–77% from QA. This enzymatic approach requires the preparation of cell-dried or membrane fractions and is, therefore, useful only for laboratory-scale synthesis.79
ASA production
Early studies on the ASA pathway in A. mediterranei focused on the production of AHBA, the precursor of the C7N units found in mitomycin and ansamycin antibiotics such as rifamycin B and ansamitocin. Purification of the last enzyme this pathway, AHBA synthase, which aromatizes aminoDHS to AHBA, and cloning of the rifK gene allowed the cloning, sequencing, and analysis of the entire 95 kb rifamycin (rif) biosynthetic gene cluster in A. mediterranei; this cluster contains seven genes rifGHJKLMN, which are involved in the biosynthesis of AHBA.9
Biosynthesis of ASA from glucose was observed in wild-type A. mediterranei ATCC 21789 using kanosamine and the iminoE4P biosynthesis and ASA pathways. The first microbial synthesis of ASA was achieved in this microorganism by overexpression of the rifI gene encoding ASA dehydrogenase. Alternatively, kanosamine synthesized by Bacillus pumilus was converted into ASA using a tandem, two-microbe route using B. pumilus and a recombinant E. coli strain for the final conversion of kanosamine to ASA (Figure 4).14,19 For commercial applications, this process requires two separate fermentations, with recovery of the kanosamine intermediate from B. pumilus cultures, which is expensive and decreases yields of ASA because of loss of kanosamine.19
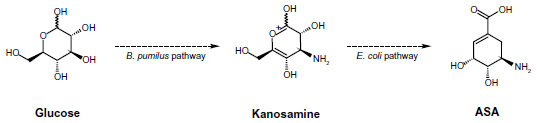 | Figure 4 Recombinant two-step pathway for ASA production. |
Different major chemical routes for the synthesis of OSP include 17-, 13-, ten-, and nine-step synthesis starting from SA.19,50 The ten-step route includes three explosive and/or toxic azide derivatives; the 17-step route is an azide-free process,19 but it is a long synthetic route, which significantly retards its use in the large-scale production of OSP.50 In the 17- and ten-step routes, approximately four of the steps are performed to add an amino group substituent at the 5-position of SA ring. ASA as a substrate for the synthesis of OSP might eliminate the requirement for those first four steps, which could significantly improve the speed and cost of the production of OSP and other oseltamivir carboxylates.19
Conclusion
SA is a key intermediate of the common biosynthetic pathway for aromatic compounds with diverse applications because it is the precursor for several pharmaceutical compounds, including antipyretics, antioxidants, anticoagulants, antithrombotics, anti-inflammatory agents, analgesic agents, or derivatives with anticancer, antibacterial, or hormonal applications. However, the most important application of SA is its use as substrate for the chemical synthesis of OSP, the effective NAI of diverse influenza viruses including stationary influenza, H5N1, and H1N1 types. The latent threat of pandemic human or H5N1 avian influenza necessitates the global production and availability of OSP. Because traditional plant sources for SA production are geographically restricted and insufficient to meet the increasing demand for SA, several successful methods have been developed by which to generate SA by biotechnological processes, applying diverse metabolic engineering strategies to obtain overproducing bacterial strains, mainly E. coli derivatives, resulting in the production of up to 70 g/L of SA with yields >0.40 mol SA/mol glucose in diverse fermentation conditions. The SA produced by microbes is then extracted and purified from fermentation broths and used as substrate for the chemical synthesis of OSP. Microbial production of ASA has only been observed in preliminary laboratory experiments, but the availability of ASA as the substrate for the OSP synthesis might significantly improve the production of the NAI or other oseltamivir carboxylates. Attractive alternative strategies have been developed for the chemical synthesis of OSP from non-SA sources;80 however, current technologies for influenza NAI synthesis demand the availability of the key aromatic intermediates SA and ASA obtained efficiently by biotechnological processes.
Acknowledgments
We thank Dr Guillermo Gosset for the critical review of this document. This work was supported by DGAPA-PAPIIT UNAM IN206812 grant.
Author contributions
All the authors participated equally in the preparation and review of this contribution.
Disclosure
The authors report no conflicts of interest in this work.
References
Herrmann KM, Weaver LM. The shikimate pathway. Annu Rev Plant Physiol Plant Mol Biol. 1999;50(1):473–503. | |
Richards TA, Dacks JB, Campbell SA, et al. Evolutionary origins of the eukaryotic shikimate pathway: gene fusions, horizontal gene transfer, and endosymbiotic replacements. Eukaryot Cell. 2006;5(9):1517–1531. | |
Blanco B, Prado V, Lence E, et al. Mycobacterium tuberculosis shikimate kinase inhibitors: design and simulation studies of the catalytic turnover. J Am Chem Soc. 2013;135(33):12366–12376. | |
Ghosh S, Chisti Y, Banerjee UC. Production of shikimic acid. Biotechnol Adv. 2012;30(6):1425–1431. | |
Floss HG. Natural products derived from unusual variants of the shikimate pathway. Nat Prod Rep. 1997;14(5):433–452. | |
Pierson LS 3rd, Pierson EA. Metabolism and function of phenazines in bacteria: impacts on the behavior of bacteria in the environment and biotechnological processes. Appl Microbiol Biotechnol. 2010;86(6):1659–1670. | |
Floss HG, Yu TW, Arakawa K. The biosynthesis of 3-amino-5-hydroxybenzoic acid (AHBA), the precursor of mC7N units in ansamycin and mitomycin antibiotics: a review. J Antibiot (Tokyo). 2011; 64(1):35–44. | |
Sappal DS, McClendon AK, Fleming JA, et al. Biological characterization of MLN944: a potent DNA binding agent. Mol Cancer Ther. 2004;3(1):47–58. | |
Arakawa K, Müller R, Mahmud T, Yu TW, Floss HG. Characterization of the early stage aminoshikimate pathway in the formation of 3-amino-5-hydroxybenzoic acid: the RifN protein specifically converts kanosamine into kanosamine 6-phosphate. J Am Chem Soc. 2002;124(36):10644–10645. | |
Rawat G, Tripathi P, Saxena RK. Expanding horizons of shikimic acid. Recent progresses in production and its endless frontiers in application and market trends. Appl Microbiol Biotechnol. 2013;97(10):4277–4287. | |
Bochkov DV, Sysolyatin SV, Kalashnikov AI, Surmacheva IA. Shikimic acid: review of its analytical, isolation, and purification techniques from plant and microbial sources. J Chem Biol. 2012;5(1):5–17. | |
Rawat G, Tripathi P, Jahan F, Saxena RK. A natural isolate producing shikimic acid: isolation, identification, and culture condition optimization. Appl Biochem Biotechnol. 2013;169(8):2290–2302. | |
Tripathi P, Rawat G, Yadav S, Saxena RK. Fermentative production of shikimic acid: a paradigm shift of production concept from plant route to microbial route. Bioprocess Biosyst Eng. 2013;36(11):1665–1673. | |
Guo J, Frost JW. Synthesis of aminoshikimic acid. Org Lett. 2004;6(10):1585–1588. | |
Light SH, Halavaty AS, Minasov G, Shuvalova L, Anderson WF. Structural analysis of a 3-deoxy-D-arabino-heptulosonate 7-phosphate synthase with an N-terminal chorismate mutase-like regulatory domain. Protein Sci. 2012;21(6):887–895. | |
Krämer M, Bongaerts J, Bovenberg R, et al. Metabolic engineering for microbial production of shikimic acid. Metab Eng. 2003;5(4):277–283. | |
Johansson L, Lidén G. Transcriptome analysis of a shikimic acid producing strain of Escherichia coli W3110 grown under carbon- and phosphate-limited conditions. J Biotechnol. 2006;126(4):528–545. | |
Muir RM, Ibáñez AM, Uratsu SL, et al. Mechanism of gallic acid biosynthesis in bacteria (Escherichia coli) and walnut (Juglans regia). Plant Mol Biol. 2011;75(6):555–565. | |
Frost JW, Guo J, inventors; Board of Trustess of Michigan State University, assignee. Synthesis of intermediates of oseltamivir carboxylates. Unites States patent US 7,977,077B2. 2011 Jul 12. | |
Estévez AM, Estévez RJ. A short overview on the medicinal chemistry of (-)-shikimic acid. Mini Rev Med Chem. 2012;12(14):1443–1454. | |
Díaz M, Ferrero M, Fernández S, Gotor V. 6-s-cis locked analogues of the steroid hormone 1alpha, 25-dihydroxyvitamin D(3). Synthesis of novel A-ring stereoisomeric 1, 25-dihydroxy-3-epi-19-nor-previtamin D(3) derivatives. J Org Chem. 2000;65(18):5647–5652. | |
Magano J. Synthetic approaches to the neuraminidase inhibitors zanamivir (Relenza) and oseltamivir phosphate (Tamiflu) for the treatment of influenza. Chem Rev. 2009;109(9):4398–4438. | |
Zhang Y, Liu A, Ye ZG, Lin J, Xu LZ, Yang SL. New approach to the total synthesis of (-)-zeylenone from shikimic acid. Chem Pharm Bull (Tokyo). 2006;54(10):1459–1461. | |
Sánchez-Abella L, Fernández S, Armesto N, Ferrero M, Gotor V. Novel and efficient syntheses of (-)-methyl 4-epi-shikimate and 4,5-epoxy-quinic and -shikimic acid derivatives as key precursors to prepare new analogues. J Org Chem. 2006;71(14):5396–5399. | |
Farrell N, Roberts JD, Hacker MP. Shikimic acid complexes of platinum. Preparation, reactivity, and antitumor activity of (R,R-1,2-diaminocyclohexane) bis(shikimate) platinum (II). Evidence for a novel rearrangement involving platinum-carbon bond formation. J Inorg Biochem. 1991;42(4):237–246. | |
Peng Y, Zhang MM, Chen ZF, et al. Synthesis, characterization, and interaction with biomolecules of platinum(II) complexes with shikimic acid-based ligands. Bioinorg Chem Appl. 2013;2013:565032. | |
Ma Y, Sun J, Xu Q, Guo Y. Inhibitory effects of shikimic acid on platelet aggragation and blood coagulation. Yao Xue Xue Bao. 2000;35(1):1–3. | |
Ma Y, Xu QP, Sun JN, Bai LM, Guo YJ, Niu JZ. Antagonistic effects of shikimic acid against focal cerebral ischemia injury in rats subjected to middle cerebral artery thrombosis. Zhongguo Yao Li Xue Bao. 1999;20(8):701–704. | |
Huang F, Xiu Q, Sun J, Hong E. Anti-platelet and anti-thrombotic effects of triacetylshikimic acid in rats. J Cardiovasc Pharmacol. 2002;39(2):262–270. | |
Tang L, Xiang H, Sun Y, et al. Monopalmityloxy shikimic acid: enzymatic synthesis and anticoagulation activity evaluation. Appl Biochem Biotechnol. 2008;158(2):408–415. | |
Wang HT, Jin HT, Sun JN, Xu QP, Guo YJ. [Experimental studies on the anti-thrombosis effect of 3,4-oxo-isopropylidene-shikimic acid]. Yao Xue Xue Bao. 2002;37(4):245–248. Chinese. | |
Ma Y, Sun JN, Xu QP, You ZL, Guo YJ. 3,4-oxo-isopropylidene-shikimic acid inhibits adhesion of polymorphonuclear leukocyte to TNF-alpha-induced endothelial cells in vitro. Acta Pharmacol Sin. 2004;25(2):246–250. | |
Xing J, Sun J, You H, Lv J, Sun J, Dong Y. Anti-inflammatory effect of 3,4-oxo-isopropylidene-shikimic acid on acetic acid-induced colitis in rats. Inflammation. 2012;35(6):1872–1879. | |
Jiang S, Singh G. Chemical synthesis of shikimic acid and its analogues. Tetrahedron. 1998;54:4697–4753. | |
Coombs GH, Müller S. Recent advances in the search for new anti-coccidial drugs. Int J Parasitol. 2002;32(5):497–508. | |
Quan N, Nie LD, Zhu RH, Shi XX, Ding W, Lu X. Total syntheses of (+)-valiolamine and (–)-1-epi-valiolamine from naturally abundant (–)-shikimic acid. Eur J Org Chem. 2013;2013(28):6389–6396. | |
Wise HM, Foeglein A, Sun J, et al. A complicated message: identification of a novel PB1-related protein translated from influenza A virus segment 2 mRNA. J Virol. 2009;83(16):8021–8031. | |
Rambaut A, Pybus OG, Nelson MI, Viboud C, Taubenberger JK, Holmes EC. The genomic and epidemiological dynamics of human influenza A virus. Nature. 2008;453(7195):615–619. | |
Labella AM, Merel SE. Influenza. Med Clin North Am. 2013; 97(4):621–645. | |
Matsuoka Y, Matsumae H, Katoh M, et al. A comprehensive map of the influenza A virus replication cycle. BMC Syst Biol. 2013;7:97. | |
Cheng VC, To KK, Tse H, Hung IF, Yuen KY. Two years after pandemic influenza A/2009/H1N1: what have we learned? Clin Microbiol Rev. 2012;25(2):223–263. | |
Sleeman K, Guo Z, Barnes J, Shaw M, Stevens J, Gubareva LV. R292K substitution and drug susceptibility of influenza A (H7N9) viruses. Emerg Infect Dis. 2013;19(9):1521–1524. | |
Yates PJ, Mehta N, Horton J, Tisdale M. Virus susceptibility analyses from a phase IV clinical trial of inhaled zanamivir treatment in children infected with influenza. Antimicrob Agents Chemother. 2013;57(4):1677–1684. | |
Ling LM, Chow AL, Lye DC, et al. Effects of early oseltamivir therapy on viral shedding in 2009 pandemic influenza A (H1N1) virus infection. Clin Infect Dis. 2010;50(7):963–969. | |
Li IW, Hung IF, To KK, et al. The natural viral load profile of patients with pandemic 2009 influenza A (H1N1) and the effect of oseltamivir treatment. Chest. 2010;137(4):759–768. | |
Abrecht S, Harrington P, Iding H, et al. The synthetic development of the anti-influenza neuraminidase inhibitor oseltamivir phosphate (Tamiflu®): a challenge for synthesis and process research. CHIMIA International Journal for Chemistry. 2004;58(9):621–629. | |
McKimm-Breschkin JL. Influenza neuraminidase inhibitors: antiviral action and mechanisms of resistance. Influenza Other Respir Viruses. 2013;7 Suppl 1:25–36. | |
Hayden FG. Newer influenza antivirals, biotherapeutics and combinations. Influenza Other Respir Viruses. 2013;7 Suppl: 63–75. | |
Butler D. Wartime tactic doubles power of scarce bird-flu drug. Nature. 2005;438(7064):6. | |
Nie LD, Shi XX, Ko KH, Lu WD. A short and practical synthesis of oseltamivir phosphate (Tamiflu) from (-)-shikimic acid. J Org Chem. 2009;74(10):3970–3973. | |
Pandemic influenza preparedness and response [webpage on the Internet]. Geneva: World Health Organization. Available from: http://www.who.int/influenza/resources/documents/pandemic_guidance_04_2009/en/. Accessed February 27, 2014. | |
Farina V, Brown JD. Tamiflu: the supply problem. Angew Chem Int Ed Engl. 2006;45(44):7330–7334. | |
McCrindle R, Overton KH, Raphael RA. 315. A stereospecific total synthesis of D-(–)-shikimic acid. J Chem Soc. 1960:1560–1565. | |
Bestmann HJ, Heid HA. Stereospecific synthesis of optically pure quinic acid and shikimic acid from D-arabinose. Angew Chem Int Ed Engl. 1971;10(5):336–337. | |
Fleet GW, Shing TK, Warr SM. Enantiospecific synthesis of shikimic acid from D-mannose: formation of a chiral cyclohexene by intramolecular olefination of a carbohydrate-derived intermediate. J Chem Soc Perkin 1. 1984:905–908. | |
Koreeda M, Teng K, Murata T. (1E,3E)-4-acetoxy-1-phenyldimethylsilyl-1,3-butadiene as a surrogate for (1E,3E)-1,4- diacetoxy-1,3-butadiene: a highly efficient synthesis of (±)-shikimic acid. Tetrahedron Lett. 1990;31(42):5997–6000. | |
Box JM, Harwood LM, Humphreys JL, Morris GA, Redon PM, Whitehead RC. Dehydration of quinate derivatives: synthesis of a difluoromethylene homologue of shikimic acid. Synlett. 2002;2002(2):358–360. | |
Kancharla PK, Doddi VR, Kokatla H, Vankar YD. A concise route to (−)-shikimic acid and (−)-5-epi-shikimic acid, and their enantiomers via Barbier reaction and ring-closing metathesis. Tetrahedron Lett. 2009;50(50):6951–6954. | |
Shin SH, Han JH, Lee SI, Ha YB, Ryu DH. Highly efficient synthesis of (-)-shikimic acid from a chiral diels-alder adduct between furan and acrylate. Bull Korean Chem Soc. 2011;32(8):2885–2886. | |
Whiting GC. Occurrence of shikimic acid in gooseberry fruits. Nature. 1957;179(4558):531. | |
Hulme AC. Quinic and shikimic acids in fruits. Qualitas Plantarum et Materiae Vegetabiles. 1958;3–4(1):468–473. | |
Avula B, Wang YH, Smillie TJ, Khan IA. Determination of shikimic acid in fruits of Illicium species and various other plant samples by LC–UV and LC–ESI–MS. Chromatographia. 2009;69(3–4):307–314. | |
Lederer I, Schulzki G, Gross J, Steffen JP. Combination of TLC and HPLC-MS/MS methods. Approach to a rational quality control of Chinese star anise. J Agric Food Chem. 2006;54(6):1970–1974. | |
Ohira H, Torii N, Aida TM, Watanabe M, Smith RL Jr. Rapid separation of shikimic acid from Chinese star anise (Illicium verum Hook. f.) with hot water extraction. Separation and Purification Technology. 2009;69(1):102–108. | |
Liu J, Zhang X, Shi Y, Zhang Q, Ma Y, Chen J. [Studies on chemical constituents of Illicium simonsii]. Zhongguo Zhong Yao Za Zhi. 2011;36(10):1311–1315. Chinese. | |
Pham AT, Malterud KE, Paulsen BS, Diallo D, Wangensteen H. DPPH radical scavenging and xanthine oxidase inhibitory activity of Terminalia macroptera leaves. Nat Prod Commun. 2011;6(8):1125–1128. | |
Huang Chusheng, Yang Xiaoyong, Liu Hongxing, He Dongmei, inventors; Guangxi Teachers Education University, assignee. Method for extracting high-pure shikimic acid from scarlet octagonal fruit. China patent 200810073502. 2008 Aug 27. | |
Usuki T, Yasuda N, Yoshizawa-Fujita M, Rikukawa M. Extraction and isolation of shikimic acid from Ginkgo biloba leaves utilizing an ionic liquid that dissolves cellulose. Chem Commun (Camb). 2011;47(38):10560–10562. | |
Case Study: Star Anise. Ottawa: ETC Group; 2012. Available from: http://www.etcgroup.org/sites/www.etcgroup.org/files/Final_CBD_Star%20Anise_case_study_TA.pdf. Accessed March 2, 2014. | |
Pandemic influenza and Tamiflu production [webpage on the Internet]. Available from: http://www.roche.com/media/events/mb051109.htm. Accessed March 2, 2014. | |
Chandran SS, Yi J, Draths KM, von Daeniken R, Weber W, Frost JW. Phosphoenolpyruvate availability and the biosynthesis of shikimic acid. Biotechnol Prog. 2003;19(3):808–814. | |
Escalante A, Calderón R, Valdivia A, et al. Metabolic engineering for the production of shikimic acid in an evolved Escherichia coli strain lacking the phosphoenolpyruvate: carbohydrate phosphotransferase system. Microb Cell Fact. 2010;9:21. | |
Rodriguez A, Martínez JA, Báez-Viveros JL, et al. Constitutive expression of selected genes from the pentose phosphate and aromatic pathways increases the shikimic acid yield in high-glucose batch cultures of an Escherichia coli strain lacking PTS and pykF. Microb Cell Fact. 2013;12:86. | |
Aguilar C, Escalante A, Flores N, et al. Genetic changes during a laboratory adaptive evolution process that allowed fast growth in glucose to an Escherichia coli strain lacking the major glucose transport system. BMC Genomics. 2012;13:385. | |
Cui YY, Ling C, Zhang YY, Huang J, Liu JZ. Production of shikimic acid from Escherichia coli through chemically inducible chromosomal evolution and cofactor metabolic engineering. Microb Cell Fact. 2014;13:21. | |
Miyata R, Ogawa K, Sakamoto K, et al, inventors; Toray Industries, Inc., applicant. Microorganism belonging to the genus citrobacter and process for producing shikimic acid. Patent EP 1092766 A1. April 18, 2001. | |
Iomantas YAV, Abalakina EG, Polanuer BM, Yampolskaya TA, Bachina TA, Kozlov YI, inventors; Ajinomoto Co, Inc., assignee. Method for producing shikimic acid. United States patent US6436664. August 20, 2002. | |
Licona-Cassani C, Lara AR, Cabrera-Valladares N, et al. Inactivation of pyruvate kinase or the phosphoenolpyruvate: sugar phosphotransferase system increases shikimic and dehydroshikimic acid yields from glucose in Bacillus subtilis. J Mol Microbiol Biotechnol. 2014;24(1):37–45. | |
Adachi O, Ano Y, Toyama H, Matsushita K. High shikimate production from quinate with two enzymatic systems of acetic acid. Biosci Biotechnol Biochem. 2006;70(10):2579–2582. | |
Ko JS, Keum JE, Ko SY. A synthesis of oseltamivir (Tamiflu) starting from D-mannitol. J Org Chem. 2010;75(20):7006–7009. | |
Cortés-Tolalpa L, Gutiérrez-Ríos RM, Martínez LM, et al. Global transcriptomic analysis of an engineered Escherichia coli strain lacking the phosphoenolpyruvate: carbohydrate phosphotransferase system during shikimic acid production in rich culture medium. Microb Cell Fact. 2014;13(1):28. | |
Chen K, Dou J, Tang S, et al. Deletion of the aroK gene is essential for high shikimic acid accumulation through the shikimate pathway in E. coli. Bioresour Technol. 2012;119:141–147. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.