Back to Journals » Clinical Ophthalmology » Volume 13
Current perspectives in Bietti crystalline dystrophy
Authors García-García GP ![]() , Martínez-Rubio M
, Martínez-Rubio M ![]() , Moya-Moya MA
, Moya-Moya MA ![]() , Pérez-Santonja JJ
, Pérez-Santonja JJ ![]() , Escribano J
, Escribano J
Received 12 March 2019
Accepted for publication 8 July 2019
Published 30 July 2019 Volume 2019:13 Pages 1379—1399
DOI https://doi.org/10.2147/OPTH.S185744
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Scott Fraser
GP García-García,1 M Martínez-Rubio,1 MA Moya-Moya,1 JJ Pérez-Santonja,1 J Escribano2,3
1Department of Ophthalmology, General University Hospital of Alicante, Alicante 03010, Spain; 2Cooperative Research Network on Ophthalmology (OftaRed), Visual and Life Quality, Instituto de Salud Carlos III, Madrid, Spain; 3Laboratory of Human Molecular Genetics, Medicine Faculty/Research Institute on Neurological Disabilities (IDINE), University of Castilla La-Mancha, Albacete 02006, Spain
Abstract: Bietti crystalline dystrophy (BCD) is a rare-inherited disease caused by mutations in the CYP4V2 gene and characterized by the presence of multiple shimmering yellow-white deposits in the posterior pole of the retina in association with atrophy of the retinal pigment epithelium (RPE) and chorioretinal atrophy. The additional presence of glittering dots located at the corneal limbus is also a frequent finding. The CYP4V2 protein belongs to the cytochrome P450 subfamily 4 and is mainly expressed in the retina and the RPE and less expressed in the cornea. The disease has its metabolic origin in the diminished transformation of fatty acid substrates into n-3 polyunsaturated fatty acids due to a dysregulation of the lipid metabolism. In this review, we provide updated insights on clinical and molecular characteristics of BCD including underlying mechanisms of BCD, genetic diagnosis, progress in the identification of causative genetic and epigenetic factors, available techniques of exploration and development of novel therapies. This information will help clinicians to improve accuracy of BCD diagnosis, providing the patient reliable information regarding prognosis and clinical prediction of the disease course.
Keywords: Bietti crystalline dystrophy, CYP4V2 gene, corneal deposits, retinal deposits
Introduction
Bietti crystalline dystrophy ((BCD), Online Mendelian Inheritance in Man (OMIM) OMIM210370) is an inherited autosomal recessive disease linked to biallelic mutations affecting the CYP4V2 gene. Professor Gian Battista Bietti first described the disorder in 1937,1 reporting three patients – including two brothers – with a pattern of retinal crystalline spots in the posterior pole, scattered conglomerations of retinal pigment, chorioretinal atrophy and corneal superficial deposits at the limbus. In addition, the author suggested the inherited and familial nature of the disease and differentiated it from other dystrophies like retinitis punctata albescens and fundus albipunctatus.
Bagolini and Ioli-Spada2 designated this pathology “Bietti´s tapetoretinal degeneration with marginal corneal dystrophy” and studied the evolution of these brothers and six additional patients in 1968, confirming the progressive and degenerative nature of their condition. Welch3 introduced the term “crystalline retinopathy” in 1977 to complete the original description, and identified the presence of lipid inclusions in fibroblasts and corneal epithelium by analyzing a corneal limbus biopsy obtained from a patient with BCD, pointing to a metabolic involvement in the genesis of the disease.
In 2000, Jiao et al4 performed genetic linkage analysis to 49 members of 10 Chinese, Japanese and European families with BCD, and identified the locus of the gene responsible on human chromosome 4q35-qter. In 2004, at a later stage, Li et al5 identified the CYP4V2 gene to be the causative for BCD, encoding a novel 525 amino acid protein member of the cytochrome P450 (family 4, subfamily IV, polypeptide 2), involved in fatty acid metabolism. The first generation and characterization of a murine model of BCD was performed by Lockhart et al6 in 2013, reproducing the presence of retinal crystalline deposits and metabolic lipid disturbances in Cyp4v3−/- knockout mice, corresponding well to BCD findings in humans.
These and other relevant studies along the years have increased our understanding of BCD disease and generated fresh perspectives and hope to patients and ophthalmologists. The aims of this report are to review relevant information about BDC and to provide current approaches on this disorder.
Epidemiology and distribution
Estimating true prevalence rates of BCD is difficult as methods of data collection vary between countries, authors and surveys. Hu7 estimated a gene frequency of 0.005 by studying the first cousin parents of an epidemiologic survey in China in 1983. In accordance with the works of Hartong et al8, Okialda et al9 and Ng et al,10 the estimated a prevalence of BCD is 1 in 67,000 individuals, affecting 21,000 patients in China and about 5000 in USA. According to our estimations, the actual prevalence of BCD in Spain may be extremely low, affecting approximately 1 in 4,500,000 subjects, ie, 10–12 cases in the whole country, based on a survey carried out in 2013 in more than 650 medical centers of Spain.11 At the same time, the disease presents worldwide distribution, and tends to be common in the east of Asia, being more prevalent in Chinese, Japanese and Korean populations. Most of the reported cases diagnosed in Europe correspond to Italian, Lebanese and Spanish patients suggesting a possible Mediterranean distribution of the disease.
Onset, staging and clinical manifestations
BCD is a progressive chorioretinal dystrophy with corneal involvement characterized by profuse yellowish sparkling deposits at the retina (Figure 1), geographical areas of atrophy of the retinal pigment epithelium (RPE) and loss of choriocapillaris, chorioretinal atrophy and crystalline deposits in peripheral cornea.
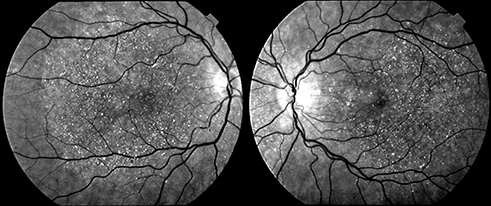 |
Figure 1 Infrared images showing a typical pattern of “starry sky” fundus with numerous tiny glittering crystal deposits throughout the entire posterior pole of a patient with BCD (both eyes). |
According to Yuzawa et al12, the illness can be classified into three stages (Figure 2):
 |
Figure 2 Color retinography of the fundus of both eyes of different patients showing different degrees of evolution of BCD disease, according to Yuzawa classification. (A) Numerous glistening crystalline dots and minimal chorioretinal damage restricted to the posterior pole of the retina, corresponding with stage 1 of Yuzawa. (B) Epithelial confluent atrophy of the posterior pole and few crystalline deposits, corresponding with stage 2 of Yuzawa. (C) Advanced choroidal sclerosis with marked chorioretinal atrophy and absence of deposits, corresponding with stage 2 of Yuzawa. Misdiagnosing with severe forms of choroideremia or retinitis pigmentosa is possible. |
Stage 1: RPE atrophy with crystalline deposits in the macular area.
Stage 2: RPE atrophy extends beyond the posterior pole. Choriocapillaris atrophy at the posterior pole. Crystalline confluent deposits in the damaged area, less in number at the advanced atrophic areas of the RPE-choriocapillaris complex.
Stage 3: RPE-choriocapillaris complex extensive atrophy and a small number of residual crystalline deposits throughout the fundus.
The first clinical manifestations most commonly appear between the second and third decade of life, but can develop from teenagers to over fourth decade, remaining the determinants of age at onset unclear. In addition, the severity of the retinal damage does not seem to correlate well with the age of debut.
During the earliest stages, patients with BCD are often asymptomatic, which difficult its diagnosis. Therefore, most early BCD cases are often diagnosed as casual findings. As BCD progresses symptoms appear slowly and painlessly, involving nyctalopia, limited peripheral vision and visual field constriction, color vision impairment, floaters, photopsias and monocular diplopia. In the late stages of BCD, severe visual damage and legal blindness are common findings. It should be kept in mind that disease evolution may be asymmetric in the two eyes, that a good visual acuity outcome may not correspond to the measure of damage in the retina and that high variability in disease presentation and evolution has been reported.11,13
Diagnosis
Corneal involvement: medical imaging techniques and clinical application
The presence of crystalline deposits in the peripheral paralimbal cornea is the main BCD clinical manifestation regarding the ocular surface, which might be explained by the moderate CYP4V2 expression in the corneal epithelium and subepithelium.14 Functional deterioration of this gene carries an increased accumulation of intracellular deposits of fatty acids in peripheral corneal and conjunctival fibroblasts and keratinocytes,15–17 involving the development of the marginal keratopathy. These subclinical alterations course with normal vision and are not essential requirements for BCD diagnosis. They are more commonly described in European than in Asian patients, although the reported frequency of marginal keratopathy is highly variable, ranging from a 75% rate of incidence in Spanish patients11 to the complete absence of keratopathy in a sample of 15 Italian patients.18 The presence of corneal crystalline deposits in the clinical reports and the identified mutations associated with them are summarized in Table 1.
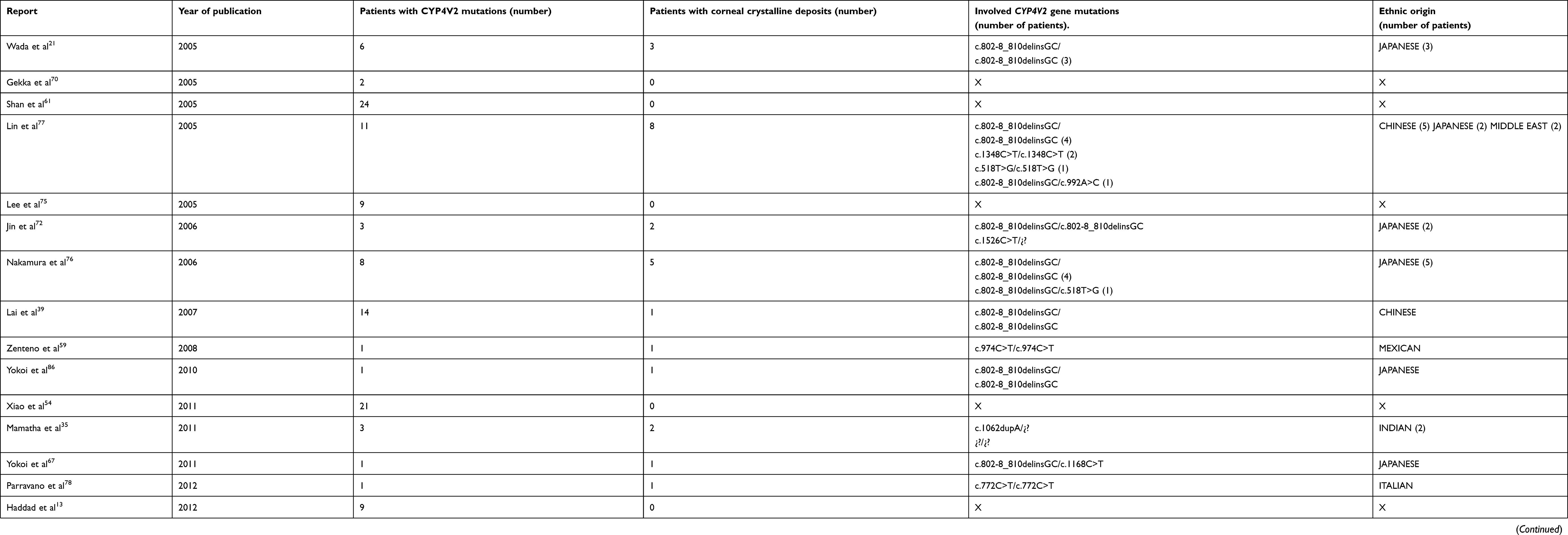 |
Table 1 Presence of corneal crystalline deposits in patients with clinical and genetic diagnosis of BCD confirmed by CYP4V2 gene analysis, according to the medical literature. |
The high quality of modern slit lamps allows the easy and quick identification of crystalline deposits throughout the entire peripheral cornea (Figure 3). Nevertheless, very subtle crystals (smaller than 15 μm) can remain undetected if a careful examination is not performed.3
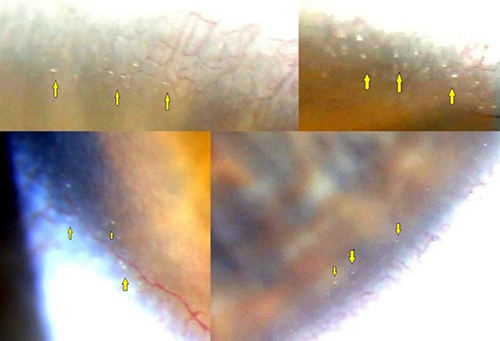 |
Figure 3 Slit-lamp biomicroscopic findings in several patients with BCD with corneal involvement. Crystalline-like corneal deposits (arrows) can be present in 360º degrees of the limbus area of both eyes. |
In vivo corneal confocal microscopy has corroborated the location of crystalline deposits in several patients with a previous slit-lamp diagnosis of BCD marginal keratopathy.11,19,20 Corneal dots were detected in the 360º of the peripheral and paracentral cornea, near the limbus, involving subepithelial and anterior stromal layers (approximately 20–40 µm long and 4–8 µm wide), with variable forms (needle, rod, pin, globular, chromosome shaped), irregular dissemination and possible centrifugal distribution (Figure 4). Specular microscopy is an accessory imaging technique that may help to identify subclinical corneal crystalline dots.21
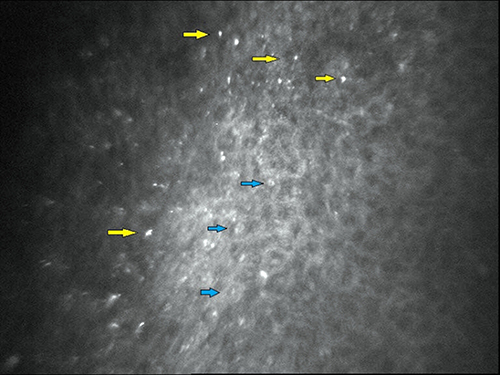 |
Figure 4 In vivo confocal microscopy image (400×400 μm) of a BCD patient (subepithelium area). The crystals (yellow arrows) were up to 24 μm in length and 16 μm in width and barely distinguishable from keratocytes nuclei (blue arrows). |
Retina involvement: medical imaging techniques and clinical application
Our understanding of structural changes and underlying retinal lesions in BCD has been highly improved by the advent of new imaging techniques. Fluorescein angiography (FA) has been traditionally considered the gold standard in the evaluation of retinal damage and clinical progression of BCD because the findings obtained with this technique clearly correlate with the severity and stage of BCD.15,16,22 In the initial phases of the disease, FA reveals marked hyperfluorescence in the posterior pole because the atrophy of the RPE generates a window defect, with a relatively intact choriocapillaris. The crystalline dots located in the central and paracentral retina accounts for the blocked choroidal fluorescence. In the middle phases of the disease, FA discloses irregular geographic hypofluorescent areas of RPE and island-like choriocapillaris, in correspondence to areas of non-perfused choriocapillaris, with generalized disturbance and atrophy of the RPE in the posterior pole and enhanced image of the choroidal vasculature. In the later stages, choroidal atrophy and deep damage of the choriocapillaris and RPE complex are present, the volume of choroidal vessels is highly diminished and the blood vasculature is mainly sclerosed15,16,22 (Figure 5). Indocyanine green angiography (ICGA) remains considered as a valuable supplement diagnosis technique of BCD, better than FA in outlining the atrophy areas and in evidencing the real level and magnitude of the choroidal circulatory damage.23,24
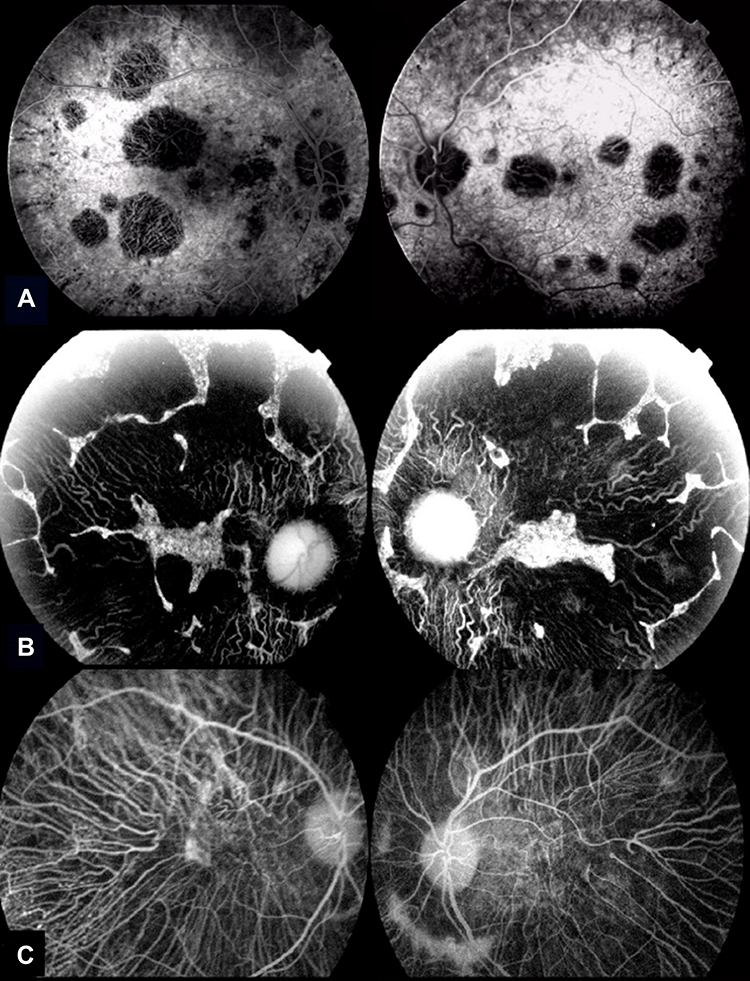 |
Figure 5 Fluorescein angiography in later stage on both eyes of different patients with genetic diagnosis of BCD. (A) Small-patched areas of atrophy of the choriocapillaris and RPE, the macular area remain relatively intact in an early stage of BCD. (B) Marked atrophy of the RPE with focal confluent areas of disappearance of the choriocapillaris. (C) Severe atrophy of the RPE with complete disappearance of the choriocapillaris. |
Early-phase ICGA shows a significant delay in the filling of the choroidal vessels in every stage of the disease. This delay increases and correlates with BCD severity. An initial damage of the parapapillary area appeared to be constant by ICGA, showing a later centrifugal pattern of involvement as BCD progresses. In the middle stages, damage of the inner choroid increases and lobular hypofluorescent lesions become evident.23,24 Late-phase ICGA also reveals a more variable pattern of hyperfluorescence in clearly delimitated choroidal areas in all stages of BCD.
However, these two procedures are invasive and time-consuming, requiring venipuncture, and are not free of risks and threats like anaphylaxis or even death related to FA and ICGA injections. In fact, its use has diminished considerably in the last years due to the development of new non-invasive high-resolution imaging techniques like spectral-domain optical coherence tomography angiography (SD-OCT-A).
Fundus auto fluorescence (FAF) is not helpful to assess and control the evolution of crystalline deposits, because these dots present neither hypo-AF nor hyper-AF nature. The peripheral retina presents a normal FAF as long as there is no damage, but in BCD the posterior pole and the paracentral retina develops hypo-AF and clearly defined confluent-patched areas corresponding to local RPE loss sections. These alterations correlate well with funduscopic and FA changes throughout the progressive evolution of BCD.25,26
SD-OCT has proven to be an effective noninvasive retinal imaging method for evaluating the morphological and functional changes in BCD. Outer retinal tubulations (ORTs) and glistening hyperreflective deposits seem to be the main SD-OCT manifestations in BCD25,27 (Figure 6). ORTs are spherical hyporeflective lesions surrounded by a halo of high reflectivity, mainly situated in the retina outer nuclear layer and more frequent in advanced stages 2 and 3 of Yuzawa.12 The mechanisms underlying ORTs remain unclear, but it has been suggested the existence of a diminished adherence of the photoreceptors outer segments to the RPE in its junction layer.26,29 This alteration can be due to a potential rearrangement and subsequent invagination of damaged photoreceptors that partially retain their function as a protection mechanism.
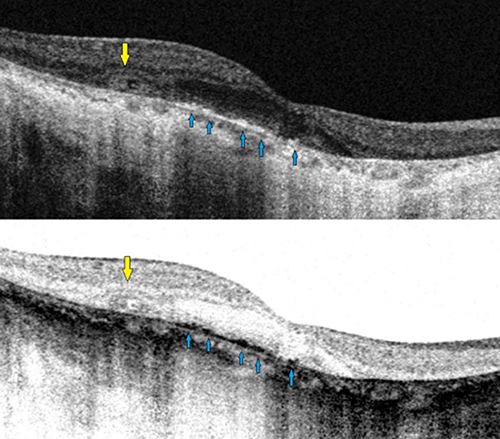 |
Figure 6 Spectral domain-optical coherence tomography (SD-OCT) image revealing both outer retinal tabulation (blue arrow) and crystalline macular deposits (yellow arrow) of a BCD individual. |
The presence of crystalline glistening deposits has been confirmed in all retinal layers by SD-OCT, mainly in the outer retina, being more numerous at the RPE-choriocapillaris level, including the Bruch membrane.20,25 According to Saatci et al28, there are three categories of glistening deposits in SD-OCT sections: 1) highly reflective spots in the inner retina, 2) reflective plaques on the top of the Bruch membrane and 3) partially encapsulated reflective plaques. There seems to be a clear correlation between funduscopy crystalline deposits and hyperreflective dots located in or on REP-Bruch membrane complex.25–30 The origin and clinical significance of the plaques, encapsulated lesions and other hyperreflective dots, including those located in the choroid, remains unclear. Accumulation of protein deposits or inflammatory cells, glial response due to retinal destruction or even artifacts, has been postulated to explain the origin of these lesions.25,28 There are three potential phases in the evolution of BCD, according to SD-OCT imaging modality as defined by Li et al.26 Structures with the appearance of druses, decrease of the interdigitating area and local disruption of the ellipsoid area are present in stage 1. Stage 2 involves the presence of isolated islands of remaining RPE in the context of a high loss of this retinal layer and damage of the outer retina. Severe macular disturbance is the main finding in stage 3. According to a recent study,31 near-infrared imaging (NIR) – a commonly performed capture modality while acquiring simultaneous OCT scans – should be a useful tool in identifying BCD crystalline retinal deposits and in differentiating them from other presents in chorioretinal dystrophies with similar phenotypes, facilitating a more efficient posterior genetic diagnosis.
The knowledge of choroidal vasculature changes in retinal dystrophies has been especially improved by the use of OCT-A, a recent tool based on motion contrast imaging. The dynamic of the retinal and choroid microvasculature in BCD can be evaluated throughout the association of the advantages of the OCT-A and the split spectrum amplitude-decorrelation angiography.
A diminished choriocapillaris blood flow appears to be present in the vast majority of the BCD patients, and the visibility of the choriocapillaris layer at the subfoveal area correlates well with the degree of function of the visual system in BCD individuals according to Miyata et al.32 These features make OCT-A a non-invasive and accessible alternative to assess the patient’s evolution. Choroidal neovascularization has occasionally been reported in patients with clinical and molecular diagnosis of BCD.26,33–35 The nature and mechanisms of this relationship remain unclear, but RPE defects and long-term friction of the Bruch’s membrane by the crystalline dots should play a reasonable role.35 There is proven efficacy of anti-Vascular Endothelial Growth Factor (VEGF) therapy in the management of this complication.34 Macular hole may also be detected in BCD and can be successfully managed by routine surgery.36
In summary, the current available imaging techniques open up many possibilities to assess retinal dystrophies, and multimodal evaluation of BCD patients is strongly recommended in the management of the disease. These combined techniques should allow us to make the diagnosis as early as possible, staging the severity and the progression of the disease and monitoring the response to potential future treatments.
Electrophysiological findings
Electrophysiological techniques are objective complementary explorations in BCD that include full-field electroretinography (ffERG) and multifocal electroretinography (mfERG) recorded according to the ISCEV protocols.37 Regarding the electrooculogram, we consider it provides no additional information if ffERG and mfERG are performed. Actually, electrophysiology provides supplementary data in the early diagnosis, allows disease severity assessment, facilitates control of progression and determines how much retinal damage has been caused. The progression of the disease may follow a similar rod – cone dystrophy pattern according to ffERG findings.37–40 Full-field ERG records range from normal in early stages to undetectable signals in the last phases of BCD. In the middle stages, the malfunction of both rod and cone mechanism involves multiple disturbances, and delayed amplitude of photopic and more evident scotopic responses should be present. The measurement of the implicit time of both responses, especially scotopic time, can be mildly delayed or even normal.40,41 mfERG assesses cone function from the central 30–40 degree field of the retina and provides additional spatial resolution and topographical evaluation not available in ffERG. This technique has become very helpful to expound the degree of responses acquired from ffERG especially in early stages of BCD, as well as to monitor the progression of the retinal central damage.42–44 The amplitudes of the mfERG are markedly attenuated compared to normal individuals in the fovea and central retina, remaining more affected than the implicit times like in ffERG. Peripheral responses may be preserved. P1 amplitudes and implicit times are markedly disturbed and N1 responses should be conserved.10,45,46
Genetic diagnosis
The classical diagnosis of BCD based on the clinical findings of typical crystalline deposits in the retina and the cornea detected by an experienced physician is, in general, useful but did not provide assurance. However, the only way to confirm a diagnosis of BCD disease is the identification of CYP4V2 gene (OMIM, *608614) mutations by genetic analysis. The DNA samples can be simply obtained from peripheral leukocytes by venipuncture or by collection of a small amount of saliva into a sterile tube. Amplification of the 11 CYP4V2 exons, including intron-exon junctions and flanking sequences, using PCR and followed by automated Sanger DNA sequencing can be performed as described by Li et al.5 The complete genetic sequence can be also obtained by next-generation techniques, although the presence of a pathogenic variant needs confirmation by Sanger sequencing, which is considered the gold standard procedure in mutation detection. The primary aims of genetic testing are to establish, confirm or exclude a clinical diagnosis of BCD. Identification and functional evaluation of the causative gene mutation, as well as confirmation or exclusion of the presence of heterozygous CYP4V2 carriers among relatives of BCD patients, are required for genetic counseling. The clinical sensitivity (proportion of positive studies if BCD is present) of the test is estimated higher than 93% and the clinical specificity (proportion of negative tests if BCD is not present) rounds 99.99%.9,47
Genetic linkage of BCD
Bietti crystalline corneoretinal dystrophy (OMIM #210370) is inherited in an autosomal recessive manner. Jiao et al4 first showed the linkage of BCD to chromosome 4q35-qter, and subsequently Li et al5 refined critical interval of the disease (4q35.1-q35.2) and identified CYP4V2 as the disease-causing gene. The CYP4V2 gene is composed of 11 exons spanning 21 kb, and encodes a 525 amino acid protein, which is the second member of the cytochrome p450, family 4 (CYP4), subfamily V and plays an important role in lipid metabolism.48
CYP4V2 protein and fatty acid metabolism
CYP4V2 is a microsomal enzyme with ω-hydroxylase activity on both saturated and polyunsaturated fatty acids (PUFAs) of medium and long-chain6,14,49 and is expressed in the vast majority of the body tissues, especially in RPE and retina, reaching a lesser degree of expression in the cornea.5,48,49 These facts supported the theory postulated by several authors which proposed lipid metabolism alteration as a major cause of BCD.3,16,49,50 Functional impairment of CYP4V2 leads to a global malfunction of the lipid metabolism system in the homozygous individuals. Lee et al48 first reported the absence of two fatty acid-binding proteins of 32 and 45 kDa in cultured lymphocytes of BCD patients. Further studies revealed that cultured lymphocytes and fibroblasts showed additional alterations in the fatty acid metabolism, including marked reduction in the transformation of fatty acid precursors into ω-3 PUFAs, decreased synthesis of eicosapentaenoic acid (20:5n-3) and docosahexaenoic acid (DHA), increased conversion of alpha-linolenic acid (ALA) into triglycerides and abnormalities in the storage of these lipids.14,17,48–50 The ω-6 biosynthesis pathway apparently was not affected.15 In serum, a reduced activity of the Δ-9-desaturase enzyme and abnormally low concentration of total monounsaturated fatty acids have been reported.16,50
The CYP4V2 enzyme is present predominantly in the endoplasmic reticulum of the human RPE cell line ARPE-19. Immunohistochemistry analyses revealed that CYP4V2 is also present in retinal outer and inner nuclear layers, retinal ganglion cells and corneal epithelial cells, in accordance with the BCD phenotype.14 In addition, the gene is expressed in different non-ocular organs such as heart, lung, liver, pancreas, kidney, brain, skeletal muscle and placenta. Interestingly, BCD patients do not show any pathological condition in these organs, suggesting the presence of compensatory enzymes which are absent in the RPE. Moreover, the exact pathogenic mechanism involved in RPE malfunction and in the subsequent damage remains unknown. Particularly interesting is the function of the CYP4V2 protein in DHA reactions. DHA is an essential constituent of the rod outer segment and plays an important role in the maturation and survival of the photoreceptors cells required for normal visual function.51 Functional CYP4V2 alteration generates a disruption of the membrane processing of lipids in RPE layer, which result in a significant focal dyslipidemia and degeneration of the photoreceptors. The endogenous conversion of DHA to resolvins, protectins and maresins52 – mediators involved in the innate anti-inflammatory modulation – suggests that defective CYP4V2 activity may also impair the resolution phase of the inflammatory process.6,53
Recently, Hata et al53 performed the induction of pluripotent stem cells from individuals who carried specific CYP4V2 mutations to generate RPE cells, in order to develop an innovative in vitro model of BCD. These cells showed vacuolated cytoplasm similar to those degenerative changes observed in individuals with BCD. In addition, the cells accumulated glucosylceramide and free cholesterol in association with lysosomal malfunction and decay of autophagy flux, involving severe impairment and cellular apoptosis. The reduction of intracellular free cholesterol might mitigate the damage caused by BCD in RPE cells, and is postulated by the authors as a potential preventive or therapeutically measure.
BCD-associated gene variants
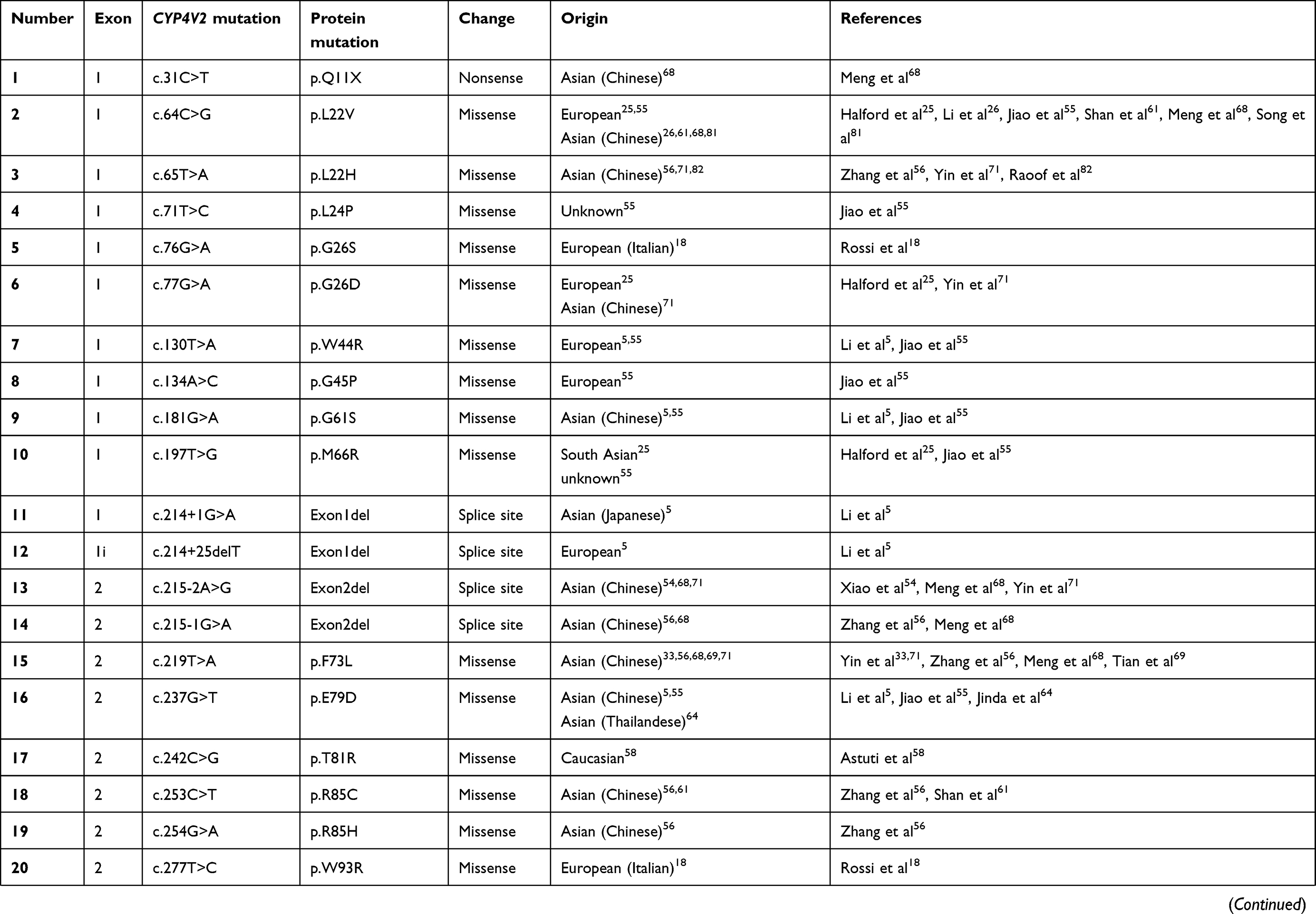
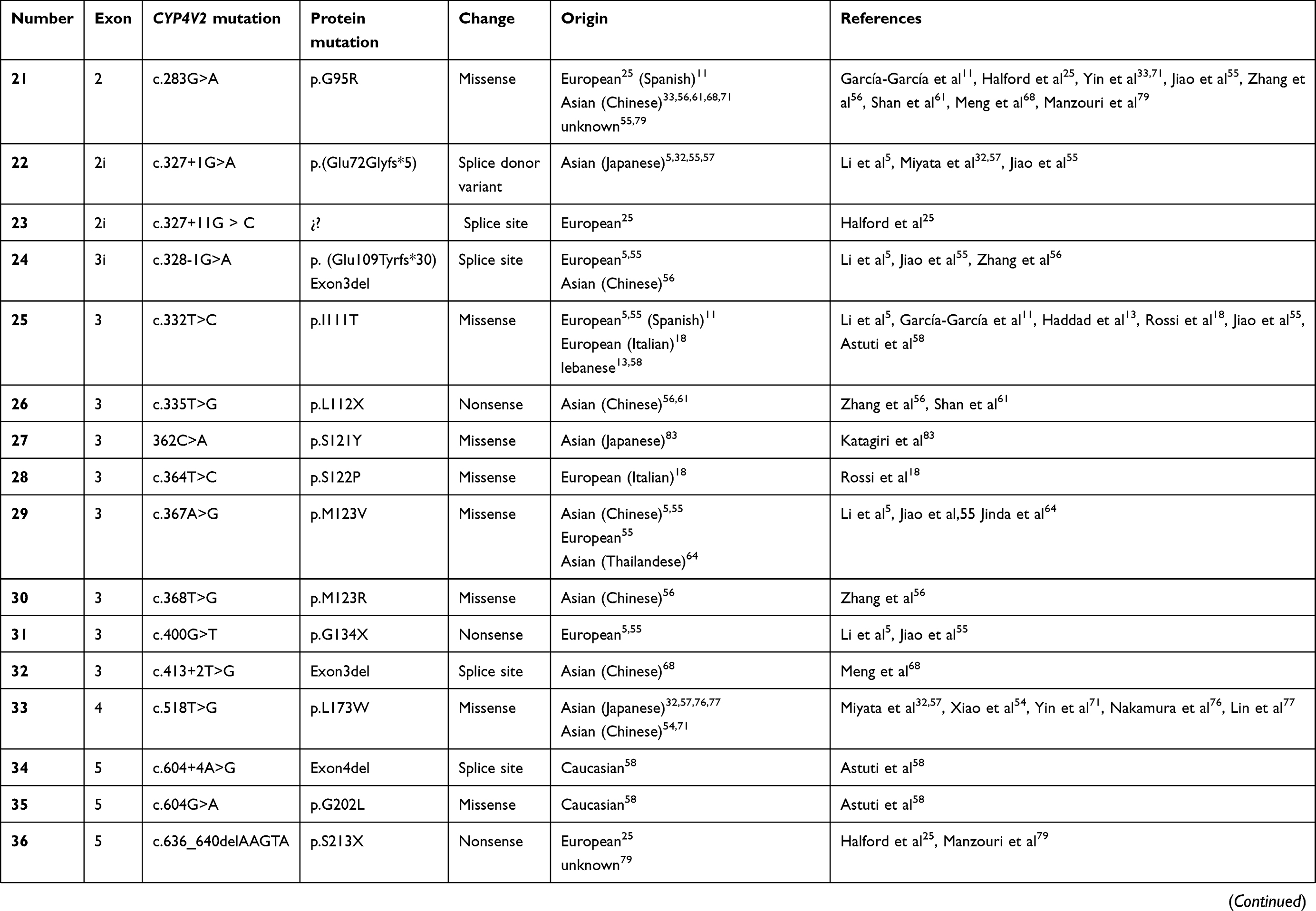
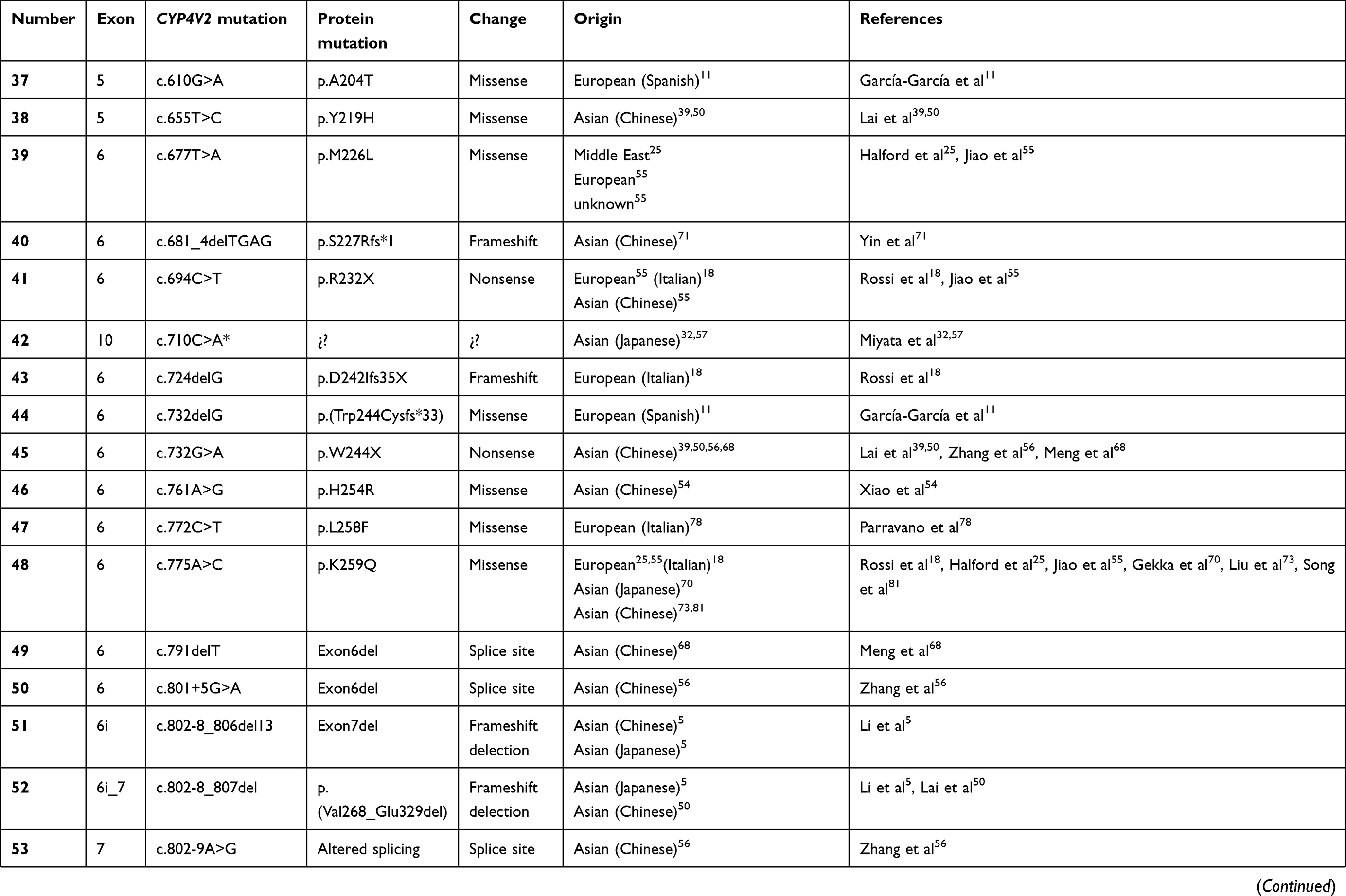
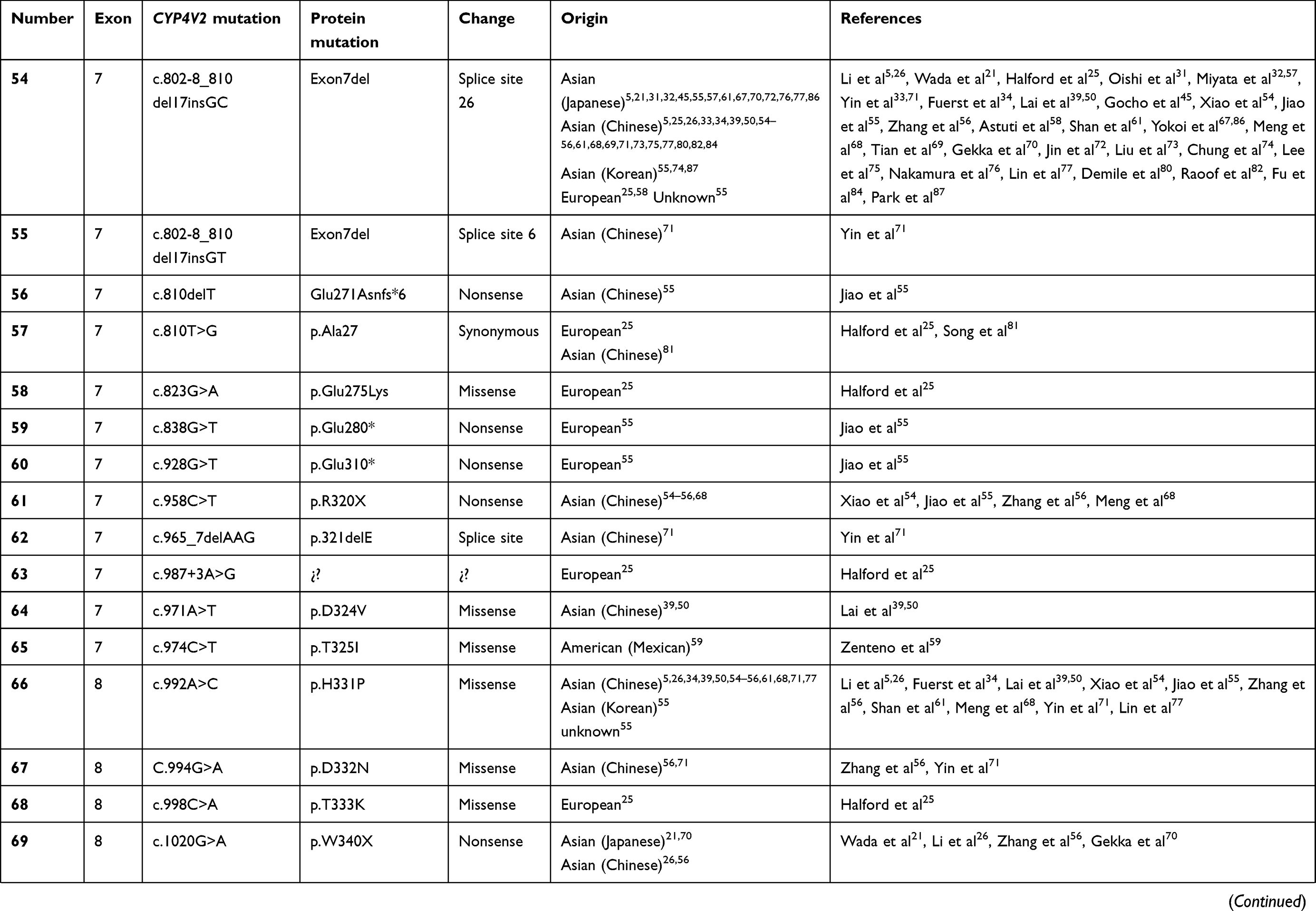
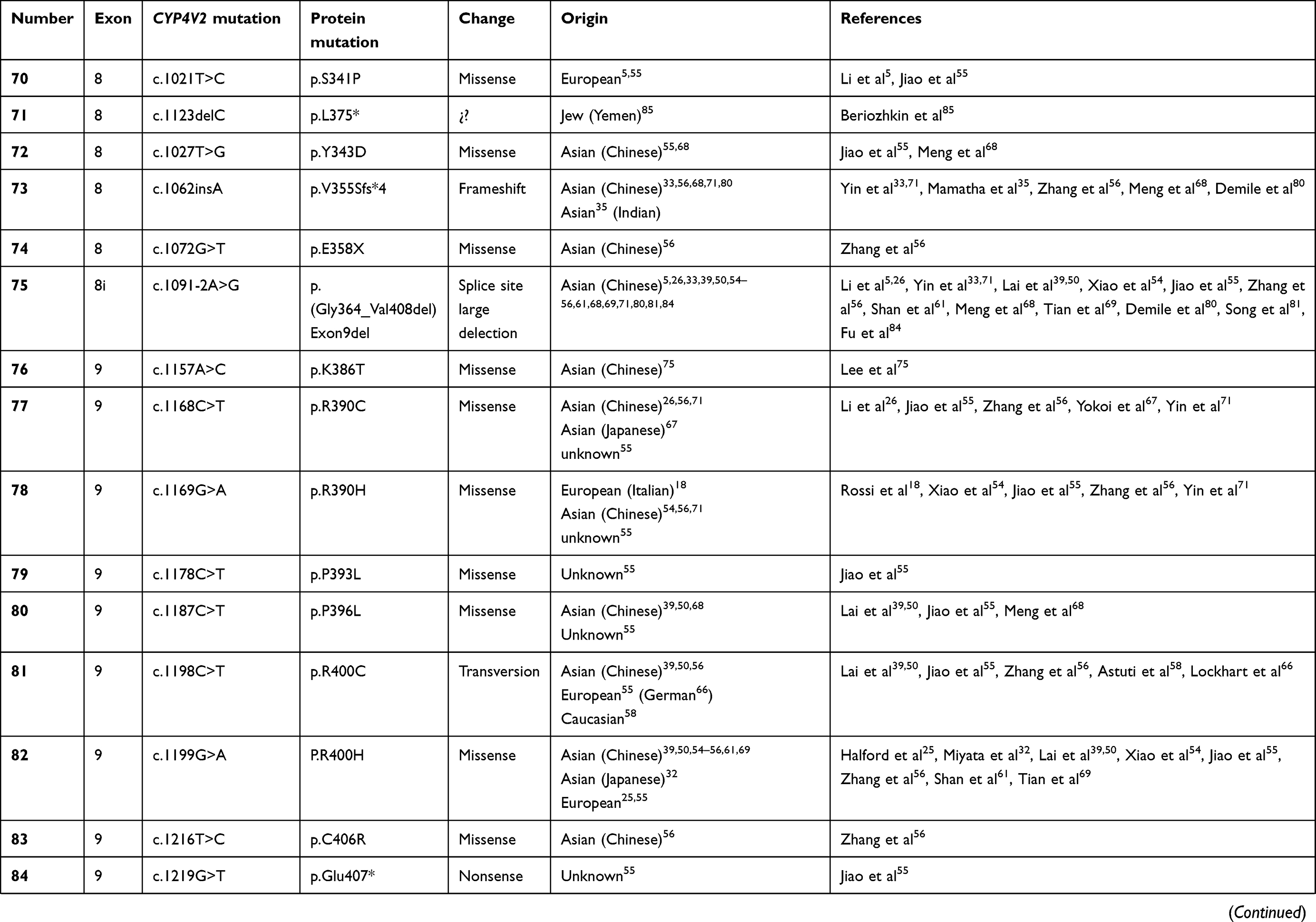
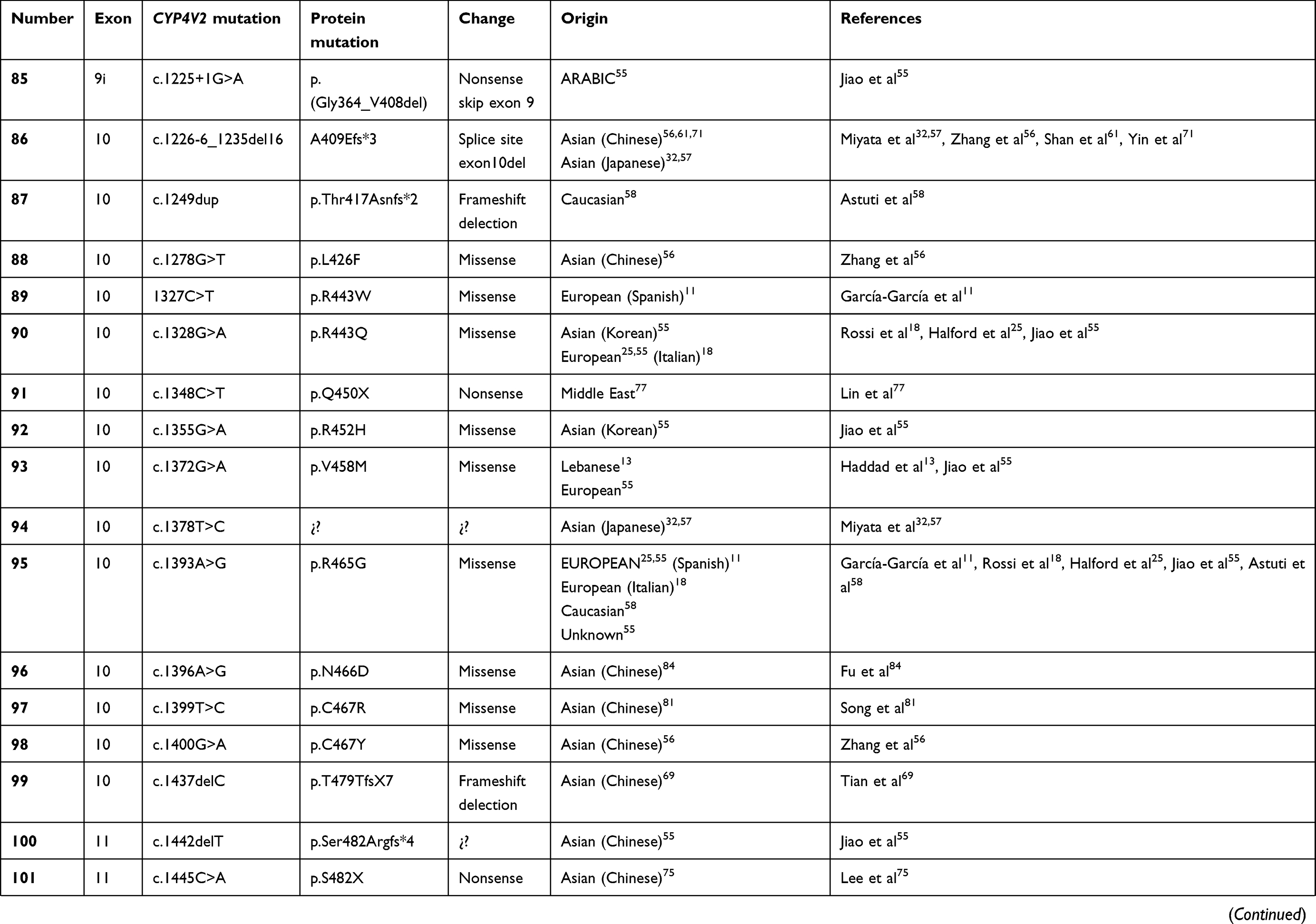
To date, over 100 disease-causing mutations in CYP4V2 gene have been reported according to previous clinical studies (Table 2). The vast majority of the mutations are missense, followed by large deletions, nonsense mutations, small insertions or deletions and splicing site variants, which are more unusual. The most frequent mutation identified is the insertion-deletion IVS6-8del17bp/insGC or c.802-8del17bp/insGC, at the junction between intron 6–exon 7 of CYP4V2 gene,54 an ancient founder mutation present in Chinese, Korean, and Japanese patients, with an allele frequency of 17.2–83.3%55,56 and undetected in European or Middle-East communities. The second most common mutations reported only in BCD East Asian patients are c.1091-2A>G [p.(Gly364_Val408del)] – mostly identified in the Chinese community56 – and c.992A>C [p.(H331P)] which is prevalent in Chinese and Korean population (Table 1). According to the literature, mutation c.518T>G [(p.L173W)] has been reported by several authors mostly in Japanese patients31,54,57 (Table 1).
 |
 |
 |
 |
 |
 |
 |
Table 2 Summary of the mutations identified in CYP4V2 gene in patients with molecular diagnosis of BCD, according to the medical literature. |
Prevalent mutations in Caucasian individuals are the missense changes c.1393A>G [p.(R465G)] and c.332T>C [p.(I111T)], widely reported in European,5,25,55 Spanish,11 Italian18 and Lebanese13,58 patients. Both of them might follow a pattern of Mediterranean distribution of the cases. This context suggests the presence of a founder effect and the potential arising of these variants in a geographically delimited Mediterranean region.11
The missense mutation c.974C>T [p.(T325I)] detected by Zenteno et al59 is to the best of our knowledge the only CYP4V2 disease-causing variant reported in Latin-American individuals.
The CYP4V2 protein structure presents a transmembrane region at the amino terminal end, followed by a globular structural domain composed of 18 α-helices connected by β-sheet and random coil structures.5 The heme group resides near the protein surface, between helices I (toward the central core) and L (toward the surface), similarly to other members of the CYP450 superfamily.5 CYP4V2 pathogenic mutations including frameshift, splicing site and nonsense changes impair this highly conserved protein folding. Missense mutations generally affect the transmembrane segment, interfering with the integration of the CYP4V2 protein into the membrane, and in the active site of the enzyme disrupting the coordination of the porphyrin ring needed for its catalytic function. The α-helix supporting the porphyrin ring may also be affected by missense changes.55 Therefore, all mutations identified in BCD patients likely lead to the loss-of-function of the enzyme.
Recently, CYP4V2 has been also detected in human hepatocellular carcinoma – being its expression significantly associated with a better disease prognosis – and in breast cancer showing a less aggressive degree of neoplasia.60 Genome-wide association research have detected the CYP4V2 c.775A>C [p. (K259Q)] variant linked with deep vein thrombosis.55,61
Both CYP4V2 intronic F11 variant CYP4V2-KLKB1-F11 secondary to rs2289252 and the rs2036914 polymorphism (rs13146272) have been also considered as risk factors associated with venous thromboembolism.62
Genotype – phenotype findings and epigenetic factors
The existence of elevated clinical variability in unrelated BCD individuals harboring the same homozygous mutation or combination of them, and the additional intra- and interfamilial variability in clinical severity suggests the presence of other factors that modulate the disease.11,18 Due to the fact that fatty acid metabolism is clearly implicated in the genesis of the disease, healthy diet and good patterns of alimentation, lipid restriction and other environmental factors are suggested to play an additional role in the final phenotypic characterization.11,18,53,63.
Regarding the most common mutations in occidental patients, c.1393A>G [p.(R465G)] is predicted to generate an aggressive phenotype of BCD, according to the bioinformatics tools that evaluate the impact of the amino acid substitutions on the architecture and functioning of CYP4V2 protein. Mutation c.332T>C [p.(I111T)] is also considered to be deleterious.18 The prediction correlates well with the clinical findings of extensive damage, rapid progression and severe course of BCD phenotype detected in a homozygous p.(Arg465Gly) Spanish patient.11 Nevertheless, there are reports of several homozygous p.(Ile111Thr) patients presenting different patterns of clinical disease ranging from slow progression, less aggressive damage and mild symptoms11,13,18 to aggressive clinical phenotype and fast rate of progression.13,58 These data suggest the potential role of environmental, epigenetic and/or additional unknown factors in the phenotypic variability observed.
Epigenetic processes including DNA methylation and chemical posttranslational alterations of the histones act coordinately to regulate gene expression and adjust physiology of healthy cells.63 Defective or absent epigenetic regulation could contribute to the presence of different patterns of phenotypic expression of BCD and the fast or slow rate progression of the disease. Identifying and targeting the defective epigenetic modulation to correct the altered mechanisms, especially in the early phases of BCD, should be the basis to develop future diagnosis tools and new clinical treatments in order to improve the quality of life of the patients.
Future perspectives in BCD
Genetic counseling for BCD patients and their heterozygous asymptomatic relatives requires detailed record of the family history, disclosing relevant information about the disease and the pattern of inheritance. Genetic testing contributes to clarify the carrier condition of different family members, and is useful to predict the degree of damage and progression rate associated with specific mutations. In addition, genetic counseling allows assessment of a person or couple’s BCD risk and the chances that their offspring will inherit CYP4V2 mutations and manifest the disease. Correct family planning requires CYP4V2 genetic testing in partners of heterozygous carriers.
The discovery of accompanying CYP4V2 mutations in patients affected by retinitis pigmentosa, Leber congenital amaurosis (LCA) and other retinal dystrophies64,65 reveals the genetic complexity of some genotypes and shows that gene panel-based genetic testing is necessary for reliable retinal dystrophy diagnosis and correct clinical diagnosis of the individuals. Interpretation of possible combinatorial phenotypic effects among different mutations coinherited in these patients is difficult and, therefore, further research is required to clarify this issue.
The creation of national and international patient registries and databases in different countries for retinal dystrophies, containing both clinical and genetic information, is key instruments to advance clinical research as well as to improve patient care and health care planning. They allow collecting enough data to achieve sufficient sample sizes for epidemiological and/or clinical research. They also allow assessment of clinical trials feasibility and facilitate the planning of appropriate clinical trials and patient enrolment. Epigenetic alterations are increasingly recognized as important players in the pathogenesis of a growing number of diseases. In this line, CYP4V2 gene activity in BCD patients has historically been thought to be influenced by these mechanisms. To the best of our knowledge, the role of epigenetic factors in BCD has not been investigated, and the reversibility of these alterations provides an excellent opportunity for research and innovation of new therapeutic options for BCD patients. Therefore, the analysis of epigenetic modifications in BCD patients and the study of dietary components, especially lipids, and traditional medicines could contribute to determine the role of epigenetics in this disease.
The recent investigation of Hata et al53 supports the possible therapeutic effect in BCD patients of intracellular free cholesterol reduction. This type of studies might provide the basis for future medical treatments of BCD. Moreover, further characterization of CYP4V2 catalytic activity and better understanding of the role of this enzyme in BCD pathogenesis may also set the basis to develop novel therapies of this disease.
Relevant advances in the treatment of hereditary dystrophies, in particular, LCA type 2 (LCA2), has revitalized the objective of achieving healing of these previously untreatable diseases. RPE65 related LCA2 was successfully treated by gene augmentation surgery using subretinal injections with voretigene neparvovec,65 a gene therapy based on an adeno-associated vector. This clinical trial led to approval by the US Food and Drug Administration of the Voretigene neparvovec-rzyl treatment.63
Currently, there are over 100 clinical trials that utilize gene therapy for treating eye disorders, and BCD is included in the Reflection Bio’s RBIO-101 program (AAV.CYP4V2), granted by the FDA, to the research and development of an AAV-based gene therapy product for treating the disease. The next step of these development programs is testing the obtained gene therapy in a human clinical trial with BCD patients.
In conclusion, this review provides insights into the current clinical diagnosis and genetic testing of BCD. Despite important advances in this field, BCD remains poorly understood, requiring further efforts to elucidate its precise molecular and biochemical alterations. The long-term goal of this research is to develop different types of personalized medical treatments based on patients’ genetic profiles, which hopefully will be achieved in the near future.
Acknowledgments
This study has been supported by research grants from the Regional Ministry of Science and Technology of the Board of the Communities of ‘Castilla-La Mancha’ (SBPLY/17/180501/000404) and the ‘Instituto de Salud Carlos III/FEDER’ (RD16/0008/0019 and PI15/01193).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Bietti G. Ueber faxmiliares Vorkommen von “Retinitis punctata albescens” (verbunden mit “dystrophis marginalis cristallinea cornea”), glitzern, des glaskorpers und anderen degenerativen augenveranderungen. Klin Monbl Augenheilkd. 1937;99:737–756.
2. Bagolini B, Ioli-Spada G. Bietti’s tapetoretinal degeneration with marginal corneal dystrophy. Am J Ophthalmol. 1968;65:53–60. doi:10.1016/0002-9394(68)91028-3
3. Welch RB. Bietti’s tapetoretinal degeneration with marginal corneal dystrophy crystalline retinopathy. Trans Am Ophthalmol Soc. 1977;75:164–179.
4. Jiao X, Munier FL, Iwata F, et al. Genetic linkage of Bietti crystalline corneoretinal dystrophy to chromosome 4q35. Am J Hum Genet. 2000;67:1309–1313. doi:10.1016/S0002-9297(07)62960-7
5. Li A, Jiao X, Munier FL, et al. Bietti crystalline corneoretinal dystrophy is caused by mutations in the novel gene CYP4V2. Am J Hum Genet. 2004;74:817–826. doi:10.1086/382659
6. Lockhart CM, Nakano M, Rettie AE, Kelly EJ. Generation and characterization of a murine model of Bietti crystalline dystrophy. Invest Ophthalmol Vis Sci. 2014;55:5572–5581. doi:10.1167/iovs.13-13717
7. Hu DN. Ophthalmic genetics in China. Ophthal Paed Genet. 1983;2:39–45.
8. Hartong DT, Berson EL, Dryja TP. Retinitis pigmentosa. Lancet. 2006;368:1795–1809. doi:10.1016/S0140-6736(06)69740-7
9. Okialda KA, Stover NB, Weleber RG et al. Bietti Crystalline Dystrophy. In: Pagon RA, Bird TD, Dolan CR et al. editors. Gene ReviewsTM. Seattle (WA) University of Washington, Seattle: 1993-2014
10. Ng DS, Lai TY, Ng TK, et al. Genetics of Bietti crystalline dystrophy. Asia Pac J Ophthalmol. 2016;4:245–252. doi:10.1097/APO.0000000000000209
11. García-García GP, Martínez-Rubio M, Moya-Moya MA, Pérez-Santonja JJ, Escribano J. Identification of novel CYP4V2 genotypes associated with Bietti crystalline dystrophy and atypical anterior segment phenotypes in Spanish patients. Acta Ophthalmol. 2018;96(7):e865–e873. doi:10.1111/aos.13768
12. Yuzawa M, Mae Y, Matsui M. Bietti’s crystalline retinopathy. Ophthalmic Paediatr Genet. 1986;7:9–20. doi:10.3109/13816818609058037
13. Haddad NM, Waked N, Bejjani R, et al. Clinical and molecular findings in three Lebanese families with Bietti crystalline dystrophy: report on a novel mutation. Mol Vis. 2012;18:1182–1188.
14. Nakano M, Kelly EJ, Wiek C, Hanenberg H, Rettie AE. CYP4V2 in Bietti’s crystalline dystrophy: ocular localization, metabolism of ω-3-polyunsaturated fatty acids, and functional deficit of the p.H331P variant. Mol Pharmacol. 2012;82:679–686. doi:10.1124/mol.112.080085
15. Wilson DJ, Weleber RG, Klein ML, et al. Bietti’s crystalline dystrophy. A clinicopathologic correlative study. Arch Ophthalmol. 1989;107:213–221. doi:10.1001/archopht.1989.01070010219026
16. Kaiser-Kupfer MI, Chan CC, Markello TC, et al. Clinical biochemical and pathologic correlations in Bietti’s crystalline dystrophy. Am J Ophthalmol. 1994;118:569–582. doi:10.1016/s0002-9394(14)72565-6
17. Lee J, Jiao X, Hejtmancik JF, et al. The metabolism of fatty acids in human Bietti crystalline dystrophy. Invest Ophthalmol Vis Sci. 2001;42:1707–1714.
18. Rossi S, Testa F, Li A, et al. Clinical and genetic features in Italian Bietti crystalline dystrophy patients. Br J Ophthalmol. 2013;97:174–179. doi:10.1136/bjophthalmol-2012-302469
19. Bozkurt B, Ozturk BT, Kerimoglu H, Irkec M, Pekel H. In vivo confocal microscopic findings of 2 patients with Bietti crystalline corneoretinal dystrophy. Cornea. 2010;29:590–593. doi:10.1097/ICO.0b013e3181be22ee
20. Toto L, Carpineto P, Parodi MB, Di Antonio L, Mastropasqua A, Mastropasqua L. Spectral domain optical coherence tomography and in vivo confocal microscopy imaging of a case of Bietti’s crystalline dystrophy. Clin Exp Optom. 2013;96:39–45. doi:10.1111/cxo.2013.96.issue-1
21. Wada Y, Itabashi T, Sato H, Kawamura M, Tada A, Tamai M. Screening for mutations in CYP4V2 gene in Japanese patients with Bietti’s crystalline corneoretinal dystrophy. Am J Ophthalmol. 2005;139:894–899. doi:10.1016/j.ajo.2004.11.065
22. Mataftsi A, Zografos L, Millá E, Secrétan M, Munier FL. Bietti’s crystalline corneoretinal dystrophy: a cross-sectional study. Retina. 2004;24:416–426. doi:10.1097/00006982-200406000-00013
23. Saatci AO, Yaman A, Öner FH, Ergin MH, Çingil G. Indocyanine green angiography in Biettils crystalline retinopathy. Can J Ophthalmol. 2002;37:346–351. doi:10.1016/S0008-4182(02)80005-9
24. Fong AM, Koh A, Lee K, Ang CL. Bietti’s crystalline dystrophy in Asians: clinical, angiographic and electrophysiological characteristics. Int Ophthalmol. 2009;29:459–470. doi:10.1007/s10792-008-9266-7
25. Halford S, Liew G, Mackay DS, et al. Detailed phenotypic and genotypic characterization of Bietti crystalline dystrophy. Ophthalmology. 2014;121:1174–1184. doi:10.1016/j.ophtha.2013.11.042
26. Li Q, Li Y, Zhang X, et al. Utilization of fundus autofluorescence, spectral domain optical coherence tomography, and enhanced depth imaging in the characterization of Bietti crystalline dystrophy in different stages. Retina. 2015;35:2074–2084. doi:10.1097/IAE.0000000000000592
27. Kojima H, Otani A, Ogino K, et al. Outer retinal circular structures in patients with Bietti crystalline retinopathy. Br J Ophthalmol. 2011;96:390–393. doi:10.1136/bjo.2010.199356
28. Saatci AO, Doruk HC, Yaman A, Öner FH. Spectral domain optical coherence tomographic findings of Bietti crystalline dystrophy. J Ophthalmol. 2014;2014:1–5. doi:10.1155/2014/739271
29. Goldberg NR, Greenberg JP, Laud K, Tsang S, Freund KB. Outer retinal tubulation in degenerative retinal disorders. Retina. 2013;33:1871–1876. doi:10.1097/IAE.0b013e318296b12f
30. Pennesi ME, Weleber RG. High-resolution optical coherence tomography shows new aspects of Bietti crystalline retinopathy. Retina. 2010;30:531–532. doi:10.1097/IAE.0b013e3181c96a15
31. Oishi A, Oishi M, Miyata M, et al. Multimodal imaging for differential diagnosis of Bietti crystalline dystrophy. Ophthalmol Retina. 2018;2(10):1071–1077. doi:10.1016/j.oret.2018.02.012
32. Miyata M, Oishi A, Hasegawa T, et al. Choriocapillaris flow deficit in Bietti crystalline dystrophy detected using optical coherence tomography angiography. Br J Ophthalmol. 2018;102(9):1208–1812. doi:10.1136/bjophthalmol-2017-311313
33. Yin H, Jin C, Fang X, et al. Molecular analysis and phenotypic study in 14 Chinese families with Bietti crystalline dystrophy. PLoS One. 2014;16(9):e94960. doi:10.1371/journal.pone.0094960
34. Fuerst NM, Serrano L, Han G, et al. Detailed functional and structural phenotype of Bietti crystalline dystrophy associated with mutations in CYP4V2 complicated by choroidal neovascularization. Ophthalmic Genet. 2016;30:1–8.
35. Mamatha G, Umashankar V, Kasinathan N, et al. Molecular screening of the CYP4V2 gene in Bietti crystalline dystrophy that is associated with choroidal neovascularization. Mol Vis. 2011;17:1970–1977.
36. Nourinia R, Dehghan MH, Fekri S. Outcome of macular hole surgery in Bietti crystalline dystrophy. J Ophthalmic Vis Res. 2017;12(3):338–341. doi:10.4103/jovr.jovr_154_15
37. Marmor MF, Zrenner E. Standard for clinical electroretinography (1999 update). International society for clinical electrophysiology of vision. Doc Ophthalmol. 1998;97:143–156.
38. Usui T, Tanimoto N, Takagi M, Hasegawa S, Abe H. Rod and cone a-waves in three cases of Bietti crystalline chorioretinal dystrophy. Am J Ophthalmol. 2001;132:395–402. doi:10.1016/S0002-9394(01)00963-1
39. Lai TY, Ng TK, Tam PO, et al. Genotype phenotype analysis of Bietti’s crystalline dystrophy in patients with CYP4V2 mutations. Invest Ophthalmol Vis Sci. 2007;48:5212–5220. doi:10.1167/iovs.07-0660
40. Sen P, Ray R, Ravi P. Electrophysiological findings in Bietti’s crystalline dystrophy. Clin Exp Optom. 2011;94:302–308. doi:10.1111/j.1444-0938.2011.00602.x
41. García-García GP, López-Garrido MP, Martínez-Rubio M, et al. Genotype-phenotype analysis of Bietti crystalline dystrophy in a family with the CYP4V2 Ile111Thr mutation. Cornea. 2013;32:1002–1008. doi:10.1097/ICO.0b013e31828a27bc
42. Hood DC. Assessing retinal function with the multifocal technique. Prog Retin Eye Res. 2000;19:607–646. doi:10.1016/S1350-9462(00)00013-6
43. Ilhan A, Yolcu U. Re: Halford et al. Detailed phenotypic and genotypic characterization of Bietti crystalline dystrophy. Ophthalmology. 2014;121:1174–1184.
44. Akıncıoğlu D, Yolcu Ü, İlhan A, Gündoğan FÇ. Objective determination of retinal function in Bietti crystalline retinopathy. Turk J Ophthalmol. 2016;46(3):144–147. doi:10.4274/tjo.02693
45. Gocho K, Kameya S, Akeo K et al. High-resolution imaging of patients with Bietti crystalline dystrophy with CYP4V2 mutation. J Ophthalmol. 2014;(1):1–11. doi:10.1155/2014/283603.
46. Lai TY, Chan WM, Lai RY, Ngai JWS, Li H, Lam DSC. The clinical applications of multifocal electroretinography: a systematic review. Surv Ophthalmol. 2007;52:61–96. doi:10.1016/j.survophthal.2006.10.005
47. Abeshi A, Bruson A, Beccari T, et al. Genetic testing for Bietti crystalline dystrophy. Eurobiotech J. 2017;1:564–615.
48. Lee J, Jiao X, Hejtmancik JF, Kaiser-Kupfer M, Chader GJ. Identification, isolation, and characterization of a 32-kDa fatty acid-binding protein missing from lymphocytes in humans with Bietti crystalline dystrophy (BCD). Mol Genet Metab. 1998;65:143–154. doi:10.1006/mgme.1998.2723
49. Nakano M, Kelly EJ, Rettie AE. Expression and characterization of CYP4V2 as a fatty acid ω-hydroxylase. Drug Metab Dispos. 2009;37:2119–2122. doi:10.1124/dmd.109.028530
50. Lai T, Chu KO, Chan KP, et al. Alterations in serum fatty acid concentrations and desaturase activities in Bietti crystalline dystrophy unaffected by CYP4V2 genotypes. Invest Ophthalmol Vis Sci. 2010;51:1092–1097. doi:10.1167/iovs.09-3665
51. Shindou H, Koso H, Sasaki J, et al. Docosahexaenoic acid preserves visual function by maintaining correct disc morphology in retinal photoreceptor cells. J Biol Chem. 2017;292:12054–12064. doi:10.1074/jbc.M117.790568
52. Spite M, Claria J, Serhan CN. Resolvins, specialized proresolving lipid mediators, and their potential roles in metabolic diseases. Cell Metab. 2014;19:21–36. doi:10.1016/j.cmet.2013.10.006
53. Hata M, Ikeda HO, Iwai S, et al. Reduction of lipid accumulation rescues Bietti’s crystalline dystrophy phenotypes. Proc Natl Acad Sci USA. 2018;115:3936–3941. doi:10.1073/pnas.1717338115
54. Xiao X, Mai G, Li S, Guo X, Zhang Q. Identification of CYP4V2 mutation in 21 families and overview of mutation spectrum in Bietti crystalline corneoretinal dystrophy. Biochem Biophys Res Commun. 2011;409:181–186. doi:10.1016/j.bbrc.2011.04.112
55. Jiao X, Li A, Jin ZB, et al. Identification and population history of CYP4V2 mutations in patients with Bietti crystalline corneoretinal dystrophy. Eur J Human Genet. 2017;25:461–471. doi:10.1038/ejhg.2016.184
56. Zhang X, Xu K, Dong B, et al. Comprehensive screening of CYP4V2 in a cohort of Chinese patients with Bietti crystalline dystrophy. Mol Vis. 2018;24:700–711.
57. Miyata M, Hata M, Ooto S, et al. Choroidal and retinal atrophy of bietti crystalline dystrophy patients with CYP4V2 mutations compared to retinitis pigmentosa patients with EYS mutations. Retina. 2017;37(6):1193–1202. doi:10.1097/IAE.0000000000001323
58. Astuti GD, Sun V, Bauwens M, et al. Novel insights into the molecular pathogenesis of CYP4V2-associated Bietti’s retinal dystrophy. Mol Genet Genomic Med. 2015;3:14–29. doi:10.1002/mgg3.109
59. Zenteno JC, Ayala-Ramirez R, Graue-Wiechers F. Novel CYP4V2 gene mutation in a Mexican patient with Bietti’s crystalline corneoretinal dystrophy. Curr Eye Res. 2008;33(4):313–318. doi:10.1080/02713680801983217
60. Eun HS, Cho SY, Lee BS, Seong I-O, Kim K-H. Profiling cytochrome P450 family 4 gene expression in human hepatocellular carcinoma. Mol Med Rep. 2018;18(6):4865–4876. doi:10.3892/mmr.2018.9526
61. Shan M, Dong B, Zhao X, et al. Novel mutations in the CYP4V2 gene associated with Bietti crystalline corneoretinal dystrophy. Mol Vis. 2005;11:738–743.
62. de Haan HG, van Hylckama Vlieg A, Lotta LA, et al. Targeted sequencing to identify novel genetic risk factors for deep vein thrombosis: a study of 734 genes. J Thromb Haemost. 2018;16(12):2432–2441. doi:10.1111/jth.14279
63. Herceg Z, Hainaut P. Genetic and epigenetic alterations as biomarkers for cancer detection, diagnosis and prognosis. Mol Oncol. 2007;1(1):26–41. doi:10.1016/j.molonc.2007.01.004
64. Jinda W, Taylor TD, Suzuki Y, et al. Whole exome sequencing in eight thai patients with leber congenital amaurosis reveals mutations in the CTNNA1 and CYP4V2 genes. Invest Ophthalmol Vis Sci. 2017;58(4):2413–2420. doi:10.1167/iovs.16-21322
65. Miraldi Utz V, Coussa RG, Antaki F, Traboulsi EI. Gene therapy for RPE65-related retinal disease. Ophthalmic Genet. 2018;39(6):671–677. doi:10.1080/13816810.2018.1533027
66. Lockhart CM, Smith TB, Yang P, et al. Longitudinal characterization of function and structure of Bietti crystalline dystrophy: report on a novel homozygous mutation in CYP4V2. Br J Ophthalmol. 2018;102(2):187–194. doi:10.1136/bjophthalmol-2016-309696
67. Yokoi Y, Sato K, Aoyagi H, Takahashi Y, Yamagami M, Nakazawa M. A novel compound heterozygous mutation in the CYP4V2 gene in a japanese patient with Bietti’s crystalline corneoretinal dystrophy. Case Rep Ophthalmol. 2011;2:296–301. doi:10.1159/000331885
68. Meng XH, Guo H, Xu HW, et al. Identification of novel CYP4V2 gene mutations in 92 Chinese families with Bietti’s crystalline corneoretinal dystrophy. Mol Vis. 2014;20:1806–1814.
69. Tian R, Wang SR, Wang J, et al. Novel CYP4V2 mutations associated with Bietti crystalline corneoretinal dystrophy in Chinese patients. Int J Ophthalmol. 2015;8:465–469.
70. Gekka T, Hayashi T, Takeuchi T, Goto-Omoto S, Kitahara K. CYP4V2 mutations in two Japanese patients with Bietti’s crystalline dystrophy. Ophthalmic Res. 2005;37:262–269. doi:10.1159/000087214
71. Yin X, Yang L, Chen N, et al. Identification of CYP4V2 mutation in 36 Chinese families with Bietti crystalline corneoretinal dystrophy. Exp Eye Res. 2016;146:154–162. doi:10.1016/j.exer.2016.03.007
72. Jin ZB, Ito S, Saito Y, Inoue Y, Yanagi Y, Nao-i N. Clinical and molecular findings in three Japanese patients with crystalline retinopathy. Jpn J Ophthalmol. 2006;50:426–431. doi:10.1007/s10384-006-0350-0
73. Liu DN, Liu Y, Meng XH, Yin ZQ. The characterization of functional disturbances in Chinese patients with Bietti’s crystalline dystrophy at different fundus stages. Graefes Arch Clin Exp Ophthalmol. 2012;250:191–200. doi:10.1007/s00417-011-1809-3
74. Chung JK, Shin JH, Jeon BR, Ki C-S, Park TK. Optical coherence tomographic findings of crystal deposits in the lens and cornea in Bietti crystalline corneoretinopathy associated with mutation in the CYP4V2 gene. Jpn J Ophthalmol. 2013;57:447–450. doi:10.1007/s10384-013-0256-6
75. Lee KY, Koh AH, Aung T, et al. Characterization of Bietti crystalline dystrophy patients with CYP4V2 mutations. Invest Ophthalmol Vis Sci. 2005;46:3812–3816. doi:10.1167/iovs.05-0378
76. Nakamura M, Lin J, Nishiguchi K, et al. Bietti crystalline corneoretinal dystrophy associated with CYP4V2 gene mutations. Adv Exp Med Biol. 2006;572:49–53.
77. Lin J, Nishiguchi KM, Nakamura M, et al. Recessive mutations in the CYP4V2 gene in East Asian and Middle Eastern patients with Bietti crystalline corneoretinal dystrophy. J Med Genet. 2005;42:e38. doi:10.1136/jmg.2004.024489
78. Parravano M, Sciamanna M, Giorno P, Boninfante A, Varano M. Bietti crystalline dystrophy: a morpho-functional evaluation. Doc Ophthalmol. 2012;124(1):73–77. doi:10.1007/s10633-011-9309-7
79. Manzouri B, Sergouniotis PI, Robson AG, Webster AR, Moore A. Bietti crystalline retinopathy: report of retinal crystal deposition in male adolescent siblings. Arch Ophthalmol. 2012;130:1470–1473. doi:10.1001/archophthalmol.2012.1567
80. Demile B, Guadie A, Cai W, et al. A clinical and genetic feature in Chinese Bietti crystalline dystrophy families with CYP4V2 mutations. Int J Adv Res. 2018;6(4):1022–1027. doi:10.21474/IJAR01/6944
81. Song Y, Mo G, Yin G. A novel mutation in the CYP4V2 gene in a Chinese patient with Bietti’s crystalline dystrophy. Int Ophthalmol. 2013;33(3):269–276. doi:10.1007/s10792-012-9686-2
82. Raoof N, Vincent AL. Novel gene mutation in a patient with Bietti crystalline dystrophy without corneal deposits. Clin Exp Ophthalmol. 2017;45(4):421–424. doi:10.1111/ceo.12871
83. Katagiri S, Hayashi T, Gekka T, Tsuneoka H. A novel homozygous CYP4V2 variant (p.S121Y) associated with a choroideremia-like phenotype. Ophthalmic Genet. 2017;38(3):286–287. doi:10.1080/13816810.2016.1193880
84. Fu Q, Wang F, Wang H, et al. Next-generation sequencing-based molecular diagnosis of a Chinese patient cohort with autosomal recessive retinitis pigmentosa. Invest Ophthalmol Vis Sci. 2013;54(6):4158–4166. doi:10.1167/iovs.13-11672
85. Beryozkin A, Shevha E, Kimchii A, et al. Whole exome sequencing reveals mutations in known retinal disease genes in 33 out of 68 Israeli families with inherited retinopathies. Sci Rep. 2015;(5):13187. doi:10.1038/srep13187
86. Yokoi Y, Nakazawa M, Mizukoshi S, Sato K, Usui T, Takeuchi K. Crystal deposits on the lens capsules in Bietti crystalline corneoretinal dystrophy associated with a mutation in the CYP4V2 gene. Acta Ophthalmol. 2010;88(5):607–609. doi:10.1111/j.1755-3768.2009.01529.x
87. Park YJ, Hwang DJ, Seong MW, Park SS, Woo SJ. Bietti crystalline retinopathy confirmed by mutation of CYP4V2 gene in a Korean patient. Korean J Ophthalmol. 2016;30(1):81–83. doi:10.3341/kjo.2016.30.1.81
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.