Back to Archived Journals » Transplant Research and Risk Management » Volume 6
Current perspectives on antibody-mediated rejection after lung transplantation
Received 25 May 2014
Accepted for publication 11 July 2014
Published 8 October 2014 Volume 2014:6 Pages 109—115
DOI https://doi.org/10.2147/TRRM.S50799
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 5
Chad A Witt, Ramsey R Hachem
Department of Internal Medicine, Division of Pulmonary and Critical Care Medicine, Washington University School of Medicine, Saint Louis, MO, USA
Abstract: The role of donor-specific antibodies (DSA) to human leukocyte antigens and the burden of antibody-mediated rejection (AMR) in lung transplantation remain enigmatic. Over the past several years, evidence has been emerging that humoral immunity plays an important role in the development of both acute and chronic lung allograft dysfunction (CLAD). Multiple case reports and case series have identified lung allograft recipients with clinical findings consistent with acute AMR. However, there is currently no widely accepted definition for AMR in lung transplantation, and this has been a significant barrier to furthering our understanding of this form of rejection. Nonetheless, the development of DSA after transplantation has consistently been identified as an independent risk factor for persistent and high-grade acute cellular rejection and CLAD. This has raised the possibility that chronic AMR may be a distinct phenotype of CLAD although evidence supporting this paradigm is still lacking. Additionally, antibodies to lung-restricted self-antigens (collagen V and K-α 1 tubulin) have been associated with primary graft dysfunction early and the development of CLAD late after transplantation, and emerging evidence underscores significant interactions between autoimmunity and alloimmunity after transplantation. There is currently an active International Society for Heart and Lung Transplantation working group that is developing an operational definition for AMR in lung transplantation. This will be critical to improve our understanding of this form of rejection and conduct clinical trials to identify optimal treatment strategies. This review will summarize the literature on DSA and AMR in lung transplantation and discuss the impact of antibodies to self-antigens on lung allograft dysfunction.
Keywords: antibody-mediated rejection, donor-specific antibodies, lung transplantation
Introduction
Over the past 30 years, lung transplantation has become the ultimate treatment for patients with end-stage lung disease. The number of lung transplant procedures has continued to increase, and more than 3,600 procedures were reported to the International Society for Heart and Lung Transplantation (ISHLT) Registry in 2011.1 Survival after lung transplantation has been slowly improving; however, long-term outcomes remain disappointing with a median survival of 6.1 years in the most recent era (June 2004 – December 2011).1 This is in contrast to allograft survival after kidney and liver transplantation where the median survival is over 9 years after deceased-donor kidney transplantation and over 12 years after living-donor kidney and deceased-donor liver transplantation.2,3 The major causes of death within the first year after lung transplantation are allograft failure and non-cytomegalovirus infections. Beyond the first year after transplantation, chronic lung allograft dysfunction (CLAD), coded in the ISHLT Registry as bronchiolitis or late allograft failure, is the leading cause of mortality, accounting for >40% of deaths.1 This clearly demonstrates that CLAD is the primary obstacle to better long-term outcomes after lung transplantation.
It is widely believed that rejection is the underlying cause of most forms of CLAD, and traditionally, solid-organ transplant rejection has been regarded as a predominantly T-cell mediated process. Accordingly, standard immunosuppressive regimens consisting of a calcineurin inhibitor, a cell cycle inhibitor, and corticosteroids targeting T-cell proliferation and function have made organ transplantation a clinical reality.2,3 However, emerging data suggest that humoral immunity may play an important and previously underappreciated role in rejection and chronic allograft dysfunction. Antibody-mediated rejection (AMR) is a widely accepted and well-defined form of rejection after kidney and heart transplantation, and the Pathology Council of the ISHLT has recently proposed a summary statement on the pathology of AMR after lung transplantation.4–7 This review will discuss the different manifestations of AMR in lung transplantation, including hyperacute rejection, acute AMR, donor-specific antibodies (DSA) to human leukocyte antigens (HLA), and antibodies to self-antigens and their role in the development of CLAD.
Hyperacute rejection
Hyperacute rejection results from circulating preexisting DSA and occurs within minutes or hours of transplantation and perfusion of the allograft. Clinically, hyperacute rejection after lung transplantation is characterized by fulminant allograft dysfunction with severe gas-exchange impairment, hemorrhagic pulmonary edema, and diffuse radiographic infiltrates.8 Preformed DSA bind to HLA on donor endothelial cells, resulting in the activation of the classical complement cascade and leading to the formation of the membrane attack complex and endothelial cell injury. The development and refinement of solid-phase antibody detection assays have significantly improved the ability to detect and identify the specificity of HLA antibodies before transplantation. Using these specificities and the potential donor’s HLA type, crossmatch results can be predicted accurately before donor organ acceptance. This virtual crossmatch improves donor selection for allosensitized patients and minimizes the risk of HLA incompatible transplants; as a result, hyperacute rejection has become rare. Nonetheless, hyperacute rejection after lung transplantation is a widely accepted form of rejection and demonstrates that circulating antibodies can severely injure the lung allograft, often resulting in allograft failure. The pathology of hyperacute rejection is notable for severe acute lung injury with diffuse microvascular thrombi, neutrophilic infiltration of the alveolar septae, capillaritis, fibrinoid necrosis, and hemorrhagic infarction.9–11 Electron microscopy, when performed, demonstrates that endothelial cells are the focal point of immunologic injury.12 Furthermore, immunostaining for immunoglobulin G or complement component 4d (C4d) illustrates an antibody and complement-mediated endothelial cell injury process.12
Outcomes after hyperacute rejection are generally poor, and allograft loss is common. Intensive treatment regimens focusing on depleting antibodies or mitigating antibody-mediated injury have been used with mixed results. Bittner et al described a case report of successful treatment of hyperacute rejection in a single lung transplant recipient with a regimen consisting of plasmapheresis, antithymocyte globulin, and cyclophosphamide.13 Dawson et al described a case report of a bilateral lung transplant recipient who developed hyperacute rejection refractory to plasma exchange, rituximab, bortezomib, and intravenous immunoglobulin (IVIG) who responded well to eculizumab.14 Rituximab, a chimeric monoclonal antibody to CD20, a cell-surface antigen expressed on mature B-cells, induces B-cell depletion through complement-dependent cytotoxicity and antibody-dependent cytotoxicity. However, plasma cells, the predominant antibody-producing cells, do not express CD20; this raises an important potential limitation to the use of rituximab in AMR. Bortezomib, a proteasome inhibitor, induces plasma cell apoptosis and offers a theoretical advantage. Eculizumab is a humanized monoclonal antibody that targets complement component 5 (C5) and inhibits the cleavage of C5 into C5a and C5b, thereby preventing formation of the membrane attack complex (C5b–C9). This first-in-class complement inhibitor is approved for use in paroxysmal nocturnal hemoglobinuria and atypical hemolytic uremic syndrome, two uncommon diseases of complement dysregulation. Further clinical investigation of its use in complement-mediated allograft injury including hyperacute rejection and acute AMR is warranted.
Acute AMR
The diagnostic criteria for lung allograft rejection were revised in 2007 and classified four forms of rejection: acute rejection; lymphocytic bronchiolitis; obliterative bronchiolitis (OB); and chronic vascular rejection.15 However, there was no consensus on the features or clinicopathologic diagnostic criteria for AMR in lung transplant recipients. The National Conference to Assess Antibody-Mediated Rejection in Solid Organ Transplantation has proposed a general paradigm of humoral responses applicable to all organs to help facilitate further investigation.16 The National Conference defined AMR according to the following criteria: circulating DSA; C4d deposition; tissue pathology; and clinical allograft dysfunction.16 C4d deposition in lung allografts has been demonstrated in cases of AMR; however, it may be nonspecific and can be seen with acute cellular rejection in the absence of DSA or other forms of lung injury. Furthermore, C4d staining and interpretation is technically difficult to perform reliably with reproducible results.17–21 The ISHLT Pathology Council summary statement concluded that the diagnosis of pulmonary AMR should require: clinical allograft dysfunction; circulating DSA; and abnormal histopathologic findings.7 Discussion of C4d staining is included in the summary statement, but not required by the ISHLT Pathology Council for the diagnosis of pulmonary AMR.7
There have been multiple case reports and case series of lung transplant recipients presenting with clinical findings consistent with AMR. Early reports included clinical presentations of allograft dysfunction presenting with diffuse alveolar hemorrhage, capillaritis, and/or C4d deposition did not identify DSA although less sensitive HLA antibody detection assays were used.22,23 In such cases, it is possible that DSA may not have be detectable in the peripheral circulation because the antibodies are adsorbed in the allograft. Furthermore, non-HLA antibodies against endothelial cells were identified in one series and there was clinical improvement with plasmapheresis implying humorally mediated injury as the mechanism of allograft dysfunction.23 In addition, this does not rule out the possibility that some of the antibodies may have been autoantibodies and autoimmunity has emerged as a risk factor for chronic allograft dysfunction.24 The role of autoimmunity in acute and chronic allograft dysfunction will be discussed in “Role of autoimmunity and antibodies to self-antigens in primary graft dysfunction and CLAD”.
Subsequently, two case reports from independent centers described similar clinicopathological features of acute AMR.25,26 In both cases, the recipient was not allosensitized prior to transplantation and had a negative direct crossmatch at the time of transplant. Both developed allograft dysfunction, capillaritis, C4d deposition, and new circulating DSA. In the first case, there was no response to high dose corticosteroids and basiliximab, but there was a good response to rituximab.25 The second patient was treated with a combination of IVIG, plasmapheresis, and rituximab with resolution of the allograft dysfunction and long-term clinical stability.26
In 2013, our center reported a case series of patients with AMR and proposed a strict definition of acute AMR in lung transplant recipients.27 This definition was based on criteria proposed by the ISHLT Pathology Council and the National Conference to Assess Antibody-Mediated Rejection in Solid Organ Transplantation. We defined acute AMR as: allograft dysfunction; circulating DSA; tissue pathology; and capillary endothelial C4d deposition (Figure 1). We conducted a retrospective cohort study at our center that included 501 lung transplant procedures from January 1, 2004–June 30, 2012. During that time, we identified 21 lung recipients meeting this stringent definition of acute AMR. All patients had diffuse, bilateral pulmonary infiltrates on imaging and other causes of acute allograft dysfunction were ruled out (Figure 2). The predominant pathology was acute lung injury, and eleven of the 21 patients had capillaritis while ten did not.
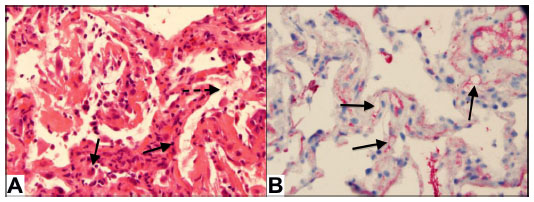 | Figure 1 Pathologic findings in patient with acute pulmonary AMR. |
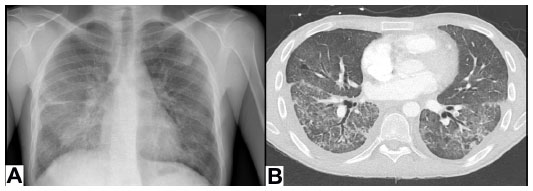 | Figure 2 Radiological findings in patient with acute pulmonary AMR. |
Based on the definition to identify cases, capillary endothelial C4d deposition was seen in all patients. AMR was diagnosed a median of 258 days after transplantation, 14 in the first year and the other seven more than 1 year after transplant. Also, 14 of the 21 patients required mechanical ventilation, and six died of refractory AMR during their index hospitalization. Of the 15 patients who survived their index episode of AMR, one had preexisting CLAD and 13 of the remaining 14 developed CLAD. Nine of the 15 patients who survived their index episode of acute AMR died during the follow-up period. Recipients who cleared the DSA had a better survival than those with persistent DSA.
During the study period, our treatment regimens were individualized, and the specific treatment depended on the clinical course; these included antithymocyte globulin, IVIG, plasmapheresis, rituximab, bortezomib, and/or eculizumab in various combinations. The high mortality and near universal development of CLAD among survivors in this cohort underscore the significant clinical impact of acute AMR in lung transplant recipients.
The optimal treatment regimen for patients with acute AMR is not currently known. Given the lack of a widely accepted definition of acute AMR after lung transplantation, it is difficult to determine from the published literature whether reported cases all had the same syndrome of allograft dysfunction. Furthermore, treatment in all cases and series has been individualized and highly dependent on the clinical course. This makes comparing the relative efficacy of different regimens impossible. Nonetheless, different combinations of plasmapheresis, IVIG, rituximab, antithymocyte globulin, bortezomib, and eculizumab have been given to treat pulmonary AMR.27–31 There is immunological rationale for combination therapy, including inhibiting DSA-mediated complement activation with eculizumab, depleting circulating DSA with plasmapheresis, decreasing antibody production by targeting plasma cells with bortezomib, and decreasing plasma cell development and activation with rituximab. These more targeted therapies are likely to be given in combination with therapies with broader spectrums of activity, including IVIG, high-dose corticosteroids, and antithymocyte globulin. Which of these therapies or combinations of therapies is most effective remains to be determined and will require randomized controlled trials with a strict definition of acute AMR in the future.
A multidisciplinary ISHLT working group, consisting of experts in pulmonary medicine, pathology, and immunology is currently reviewing the published literature and professional experiences to develop a definition for AMR after lung transplantation. This will standardize the nomenclature and facilitate the necessary clinical and immunological research to better understand AMR and ultimately identify effective preventative and treatment strategies.
DSA and CLAD
Traditionally, OB has been the histologic hallmark of chronic lung allograft rejection. However, histologic confirmation of OB is difficult because of the patchy nature of the disease and the small samples obtained with transbronchial lung biopsies, so bronchiolitis obliterans syndrome (BOS), defined and staged according to changes in spirometry, has served as the clinical surrogate for OB. More recently, there has been a paradigm shift with the introduction of the more encompassing term “chronic lung allograft dysfunction,” which includes BOS and restrictive allograft syndrome (RAS).32 There has been a strong association between DSA and BOS, but it remains unclear if DSA play a role in RAS development.
An association between the development of HLA antibodies and BOS has been recognized since the late 1990s, and more recent clinical data linked HLA antibodies, and DSA in particular, with persistent and high-grade acute cellular rejection and lymphocytic bronchiolitis.33–37 In addition, a recent large cohort analysis identified the development of HLA antibodies as an independent risk factor for mortality after transplant.38 Despite these clinical data, it remains unclear if HLA antibodies cause BOS or if they are an epiphenomenon of the cellular immune response. However, in vitro and animal studies suggest that HLA (and major histocompatibility complex [MHC]) antibodies play a pathogenic role; HLA antibodies have been shown to induce airway epithelial cell proliferation, lead to the release of fibrogenic growth factors, and result in epithelial cell apoptosis.39 Additionally, MHC antibodies have been shown to induce airway obliteration when systemically administered in a murine heterotopic tracheal transplant model and cause small airway obliteration when delivered intrabronchially into native lungs.40,41 Given the experimental and clinical data suggesting a role for HLA antibodies in the development of BOS and CLAD, there has been an intensified focus on the role of these antibodies in lung transplant recipients.
Our center examined the outcomes of lung transplant recipients who developed DSA and the response to treatment with either IVIG or IVIG and rituximab.37 In this series of 116 recipients, 65 (56%) developed de novo DSA during the study period. There was no statistically significant difference in the incidence of acute cellular rejection, lymphocytic bronchiolitis, or BOS between those who developed DSA and were treated and those who did not develop DSA. However, since this was not a randomized controlled trial, it is not clear if this finding reflects a treatment effect. Nonetheless, this study demonstrated that patients who had successful depletion of DSA (ie, DSA was no longer detectable after treatment) had greater freedom from BOS and better survival than those who had persistent DSA.
In a recent large single center cohort study, DSA development was a significant risk factor for mortality but not BOS.38 In contrast, non-DSA HLA antibodies were significant risk factors for both BOS and mortality.38 Reasons for the lack of association between DSA and BOS in this study are not clear, but the results demonstrate that non-DSA HLA antibodies can also increase the risk of BOS. In fact, pretransplant non-DSA HLA antibodies and major histocompatibility complex (MHC) class I-related chain A antibodies have been associated with an increased risk of BOS.42 Clearly, additional studies and randomized controlled trials are needed to better understand the role of HLA and non-HLA antibodies in BOS development and to critically examine the potential benefit of DSA depletion in clinically stable lung transplant recipients. Of note, a multicenter randomized controlled trial examining the efficacy of rituximab as induction immunosuppression in pediatric lung transplant recipients is being planned by the Clinical Trials in Organ Transplantation in Children network.
In addition, since CLAD is the leading cause of death beyond the first year after lung transplantation and its pathogenesis remains unclear, further investigation into the potential role of DSA in the development of a chronic form of AMR is indicated. It is possible that RAS may represent chronic AMR; however, data linking RAS to humoral immune responses are lacking to date. Clearly, this is an area of ongoing investigation.
Role of autoimmunity and antibodies to self-antigens in primary graft dysfunction and CLAD
Autoimmunity is the hallmark of systemic autoimmune diseases, such as rheumatoid arthritis, systemic lupus erythematosus, and the vasculitides. Many of these diseases are thought to result from a loss of immunologic tolerance and the development of immune responses to self-antigens.43 Antibodies to myosin, vimentin, and heat-shock proteins after heart transplantation and to angiotensin type 1 receptor after kidney transplantation have been associated with allograft dysfunction.44–47 More recent studies have demonstrated a potential role for autoimmunity in both primary graft dysfunction and BOS. For example, peripheral blood mononuclear cells taken from lung transplant recipients have been found to be reactive against self-antigens, demonstrating the presence of T-cell mediated autoimmunity.48
The two self-antigens that have received the most attention in lung transplant recipients are type V collagen (Col V) and K-α 1 tubulin. Col V and K-α 1 tubulin are both expressed on airway epithelial cells, and antibodies against these self-antigens have been associated with primary graft dysfunction and BOS.49–51 Furthermore, the development of DSA has been associated with the development of antibodies to self-antigens, and it has been proposed that alloimmune responses in the form of DSA may induce an autoimmune response and contribute to the pathogenesis of BOS.52 It has also been demonstrated that lung transplant recipients who were treated with antibody-directed therapy and cleared the antibodies to self-antigens are significantly less likely to develop BOS compared to those who had persistent antibodies to self-antigens.24 The precise mechanism by which autoimmunity contributes to acute and chronic allograft dysfunction and how to use these emerging data clinically remains an area of ongoing investigation.
Conclusion
CLAD remains the major cause of death in lung transplant recipients beyond the first year after transplantation. Although T-cell inhibition has been the primary goal of immunosuppression to prevent allograft rejection, it is becoming increasingly evident that AMR and the development of DSA and subsequent CLAD play a significant role in allograft and patient survival. Additionally, antibodies to self-antigens appear to play a role in the development of CLAD. As the understanding of the pathophysiology and clinical science advances, further investigations into treatment strategies for antibody depletion and immune modulation will be possible. This is crucial to identify strategies to improve outcomes after lung transplantation.
Disclosure
The authors report no conflicts of interest in this work.
References
Yusen RD, Christie JD, Edwards LB, et al; International Society for Heart and Lung Transplantation. The Registry of the International Society for Heart and Lung Transplantation: Thirtieth Adult Lung and Heart-Lung Transplant Report – 2013; focus theme: age. J Heart Lung Transplant. 2013;32(10):965–978. | |
Bhorade SM, Stern E. Immunosuppression for lung transplantation. Proc Am Thorac Soc. 2009;6(1):47–53. | |
Durrbach A, Francois H, Beaudreuil S, Jacquet A, Charpentier B. Advances in immunosuppression for renal transplantation. Nat Rev Nephrol. 2010;6(3):160–167. | |
Berry GJ, Angelini A, Burke MM, et al. The ISHLT working formulation for pathologic diagnosis of antibody-mediated rejection in heart transplantation: evolution and current status (2005–2011). J Heart Lung Transplant. 2011;30(6):601–611. | |
Sis B, Mengel M, Haas M, et al. Banff ‘09 meeting report: antibody mediated graft deterioration and implementation of Banff working groups. Am J Transplant. 2010;10(3):464–471. | |
Solez K, Colvin RB, Racusen LC, et al. Banff 07 classification of renal allograft pathology: updates and future directions. Am J Transplant. 2008;8(4):753–760. | |
Berry G, Burke M, Andersen C, et al. Pathology of pulmonary antibody-mediated rejection: 2012 update from the Pathology Council of the ISHLT. J Heart Lung Transplant. 2013;32(1):14–21. | |
Hachem R. Antibody-Mediated Lung Transplant Rejection. Curr Respir Care Rep. 2012;1(3):157–161. | |
Choi JK, Kearns J, Palevsky HI, et al. Hyperacute rejection of a pulmonary allograft. Immediate clinical and pathologic findings. Am J Respir Crit Care Med. 1999;160(3):1015–1018. | |
Frost AE, Jammal CT, Cagle PT. Hyperacute rejection following lung transplantation. Chest. 1996;110(2):559–562. | |
Masson E, Stern M, Chabod J, et al. Hyperacute rejection after lung transplantation caused by undetected low-titer anti-HLA antibodies. J Heart Lung Transplant. 2007;26(6):642–645. | |
Campo-Cañaveral de la Cruz JL, Naranjo JM, Salas C, Varela de Ugarte A. Fulminant hyperacute rejection after unilateral lung transplantation. Eur J Cardiothorac Surg. 2012;42(2):373–375. | |
Bittner HB, Dunitz J, Hertz M, Bolman MR 3rd, Park SJ. Hyperacute rejection in single lung transplantation – case report of successful management by means of plasmapheresis and antithymocyte globulin treatment. Transplantation. 2001;71(5):649–651. | |
Dawson KL, Parulekar A, Seethamraju H. Treatment of hyperacute antibody-mediated lung allograft rejection with eculizumab. J Heart Lung Transplant. 2012;31(12):1325–1326. | |
Stewart S, Fishbein MC, Snell GI, et al. Revision of the 1996 working formulation for the standardization of nomenclature in the diagnosis of lung rejection. J Heart Lung Transplant. 2007;26(12):1229–1242. | |
Takemoto SK, Zeevi A, Feng S, et al. National conference to assess antibody-mediated rejection in solid organ transplantation. Am J Transplant. 2004;4(7):1033–1041. | |
Magro CM, Pope Harman A, Klinger D, et al. Use of C4d as a diagnostic adjunct in lung allograft biopsies. Am J Transplant. 2003;3(9):1143–1154. | |
Westall GP, Snell GI, McLean C, Kotsimbos T, Williams T, Magro C. C3d and C4d deposition early after lung transplantation. J Heart Lung Transplant. 2008;27(7):722–728. | |
Ionescu DN, Girnita AL, Zeevi A, et al. C4d deposition in lung allografts is associated with circulating anti-HLA alloantibody. Transpl Immunol. 2005;15(1):63–68. | |
Roden AC, Maleszewski JJ, Yi ES, Jenkins SM, Aubry MC. Reproducibility of C4d by Immunofluorescence and Immunohistochemistry in Routine Lung Allograft Biopsies. J Heart Lung Transplant. 2014;33(4):S113. Available from: http://www.jhltonline.org/article/S1053-2498(14)00034-5/fulltext. Accessed September 5, 2014. | |
Yousem SA, Zeevi A. The histopathology of lung allograft dysfunction associated with the development of donor-specific HLA alloantibodies. Am J Surg Pathol. 2012;36(7):987–992. | |
Badesch DB, Zamora M, Fullerton D, et al. Pulmonary capillaritis: a possible histologic form of acute pulmonary allograft rejection. J Heart Lung Transplant. 1998;17(4):415–422. | |
Magro CM, Deng A, Pope-Harman A, et al. Humorally mediated posttransplantation septal capillary injury syndrome as a common form of pulmonary allograft rejection: a hypothesis. Transplantation. 2002;74(9):1273–1280. | |
Hachem RR, Tiriveedhi V, Patterson GA, Aloush A, Trulock EP, Mohanakumar T. Antibodies to K-α 1 tubulin and collagen V are associated with chronic rejection after lung transplantation. Am J Transplant. 2012;12(8):2164–2171. | |
Astor TL, Galantowicz M, Phillips A, Palafox J, Baker P. Pulmonary capillaritis as a manifestation of acute humoral allograft rejection following infant lung transplantation. Am J Transplant. 2009;9(2):409–412. | |
Morrell MR, Patterson GA, Trulock EP, Hachem RR. Acute antibody-mediated rejection after lung transplantation. J Heart Lung Transplant. 2009;28(1):96–100. | |
Witt CA, Gaut JP, Yusen RD, et al. Acute antibody-mediated rejection after lung transplantation. J Heart Lung Transplant. 2013;32(10):1034–1040. | |
Astor TL, Weill D, Cool C, Teitelbaum I, Schwarz MI, Zamora MR. Pulmonary capillaritis in lung transplant recipients: treatment and effect on allograft function. J Heart Lung Transplant. 2005;24(12):2091–2097. | |
Daoud AH, Betensley AD. Diagnosis and treatment of antibody mediated rejection in lung transplantation: a retrospective case series. Transpl Immunol. 2013;28(1):1–5. | |
Lobo LJ, Aris RM, Schmitz J, Neuringer IP. Donor-specific antibodies are associated with antibody-mediated rejection, acute cellular rejection, bronchiolitis obliterans syndrome, and cystic fibrosis after lung transplantation. J Heart Lung Transplant. 2013;32(1):70–77. | |
Jackups R, Canter C, Sweet SC, Mohanakumar T, Morris GP. Measurement of donor-specific HLA antibodies following plasma exchange therapy predicts clinical outcome in pediatric heart and lung transplant recipients with antibody-mediated rejection. J Clin Apher. 2013;28(4):301–308. | |
Verleden GM, Raghu G, Meyer KC, Glanville AR, Corris P. A new classification system for chronic lung allograft dysfunction. J Heart Lung Transplant. 2014;33(2):127–133. | |
Girnita AL, McCurry KR, Iacono AT, et al. HLA-specific antibodies are associated with high-grade and persistent-recurrent lung allograft acute rejection. J Heart Lung Transplant. 2004;23(10):1135–1141. | |
Sundaresan S, Mohanakumar T, Smith MA, et al. HLA-A locus mismatches and development of antibodies to HLA after lung transplantation correlate with the development of bronchiolitis obliterans syndrome. Transplantation. 1998;65(5):648–653. | |
Smith MA, Sundaresan S, Mohanakumar T, et al. Effect of development of antibodies to HLA and cytomegalovirus mismatch on lung transplantation survival and development of bronchiolitis obliterans syndrome. J Thorac Cardiovasc Surg. 1998;116(5):812–820. | |
Palmer SM, Davis RD, Hadjiliadis D, et al. Development of an antibody specific to major histocompatibility antigens detectable by flow cytometry after lung transplant is associated with bronchiolitis obliterans syndrome. Transplantation. 2002;74(6):799–804. | |
Hachem RR, Yusen RD, Meyers BF, et al. Anti-human leukocyte antigen antibodies and preemptive antibody-directed therapy after lung transplantation. J Heart Lung Transplant. 2010;29(9):973–980. | |
Snyder LD, Wang Z, Chen DF, et al. Implications for human leukocyte antigen antibodies after lung transplantation: a 10-year experience in 441 patients. Chest. 2013;144(1):226–233. | |
Jaramillo A, Smith CR, Maruyama T, Zhang L, Patterson GA, Mohanakumar T. Anti-HLA class I antibody binding to airway epithelial cells induces production of fibrogenic growth factors and apoptotic cell death: a possible mechanism for bronchiolitis obliterans syndrome. Hum Immunol. 2003;64(5):521–529. | |
Maruyama T, Jaramillo A, Narayanan K, Higuchi T, Mohanakumar T. Induction of obliterative airway disease by anti-HLA class I antibodies. Am J Transplant. 2005;5(9):2126–2134. | |
Fukami N, Ramachandran S, Saini D, et al. Antibodies to MHC class I induce autoimmunity: role in the pathogenesis of chronic rejection. J Immunol. 2009;182(1):309–318. | |
Lyu DM, Grazia TJ, Benson AB, Cagle LR, Freed BM, Zamora MR. Pre-transplant presence of antibodies to MICA and HLA class I or II are associated with an earlier onset of bronchiolitis obliterans syndrome in lung transplant recipients. Clin Transpl. 2012:237–246. | |
Weber DJ, Wilkes DS. The role of autoimmunity in obliterative bronchiolitis after lung transplantation. Am J Physiol Lung Cell Mol Physiol. 2013;304(5):L307–L311. | |
Jurcevic S, Ainsworth ME, Pomerance A, et al. Antivimentin antibodies are an independent predictor of transplant-associated coronary artery disease after cardiac transplantation. Transplantation. 2001;71(7):886–892. | |
Morgun A, Shulzhenko N, Unterkircher CS, et al. Pre- and post-transplant anti-myosin and anti-heat shock protein antibodies and cardiac transplant outcome. J Heart Lung Transplant. 2004;23(2):204–209. | |
Dragun D, Müller DN, Bräsen JH, et al. Angiotensin II type 1-receptor activating antibodies in renal-allograft rejection. N Engl J Med. 2005;352(6):558–569. | |
Reinsmoen NL, Lai CH, Heidecke H, et al. Anti-angiotensin type 1 receptor antibodies associated with antibody mediated rejection in donor HLA antibody negative patients. Transplantation. 2010;90(12):1473–1477. | |
Burlingham WJ, Love RB, Jankowska-Gan E, et al. IL-17-dependent cellular immunity to collagen type V predisposes to obliterative bronchiolitis in human lung transplants. J Clin Invest. 2007;117(11):3498–3506. | |
Iwata T, Chiyo M, Yoshida S, et al. Lung transplant ischemia reperfusion injury: metalloprotease inhibition down-regulates exposure of type V collagen, growth-related oncogene-induced neutrophil chemotaxis, and tumor necrosis factor-alpha expression. Transplantation. 2008;85(3):417–426. | |
Goers TA, Ramachandran S, Aloush A, Trulock E, Patterson GA, Mohanakumar T. De novo production of K-alpha1 tubulin-specific antibodies: role in chronic lung allograft rejection. J Immunol. 2008;180(7):4487–4494. | |
Bharat A, Saini D, Steward N, et al. Antibodies to self-antigens predispose to primary lung allograft dysfunction and chronic rejection. Ann Thorac Surg. 2010;90(4):1094–1101. | |
Saini D, Weber J, Ramachandran S, et al. Alloimmunity-induced autoimmunity as a potential mechanism in the pathogenesis of chronic rejection of human lung allografts. J Heart Lung Transplant. 2011;30(6):624–631. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.