Back to Journals » International Journal of Nanomedicine » Volume 13
Curcumin-loaded galactosylated BSA nanoparticles as targeted drug delivery carriers inhibit hepatocellular carcinoma cell proliferation and migration
Authors Huang Y, Hu L, Huang S, Xu W, Wan J, Wang D, Zheng G ![]() , Xia Z
, Xia Z
Received 17 August 2018
Accepted for publication 6 November 2018
Published 6 December 2018 Volume 2018:13 Pages 8309—8323
DOI https://doi.org/10.2147/IJN.S184379
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Linlin Sun
Yike Huang,1 Lu Hu,2 Shan Huang,1 Wanjun Xu,1 Jingyuan Wan,2 Dandan Wang,1 Guocan Zheng,3 Zhining Xia1
1Department of Pharmaceutical Analysis, School of Pharmaceutical Sciences and Innovative Drug Research Centre, Chongqing University, Chongqing, China; 2Department of Pharmacology, Chongqing Key Laboratory of Biochemistry and Molecular Pharmacology, School of Pharmacy, Chongqing Medical University, Chongqing, China; 3Analytical and Testing Center, Chongqing University, Chongqing, China
Background: The main objective of this study was to develop novel BSA nanoparticles (BSA NPs) for improving the bioavailability of curcumin as an anticancer drug, and those BSA NPs were galactosylated for forming the curcumin-loaded galactosylated BSA nanoparticles (Gal-BSA-Cur NPs), thus enhancing their ability to target asialoglycoprotein receptor (ASGPR) overexpressed on hepatocellular carcinoma (HCC) cells.
Materials and methods: Gal-BSA-Cur NPs were prepared by the desolvation method and showed a spherical shape and well distribution with the average particle size of 116.24 nm.
Results: In vitro drug release assay exhibited that Gal-BSA-Cur NPs had higher release rates and improved the curcumin solubility. Cell uptake studies confirmed that Gal-BSA-Cur NPs could selectively recognize receptors on the surface of HCC (HepG2) cells and improve internalization ability of drug compared with BSA NPs-loaded curcumin (BSA-Cur NPs), which might be due to high affinity to galactose. Further, the effects of Gal-BSA-Cur NPs were evaluated by cytotoxicity assay, crystal violet assay, cell apoptosis assay, and wound healing assay, respectively, which revealed that Gal-BSA-Cur NPs could inhibit HepG2 cells proliferation, induce cell apoptosis, and inhibit cell migration.
Conclusion: Immunofluorescence staining has proved that the effects of Gal-BSA-Cur NPs related to the suppression of the nuclear factor κB-p65 (NF-κB-p65) expression in HepG2 cell nucleus. Therefore, these results indicate that novel Gal-BSA-Cur NPs are potential candidates for targeted curcumin delivery to HCC cells.
Keywords: albumin, curcumin, nanoparticles, galactosylated, hepatocellular carcinoma
Introduction
Hepatocellular carcinoma (HCC) is the third cause of cancer-related deaths worldwide and represents a major global health problem, with exceedingly high mortality over recent decades in many countries.1–3 The high mortality of HCC should be due to late diagnosis4,5 and limited treatment.6 Pharmacotherapies are the common palliative strategies for clinical treatment.7 However, they have many drawbacks such as low selectivity, dose-limiting toxicity, and unfavorable adverse effects.8 Thus, it is a necessary requirement to develop effective strategies. Targeted drug delivery, one advanced method for clinical agents, can improve the pharmacological profiles of anticancer drugs and enrich drugs to the targeted site. The asialoglycoprotein receptor (ASGPR) is one of the most studied targets to selectively deliver anticancer drugs to HCC. It is primarily overexpressed on hepatocytes surfaces, while poorly distributed in extrahepatic tissues.9 ASGPR facilitates internalization by clathrin-mediated endocytosis with minimal concerns of toxicity and shows high affinity for carbohydrates, including galactose and N-acetylgalactosamine, which can enhance cellular uptake.10 Galactosylated targeted drug delivery has been widely used for antitumor activity of HCC (HepG2) cells in vitro because of ready availability of the specific ASGPR ligand galactose.11,12
Curcumin (Cur) is a polyphenolic compound extracted from the rhizomes of Curcuma longa. It is particularly attractive for its antioxidant, antimicrobial, anti-inflammatory, and anticancer effects.13–15 Moreover, it has been reported that Cur has no adverse drug reaction, and the treatment is safe. Its anticancer activity is also showed by capturing various cancer cells and inducing apoptosis. However, the poor solubility in water and low bioavailability of Cur have limited access of sufficient concentration for pharmacologic effects in tissues.16
Nanoparticles (NPs) as drug delivery systems offer many benefits due to their high internalization efficiencies. Among various NPs, BSA nanoparticles (BSA NPs), versatile protein carriers for drug delivery have been shown to be non-toxic, non-immunogenic, low cost, biocompatible, and ease to be metabolized in vivo and solubility in water.17 Thus, BSA NPs are widely applied in the pharmaceutical applications. Nab-paclitaxel, the first drug-loaded serum albumin NPs have been approved and marketed.18 Most importantly, BSA NPs has many functional carboxylic and amino groups on their surface, so that they own high covalent binding ability of compounds, including Cur and other flavonoids.19–21 For Cur-loaded NPs, they could reduce the required therapeutic dosages and toxicity, provide Cur-targeted delivery to the special sites, and enhance the bioavailability.22
To improve more bioavailability of Cur, the ASGPR receptor-mediated targeting delivery has been designed in this study. Curcumin-loaded galactosylated BSA nanoparticles (Gal-BSA-Cur NPs) have been developed. Physicochemical characteristics of Gal-BSA-Cur NPs were evaluated, and their entrapment efficiency (EE) and drug loading (DL) capacity were calculated. Release behavior in vitro of Gal-BSA-Cur NPs was proved. Tumor cell targeting efficiency of Gal-BSA-Cur NPs was determined. Furthermore, HepG2 cells were selected to investigate cell proliferation, cell apoptosis, and cell migration. In addition, the nuclear factor κB-p65 (NF-κB-p65)-mediated mechanism of HepG2 cells in Gal-BSA-Cur NPs group was demonstrated.
Materials and methods
Materials
Cur (purity ≥98%) and sodium cyanoborohydride were obtained from Adamas Reagent Co., Ltd. (Shanghai, China). D (+) Galactose, 4-O-β-d-galactopyranos, BSA (purity ≥98%), tetrazolium salt (MTT), dimethyl sulfoxide (DMSO), DAPI, and trypsin solution (0.25%) were all provided by Solarbio Science & Technology Co., Ltd. (Beijing, China). FBS was purchased from Lonsa Science Srl Co., Ltd. (Shanghai, China). DMEM with high glucose and penicillin–streptomycin solution was supplied by Thermo Fisher Scientific Biological Chemical Co., Ltd. (Beijing, China). Glutaraldehyde was obtained from Chengdu Kelong Chemical Co., Ltd. (Chengdu, China). All the other reagents were of analytical grade.
Preparation and characterization of galactosylated BSA (Gal-BSA)
Gal-BSA was prepared by a reductive amination technique as described previously.23 Briefly, 132 mg of BSA in 10.0 mL of phosphate buffer (0.2 M, pH 7.4) was added to 360 mg 4-O-β-d-galactopyranos and then placed for 2 hours at room temperature. The mixture solution with 310 mg sodium cyanoborohydride reacted for 70 hours in a thermostat water bath at 37°C. After the scheduled reaction time, the solution was added to a dialysis pocket placed in water. The product was finally dried in a vacuum freezing drying oven.
The matrix-assisted laser desorption/ionization time of flight mass spectrometry (MALDI-TOF MS) was used to determine the mass increase of BSA-modified galactose.24,25 The samples were dissolved with pure water to a concentration of 1 mg/mL. The samples were diluted 1:1 (v/v) in matrix solution containing 15 mg/mL sinapic acid in 50% acetonitrile and 0.1% trifluoroacetic acid. An aliquot (1 μL) was spotted onto a target steel plate and air dried. The mass spectrum was recorded in the reflector mode. Ladder-desorbed positive ions were performed after an acceleration voltage of 20 kV in the linear mode. External calibration was analyzed by standard proteins: BSA (66,430 Da). Repeated individual spectra (n=100) were averaged to obtain a mass spectrum.
Mass to charge ratio (m/z; equaling to molecular weight [MW]) of BSA and Gal-BSA was obtained. The number (N) of galactose conjugated with BSA molecule was calculated as follows (326 is MW of lactose residue-connected BSA):
|
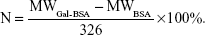
SDS-PAGE was performed according to the method described by Weber and Osborn26 and Laemmli.27 A 4% stacking and 10% resolving gels were used in vertical gel electrophoresis unit (Bio-Rad, USA). 2 mg/mL of the sample was mixed with loading buffer containing 2% (v/v) β-mercaptoethanol and heated in a boiling water bath for 10 minutes. The prepared sample was loaded on the gel, and then electrophoresis was carried out at a constant voltage of 110 V for 60 minutes. The gel was subsequently stained with Coomassie brilliant blue G250 for 1 hour and washed in a solution of methanol/acetic acid/dH2O (20/10/70, v/v/v). Finally, protein bands on the gel were visualized by Bio-Rad ChemiDoc imaging system.
Synthesis of Gal-BSA-Cur NPs
Gal-BSA-Cur NPs were synthesized using the desolvation method.28–30 In principle, 100 mg Gal-BSA in 4.0 mL of purified water was titrated to pH 7.4. 15 mg of Cur was dissolved in 6 mL ethanol, and then the Cur solution was added dropwise to Gal-BSA solution at the rate of 1 mL/min under magnetic stirring of 500 rpm. The NPs were transformed by continued stirring for 6 hours under room temperature, followed by cross-linking with the addition of 8% glutaraldehyde solution of 25 μL. The resulting NPs were purified by repeated centrifugation at 12,000 rpm for 30 minutes. Finally, the NPs were redispersed to 1 mL ultrapure water and freeze dried for 72 hours.
Otherwise, BSA NPs-loaded curcumin (BSA-Cur NPs) and blank Gal-BSA NPs were obtained as described earlier. The differences were follows. For the former, 100 mg of BSA was only dissolved, instead of 100 mg Gal-BSA. For the later, ethanol without Cur was added dropwise to Gal-BSA solution.
Gal-BSA-Cur NPs morphology, size distribution, and zeta potential analysis
The morphology of the Gal-BSA-Cur NPs was observed using ZEISS MERLIN VP Compact scanning electron microscope (SEM; Zeiss, Oberkochen, Germany). The average particle size and zeta potential of the NPs were evaluated using a NanoBrook Omni Particle Size & Zeta Potential Analyzer (173 plus; Brookhaven, USA). All measurements were determined in triplicate.
Fourier transform infrared spectroscopy (FT-IR) and differential scanning calorimetry (DSC) analysis
The FT-IR spectra of Cur, Gal, BSA, BSA-Cur NPs, Gal-BSA-Cur NPs, and blank NPs were obtained using an FT-IR spectrometer (TENSOR 27; Bruker, USA) to investigate the successful synthesis of Gal-BSA-Cur NPs and BSA-Cur NPs. All samples were mixed thoroughly with KBr and then pressed into pellets. The pellets were measured in the range of (4,000–400) cm−1.
DSC thermal properties of BSA, Cur, BSA-Cur NPs, and Gal-BSA-Cur NPs were confirmed using a thermal analysis system (DSC404 F3; NETZSCH, Germany). Briefly, samples were sealed in an aluminum pan and heated over a temperature range of 25°C–260/400°C in presence of nitrogen at a constant heating rate of 10°C/min.
EE capacity and DL capacity of Cur in Gal-BSA-Cur NPs and BSA-Cur NPs
EE and DL were examined by centrifugation at 10,000 rpm for 30 minutes. The free Cur in supernatant was determined through the absorbance at 430 nm by an ultraviolet spectrophotometer (UV-2600; Shimadzu, Japan). EE and DL were calculated using the following equations:
|
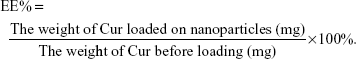
|
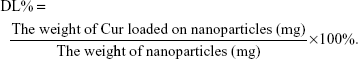
In vitro drug release study
In vitro release of Cur from free Cur, BSA-Cur NPs, and Gal-BSA-Cur NPs was evaluated by using dialysis bag technique with a MW cutoff of 8,000–14,000. 1 mL of 0.5 mg/mL free Cur or Gal-BSA-Cur NPs suspension was added into a dialysis bag. The bag was immersed in 25 mL of 0.1 M PBS (pH 7.4) and incubated at 37°C with the shake speed of 100 rpm. At set intervals, 4 mL of sample was taken and replaced with fresh dissolution medium. The samples were quantified using an ultraviolet-visible spectrophotometer (UV-2600; Shimadzu) at 430 nm. Each experiment was conducted in triplicate.
Cell culture
The HepG2 cell line was purchased from Chongqing Key Laboratory of Biochemistry and Molecular Pharmacology (Chongqing, China). The cells were cultured in a 5% CO2 humidified incubator at 37°C with a normal culture medium consisting of DMEM with high glucose medium, 10% FBS, and 1% penicillin–streptomycin solution. No ethics statement was required from the institutional review board for the use of this cell line.
In vitro cellular uptake assay
ASGPR-mediated cellular of Gal-BSA-Cur NPs was evaluated in HepG2 cells, which were known to overexpress the ASGPR. Qualitative analyses of cellular internalization and cell targeting evaluation of Gal-BSA-Cur NPs were evaluated by fluorescence microscopy (ECLIPSE Ti; Nikon, Japan), whereas quantitative analyses of Gal-BSA-Cur NPs were carried out by HPLC (LC-20A; Shimazdu).
HepG2 cells (5×104 cells/well) were seeded into the 24-well plates. After the confluence and morphology were checked, drugs (50 μmol/L) were added to each well and co-incubated with the cells for 24 hours. At the end of the incubation time, the cells were washed three times and fixed with 4% formaldehyde for 15 minutes. To label the cell nucleus, the cells were further stained with 1 μg/mL DAPI. Finally, the cells were observed by fluoresce microscopy.
To further assess whether NPs are internalized in HepG2 via the mechanism that involves ASGPR-dependent recognition of galactose residues in asialoglycoproteins, the cells were pretreated with free galactose (50 mM) for 1 hour, then the Gal-BSA-Cur NPs were added, and the cells were cultured for another 4 hours. After that, the subsequent process was treated as that mentioned earlier.
HepG2 cells (5×104 cells/well) were cultured in six-well plates. The cells were incubated with 50 μmol/L of Cur, BSA-Cur NPs, Gal-BSA-Cur NPs, and Gal-BSA-Cur NPs+Gal for 4 hours and washed three times. Then, the cells were collected and suspended in 200 μL ultrapure water, frozen at −80°C three times. After centrifugation (14,000× g, 20 minutes), 1.5 mL of ethyl acetate was added to the supernatant and vortexed. The mixture solution was centrifuged, and the precipitate was dried in a vacuum. Aliquots of the samples were measured by HPLC as described using InertSustain® C18 column (15 cm×4.5 mm×5 μm; GL Sciences Inc., Tokyo, Japan) with the mobile phase of methanol/water/phosphoric acid (60/39.7/0.3, v/v) at the flow rate of 1.0 mL/min.
Cell cytotoxicity assay
In vitro cell cytotoxicity studies of Cur, BSA-Cur NPs, and Gal-BSA-Cur NPs against cancer cells were analyzed by MTT assay. Briefly, HepG2 cells were seeded in 96-well culture plates at a density of 5,000 cells per well. After the cells reached 70%–80% confluence, they were treated with the prepared samples with different concentrations of 25, 50, 75, 100, and 150 μmol/L and incubated for 24 hours. The control group was treated with empty medium solution (without any samples). After incubation, 20 μL of MTT solution (5 mg/mL) was added to each well, followed by incubation at 37°C for 4 hours. Then, the medium was removed, and the formazan crystals were dissolved with 150 μL of DMSO by gently shaking the plates for 10 minutes. Finally, the solubilized formazan was analyzed with a microplate spectrophotometer (iMark; Bio-Rad Laboratories Inc., Hercules, CA, USA) at a wavelength of 490 nm. Cytotoxicity was calculated as follows:
|
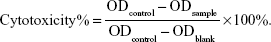
By crystal violet assay, the effects of Cur, BSA-Cur NPs, and Gal-BSA-Cur NPs on the proliferation of HepG2 cells were also explored. Briefly, cells were seeded in 24-well culture plate at a density of 5×104 cells per well and grown to 70%–80% confluence. After incubation for 24 hours with different concentrations of the three drugs mentioned earlier, cells were incubated with 2.5 mg/mL crystal violet in a 10% (v/v) solution of methanol/ultrapure water at room temperature for 15 minutes and washed three times with pure water. Images of the cells were captured.
Cell apoptosis assay
Flow cytometry (FCM) was performed to investigate the apoptosis in HepG2 cells. The cells (5×105 cells/well) were seeded into a six-well plate. After treatment with 50 μmol/L of Cur, BSA-Cur NPs, Gal-BSA-Cur NPs, and Gal-BSA-Cur NPs with free galactose for 24 hours, the cells were washed three times with PBS and were harvested by centrifugation at 1,000 rpm for 5 minutes. Then, cells in the sediment were resuspended in 1× PBS solution and stained with annexin V-fluorescein isothiocyanate (FITC-V) and propidium iodide (PI). Finally, the apoptotic cells were measured by an FCM instrument (CytoFLEX; Beckman Coulter, USA).
Wound healing assays
For wound healing assay, HepG2 cells were seeded into the six-well plate and allowed to grow to 100% confluence. The cell monolayers were carefully wounded with a sterile toothpick and washed with PBS. Then, the wounded cell monolayers were co-cultured for 24 hours with the different treatments (50 μmol/L). The control group was co-cultured for the same time with empty medium solution. Images were obtained by microscope to observe the distribution of the cells at the scratch zone. Through ImageJ software, scratch area was obtained, and the percentage of wound closure was evaluated as the parameter of the degree of wound closure.
Immunofluorescence (IF) analysis
HepG2 cells were seeded in 24-well plates and treated with 50 μM Gal-BSA-Cur NPs and 50 μM Cur for 24 hours. The control group (empty medium solution) was treated as mentioned above. After that, the cells were fixed with 4% paraformaldehyde and pretreated with 0.05% Triton X-100 at room temperature. The cells were incubated with primary antibody NF-κB P65 (1:100) from Beyotime Institute of Biotechnology (Shanghai, China) and then incubated with DyLight 594 secondary antibody against rabbit IgG. Further, the cells were stained with DAPI and imaged.
Statistical analyses
Data are expressed as mean±standard error of the mean. Statistical data were analyzed by the Student’s t-test at the significance level of P<0.05.
Results and discussion
Preparation and characterization of Gal-BSA-Cur NPS
The synthesis scheme of Gal-BSA-Cur NPs is shown in Figure 1. First, Gal-BSA was prepared via a reductive amination reaction with the alkylation of the 6-amino group of lysine of BSA. Subsequently, Cur was conjugated to Gal-BSA using the desolvation method via covalent binding ability to form Gal-BSA-Cur NPs. The synthesized Gal-BSA-Cur NPs have the potential to specifically deliver Cur to HepG2 cells.
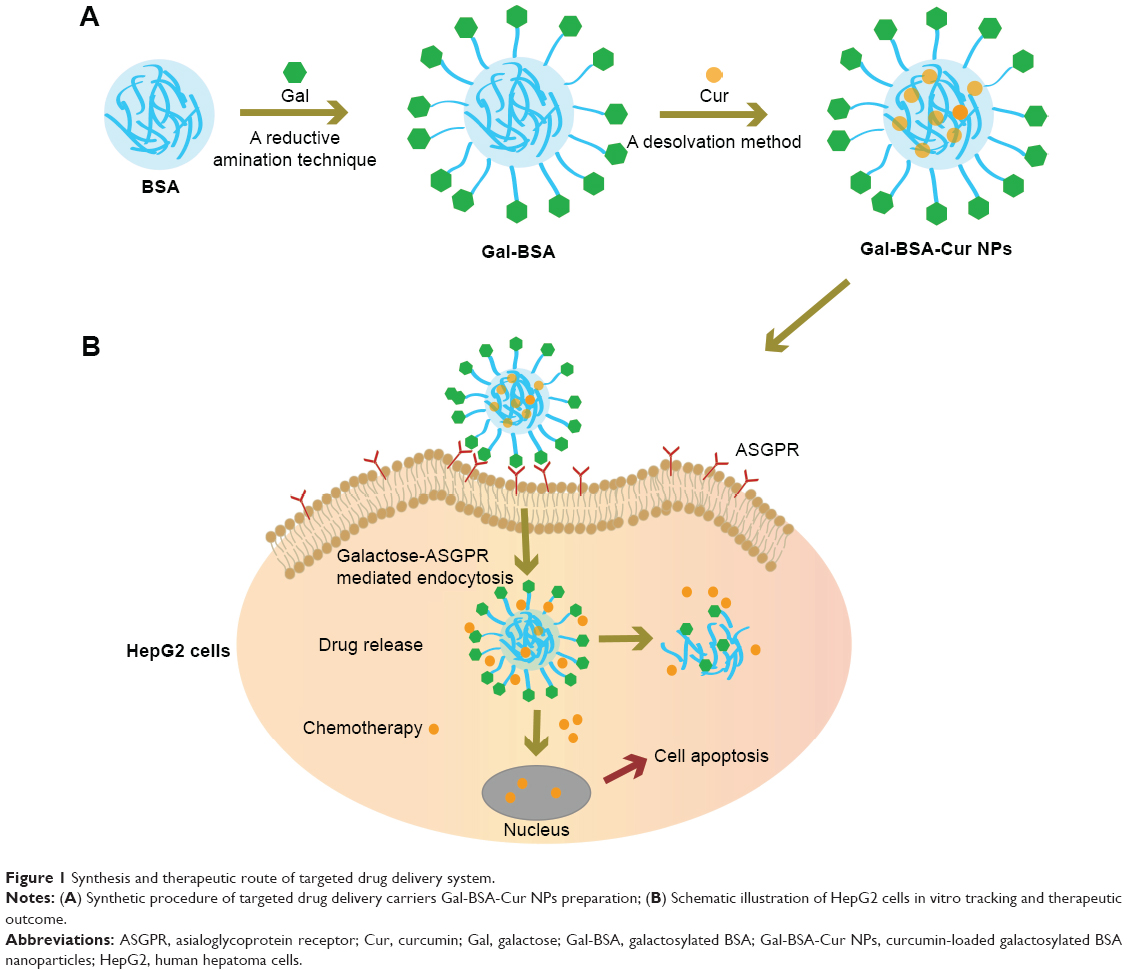 | Figure 1 Synthesis and therapeutic route of targeted drug delivery system. |
As shown in Figure 2, the MW of BSA and Gal-BSA was estimated with SDS-PAGE. A major band around 66 kDa with high density was observed for BSA in Lane 1 (Figure 2, L1). The MW of BSA was consistent with the report using electrospray ionization mass spectrometry.31 The new band of Gal-BSA was around 70 kDa (Figure 2, L2–L4, respectively), which was higher than that of BSA. The results show that Gal has been conjugated to BSA. Compared to different glycated BSA synthesized by the Maillard reaction,32 Gal-BSA was completely obtained without BSA by using a reductive amination method. In, addition, the increased MW of Gal-BSA may relate to the numbers of galactose (MW=180.16) conjugated to BSA.
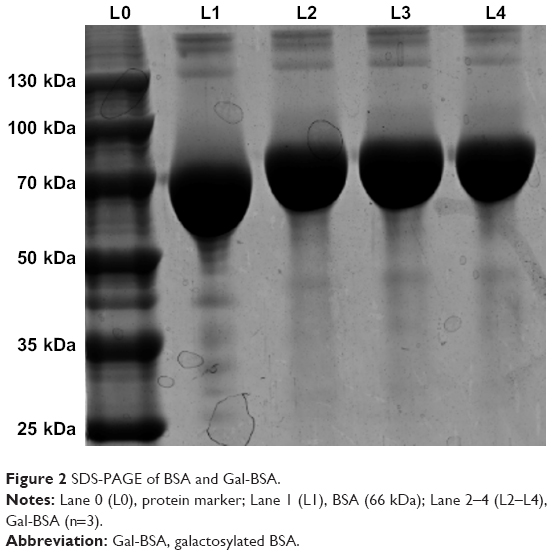 | Figure 2 SDS-PAGE of BSA and Gal-BSA. |
According to the FT-IR sepctra (Figure 3) of BSA, Gal, and Gal-BSA, the adsorption peaks of BSA (Figure 3A) at 1,658 cm−1, 1,539 cm−1, and 1,243 cm−1 were attributed to flexural vibration adsorption of amide I (−NH2), amide II (−NH−), and amide III (C−N), respectively. The broad peak at 3,381 cm−1 of Gal (Figure 3B) related to the aromatic sugar group with O−H as the main functional group. The peaks at 2,932 cm−1 pinpointed the C−H stretching, which prevailed in galactose in addition. As shown in Figure 3C, all the characteristic absorption peaks of Gal could be found in the FT-IR spectra of the galactose-conjugated BSA.
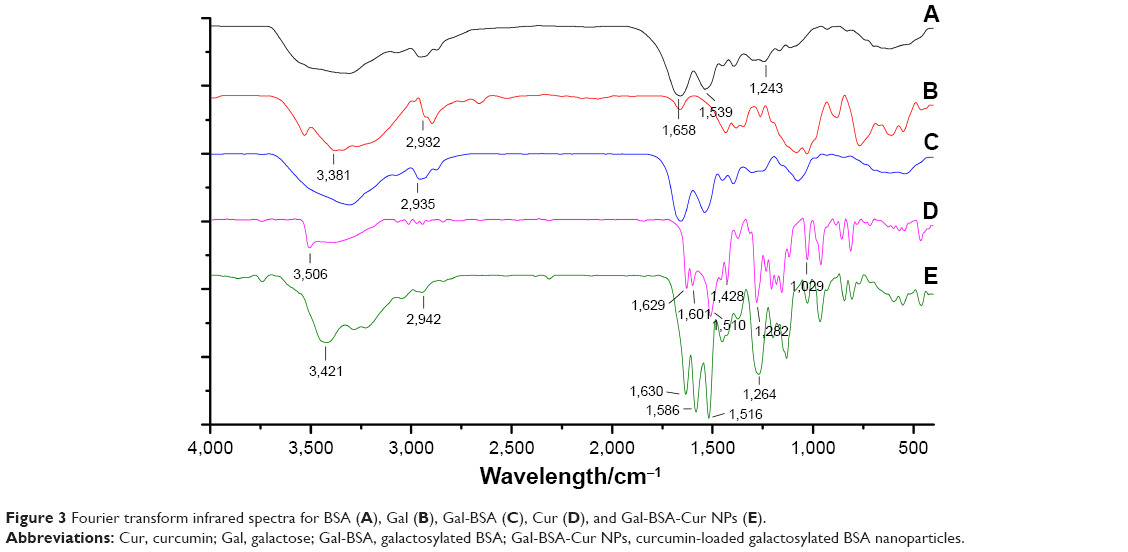 | Figure 3 Fourier transform infrared spectra for BSA (A), Gal (B), Gal-BSA (C), Cur (D), and Gal-BSA-Cur NPs (E). |
Further to quantify the number of galactose bonded to BSA in Gal-BSA, the MWs of BSA and Gal-BSA conjugates were rapidly determined by MALDI-TOF MS. Figure 4 shows the molecular mass ion peaks of BSA and Gal-BSA observed at 66,339.48 and 74,400.32 m/z, respectively. The major mass ion peak shifted after BSA was galactosylated. Based on the MWs 326 for galactose residues moieties, it could be calculated that about 25 molecules of Gal have been conjugated with BSA by triplicate measurements (Table 1). The galactose density on Gal-BSA might control the targeting efficiency.33 When the galactose numbers conjugated to BSA is more than around 30 molecules, the affinity with ASGPR was equal to endogenous ligand asialoorosomucoid (ASORO).34 On the other hand, to keep some amino groups and acid groups of BSA that will further conjugate with Cur, the galactose number should be less than 30. Our results with galactose of 25 were available. Therefore, the number of galactose is suitable for further studies.
 | Figure 4 Mass spectra of BSA (A) and Gal-BSA (B). |
 | Table 1 The galactose numbers conjugated on BSA |
According to SEM images (Figure 5), the spherical Gal-BSA-Cur NPs, with uniform size distribution were obtained. As shown in Table 2, the mean diameter of Gal-BSA-Cur NPs was 116.24±2.75 nm, whereas for BSA-Cur NPs, the mean diameter was 280.56±5.24 nm. This significant difference in particle size might be due to the pH in dissolved solution. In this study, Gal-BSA solution has higher pH than that of BSA solution (their pH are about 7 and 8, respectively). Thus, the particle diameter of Gal-BSA-Cur NPs (formed by Gal-BSA) is smaller than that of BSA-Cur NPs (formed by BSA). The results are consistent with references. Langer et al28 has mentioned that higher pH formed by dissolution of human serum albumin could lead to the production of smaller NPs. Li et al35 reported, with pH increasing from 7.0 to 9.0, that there is a decrease on the average particle diameter of BSA NPs. The acidity and alkaline of reaction solution may lead to convolute of BSA molecules and further fasciculation of the NPs. So the average particle diameter of Gal-BSA-Cur NPs was larger than that of BSA-Cur NPs. For galactose, as a hydrophilic molecule, it can stabilize a larger surface area.36 In addition, the NPs with the mean particle size at the range of 100–150 nm would show an enhanced permeability and retention (EPR) effect, which improve the uptake ability of drug-loaded NPs for tumors. Thus, the Gal-BSA-Cur NPs might possess great potential as drug-loaded delivery system. The polydispersity index (PDI) values, less than 0.3, revealed their relatively narrow size distributions.
 | Figure 5 Scanning electronic micrographs of Gal-BSA-Cur NPs. |
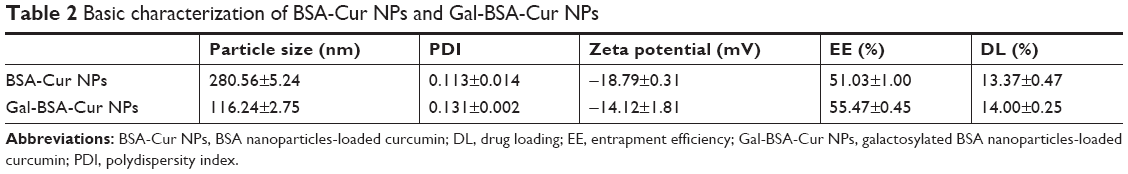 | Table 2 Basic characterization of BSA-Cur NPs and Gal-BSA-Cur NPs |
The zeta potential is directly related with the surface charge of NPs. The zeta potentials for Gal-BSA-Cur NPs and BSA-Cur NPs were measured to be −14.12±1.81 and −18.79±0.31 mV, respectively (Table 2). The solubility of Gal-BSA-Cur NPs after galactosylated has been increased by comparing to the BSA-Cur NPs, since galactose is hydrophilic.
To identify whether the Gal-BSA-Cur NPs are successfully synthesized, FT-IR method has been used. The spectrum of Cur (Figure 3D) in our experiment is consistent with the standard FT-IR spectra. With respect to the spectrum of Gal-BSA-Cur NPs shown in Figure 3E, the related peak to Cur, a shift from 3,506 to 3,421 cm−1, was observed, and the valley of 3,421 cm−1 became wider, thereby indicating an enhanced hydrogen bonding.37,38 Meanwhile, the peaks of Cur at 1,629, 1,601, and 1,510 cm−1 slightly shifted to 1,630, 1,586, and 1,516 cm−1, respectively. In addition, the absorption peak of Amide III at 1,243 cm−1 shifted to 1,264 cm−1. The peak at 2,935 cm−1 of hydrogen bonding in Gal-BSA shifted to 2,942 cm−1. Some reports the present results of conjugation between BSA and Cur,20,39 which indicates the participation of functional groups in intermolecular interactions in Gal-BSA-Cur NPs.
The thermal behavior and transition temperature depend on the structural and functional groups.25 The thermograms of BSA, Cur, BSA-Cur NPs, and Gal-BSA-Cur NPs are illustrated in Figure 6. The thermogram of BSA (Figure 6A) revealed an endothermic peak at 225°C. Cur (Figure 6B) showed a sharp endothermic peak corresponding to its melting point at 178°C. Two melting point peaks of BSA and Cur were observed on one curve for physical mixture of BSA and Cur (Figure 6C). In the case of BSA-Cur NPs, the Cur melting point peak disappeared and the BSA curve showed a slight endotherm with the peak temperature shifted to a higher temperature when compared to BSA and Cur. The result is similar to the reports by Liu and Jing,25 which indicates that the BSA-Cur NPs have formed a more organized and stable structure.
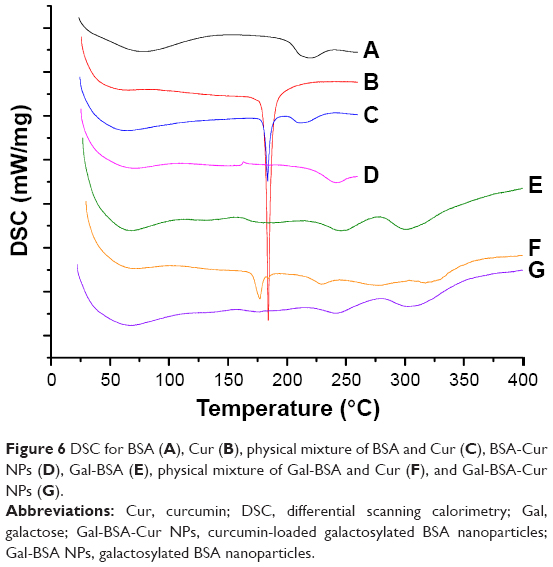 | Figure 6 DSC for BSA (A), Cur (B), physical mixture of BSA and Cur (C), BSA-Cur NPs (D), Gal-BSA (E), physical mixture of Gal-BSA and Cur (F), and Gal-BSA-Cur NPs (G). |
The endothermic peaks of Gal-BSA (Figure 6E) were detected at 248°C and 300°C. The peak of physical mixture of Gal-BSA and Cur (Figure 6F) was obtained at approximately 178°C. In the curve of Gal-BSA-Cur NPs (Figure 6G), no peak at approximately 178°C was observed, which indicates that the Cur does not exist in independent form inside the NPs. Moreover, Gal-BSA-Cur NPs showed higher point temperature than BSA-Cur NPs, which means that the more energy was needed to melt the NPs, evidencing the strong interaction between the Cur and Gal-BSA. Hence, the aforementioned results imply the successful synthesis of Gal-BSA-Cur NPs and BSA-Cur NPs. In addition, Gal-BSA-Cur NPs are first obtained as nanomaterials.
Assessment of EE and drug-loading capacity of Cur in Gal-BSA-Cur NPs and BSA-Cur NPs
Table 2 lists the calculated EE and DL, which are considered as basic parameters of NPs. EE of BSA-Cur NPs and Gal-BSA-Cur NPs were 51.03%±1.00% and 55.47%±0.45%, respectively. DL of BSA-Cur NPs and Gal-BSA-Cur NPs were 13.37%±0.47% and 14.00%±0.25%, respectively.
In vitro drug release
In general, in vitro drug release has an influence on the rate of drug release and release mechanism from nanosize carriers.40 In the present experiment, BSA-Cur NPs and Gal-BSA-Cur NPs were investigated on the Cur release behavior.
Figure 7 shows the release profiles of Cur, BSA-Cur NPs, and Gal-BSA-Cur NPs. For control, the release rate of Cur was slow because of its poor water solubility. In contrast, the release rates of BSA-Cur NPs and Gal-BSA-Cur NPs dramatically increased, which indicates that the Cur solubility of two NPs could effectively improve. The plot for the two NPs also shows that they had a gradually fast release rate before 72 hours, followed by a slowdown until the cumulative release reached the levels of 58% and 79% at 96 hours. Through this result, we speculated that Cur may release in bloodstream (which is neutral) for a long time. Moreover, Gal-BSA-Cur NPs exhibited higher release rate than that of BSA-Cur NPs, which can be attributed to the release capability provided by the galactose moiety.
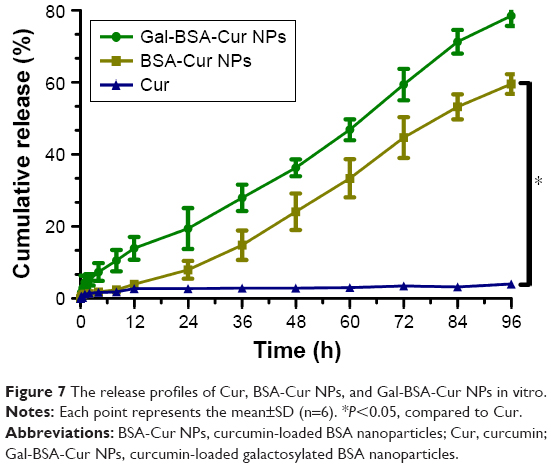 | Figure 7 The release profiles of Cur, BSA-Cur NPs, and Gal-BSA-Cur NPs in vitro. |
Cellular uptake and competitively binding assay
To evaluate the ASGPR-targeting ability of Gal-BSA-Cur NPs, HepG2 cells with high expression of ASGPR on the cell surface were selected (ASGPR+ cells), while human embryonic kidney 293 (HEK293) cells were used as a negative control (ASGPR– cells).41 NPs that target cancer cells could release drugs inside cells and enhance the therapeutic ability cancer. After 3 hours of incubation with samples, the green fluorescence properties of Cur provide qualitative analysis of biodistribution in HepG2 and HEK293. The localization was confirmed by nuclear staining with DAPI, which was observed by fluoresce microscopy as shown in Figure 8.
 | Figure 8 Fluorescence microscopy images of cells incubated with 50 μmol/L of Cur, BSA-Cur NPs, Gal-BSA-Cur NPs, and Gal-BSA-Cur NPs+Gal for 3 hours. |
The green fluorescent intensity of Gal-BSA-Cur NPs is noticeably more in HepG2 cells compared to that of Cur, which demonstrates that galactose moieties of Gal-BSA-Cur NPs significantly facilitate the cell endocytosis. When HepG2 cells were pretreated with galactose, the amounts of Gal-BSA-Cur NPs uptaken by the cells was decreased. It was mainly because the free galactose as a competitive binder hindered the binding of Gal loaded on the Gal-BSA-Cur NPs to their receptors. Moreover, when HepG2 cells were incubated with Gal-BSA-Cur NPs, very weak fluorescence with HEK293 cells was observed. Hence, the galactose residues displayed on the surface of Gal-BSA-Cur NPs improved cell uptake ability via ASGPR-mediated endocytosis. In addition, the fluorescence intensity of BSA-Cur NPs was stronger than that of Cur, which may attribute to BSA easily binding to the cytomembrane and improve permeability. Similar to BSA-Cur NPs, Gal-BSA-Cur NPs own the same BSA molecule, which indicates that the two molecules of galactose and BSA have effect on Cur delivery into HepG2 cells.
Meanwhile, the intracellular Cur concentrations uptaken by HepG2 cells were quantitatively analyzed. As shown in Figure 9, the intracellular curcumin concentrations in three groups of Cur, BSA-Cur NPs and Gal-BSA-Cur NPs were 46.8±14.8, 133.6±7.9, and 240.0±18.2 ng/105 cells, respectively. The results indicate that the Cur concentration of Gal-BSA-Cur NPs was dramatically higher than that of non-galactose-conjugated BSA-Cur NPs and Cur probably because of the specific binding between galactose and its receptor. Further, the Gal-BSA-Cur NPs+Gal group pretreated with free galactose (50 mM) in advance showed that Cur concentration declined slightly to 219.0±13.0 ng/105 cells compared with the Gal-BSA-Cur NPs group. Therefore, all results strongly support that the galactose-mediated endocytosis plays an important role in the cellular uptake of Gal-BSA-Cur NPs.
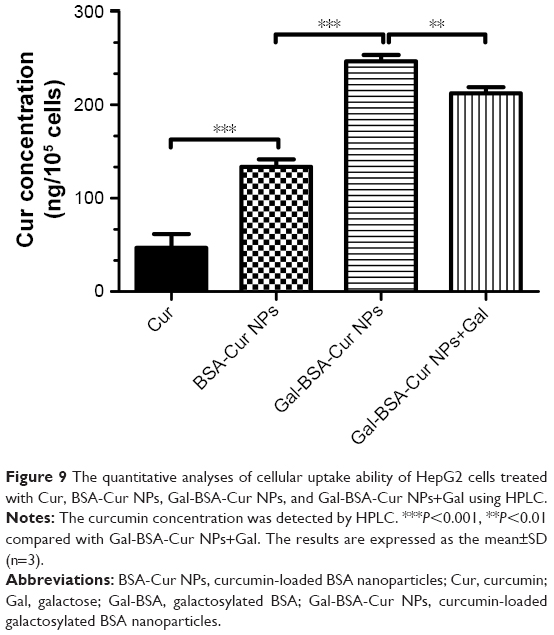 | Figure 9 The quantitative analyses of cellular uptake ability of HepG2 cells treated with Cur, BSA-Cur NPs, Gal-BSA-Cur NPs, and Gal-BSA-Cur NPs+Gal using HPLC. |
Cytotoxicity
HepG2 cancer cells were used as a model to evaluate the cytotoxicity of Gal-BSA-Cur NPs as a targeted anticancer drug. As shown in Figure 10, the blank BSA NPs and Gal-BSA NPs of 150 μmol/L, which is the maximum concentration used by previous research and references, had no obvious cytotoxicity on the growth of the HepG2 cell lines, which proves that BSA NPs and Gal-BSA NPs are highly biocompatible and do not have toxic effect to the HepG2 cells. Further, HepG2 cells were, respectively, incubated with different concentrations of Cur, BSA-Cur NPs, and Gal-BSA-Cur NPs for 24 hours (Figure 11). The cell inhibition rate of BSA-Cur NPs was higher than that of Cur, which may relate to the amounts of Cur in the cells. Gal-BSA-Cur NPs exhibited higher inhibition rate than that of BSA-Cur NPs, indicating that the Gal-BSA-Cur NPs showed stronger inhibition effect for tumor cells. It is worth noting that the inhibition effect of Gal-BSA-Cur NPs could be related to the target function of 25 numbers of galactose. Thus, galactose number and Cur dose may be the key factors for cytotoxicity of tumor cells. The IC50 values of Gal-BSA-Cur NPs (45.97±0.023 μmol/L) in HepG2 were significantly lower than that of Cur (111.60±0.020 μmol/L) and BSA-Cur NPs (89.19±0.022 μmol/L), showing that Gal-BSA-Cur NPs strongly inhibit HepG2 cell proliferation.
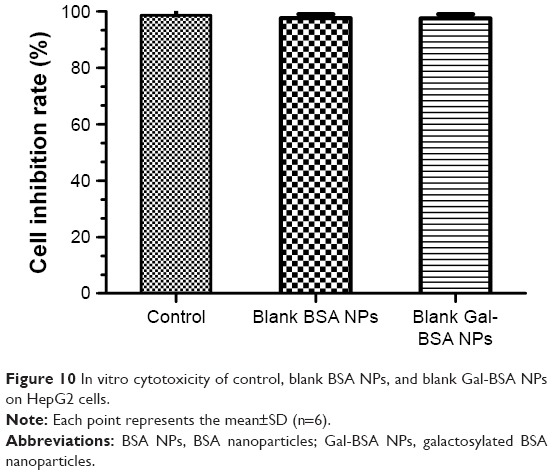 | Figure 10 In vitro cytotoxicity of control, blank BSA NPs, and blank Gal-BSA NPs on HepG2 cells. |
 | Figure 11 The vitro cytotoxicity of Cur, BSA-Cur NPs, and Gal-BSA-Cur NPs on HepG2 cells. |
Furthermore, we used a crystal violet assay to evaluate the Gal-BSA-Cur NPs on HepG2 cell viability (Figure 12). Compared with the control, the amounts of viable cells of three groups (Cur, BSA-Cur NPs, and Gal-BSA-Cur NPs) were less. At the various drug concentrations, the Gal-BSA-Cur NPs had less cells than that of Cur and BSA-Cur. The results revealed that all three treatment groups of Cur, BSA-Cur NPs, and Gal-BSA-Cur NPs could inhibit the proliferation of the tumor cell lines. Gal-BSA-Cur NPs also display a higher inhibition ability than Cur and BSA-Cur NPs.
 | Figure 12 Crystal violet photographs of HepG2 cells treated with different concentrations of Cur, BSA-Cur NPs, and Gal-BSA-Cur NPs. |
Cell apoptosis assay
The Cur, BSA-Cur NPs, and Gal-BSA-Cur NPs of 50 μmol/L were added into the HepG2 cell media and incubated for 24 hours, and cell apoptosis was detected by FCM (Figure 13). Compared with the control, the cells treated with Gal-BSA-Cur NPs had higher apoptotic rate (62.54%) than that of Cur (23.2%) and Cur-BSA NPs (31.84%), suggesting that the Gal-BSA-Cur NPs could better deliver lipophilic Cur into the cells. Further, Gal-BSA-Cur NPs may show the ability of inducing apoptosis to inhibit HepG2 cells, which is consistent with the previous results. In addition, the competitive binding experiment was conducted. The 50 mM galactose was pretreated for 2 hours before Gal-BSA-Cur NPs were added in the cell media. The results show that apoptotic cells were continuously declined by 20.95% than that of Gal-BSA-Cur NPs. This suggests that Gal-BSA-Cur NPs targeted ASGPR on the HepG2 cells.
 | Figure 13 Cell apoptosis effect of blank, Cur, BSA-Cur NPs, Gal-BSA-Cur NPs, and Gal-BSA-Cur NPs+Gal on HepG2 cells. |
Cell migration assay
Whether Gal-BSA-Cur NPs has the potential to inhibit HepG2 cell migration has been shown in Figure 14, the different groups showed different cell migration distance by wound healing assay of Cur, BSA-Cur NPs, and Gal-BSA-Cur NPs. The percentage of wound closure (Gal-BSA-Cur NPs group) was lower than that of Cur and BSA-Cur NPs When Gal-BSA-Cur NPs was pretreated with Gal, the percentage of wound closure was higher. The results indicate that Gal-BSA-Cur NPs could inhibit the migration of HepG2 cells, thereby substantiating its tumor-suppressive role in tumor.
 | Figure 14 Cell migration effect of HepG2 cancer cells pretreated with Cur, BSA-Cur NPs, Gal-BSA-Cur NPs, and Gal-BSA-Cur NPs+Gal. (A) The cell migration was determined by wound healing assay after treatment for 24 hours. (B) The quantitative analyses of cell migration. |
Gal-BSA-Cur NPs suppress the expression and activity of NF-κB-p65 in HepG2 cells
The expression and activity of NF-κB-p65 in HepG2 cells were detected by IF staining. Cells were treated with 50 μmol/L Gal-BSA-Cur NPs for 24 hours. Figure 15 shows that in the control group the robust expression of p65 protein located in the nucleus, while red fluorescence intensity of the p65 protein in the Gal-BSA-Cur NPs groups was weak, which indicates that Gal-BSA-Cur NPs are able to inhibit the expression of NF-κB-p65. Thus, Gal-BSA-Cur NPs may induce apoptosis by inhibiting NF-κB-p65 expression.
 | Figure 15 Effect of Cur and Gal-BSA-Cur NPs on the NF-κB-p65 protein. (A) Fluoresce microscopy images of cells. (B) Quantitative analyses of the NF-κB-p65 protein expression. |
Conclusion
In this study, a targeted drug-loaded NPs delivery carrier was successfully obtained, which is non-toxic, is biocompatible, and has high DL capacity to deliver anticancer drugs into HCC cells. Gal-BSA-Cur NPs showed high release effect and good bioavailability in vitro. The targeting ability of Gal-BSA-Cur NPs has been confirmed that the NPs with galactose moiety were much internalized by HepG2 cells (ASGPR+ cells) than BSA-Cur NPs with no galactose molecules. This study verified that the entrance of Gal-BSA-Cur NPs into HepG2 cells was supported by ASGPR receptor located on the cell surface. With the pretreatment of galactose in HepG2 cells, the amount of Cur absorbed into the cells for Gal-BSA-Cur NPs declined. Moreover, the effects of Gal-BSA-Cur NPs were evaluated that the NPs could inhibit the proliferation of HepG2 cells, induce the cell apoptosis, and inhibit the cell migration. The effects of Gal-BSA-Cur NPs on HepG2 were associated with the inactivation the protein level of NF-κB-p65. These results validate that Gal-BSA-Cur NPs could be a more effective targeted drug for HepG2 cells, which elucidates the mechanism of ASGPR receptor-mediated endocytosis. The Cur-loaded Gal-BSA NPs may be a potential novel therapeutic platform for HCC treatment via hepatocyte targeting.
Acknowledgment
This work was supported by the National Natural Science Foundation of China (No 21675016).
Disclosure
The authors report no conflicts of interest in this work.
References
Bertuccio P, Turati F, Carioli G, et al. Global trends and predictions in hepatocellular carcinoma mortality. J Hepatol. 2017;67(2):302–309. | ||
Jemal A, Center MM, Desantis C, Ward EM. Global patterns of cancer incidence and mortality rates and trends. Cancer Epidemiol Biomarkers Prev. 2010;19(8):1893–1907. | ||
Siegel RL, Miller KD, Jemal A. Colorectal Cancer Mortality Rates in Adults Aged 20 to 54 Years in the United States, 1970–2014. JAMA. 2017;318(6):572–574. | ||
Bruix J, Reig M, Sherman M. Evidence-Based Diagnosis, Staging, and Treatment of Patients With Hepatocellular Carcinoma. Gastroenterology. 2016;150(4):835–853. | ||
Park JW, Chen M, Colombo M, et al. Global patterns of hepatocellular carcinoma management from diagnosis to death: the BRIDGE Study. Liver Int. 2015;35(9):2155–2166. | ||
Raza A, Sood GK. Hepatocellular carcinoma review: current treatment, and evidence-based medicine. World J Gastroenterol. 2014;20(15):4115–4127. | ||
Dutta R, Mahato RI. Recent advances in hepatocellular carcinoma therapy. Pharmacol Ther. 2017;173:106–117. | ||
Waidmann O, Trojan J. Novel drugs in clinical development for hepatocellular carcinoma. Expert Opin Investig Drugs. 2015;24(8):1075–1082. | ||
D’Souza AA, Devarajan PV. Asialoglycoprotein receptor mediated hepatocyte targeting – strategies and applications. J Control Release. 2015;203:126–139. | ||
Turato C, Balasso A, Carloni V, et al. New molecular targets for functionalized nanosized drug delivery systems in personalized therapy for hepatocellular carcinoma. J Control Release. 2017;268:184–197. | ||
Fu L, Sun C, Yan L. Galactose targeted pH-responsive copolymer conjugated with near infrared fluorescence probe for imaging of intelligent drug delivery. ACS Appl Mater Interfaces. 2015;7(3):2104–2115. | ||
Singh B, Jang Y, Maharjan S, et al. Combination therapy with doxorubicin-loaded galactosylated poly(ethyleneglycol)-lithocholic acid to suppress the tumor growth in an orthotopic mouse model of liver cancer. Biomaterials. 2017;116:130–144. | ||
Nelson KM, Dahlin JL, Bisson J, Graham J, Pauli GF, Walters MA. The Essential Medicinal Chemistry of Curcumin. J Med Chem. 2017;60(5):1620–1637. | ||
Kunnumakkara AB, Bordoloi D, Padmavathi G, et al. Curcumin, the golden nutraceutical: multitargeting for multiple chronic diseases. Br J Pharmacol. 2017;174(11):1325–1348. | ||
Vallianou NG, Evangelopoulos A, Schizas N, Kazazis C. Potential anticancer properties and mechanisms of action of curcumin. Anticancer Res. 2015;35(2):645–651. | ||
Bar-Sela G, Epelbaum R, Schaffer M. Curcumin as an anti-cancer agent: review of the gap between basic and clinical applications. Curr Med Chem. 2010;17(3):190–197. | ||
Yu Z, Yu M, Zhang Z, Hong G, Xiong Q. Bovine serum albumin nanoparticles as controlled release carrier for local drug delivery to the inner ear. Nanoscale Res Lett. 2014;9(1):343. | ||
Fabi A, Giannarelli D, Malaguti P, et al. Prospective study on nanoparticle albumin-bound paclitaxel in advanced breast cancer: clinical results and biological observations in taxane-pretreated patients. Drug Des Devel Ther. 2015;9:6177–6183. | ||
Nosrati H, Sefidi N, Sharafi A, Danafar H, Kheiri Manjili H, Manjili HK, Albumin BS. Bovine Serum Albumin (BSA) coated iron oxide magnetic nanoparticles as biocompatible carriers for curcumin-anticancer drug. Bioorg Chem. 2018;76:501–509. | ||
Fu C, Ding C, Sun X, Fu A. Curcumin nanocapsules stabilized by bovine serum albumin-capped gold nanoclusters (BSA-AuNCs) for drug delivery and theranosis. Mater Sci Eng C Mater Biol Appl. 2018;87:149–154. | ||
Fu L, Sun Y, Ding L, et al. Mechanism evaluation of the interactions between flavonoids and bovine serum albumin based on multi-spectroscopy, molecular docking and Q-TOF HR-MS analyses. Food Chem. 2016;203:150–157. | ||
Lee WH, Loo CY, Young PM, Traini D, Mason RS, Rohanizadeh R. Recent advances in curcumin nanoformulation for cancer therapy. Expert Opin Drug Deliv. 2014;11(8):1183–1201. | ||
Stowell CP, Lee VC. Neoglycoproteins: the preparation and application of synthetic glycoproteins. Adv Carbohydr Chem Biochem. 1980;37:225–281. | ||
Wa C, Cerny RL, Clarke WA, Hage DS. Characterization of glycation adducts on human serum albumin by matrix-assisted laser desorption/ionization time-of-flight mass spectrometry. Clin Chim Acta. 2007;385(1–2):48–60. | ||
Liu J, Jing H. Glycation of bovine serum albumin with monosaccharides inhibits heat-induced protein aggregation. RSC Adv. 2016;6(116):115183–115188. | ||
Weber K, Osborn M. The reliability of molecular weight determinations by dodecyl sulfate-polyacrylamide gel electrophoresis. J Biol Chem. 1969;244(16):4406–4412. | ||
Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227(5259):680–685. | ||
Langer K, Balthasar S, Vogel V, Dinauer N, von Briesen H, Schubert D. Optimization of the preparation process for human serum albumin (HSA) nanoparticles. Int J Pharm. 2003;257(1–2):169–180. | ||
Li J, Chen T, Deng F, et al. Synthesis, characterization, and in vitro evaluation of curcumin-loaded albumin nanoparticles surface-functionalized with glycyrrhetinic acid. Int J Nanomedicine. 2015;10:5475–5487. | ||
Weber C, Kreuter J, Langer K. Desolvation process and surface characteristics of HSA-nanoparticles. Int J Pharm. 2000;196(2):197–200. | ||
Hirayama K, Akashi S, Furuya M, Fukuhara K. Rapid confirmation and revision of the primary structure of bovine serum albumin by ESIMS and Frit-FAB LC/MS. Biochem Biophys Res Commun. 1990;173(2):639–646. | ||
Jing H, Kitts DD. Chemical characterization of different sugar-casein Maillard reaction products and protective effects on chemical-induced cytotoxicity of Caco-2 cells. Food Chem Toxicol. 2004;42(11):1833–1844. | ||
Kawakami S, Hashida M. Glycosylation-mediated targeting of carriers. J Control Release. 2014;190:542–555. | ||
Nishikawa M, Miyazaki C, Yamashita F, Takakura Y, Hashida M. Galactosylated proteins are recognized by the liver according to the surface density of galactose moieties. Am J Physiol. 1995;268(5 Pt 1):G849–G856. | ||
Li C, Zhang D, Guo H, et al. Preparation and characterization of galactosylated bovine serum albumin nanoparticles for liver-targeted delivery of oridonin. Int J Pharm. 2013;448(1):79–86. | ||
Li J, Yu S, Yao P, Jiang M. Lysozyme-dextran core-shell nanogels prepared via a green process. Langmuir. 2008;24(7):3486–3492. | ||
Bourassa P, Kanakis CD, Tarantilis P, Pollissiou MG, Tajmir-Riahi HA. Resveratrol, genistein, and curcumin bind bovine serum albumin. J Phys Chem B. 2010;114(9):3348–3354. | ||
Yu H, Nguyen MH, Cheow WS, Hadinoto K. A new bioavailability enhancement strategy of curcumin via self-assembly nano-complexation of curcumin and bovine serum albumin. Mater Sci Eng C Mater Biol Appl. 2017;75:25–33. | ||
Salehiabar M, Nosrati H, Javani E, et al. Production of biological nanoparticles from bovine serum albumin as controlled release carrier for curcumin delivery. Int J Biol Macromol. 2018;115:83–89. | ||
Chan JM, Zhang L, Yuet KP, et al. PLGA-lecithin-PEG core-shell nanoparticles for controlled drug delivery. Biomaterials. 2009;30(8):1627–1634. | ||
Ye Z, Wu WR, Qin YF, et al. An integrated therapeutic delivery system for enhanced treatment of hepatocellular carcinoma. Adv Funct Mater. 2018;28(18):1706600–1706613. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.