Back to Journals » Journal of Experimental Pharmacology » Volume 7
Current knowledge on the neuroprotective and neuroregenerative properties of citicoline in acute ischemic stroke
Authors Martynov M, Gusev E
Received 30 June 2015
Accepted for publication 11 August 2015
Published 1 October 2015 Volume 2015:7 Pages 17—28
DOI https://doi.org/10.2147/JEP.S63544
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Bal Lokeshwar
Mikhail Yu Martynov, Eugeny I Gusev
Department of Neurology, Neurosurgery and Medical Genetics, Russian National Research Medical University, Moscow, Russia
Abstract: Ischemic stroke is one of the leading causes of long-lasting disability and death. Two main strategies have been proposed for the treatment of ischemic stroke: restoration of blood flow by thrombolysis or mechanical thrombus extraction during the first few hours of ischemic stroke, which is one of the most effective treatments and leads to a better functional and clinical outcome. The other direction of treatment, which is potentially applicable to most of the patients with ischemic stroke, is neuroprotection. Initially, neuroprotection was mainly targeted at protecting gray matter, but during the past few years there has been a transition from a neuron-oriented approach toward salvaging the whole neurovascular unit using multimodal drugs. Citicoline is a multimodal drug that exhibits neuroprotective and neuroregenerative effects in a variety of experimental and clinical disorders of the central nervous system, including acute and chronic cerebral ischemia, intracerebral hemorrhage, and global cerebral hypoxia. Citicoline has a prolonged therapeutic window and is active at various temporal and biochemical stages of the ischemic cascade. In acute ischemic stroke, citicoline provides neuroprotection by attenuating glutamate exitotoxicity, oxidative stress, apoptosis, and blood–brain barrier dysfunction. In the subacute and chronic phases of ischemic stroke, citicoline exhibits neuroregenerative effects and activates neurogenesis, synaptogenesis, and angiogenesis and enhances neurotransmitter metabolism. Acute and long-term treatment with citicoline is safe and in most clinical studies is effective and improves functional outcome.
Keywords: ischemic stroke, neuroprotection, neuroregeneration, cell membranes, oxidative stress
Introduction
Stroke is one of the leading causes of mortality and long-lasting disability.1 Almost one-fourth of the patients die within the first year after insult,2 and among the survivors nearly one-third have restrictions in daily activities and require medical and social assistance.1,3
Stroke is classified into ischemic stroke, intracerebral hemorrhage, subarachnoid hemorrhage, and cerebral venous sinus thrombosis. Ischemic stroke is the most prevalent type and accounts for 75%–85% of all the strokes.
Current concepts in pathophysiology of cerbral ischemia
Originally, the concept of ischemic core and penumbra was introduced by Astrup et al.4 The last few decades have seen major advances in the understanding of the pathophysiology of cerebral ischemia. Recent data take into account that during cerebral ischemia the entire neurovascular unit is involved and that the temporal and spatial evolution of ischemic injury is influenced by many factors, which include age, vascular risk factors, comorbidities, site and diameter of the occluded vessel, adequacy of collateral blood flow, systolic blood pressure (BP) and perfusion pressure, glucose metabolism, hematocrit, and other parameters.5–7
After the interruption of blood flow, ischemic injury develops heterogeneously; in the early minutes and hours after the onset of ischemia, the core may contain multiple sites of injury, which del Zoppo et al7 characterized as “mini-cores”, that are surrounded by “mini-penumbras”, which with time, depending on the systemic blood pressure, local perfusion, and metabolic needs, transform into homogeneous necrotic core.8 In the ischemic core, cerebral blood flow (CBF), cerebral blood volume (CBV), and local cerebral metabolic rates of oxygen (CMRO2) and glucose (CMRglc) are severely decreased.9 Experimental and clinical studies indicate that the ischemic threshold for progression to infarction is 5–8 mL 100 g−1 min−1 within the first hours after stroke onset, but rises progressively to reach ~12–25 mL 100 g− min−1 if CBF is not restored and metabolic needs increase.10,11
The tissue surrounding the core of ischemic lesion is divided based on perfusion threshold into “penumbra” and “oligemia”. Baron et al6 defined penumbra as “a hypoperfused and functionally impaired ischemic tissue which is at risk for infarction”. In the penumbra region, blood flow is decreased to ~12–25 mL 100 g−1 min−1, the oxygen extraction fraction and local CMRglc are increased, oxygen metabolism is preserved relative to CBF, and the CBV is either normal or elevated.8,11 The resulting elevated CMRglc/CBF ratio reflects various degrees of metabolism–blood flow dissociation.12 The extent and temporal evolution of penumbra varies, and according to most experimental and clinical studies brain tissue remains viable for hours after stroke.5 Electrophysiologically, the penumbra region is characterized by spontaneously occurring peri-infarct depolarizations (PID), which bear a resemblance to spreading depression of cortical electrical activity. In the penumbra region, the transient hyperperfusion in response to PID is diminished or absent and recovery from depolarization state is delayed.13 Repeated PIDs lead to depletion of ATP storage14 and have an adverse effect on microcirculation with progressive local metabolism–blood flow uncoupling15 and may lead to necrosis.16
The oligemic area represents a region with a reduced blood flow (~25–35 mL 100 g−1 min−1), normal oxygen consumption, and elevated CBV and oxygen extraction fraction. In most cases, the oligemic area is not at risk of infarction; however, prolonged systemic hypotension, increased intracranial pressure, decreased perfusion pressure, and hyperglycemia may aggravate perfusion and metabolic deficit to penumbra levels.6
Penumbra as a therapeutic target
Penumbra is considered as the primary target for therapy. Several approaches have been proposed for the recovery of brain cells during cerebral ischemia.17–20 Restoration of blood flow by thrombolysis or mechanical thrombus extraction during the first few hours of ischemic stroke, which is one of the most effective treatments, leads to a better functional and clinical outcome.17 Presently, 5%–20% of patients with acute ischemic stroke are treated with thrombolysis and/or thromboextraction.18
The other direction of treatment of acute cerebral ischemia, which has been explored in numerous experimental and clinical studies, that is potentially applicable to most of the patients with ischemic stroke, including the pre-hospital stage, is neuroprotection. Initially, neuroprotection was mainly targeted at protecting gray matter and interrupting a certain biochemical event. Although most of the neuroprotective agents proved to be effective in reducing infarct size and improving functional outcome in experimental studies, the translation to clinical trials was mainly disappointing.19 During the past few years, the transition from a purely neuron-oriented view toward a more integrative approach, which includes salvaging the whole neurovascular unit, is observed, and multimodal drugs, which are targeted at various temporal and biochemical stages of ischemic cascade, are tested in experimental and clinical trials.20
Citicoline
Citicoline, also called as cytidine diphohphate (CDP)-choline, is a natural compound and an intermediary in the biosynthesis of phosphatidylcholine – the structural phospholipid of cell membranes.21 Citicoline is composed of two molecules: cytidine and choline. Cytidine and choline separately pass through the blood–brain barrier (BBB), enter brain cells, and act as substrates for intracellular synthesis of CDP-choline.22,23
Experimental and clinical data
Citicoline has been shown to be effective in a variety of experimental and clinical disorders of the central nervous system (CNS), including acute and chronic cerebral ischemia, intracerebral hemorrhage, global cerebral hypoxia, and neurodegenerative diseases.24–28 Pharmacological actions of citicoline in the CNS are pleiotropic, affect various cell structures and biochemical pathways, and include neuroprotective and neuroregenerative effects.
In experimental models of focal or global cerebral ischemia, citicoline has been proven to have an extended therapeutic window and exhibit effects at several stages of the ischemic cascade.29–31 Citicoline alone or in combination with r-tPA or neuroprotective drugs improved neurological functions and reduced infarct size.32,33 Moreover, it has been shown that the effects of citicoline vary with dose, and that larger doses provide greater reduction of infarct volume34 and better improvement of neurological symptoms.35 In a meta-analysis, Bustamante et al36 summarized the results of 14 experimental studies on ischemic stroke (522 animals, 280 in the citicoline and 242 in the control group). Citicoline reduced the infarct volume compared with the control group both in permanent (25.4%, 95% confidence interval [CI]: 17.6%–33.3%) and transient (30.2%, 95% CI: 15.3%–45.1%) ischemia models and seemed to be equally effective at low (≤250 mg) and high (>250 mg) doses (27.4%, 95% CI: 14.6%–40.2% and 27.4%, 95% CI: 18.2%–36.5%, respectively). Also, the efficacy of citicoline in reducing infarct volume was better when the drug was administered in multiple doses than in a single dose (31.1%, 95% CI: 18.8%–43.5% vs. 22.6%, 95% CI: 14.1%–31.1%, P<0.0001). Combined therapy with other drugs led to a greater decrease in infarct volume: 40.2% (95% CI: 27.3%–53.1%) reduction in cotreated animals vs 24.0% (95% CI: 16.0%–31.6%) reduction in monotreated animals. Data from neurological outcome were obtained in 176 animals (104 citicoline-treated animals and 72 controls). Citicoline administration led to a better neurological recovery (20.2% compared with the control group, 95% CI: 6.8%–33.7%).
In most of the clinical studies,37–40 citicoline has shown effectiveness in reducing neurological symptoms and disability (Table 1). Davalos et al41 in a meta-analysis of four studies (1,372 patients, 789 in citicoline group and 583 in placebo group) demonstrated that citicoline significantly improved global recovery (BI ≥95, mRS ≤1, and NIHSS ≤1) at 3 months with the best outcome in the 2,000 mg group. Global recovery was also achieved in a subgroup of patients not treated with r-tPA (1,246 patients, odds ratio [OR]: 1.35, 95% CI: 1.10–1.65). Treatment with citicoline also increased the probability of recovering full daily living activities (BI ≥95) and functional capacity (mRS ≤1) by 29% (95% CI: 3%–62%) and 42% (95% CI: 8%–88%), respectively. Saver42 assessed ten placebo controlled clinical trials in patients with ischemic stroke or spontaneous intracerebral hemorrhage. Among 1,921 patients with ischemic stroke, verified by computed chromatography (CT)/magnetic resonance imaging (MRI) (five trials), 1,100 received citicoline and 821 were placed in placebo group. Patients treated with citicoline showed significant reduction in the frequency of death or dependency at follow-up: 57.4% events in citicoline group vs 65.7% in placebo group, χ2=13.22, OR: 0.70, 95% CI: 0.58–0.85, P=0.00027. Moreover, analysis confined to four largest trials37–40 with a more homogenous group of patients also yielded a significant positive effect of citicoline: χ2=17.66, OR: 0.66, 95% CI: 0.55–0.80, P=0.00027. In another analysis, 4,191 patients with acute ischemic stroke received oral citicoline (500–4,000 mg/day) for a period of at least 6 weeks, and 125 patients continued the treatment for 12 weeks.26 Citicoline administration led to a dose-dependent improvement in neurological deficit measured by NIHSS, BI, and mRS. In addition, better improvement was observed in patients with extended treatment.
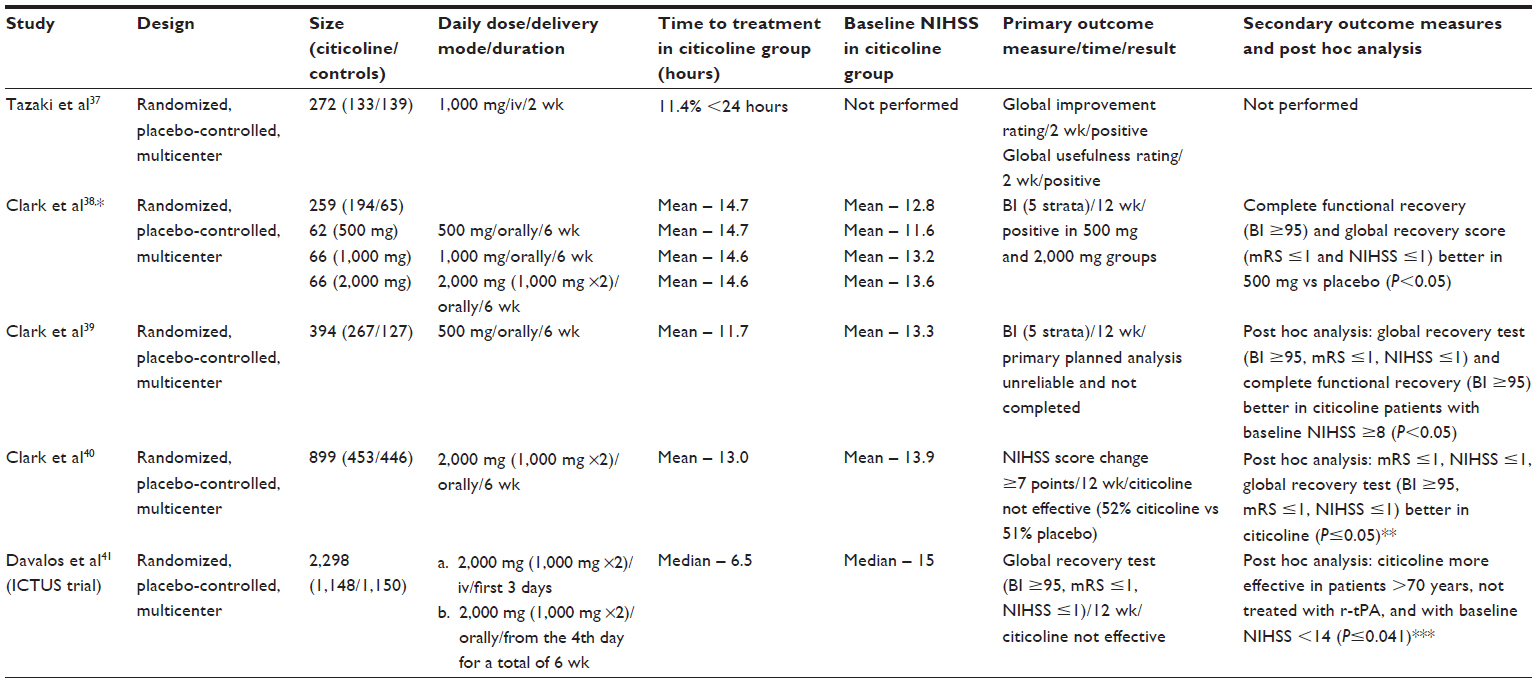 | Table 1 Major clinical trials of citicoline in ischemic stroke |
The ICTUS trial
One of the most recent multicenter studies – the ICTUS trial – showed neutral results (Table 1).43 In this trial, patients were randomly assigned to citicoline or placebo group. The primary end point at 90 days was a global recovery test that combined NIHSS (≤1), mRS (≤1), and Barthel Index (≥95). The secondary outcome measures included separate analyses of NIHSS, BI, and mRS, comparison of the mRS scores between groups, and the absolute difference in the NIHSS scores between baseline and 90 days. The trial was stopped prematurely after the third interim analysis (complete analysis of 2,078 patients) for failure to produce positive results.
However, in spite of absence of positive results, several aspects of this trial deserve further attention:
- The absolute difference in the NIHSS between baseline and 90 days (raw average improvement) in the per-protocol study was -2.18 in the citicoline group compared with -0.91 in the placebo group (effect size 1.26, 95% CI: 0.99–2.53, P=0.051).
- Patients with less severe strokes (NIHSS =8–14) appeared to respond better to the citicoline treatment. A subgroup analysis (intention-to-treat analysis [ITTA] and per-protocol analysis [PPA]) demonstrated more favorable effects of citicoline in patients with less severe strokes (ITTA, P=0.0209; PPA, P=0.0043). Generally, patients in the ICTUS trial had more severe strokes than in the previous studies: median NIHSS in the ICTUS trial was 15 compared with 14 in the meta-analysis by Davalos et al.42 Moreover, in the meta-analysis, median NIHSS score in patients with 2,000 mg citicoline was only 13.42
- Citicoline also led to a better outcome in patients not treated with r-tPA (ITTA, P=0.0413; PPA, P=0.0956). In the ICTUS trial, almost 50% of the patients in both groups received thrombolysis compared with 12.6% in the group with 2,000 mg citicoline in the meta-analysis42 and 5% –20% in a standard stroke practice.18 High percentage of thrombolyses in the ICTUS trial may have biased the results, leading to a maximal functional recovery due to r-tPA and attenuating effects by citicoline.
- The investigators also reported that the effect of citicoline seemed to be more beneficial in patients older than 70 years compared with those aged ≤70 years (ITTA, P=0.0014; PPA, P<0.0001). This finding may deserve a further analysis, since age-dependent modification in metabolism was observed for drugs acting on central nervous and cardiovascular systems.44
Main mechanisms of action
Neuroprotective action
The effectiveness of citicoline in acute ischemic stroke may be due to its pleiotropic neuroprotective functions (Table 2) with stabilization of cell membranes,22,23,45 attenuation of glutamate exitotoxicity,34 oxidative stress,46–48 apoptosis,35 and endothelial barrier dysfunction.49
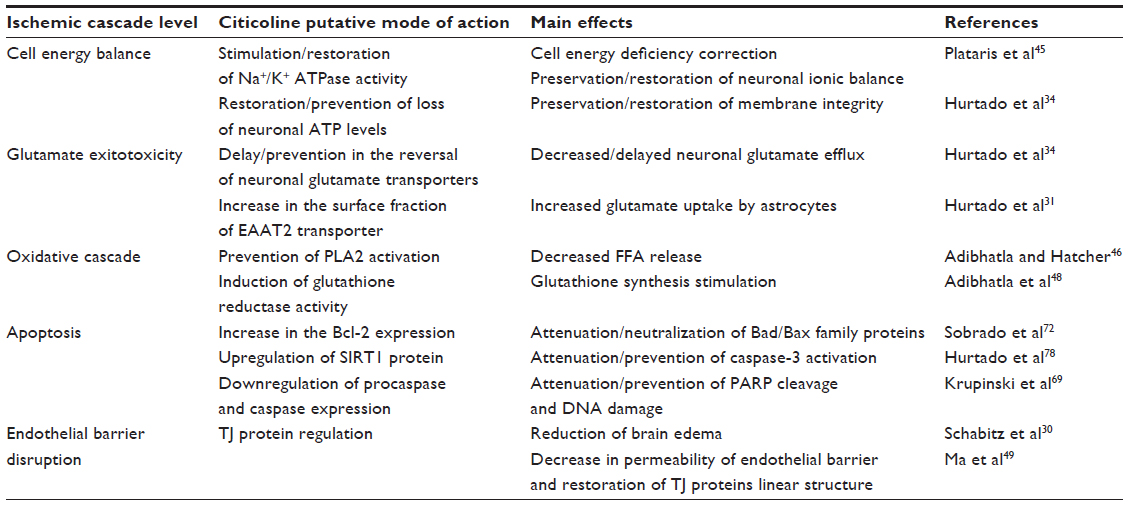 | Table 2 Main neuroprotective effects of citicoline |
Glutamate exitotoxicity
Within a few minutes after the onset of cerebral ischemia in the core of ischemic injury, ATP-dependent ion pumps fail and intracellular Na+ increases, resulting in the release of glutamate into the extracellular space.14 Citicoline protects the membranes by inhibiting glutamate release. Hurtado et al34 in a rat model of combined CCA and MCA occlusion demonstrated that citicoline inhibited ischemia-induced increase in glutamate concentrations. In addition, incubation of cortical neurons with citicoline prevented glutamate release induced by oxygen and glucose deprivation (OGD).34 Pretreatment of cerebellar granule neurons with CDP-choline prior to glutamate administration led to a dose- and time-dependent reduction of glutamate-induced excitotoxicity.50
Glutamate clearance is regulated by excitatory amino acid transporters (EAATs), and citicoline may modulate glutamate actions through induction of EAATs. At present, five EAATs have been cloned and characterized. Most of them are tissue-specific,51 and downregulation of glutamate transporters may lead to neuronal death during ischemia.52 EAAT2 is a glutamate transporter responsible for up to 90% of all the glutamate transport.53 Positive clinical effect of focal overexpression of EAAT2 in reducing ischemia-induced glutamate overflow and decreasing apoptotic cell death was observed in a study by Harvey et al.54 Hurtado et al34 in a model of OGD test reported an increased EAAT2 expression and glutamate uptake in rat astrocytes. Administration of CDP-choline in a model of MCA occlusion induced EAAT2 translocation to the membrane, increased EAAT2 association to lipid rafts, and thus intensified glutamate uptake compared with saline-treated animals.31
Oxidative damage
After cerebral ischemia, increased levels of reactive oxygen species are generated, which cause injury through destruction of nucleic acids, proteins, and lipids, and/or by disrupting gene expression and cellular signaling.55 Phospholipase A2 (PLA2) and arachidonic acid (AA) play a key role in post-ischemic oxidative stress.56 During acute ischemia, cytosolic Ca2+-dependent PLA2 is activated by release of glutamate and elevation of intracellular Ca2+,57 resulting in hydrolysis of phospholipids and release of free fatty acids (FFA).58 Free AA accumulates inside the cells, promotes the formation of reactive oxygen species, and amplifies the oxidative damage.58
Citicoline may provide neuroprotection by preventing the activation of PLA2 and AA release and by modulating glutathione synthesis through choline. In a model of transient forebrain ischemia in gerbils, citicoline stabilized cell membranes, attenuated activation of PLA2 in membrane and mitochondrial fractions, and decreased AA and release of other FFA after reperfusion.46 Trovarelli et al59 demonstrated that intraventricular preconditioning with CDP-choline in a model of brain ischemia in gerbils resulted in a partial restoration of brain phosphatidylcholine concentration and attenuated the release of FFA including AA. Rao et al60 in a model of transient forebrain ischemia observed that CDP-choline given immediately after the onset of ischemia and at 3 hours reperfusion significantly restored the phosphatidylcholine, sphingomyelin, and cardiolipin levels. Oral administration of CDP-choline in a model of brain cryogenic injury inhibited activation of PLA2 and prevented destruction of membrane phospholipids.61 Cytidine and choline, the breakdown products of citicoline, may enhance the incorporation of FFA – including AA – into phosphatidylcholine and other major phospholipids. Addition of equimolar concentrations of cytidine and choline to [3H]-arachidonic acid dose-dependently increased the accumulations of [3H]-phosphatidylcholine, [3H]-phosphatidylinositol, and [3H]-phosphatidylethanolamine,47 thus removing the free AA, which is as a potential source of oxidative damage. In another study, citicoline administration stimulated reincorporation of AA in phosphatidylcholine and thus reduced oxidative damage.60
Citicoline through choline–S-adenosyl-L-methionine pathway may modulate synthesis of glutathione.62 Adibhatla et al48 reported that serial intraperitoneal citicoline administrations in doses 500 mg/kg in a gebril model of transient forebrain ischemia significantly increased total glutathione and glutathione reductase activity compared with the ischemia/saline group. Citicoline treatment also decreased the glutathione oxidation ratio at 6 hours, 1 and 3 days after stroke.
Apoptosis
Apoptosis is an orderly and energy-dependent process of programmed cell death.63 In experimental and clinical ischemic stroke, apoptotic changes are mainly confined to penumbra.64,65 Cerebral ischemia triggers both apoptotic pathways: the intrinsic or mitochondrial pathway, initiated by internal events, and extrinsic pathway, initiated by external events.66
Several reports indicate that citicoline may attenuate/prevent apoptosis by influencing both pathways, while CDP-choline deficiency may induce apoptotic process.50,67,68 A recent study also suggests also suggest that during apoptosis, cytidine and choline incorporation into cells and intercellular CDP-choline resynthesis may be blocked.68 Mir et al50 demonstrated that 6-hour preconditioning of cerebellar granule neurons with 100 microM CDP-choline solution significantly decreased the number of apoptotic neurons caused by glutamate excitotoxicity. Krupinski et al69 in a rat model of permanent MCA occlusion showed a significant reduction in immunoreactive cells for procaspases-1, -2, -3, -6, and -8 in the ischemic area of citicoline-treated animals compared with sham-operated animals. Also, the number of cells expressing nuclear DNA fragmentation, cleaved caspase-3, and caspase-cleaved products of poly (ADP-ribose) polymerase were significantly reduced in the penumbra area of animals treated with citicoline. Preincubation of human brain microvascular endothelial cells (hCMEC/D3) with citicoline significantly decreased the number of apoptotic cells after ischemia.70 Sahin et al71 analyzed the protective effects of citicoline alone or in combination with hypothermia. After 2 hours of MCA occlusion, both groups showed reduced number of apoptotic cells compared with saline-treated animals. In another study in a model of bilateral CCA ligation and MCA occlusion, intraperitoneal administration of citicoline for 7 days significantly decreased apoptosis measured by TUNEL positive cells and increased the Bcl-2 expression within the border zone of necrotic core.72 In rats subjected to serial transient focal cerebral ischemia, repeated administration of CDP-choline significantly reduced caspase-3-positive neurons.35 Citicoline may also influence the apoptotic pathways in cerebral ischemia through the preservation of glutathione synthesis48 and prevention of ceramide generation.73
LRP is a multifunctional protein expressed in neurons and astrocytes, and recent data suggest its scavenging role during phagocytosis of myelin debris,74 as well as during apoptotic75 and necrotic76 cell death. Prolonged citicoline administration in a model of permanent MCA occlusion significantly downregulated LRP expression in the peri-infarct (PI) region compared with saline-treated animals,77 suggesting decreased apoptosis, necrosis, and myelin degradation.
Of interest is also a recent finding that citicoline may upregulate Sirtuin 1 (SIRT1) expression in cultured neurons.78 Mammalians have seven proteins of the Sirtuin family (SIRT1–7). SIRT1 is a protein with the highest sequence homology to Sir2 (silent information regulator 2), and plays an important role in mediating cell cycle and apoptosis, as well as in anti-inflammatory and antioxidant responses.79 SIRT1 induction may attenuate expression of p53 and caspase-3.80 Citicoline administration (0.2 or 2.0 g/kg) in a model of permanent MCA occlusion significantly increased SIRT 1 levels and led to a reduction in infarct volume in Fisher rats, but failed to upregulate SIRT1 expression and decrease infarct volume in SIRT1-deficient mice.78
Blood–brain barrier
The BBB is characterized by the presence of tight cell–cell junctions and a lack of fenestrations at the level of cerebral microvascular endothelium. Tight cell–cell junctions are constituted by proteins belonging to occludin, claudin, and membrane-associated guanylate kinase-like proteins (zonula occludens [ZO]-1, -2, -3) family, as well as by other accessory proteins and adhesion molecules.81 The permeability of the BBB depends on the integrity of the tight junction (TJ) proteins.82 Occludin is expressed in the cerebral endothelial cells81 and its cytoplasmic C-terminal domain is involved in the association with cytoskeleton via ZO-1 and -2 proteins.83 Claudin-5 and ZO-1 are also important components for the integrity of cell barrier. Claudin-5 can reduce the permeability assessed by dextran.84 ZO-1 links transmembrane and skeleton proteins, and this interaction is important to the stability and function of the TJs.85 ZO-1 may also act as a signaling molecule that translates the state of the TJ to the interior of the cell and vice versa.
Ischemic stroke leads to a BBB disruption and increased permeability. Experimental and, to a lesser degree, clinical studies suggest that in ischemic stroke BBB disruption is a dynamic process, and the initial breakdown in most cases is followed by a partial BBB recovery, before the second increase in BBB permeability occurs.86,87 Dysregulation of the TJ proteins plays an important role in BBB permeability in stroke pathogenesis. In experimental stroke caused by MCA occlusion, a decrease in expression of claudin-5, occludin, and ZO-1, which paralleled increased BBB permeability, was observed.86 Schreibelt et al88 demonstrated that activation of oxidative metabolism alters BBB integrity due to cytoskeleton rearrangements, and redistribution and disappearance of claudin-5 and occludin.
Several previous studies indicated that citicoline administration may attenuate brain edema, possibly through BBB stabilization. Schabitz et al30 evaluated the effect of prolonged treatment with citicoline in a rat model of temporary MCA ischemia. Intraperitoneal administration of citicoline significantly reduced perifocal edema. Rao et al89 reported the reduction of brain edema induced by transient forebrain ischemia in gerbils treated with citicoline. Citicoline was also dose-dependently effective in reducing brain edema and BBB permeability in the animal model of traumatic brain injury.90 Recently, it has been shown that LRP-1 may modulate BBB permeability, and the association of tPA with LRP-1 leads to increase in vascular permeability and BBB opening.91 Citicoline administration, started 30 minutes after MCA occlusion in rats, resulted in decreased brain edema, downregulation of LRP-1, and restoration of endothelial barrier function.77
In a recent study, Ma et al49 in an OGD and hypoxia endothelial barrier breakdown model (human umbilical vein endothelial cells [HUVEC] and brain microvascular endothelial cells [bEnd.3s]) examined the effect of citicoline on endothelial permeability and expressions of ZO-1, claudin-5, and occludin. Both OGD and hypoxia increased the permeability of the BBB, while citicoline dose-dependently decreased FITC-dextran endothelial permeability at low (0.1 mmol/L) and high (1.0 mmol/L) doses. Hypoxia induced significant decrease in mRNA and protein levels, as well as disappearance of normal linear structure of ZO-1, occludin, and claudin-5 in HUVECs and bEnd.3s cells.’ After citicoline treatment, the expression of all the above-mentioned TJ proteins was upregulated, and linear distribution of proteins was restored.
Neuroregenerative action
In the subacute and chronic phases of stroke, citicoline may exhibit neuroregenerative effects (Table 3), by activating neurogenesis, synaptogenesis, and angiogenesis,70,92 and modulate neurotransmitter metabolism93 and brain bioenergetics.94
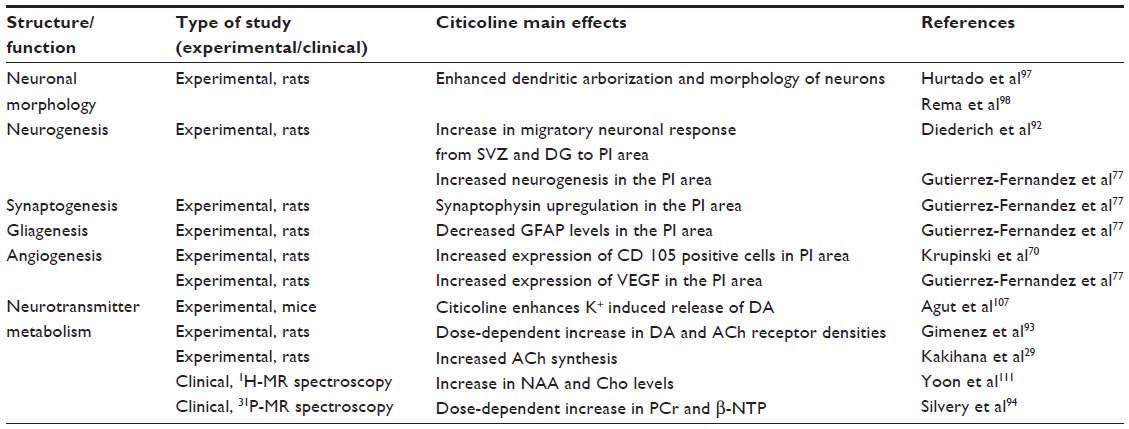 | Table 3 Main neuroregenerative/neurorestorative effects of citicoline |
Neurogenesis, synaptogenesis, and angiogenesis
Stroke induces gene profile changes associated with neurogenesis, synaptogenesis, and angiogenesis.95
Citicoline may stimulate recovery after stroke by modulating brain cell differentiation. In a model of photothrombotic stroke,92 significant increase in newborn neurons 28 days after ischemia was observed in the subventricular zone (SVZ), dentate gyrus, and PI area of citicoline-treated animals (100 mg/kg for 10 days) compared with controls. Citicoline treatment also led to a significantly higher percentage of BrdU/NeuN-positive cells in the dentate gyrus, SVZ, and PI area. In addition, in the citicoline group the migration of neuronal precursor cells from the SVZ to the PI area was more active than in saline-treated animals. Gutierrez-Fernandez et al77 in a model of permanent MCA occlusion in rats also demonstrated that early (30 minutes after occlusion) and prolonged (14 days) treatment with CDP-choline in doses of 500 mg/kg significantly increased the number of BrdU-positive cells, indicating active neurogenesis in the PI region.
Citicoline may facilitate recovery by enhancing synaptic growth and synaptogenesis. Citicoline administration led to the upregulation of synaptophysin in the penumbra region,77 which could be indicative of increased synaptic activity, since synaptophysin regulates the recovery of recycling vesicle pool and facilitates synaptic vesicle endocytosis.96 Administration of CDP-choline for 28 days, initiated 24 hours after MCA occlusion, enhanced dendritic complexity and spine density in pyramidal cells (layer V) in the undamaged areas of the motor cortex compared with the saline group.97 Analysis of dendritic integrity in the immediate border zone in the PI region revealed a difference between citicoline-treated animals and controls with the tendency to significant values (P=0.082).92 Treatment of primary neuronal cultures from somatosensory cortex with CDP-choline increased the neurite length, branch points, and total area occupied by neurons.98 Also, daily administration of CDP-choline from conception (maternal ingestion) to postnatal day 60 to Long–Evans rats resulted in a significant increase in length and branch points of apical and basal dendrites in the pyramidal neurons of somatosensory cortex layers 2, 3, and 5.98
Angiogenesis is a key feature of the post-stroke neurovascular remodeling, and newly formed vessels may provide a scaffold for the migration of neural cells toward PI area.99 Krupinski et al70 demonstrated that citicoline induced angiogenesis by improving survival of vascular/human brain microvessel endothelial cells (hCMEC/D3) through pathways involving phosphor-extracellular signal-regulated kinase (ERK1/2) and insulin receptor substrate-1 (IRS-1). In hCMEC/D3 citicoline stimulated the expression of ERK1/2. Also, the expression of IRS-1 – a modulator of differentiation of vascular endothelial cells100 – was increased in citicoline-treated hCMEC/D3 cells. Treatment with citicoline increased the number of CD31-positive microvessels and significantly increased the number of CD105-positive (active) microvessels in the PI area by 21st day post-stroke.70
Vascular endothelial growth factor (VEGF) is an angiogenic factor that plays a key role in vascular homeostasis.101 In rats with permanent MCA occlusion and treated with citicoline, increased expression of VEGF compared with saline-treated animals was observed.77 The upregulation of VEGF may be linked to activation of IRS-1, which contributes significantly to VEGF expression.102
Endothelial progenitor cells (EPC) augment collateralization, increase capillary density, and enhance neurorepair and neurogenesis after experimental cerebral ischemia,103,104 and a higher increment in EPC during the first week in patients with lacunar stroke is associated with a better functional outcome.105 Administration of 2,000 mg of citicoline from the first day of ischemic lacunar stroke led to a significant increase in circulating EPC by day 7 compared with nontreated patients106 and better clinical outcome.
Modulation of neurotransmitter release
Post-stroke recovery is modulated by neurotransmitters. Dopamine (DA) and acetylcholine (ACh) influence emotional, cognitive, and motor responses. Citicoline promotes changes in brain DA receptors and modulates the release of DA.107 Ischemic stroke mainly occurs in the elderly population, and aging is associated with a decrease in the number of DA and ACh receptors.93 Administration of citicoline for 7 months in doses of 100 mg/kg or 500 mg/kg per day to aging mice led to a partial recovery of receptor function and density. Treated animals displayed a dose-dependent increase in DA and ACh receptor densities, while control animals showed a significant reduction in DA and ACh receptor densities.93 Radad et al108 demonstrated that CDP-choline may reduce dopaminergic cell loss induced by MMP (+) and glutamate in primary mesencephalic cell culture. In another study, addition of various concentrations of citicoline breakdown product, choline, to the rat striatum and cerebellum slices differentially stimulated basal and evoked ACh release, indicating that free choline may be used as a source of ACh synthesis.109 Kakihana et al29 in a model of transient bilateral occlusion of common carotid arteries (after permanent occlusion of both vertebral arteries) observed that CDP-choline administration in doses of 250 mg/kg accelerated glucose utilization and restored the synthesis of acetylcholine. N-acetyl-aspartate (NAA) is a major metabolite located primarily in pyramidal neurons and axons. NAA is involved in neuronal plasticity, axon–glia signaling, mitochondrial energy, and myelin lipid metabolism and is viewed as a surrogate marker of integrity and viability of neural tissue.110 Yoon et al111 in a 1H MR spectroscopy study observed that citicoline administered for 4 weeks at a dose of 2,000 mg/day steadily increased prefrontal NAA and Cho levels after second and fourth weeks of treatment.
Citicoline may also enhance frontal lobe bioenergetics and in this way activate attention, memory tasks, and speed information processing. Silvery et al94 in a MR spectroscopy study (31P) established that oral administration of 500 or 2,000 mg of citicoline for 6 weeks to 16 healthy men and women led to a region-specific increase in the amount of β-NTP (primarily reflects concentrations of ATP) and PCr (reflects the high-energy phosphate buffer stores), and in membrane phospholipids in frontal lobes (anterior cingulated cortex).
Conclusion
Experimental and clinical data support the evidence that citicoline is a multimodal drug with prolonged therapeutic window and neuroprotective and neuroregenerative effects. Citicoline acts at various temporal and biochemical stages of the ischemic cascade and attenuates glutamate exitotoxicity, oxidative stress, apoptosis, and BBB dysfunction. Citicoline also modulates neurotransmitter metabolism; enhances neuroplasticity; and activates neurogenesis, synaptogenesis, and angiogenesis. Acute and long-term treatment with citicoline is safe and in most clinical studies is effective and improves functional outcome. However, further experimental research with lower citicoline doses and stroke models corresponding to clinical practice, as well as adequately planned clinical trials, are needed.
Disclosure
No sources of funding were used in the preparation of this review. The authors report no conflicts of interest in this work.
References
Feigin VL, Forouzanfar MH, Krishnamurthi R, Mensah GA, et al. Global burden of diseases, injuries, and risk factors study 2010 (GBD 2010) and the GBD Stroke experts group. Global and regional burden of stroke during 1990–2010: findings from the Global burden of disease study 2010. Lancet. 2014;383(9913):245–254. | |
Saposnik G, Kapral MK, Liu Y, et al; Investigators of the Registry of the Canadian Stroke Network; Stroke Outcomes Research Canada (SORCan) Working Group. IScore: a risk score to predict death early after hospitalization for an acute ischemic stroke. Circulation. 2011;123:739–749. | |
Skolarus LE, Burke JF, Brown DL, Freedman VA. Understanding stroke survivorship: expanding the concept of poststroke disability. Stroke. 2014;45:224–230. | |
Astrup J, Siesjö BK, Symon L. Thresholds in cerebral ischemia – the ischemic penumbra. Stroke. 1981;12:723–725. | |
Marchal G, Beaudouin V, Rioux P, et al. Prolonged persistence of substantial volumes of potentially viable brain tissue after stroke: a correlative PET-CT study with voxel-based data analysis. Stroke. 1996;27:599–606. | |
Baron JC. Mapping the ischemic penumbra with PET: a new approach. Brain. 2001;124(Pt 1):2–4. | |
del Zoppo GJ, Sharp FR, Heiss WD, Albers GW. Heterogeneity in the penumbra. J Cereb Blood Flow Metab. 2011;31:1836–1851. | |
Heiss WD, Huber M, Fink GR, et al. Progressive derangement of peri-infarct viable tissue in ischemic stroke. J Cereb Blood Flow Metab. 1992;12:193–203. | |
Marchal G, Benali K, Iglesias S, Viader F, Derlon JM, Baron JC. Voxel-based mapping of irreversible ischemic damage with PET in acute stroke. Brain. 1999;122(Pt 12):2387–2400. | |
Baron JC, Marchal G. Ischemic core and penumbra in human stroke. Stroke. 1999;30:1150–1153. | |
Kaufmann AM, Firlik AD, Fukui MB, Wechsler LR, Jungries CA, Yonas H. Ischemic core and penumbra in human stroke. Stroke. 1999;30:93–99. | |
Back T, Zhao W, Ginsberg MD. Three-dimensional image analysis of brain glucose metabolism-blood flow uncoupling and its electrophysiological correlates in the acute ischemic penumbra following middle cerebral artery occlusion. J Cereb Blood Flow Metab.1995;15:566–577. | |
Back T, Kohno K, Hossmann KA. Cortical negative DC deflections following middle cerebral artery occlusion and K+/Cl--induced spreading depression: effect on blood flow, tissue oxygenation, and electroencephalogram. J Cereb Blood Flow Metab. 1994;14:12–19. | |
Hossmann KA. Periinfarct depolarizations. Cerebrovasc Brain Metab Rev. 1996;8(3):195–208. | |
Pinard E, Nallet H, MacKenzie ET, Seylaz J, Roussel S. Penumbral microcirculatory changes associated with peri-infarct depolarizations in the rat. Stroke. 2002;33:606–612. | |
Busch E, Gyngell ML, Eis M, Hoehn-Berlage M, Hossmann KA. Potassium-induced cortical spreading depressions during focal cerebral ischemia in rats: contribution to lesion growth assessed by diffusion-weighted NMR and biochemical imaging. J Cereb Blood Flow Metab. 1996;16:1090–1099. | |
Jauch EC, Saver JL, Adams HP Jr, et al; American Heart Association Stroke Council; Council on Cardiovascular Nursing; Council on Peripheral Vascular Disease; Council on Clinical Cardiology. Guidelines for the early management of patients with acute ischemic stroke: a guideline for healthcare professionals from the American Heart Association/American Stroke Association. Stroke. 2013;44:870–947. | |
van Wijngaarden JDH, Dirks M, Huijsman R, Niessen LW, Fabbricotti IN, Dippel DWJ; Promoting acute thrombolysis for ischemic stroke (PRACTISE) investigators. Hospital rates of thrombolysis for acute ischemic stroke: the influence of organizational culture. Stroke. 2009;40:3390–3392. | |
Ginsberg MD. Neuroprotection for ischemic stroke: past, present and future. Neuropharmacology. 2008;55:363–389. | |
Lapchak PA. Emerging therapies: pleiotropic multi-target drugs to treat stroke victims. Transl Stroke Res. 2011;2:129–135. | |
Kennedy EP, Weiss SB. The functions of cytidine coenzymes. J Biol Chem. 1956;222:193–214. | |
G-Coviella IL, Wurtman RJ. Enhancement by cytidine of membrane phospholipid synthesis. J Neurochem. 1992;59:338–343. | |
Savci V, Wurtman RJ. Effect of cytidine on membrane phospholipid synthesis in rat striatal slices. J Neurochem. 1995;64:378–384. | |
Shuaib A, Yang Y, Li Q. Evaluating the efficacy of citicoline in embolic ischemic stroke in rats: neuroprotective effects when used alone or in combination with urokinase. Exp Neurol. 2000;161:733–739. | |
Secades JJ, Alvarez-Sabin J, Rubio F, Lozano R, Davalos A, Castillo J; Trial Investigators. Citicoline in intracerebral hemorrhage: a double-blind, randomized, placebo-controlled, multi-centre pilot study. Cerebrovasc Dis. 2006;21:380–385. | |
Cho HJ, Kim YJ. Efficacy and safety of oral citicoline in acute ischemic stroke: drug surveillance study in 4,191 cases. Methods Find Exp Clin Pharmacol. 2009;31:171–176. | |
Davalos A, Secades J. Citicoline: preclinical and clinical update 2009–2010. Stroke. 2011;42:S36–S39. | |
Alvarez-Sabin J, Ortega G, Jacas C, et al. Long-term treatment with citicoline may improve post-stroke vascular cognitive impairment. Cerebrovasc Dis. 2013;35:146–154. | |
Kakihana M, Fukuda N, Suno M, Nagaoka A. Effects of CDP-choline on neurologic deficits and cerebral glucose metabolism in a rat model of cerebral ischemia. Stroke. 1988;19:217–222. | |
Schabitz WR, Weber J, Takano K, Sandage BW, Locke KW, Fisher M. The effects of prolonged treatment with citicoline in temporary experimental focal ischemia. J Neurol Sci. 1996;138:21–25. | |
Hurtado O, Pradillo JM, Fernandez-Lopez D, et al. Delayed post-ischemic administration of CDP-choline increases EAAT2 association to lipid rafts and affords neuroprotection in experimental stroke. Neurobiol Dis. 2008;29:123–131. | |
de Lecinana AM, Gutierrez M, Roda JM, Carceller F, Diez-Tejedor E. Effect of combined therapy with thrombolysis and citicoline in a rat model of ischemic stroke. J Neurol Sci. 2006;247(2):121–129. | |
Onal MA, Li F, Tatlisumak T, Locke KW, Sandage BW, Fisher M. Synergistic effects of citicoline and MK-801 in temporary experimental focal cerebral ischemia in rats. Stroke. 1997;28:1060–1065. | |
Hurtado O, Moro MA, Cardenas A, et al. Neuroprotection afforded by prior citicoline administration in experimental brain ischemia: Effects on glutamate transport. Neurobiol Dis. 2005;18:336–345. | |
Takasaki K, Uchida K, Fujikawa R, et al. Neuroprotective effects of citidine-5-diphosphocholine on impaired spatial memory in a rat model of cerebrovascular dementia. J Pharmacol Sci. 2011;116:232–237. | |
Bustamante A, Giralt D, Garcia-Bonilla L, Campos M, Rosell A, Montaner J. Citicoline in pre-clinical animal models of stroke: a meta-analysis shows the optimal neuroprotective profile and the missing steps for jumping into a stroke clinical trial. J Neurochem. 2012;123:217–225. | |
Tazaki Y, Sakai F, Otomo E, Kutsuzawa, et al. Treatment of acute cerebral infarction with a choline precursor in a multicenter double-blind placebo-controlled study. Stroke. 1988;19:211–216. | |
Clark WM, Warach SJ, Pettigrew LC, Gammans RE, Sabounjian LA. A randomized dose-response trial of citicoline in acute ischemic stroke patients. Citicoline Stroke Study Group. Neurology. 1997;49(3):671–678. | |
Clark WM, Williams BJ, Selzer KA, Zweifler RM, Sabounjian LA, Gammans RE; Citicoline Stroke Study Group. A randomized efficacy trial of citicoline in patients with acute ischemic stroke. Stroke. 1999;30:2592–2597. | |
Clark WM, Wechsler LR, Sabounjian LA, Schwiderski UE; Citicoline Stroke Study Group. A phase III randomized efficacy trial of 2000 mg citicoline in acute ischemic stroke patients. Neurology. 2001; 57(9):1595–1602. | |
Davalos A, Castillo J, Alvarez-Sabin J, et al. Oral citicoline in acute ischemic stroke: an individual patient data pooling analysis of clinical trials. Stroke. 2002;33:2850–2857. | |
Saver JL. Citicoline: update on a promising and widely available agent for neuroprotection and neurorepair. Rev Neurol Dis. 2008;5:167–177. | |
Davalos A, Alvarez-Sabin J, Castillo J, et al; International Citicoline Trial on acUte Stroke (ICTUS) trial investigators. Citicoline in the treatment of acute ischemic stroke: an international, randomized, multicentre, placebo-controlled study (ICTUS trial). Lancet. 2012;380:349–357. | |
Trifirò G, Spina E. Age-related changes in pharmacodynamics: focus on drugs acting on central nervous and cardiovascular systems. Curr Drug Metab. 2011;12(4):542–549. | |
Plataras C, Tsakiris S, Angelogianni P. Effect of CDP-choline on brain acetylcholinesterase and Na(+), K(+)-ATPase in adult rats. Clin Biochem. 2000;33:351–357. | |
Adibhatla RM, Hatcher JF. Citicoline decreases phospholipase A2 stimulation and hydroxyl radical generation in transient cerebral ischemia. J Neurosci Res. 2003;73:308–315. | |
Knapp S, Wurtman RJ. Enhancement of free fatty acid incorporation into phospholipids by choline plus cytidine. Brain Res. 1999;822:52–59. | |
Adibhatla RM, Hatcher JF, Dempsey RJ. Effect of citicoline on phospholipids and glutathione levels in transient cerebral ischemia. Stroke. 2001;32:2376–2381. | |
Ma X, Zhang H, Pan Q, et al. Hypoxia/aglycemia-induced endothelial barrier dysfunction and tight junction protein down-regulation can be ameliorated by Citicolone. PLoS One. 2013;8:e82604. | |
Mir C, Clotet J, Aledo R, et al. CDP-choline prevents glutamate-mediated cell death in cerebellar granule neurons. J Mol Neurosci. 2003;20:53–60. | |
Rothstein JD, Martin L, Levey AI, Dykes-Hoberg M, et al. Localization of neuronal and glial glutamate transporters. Neuron. 1994;13:713–725. | |
Rao VLR, Rao AM, Dogan A, et al. Glial glutamate transporter GLT-1 down-regulation precedes delayed neuronal death in gerbil hippocampus following transient global cerebral ischemia. Neurochem Int. 2000;36:531–537. | |
Kim K, Lee S-G, Kegelman TP, et al. Role of excitatory amino acid transporter-2 (EAAT2) and glutamate in neurodegeneration: opportunities for developing novel therapeutics. J Cell Physiol. 2011;226:2484–2493. | |
Harvey BK, Airavaara M, Hinzman J, et al. Targeted over-expression of glutamate transporter 1 (GLT-1) reduces ischemic brain injury in a rat model of stroke. PLoS One. 2011;6(8):e22135. | |
Chan PH. Reactive oxygen radicals in signaling and damage in the ischemic brain. J Cereb Blood Flow Metab. 2001;21:2–14. | |
Kishimoto K, Li RC, Zhang J, et al. Cytosolic phospholipase A2 alpha amplifies early cyclooxygenase-2 expression, oxidative stress and MAP kinase phosphorolation after cerebral ischemia in mice. J Neuroinflammation. 2010;7:42. | |
Kim DK, Rordorf G, Nemenoff RA, Koroshetz WJ, Bonventre JV. Glutamate stably enhances the activity of two cytosolic forms of phospholipase A2 in brain cortical cultures. Biochem J. 1995;310(Pt 1):83–90. | |
Katsuki H, Okuda S. Arachidonic acid as a neurotoxic and neurotrophic substance. Prog Neurobiol. 1995;46:607–636. | |
Trovarelli G, de Medio GE, Dorman RV, Piccinin GL, Horrocks LA, Porcellati G. Effect of cytidine diphosphate choline (CDP-choline) on ischemia-induced alterations of brain lipid in the gerbil. Neurochem Res. 1981;6:821–833. | |
Rao AM, Hatcher JF, Dempsey RJ. Lipid alterations in transient forebrain ischemia: possible new mechanisms of CDP-choline neuroprotection. J Neurochem. 2000;75:2528–2535. | |
Arrigoni E, Averet N, Cohadon F. Effects of CDP-choline on phospholipase A2 and cholinephosphotransferase activities following a cryogenic brain injury in the rabbit. Biochem Pharmacol. 1987;36:3697–3700. | |
de la Cruz JP, Villalobos MA, Cuerda MA, Guerrero A, Gonzalez-Correa JA, Sanchez De La Cuesta F. Effects of S-adenosyl-L-methionine on lipid peroxidation and glutathione levels in rat brain slices exposed to re-oxygenation after oxygen-glucose deprivation. Neurosci Lett. 2002;318:103–107. | |
Kerr JFR, Wyllie AH, Curriet AR. Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br J Cancer. 1972;26:239–257. | |
Mitsios N, Gaffney J, Krupinski J, et al. Expression of signaling molecules associated with apoptosis in human ischemic stroke tissue. Cell Biochem Biophys. 2007;47:73–86. | |
Ramos-Cejudo J, Gutierrez-Fernandez M, Rodriguez-Frutos B, et al. Spatial and temporal gene expression differences in core and peri-infarct areas in experimental stroke: a microarray analysis. PLoS One. 2012;7(12):e52121. | |
Broughton BRS, Reutens DC, Sobey CG. Apoptotic mechanisms after cerebral ischemia. Stroke. 2009;40:e331–e339. | |
Yen CL, Mar MH, Zeisel SH. Choline deficiency-induced apoptosis in PC12 cells is associated with diminished membrane phosphatidylcholine and sphingomyelin, accumulation of ceramide and diacylglycerol, and activation of a caspase. FASEB J. 1999;13:135–142. | |
Morton CC, Aitchison AJ, Gehrig K, Ridgway ND. A mechanism for suppression of the CDP-choline pathway during apoptosis. J Lipid Res. 2013;54:3373–3384. | |
Krupinski J, Ferrer I, Barrachina M, Secades JJ, Mercadal J, Lozano R. CDP-choline reduces pro-caspase and cleaved caspase-3 expression, nuclear DNA fragmentation, and specific PARP-cleaved products of caspase activation following middle cerebral artery occlusion in the rat. Neuropharmacology. 2002;42:846–854. | |
Krupinski J, Abudawood M, Matou-Nasri S, et al. Citicoline induces angiogenesis improving survival of vascular/human brain microvessel endothelial cells through pathways involving ERK1/2 and insulin receptor substrate-1. Vasc Cell. 2012;4:20. | |
Sahin S, Alkan T, Temel SG, Tureyen K, Tolunay S, Korfali E. Effects of citicoline used alone and in combination with mild hypothermia on apoptosis induced by focal cerebral ischemia in rats. J Clin Neurosci. 2010;17:227–231. | |
Sobrado M, Lopez MG, Carceller F, Garcia AG, Roda JM. Combined nimodipine and citicoline reduce infarct size, attenuate apoptosis and increase bcl-2 expression after focal cerebral ischemia. Neuroscience. 2003;118:107–113. | |
Haimovitz-Friedman A, Kolesnik RN, Fuks Z. Ceramide signaling in apoptosis. Brit Med Bull. 1997;53:539–553. | |
Gaultier A, Wu X, Le Moan N, et al. Low-density lipoprotein receptor-related protein 1 is an essential receptor for myelin phagocytosis. J Cell Sci. 2009;122:1155–1162. | |
Gardai SJ, McPhillips KA, Frasch SC, et al. Cell-surface calreticulin initiates clearance of viable or apoptotic cells through trans-activation of LRP on the phagocyte. Cell. 2005;123:321–334. | |
Fernandez-Castaneda A, Arandjelovic S, Stiles TL, et al. Identification of the low density lipoprotein (LDL) receptor-related protein-1 interactome in central nervous system myelin suggests a role in the clearance of necrotic cell debris. J Biol Chem. 2013;288:4538–4548. | |
Gutierrez-Fernandez M, Rodriguez-Frutos B, Fuentes B, et al. CDP-choline treatment induces brain plasticity markers expression in experimental animal stroke. Neurochem Int. 2012;60:310–317. | |
Hurtado O, Hernandez-Jimenez M, Zarruk JG, et al. Citicoline (CDP-choline) increases Sirtuin1 expression concomitant to neuroprotection in experimental stroke. J Neurochem. 2013;126:819–826. | |
Zhang T, Kraus WL. SIRT1-dependent regulation of chromatin and transcription: linking NAD(+) metabolism and signaling to the control of cellular functions. Biochim Biophys Acta. 2010;1804:1666–1675. | |
Ye J, Liu Z, Wei J, et al. Protective effect of SIRT1 on toxicity of microglial-derived factors induced by LPS to PC12 cells via the p53-caspase-3-dependent apoptotic pathway. Neurosci Lett. 2013;553:72–77. | |
Hawkins BT, Abbruscato TJ, Egleton RD, et al. Nicotine increases in vivo blood-brain barrier permeability and alters cerebral microvascular tight junction protein distribution. Brain Res. 2004;1027:48–58. | |
Strazielle N, Ghersi-Egea JF. Physiology of blood-brain interfaces in relation to brain disposition of small compounds and macromolecules. Mol Pharm. 2013;15:1473–1491. | |
Fanning AS, Jameson BJ, Jesaitis LA, Anderson JM. The tight junction protein ZO-1 establishes a link between the transmembrane protein occludin and the actin cytoskeleton. J Biol Chem. 1998;273:29745–29753. | |
Inamura A, Adachi Y, Inoue T, et al. Cooling treatment transiently increases the permeability of brain capillary endothelial cells through translocation of claudin-5. Neurochem Res. 2013;38:1641–1647. | |
Xia YP, He QW, Li YN, et al. Recombinant human sonic hedgehog protein regulates the expression of ZO-1 and occludin by activating angiopoetin-1 in stroke damage. PLoS One. 2013;8:e68891. | |
Jiao H, Wang Z, Liu Y, Wang P, Xue Y. Specific role of tight junction proteins claudin-5, occludin, and ZO-1 of the blood-brain barrier in a focal cerebral ischemic insult. J Mol Neurosci. 2011;44:130–139. | |
Knowland D, Arac A, Sekiguchi K, et al. Stepwise recruitment of transcellular and paracellular pathways underlies blood-brain barrier breakdown in stroke. Neuron. 2014;82:603–617. | |
Schreibelt G, Kooij G, Reijerkerk A, et al. Reactive oxygen species alter brain endothelial tight junction dynamics via RhoA, PI3 kinase, and PKB signaling. FASEB J. 2007;21:3666–3676. | |
Rao AM, Hatcher JF, Dempsey RJ. CDP-choline: neuroprotection in transient forebrain ischemia of gerbils. J Neurosci Res. 1999;58:697–705. | |
Baskaya MK, Dogan A, Rao AM, Dempsey RJ. Neuroprotective effects of citicoline on brain edema and blood-brain barrier breakdown after traumatic brain injury. J Neurosurg. 2000;92:448–452. | |
Yepes M, Sandkvist M, Moore EG, Bugge TH, Strickland DK, Lawrence DA. Tissue-type plasminogen activator induces opening of the blood-brain barrier via the LDL receptor-related protein. J Clin Invest. 2003;112:1533–1540. | |
Diederich K, Frauenknecht K, Minnerup J, et al. Citicoline enhances neuroregenerative processes after experimental stroke in rats. Stroke. 2012;43:1931–1940. | |
Gimenez R, Raich J, Aguilar J. Changes in brain striatum dopamine and acetylcholine receptors induced by chronic CDP-choline treatment of aging mice. Br J Pharmacol. 1991;104:575–578. | |
Silvery MM, Dikan J, Ross AJ, et al. Citicoline enhances frontal lobe bioenergetics as measured by phosphorus magnetic resonance spectroscopy. NMR Biomed. 2008;10:1066–1075. | |
Liu XS, Zhang ZG, Zhang RL, et al. Stroke induces gene profile changes associated with neurogenesis and angiogenesis in adult subventricular zone progenitor cells. J Cereb Blood Flow Metab. 2007;27:564–574. | |
Kwon SE, Chapman ER. Synaptophysin regulates the kinetics of synaptic vesicle endocytosis in central neurons. Neuron. 2011;70:847–854. | |
Hurtado O, Cardenas A, Pradillo JM, et al. A chronic treatment with CDP-choline improves functional recovery and increases neuronal plasticity after experimental stroke. Neurobiol Dis. 2007;26:105–111. | |
Rema V, Bali KK, Ramachandra R, Chugh M, Darokhan Z, Chaudhary R. Cytidine-5-diphosphocholine supplement in early life induces stable increase in dendritic complexity of neurons in the somatosensory cortex of adult rats. Neuroscience. 2008;155:556–564. | |
Kojima T, Hirota Y, Ema M, et al. Subventricular zone-derived neural progenitor cells migrate along a blood vessel scaffold toward the post-stroke striatum. Stem Cells. 2010;28:545–554. | |
Rosell A, Morancho A, Navarro-Sobrino M, Martinez-Saez E, et al. Factors secreted by endothelial progenitor cells enhance neurorepair responses after cerebral ischemia in mice. PLoS One. 2013;8(9):e73244. | |
Martin KA, Merenick BL, Ding M, et al. Rapamycin promotes vascular smooth muscle cell differentiation through insulin receptor substrate-1/phosphatidylinositol 3-kinase/Akt2 feedback signaling. J Biol Chem. 2007;282:36112–36120. | |
Ferrara N. Role of vascular endothelial growth factor in regulation of physiological angiogenesis. Am J Physiol Cell Physiol. 2001;280:C1358–C1366. | |
Jiang ZY, He Z, King BL, et al. Characterization of multiple signaling pathways of insulin in the regulation of vascular endothelial growth factor expression in vascular cells and angiogenesis. J Biol Chem. 2003;278:31964–31971. | |
Hecht N, Schneider UC, Czabanka M, et al. Endothelial progenitor cells augment collateralization and hemodynamic rescue in a model of chronic cerebral ischemia. J Cereb Blood Flow Metab. 2014;34:1297–1305. | |
Sobrino T, Hurtado O, Moro MA, et al. The increase of circulating endothelial progenitor cells after acute ischemic stroke is associated with good outcome. Stroke. 2007;38:2759–2764. | |
Sobrino T, Rodriguez-Gonzalez R, Blanco M, et al. CDP-choline treatment increases circulating endothelial progenitor cells in acute ischemic stroke. Neurol Res. 2011;33:572–577. | |
Agut J, Ortiz JA, Wurtman RJ. Cytidine (5′)diphosphocholine modulates dopamine K(+)-evoked release in striatum measured by microdialysis. Ann N Y Acad Sci. 2000;920:332–335. | |
Radad K, Gille G, Xiaojing J, Durany N, Rausch WD. CDP-choline reduces dopaminergic cell loss induced by MMP (+) and glutamate in primary mesencephalic cell culture. Int J Neurosci. 2007;117:985–998. | |
Ulus IH, Wurtman RJ, Mauron C, Blusztajn JK. Choline increases acetylcholine release and protects against the stimulation-induced decrease in phosphatide levels within membranes of rat corpus striatum. Brain Res. 1989;484:217–227. | |
Moffett JR, Ross BA, Arun P, Madhavarao CN, Namboodiri AM. N-Acetylaspartate in the CNS: from neurodiagnostics to neurobiology. Prog Neurobiol. 2007;81:89–131. | |
Yoon SJ, Lyoo IK, Kim HJ, et al. Neurochemical alterations in methamphetamine-dependent patients treated with cytidine-5′-diphosphate choline: a longitudinal proton magnetic resonance spectroscopy study. Neuropsychopharmacology. 2010;35(5):1165–1173. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.