Back to Journals » Clinical Pharmacology: Advances and Applications » Volume 9
CRTH2 antagonists in asthma: current perspectives
Authors Singh D, Ravi A ![]() , Southworth T
, Southworth T
Received 6 October 2017
Accepted for publication 8 November 2017
Published 15 December 2017 Volume 2017:9 Pages 165—173
DOI https://doi.org/10.2147/CPAA.S119295
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Arthur E. Frankel
Dave Singh, Arjun Ravi, Thomas Southworth
Division of Infection, Immunity and Respiratory Medicine, The Medicines Evaluation Unit, Manchester Academic Health Science Centre, Manchester University NHS Foundation Trust, The University of Manchester, Manchester, UK
Abstract: Chemoattractant receptor-homologous molecule expressed on TH2 cells (CRTH2) binds to prostaglandin D2. CRTH2 is expressed on various cell types including eosinophils, mast cells, and basophils. CRTH2 and prostaglandin D2 are involved in allergic inflammation and eosinophil activation. Orally administered CRTH2 antagonists are in clinical development for the treatment of asthma. The biology and clinical trial data indicate that CRTH2 antagonists should be targeted toward eosinophilic asthma. This article reviews the clinical evidence for CRTH2 involvement in asthma pathophysiology and clinical trials of CRTH2 antagonists in asthma. CRTH2 antagonists could provide a practical alternative to biological treatments for patients with severe asthma. Future perspectives for this class of drug are considered, including the selection of the subgroup of patients most likely to show a meaningful treatment response.
Keywords: CRTH2, clinical trial, eosinophilic asthma, prostaglandin D2
Introduction
Asthma is characterized by variable airflow obstruction, bronchial hyperreactivity, and airway inflammation. The presence of allergy is common in asthma patients and can cause bronchoconstriction and promote chronic airway inflammation.1 However, many asthma patients have no evidence of allergy.2 The heterogeneous nature of asthma means that a “one size fits all” approach to pharmacotherapy is unlikely to be successful. The development of novel asthma treatments requires an individualized approach, where medicines are targeted toward subgroups of patients with distinct characteristics who are most likely to benefit.
A clinical phenotyping approach has been advocated in order to identify patient subgroups with clinical characteristics that are associated with a treatment response or prognosis.3 Endotyping is the identification of a patient subgroup defined by the presence of a biological mechanism.3 Recently, biological treatments for asthma have been developed that use biomarkers to identify patients with specific mechanisms (i.e., endotypes).4 The use of both clinical phenotype information and biomarkers to select patients for novel anti-inflammatory treatments aligns to the precision medicine strategy that takes an individualized approach to pharmacotherapy to optimize the benefit versus risk ratio.5
Our understanding of the complex nature of inflammation in asthma has evolved far beyond the simple dichotomy of allergic versus nonallergic asthma. The subset of lymphocytes called T-helper 2 cells were thought to drive allergic inflammation, leading to the term “TH2 inflammation” being associated with asthma.6 We recognized that many cytokines involved in allergic inflammation are also released from other cell types, including the recently identified innate lymphoid cells (ILCs).7 This has led to a change in terminology to the more general “T2” inflammation.8 Furthermore, T2 inflammation, such as eosinophilic inflammation, can exist in the absence of allergy.9 To add further complexity, non-T2 inflammation can also contribute to asthma pathophysiology, such as through the IL-17 cytokine family, which is associated with neutrophilic inflammation.10
The most commonly used pharmacological treatments for asthma are inhaled beta-agonists and inhaled corticosteroids (ICS), which provide bronchodilator and anti-inflammatory effects, respectively. Combination inhalers containing ICS plus a long-acting beta-agonist (ICS/LABA) have shown greater clinical efficacy than ICS alone, providing a treatment option that is widely used for many asthma patients.11 However, there is an unmet medical need, as many patients remain poorly controlled while taking ICS/LABA combinations. The use of long-acting muscarinic antagonists provides an additional bronchodilator option for these patients.12 Monoclonal antibodies targeting eosinophilic inflammation and T2 inflammation have been developed for asthma,13 but there is a need for additional novel anti-inflammatory treatments.
Chemoattractant receptor-homologous molecule expressed on TH2 cells (CRTH2) is a G-protein coupled receptor that binds to the ligand prostaglandin D2 (PGD2).14 There is evidence from in vitro studies, as well as animal and human investigations, that CRTH2 is involved in allergic and eosinophilic inflammation.15–17 A number of orally administered CRTH2 antagonists have been developed for the treatment of asthma in recent years.16,18 This article reviews the evidence for the involvement of the CRTH2 pathway in asthma and the results of clinical trials of CRTH2 antagonists in asthma patients. We also consider future perspectives for this class of drug, including considerations of which asthma subgroup is most likely to show a clinically meaningful treatment response, and whether biomarkers can be used to identify these patients.
PGD2–CRTH2 biology
Arachidonic acid metabolism by cyclooxygenase enzymes and, subsequently, prostaglandin synthases leads to the production of prostaglandins.19 PGH2 is converted to PGD2 by PGD2 synthase in various cell types including mast cells and leukocytes.20 Mast cells are an important source of PGD2 in tissues,21,22 with lower levels produced by TH2 lymphocytes,23 dendritic cells,24 and eosinophils.25 PGD2 undergoes rapid metabolism, with a short half-life of ~30 min in the circulation.26 The main products of PGD2 metabolism are Δ12PGJ2 and 9α11βPGF2, which also have agonist effects at PGD2 receptors.27,28
The biological effects of PGD2 are mediated by three G-protein-coupled receptors: CRTH2 (which is also called the D prostanoid receptor 2[DP2 receptor]), DP1, and T prostanoid (TP) receptors. The interaction of PGD2 and DP1 increases smooth muscle relaxation, vasodilation, vascular permeability, and epithelial CCL22 production, all of which may assist in the recruitment of leukocytes to the sites of inflammation, as well as in the inhibition of TH1 development and function.29 CRTH2 activation increases intracellular calcium levels and reduces intracellular cyclic adenosine monophosphate levels, and activates various signaling pathways including phospholipase C, phosphatidylinositol 3-kinase, and p38 mitogen-activated kinase.30 CRTH2 is expressed by TH2 cells,31 eosinophils, basophils,32 epithelial cells,33 and innate lymphoid type 2 cells (ILC2).34 Mast cells also express CRTH2, but only internally, and treatment with PGD2 does not induce CRTH2-dependent changes in Ca2+, suggesting the receptor has a different function to that in other inflammatory cells.35
The PGD2–CRTH2 interaction is strongly implicated in allergic inflammation (summarized in Figure 1). The TH2 cells orchestrate the allergic inflammatory response by releasing mediators such as IL-4, IL-5, and IL-13 that promote the recruitment and activation of inflammatory cells.36 Also, TH2 cells demonstrate increased CRTH2 expression compared with other lymphocyte subtypes.22 Furthermore, TH2 cells migrate toward PGD2 in vitro, and this chemotaxis is blocked by CRTH2 antagonism.22 PGD2–CRTH2 signaling is also involved in the recruitment and activation of eosinophils and basophils.22,37,38 There is recent evidence that ILC2 cells play a role in airway inflammation in asthma by secreting T2 cytokines. ILC2 cells also show CRTH2-dependent migration after exposure to PGD2.39 Animal studies have also shown a role for CRTH2 in allergic lung inflammation, with PGD2 treatment causing significantly increased eosinophilic lung inflammation in ovalbumin-challenged mice.40
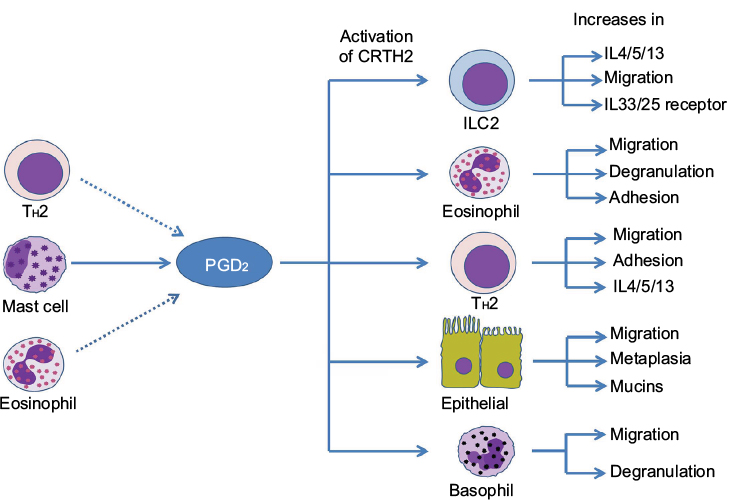 | Figure 1 Effects of PGD2–CRTH2 signaling in asthma. Notes: PGD2 is predominantly released from mast cells following allergen stimulation; other cells such as eosinophils and TH2 cells may also contribute to PGD2 levels. Interaction of PGD2 with CRTH2 stimulates the recruitment of T2-associated cells to the airways and release of associated cytokines, as well as eosinophil and basophil degranulation and epithelial metaplasia. Abbreviations: CRTH2, chemoattractant receptor-homologous molecule on T helper type 2 cells; ILC2, innate lymphoid type 2 cells; IL, interleukin; PGD2, prostaglandin D2; TH2, T-helper type 2 cell. |
Evidence for increased PGD2–CRTH2 activity in asthma
It is not clear if PGD2 levels in bronchoalveolar lavage (BAL) are increased in patients with mild asthma compared to healthy subjects as reports are contradictory.41–43 However, there is evidence that PGD2 levels increase in more severe disease, as BAL PGD2 levels are increased in severe asthma patients compared with mild and moderate asthma patients.15,44
Gene and protein expressions of hematopoietic prostaglandin synthase (HPGDS; responsible for PGD2 formation) are elevated in the bronchial epithelium of patients with moderate and severe asthma, compared with healthy subjects.15 Furthermore, the source of HPGDS appeared to be mast cells embedded in the epithelium, as HPGDS levels correlated with mast cell tryptase. Increased PGD2 levels are observed in severe asthma patients taking high-dose ICS despite the known effect of ICS in terms of reducing the number of PGD2-producing mast cells in bronchial tissue.44
Levels of PGD2 are rapidly released following allergen challenge, with a 150-fold increase in BAL levels within 9 min of exposure.42,45 Levels probably return to normal within a short period of time as no increase in PGD2 was seen in sputum samples collected 24 h post allergen exposure.46 Ex vivo studies using human lung tissue have demonstrated that allergen induction of PGD2 is dependent on mast cell activation.47
Mutalithas et al investigated the expression of CRTH2 by T lymphocytes in the blood and BAL fluid of asthma patients and healthy controls using flow cytometry.48 The proportion of T-cells expressing CRTH2 was low in both blood and BAL. The numbers of CRTH2+ blood lymphocytes were similar in healthy and asthma subjects, but there was a significant increase in the number of BAL CRTH2+ T-cells in patients with asthma (2.3% versus 0.3%, p<0.05). In both blood and BAL, TH2 cells producing either IL-4 or IL-13 showed greater expression of CRTH2 than IFNγ producing TH1 cells. Based on the results of this relatively small study (n=11 asthma patients and n=7 healthy subjects), the authors proposed that the low expression levels indicated a limited role of CRTH2 in the control of lymphocyte activity in asthma and that CRTH2 antagonism may not diminish T-cell recruitment to the asthma lung.
Fajt et al reported a larger bronchoscopy study involving mild asthma (steroid naïve; n=11), moderate asthma (using ICS; n=22), and severe asthma patients (n=46), along with a healthy control group (n=33).15 In this study, the highest CRTH2 expression levels in BAL cells were observed in severe asthma patients, measured through gene expression and immunocytochemical studies. Interestingly, there were also associations between PGD2, HPGDS, and CRTH2 expression levels and worse clinical outcomes, including exacerbations and asthma control. Stinson et al also reported increased numbers of CRTH2+ inflammatory cells in severe asthma patients compared with healthy subjects, this time in the submucosa of bronchial biopsies.33
Bronchial epithelial cells are known to express CRTH2, and in vitro studies using primary human cells have shown that activation of CRTH2 induces epithelial cell migration, mucin production, and metaplasia.33 Stinson et al showed that the number of CRTH2+ epithelial cells decreased with asthma severity and that this reduction in CRTH2 expression was specifically related to areas of the epithelium that were undergoing metaplasia, which was increased in biopsies from more severe patients.33 It is yet unclear if the increase in metaplasia in severe asthma is induced by PGD2 or if such an activation results in a reduction in CRTH2 expression. PGD2 activation in other cell types, such as TH2 cells, is known to downregulate CRTH2 expression.49
Overall, these studies in asthma have shown a pattern of increased PGD2 and CRTH2 expressions in more severe asthma. CRTH2 expression has also been shown to be elevated in other allergic diseases.50 Blood eosinophils from atopic kerato-conjunctivitis patients express higher levels of CRTH2 than those from healthy subjects, and these cells showed enhanced migration toward PGD2 and its stable metabolite 13,14-dihydro-15-keto-PGD2, which could explain the increased number of eosinophils in atopic tissue.51
The effects of CRTH2 antagonists in asthma
Small-molecule antagonists of the CRTH2 receptor designed for oral administration have been synthesized, with some showing sufficient potency in preclinical studies to warrant evaluation in asthma clinical trials.52 Clinical trials of CRTH2 antagonists on asthma clinical end points are summarized in Table 1. The first CRTH2 antagonist to be evaluated in patients with asthma was OC000459. A 4-week, placebo-controlled parallel group study was conducted in 132 patients who were not using ICS.16 There was an improvement in FEV1 with OC000459 (200 mg administered twice daily) compared to placebo in the “full analysis population” (7.1% versus 4.3%, respectively). This difference was not statistically significant, but was significant in the “per protocol population” which included only patients who completed the study with valid spirometry measurements (9.8% [210 mL] versus 1.8% [30 mL] improvement; p=0.037). Significant improvements in quality of life and nighttime symptoms were observed with OC000459 compared with placebo in populations in both analyses. Induced sputum was conducted in a subset (n=27), and it showed a significant reduction in eosinophil counts with active treatment, but not placebo. This study provided the first clinical evidence that CRTH2 antagonism could benefit patients with asthma, targeting eosinophilic inflammation.
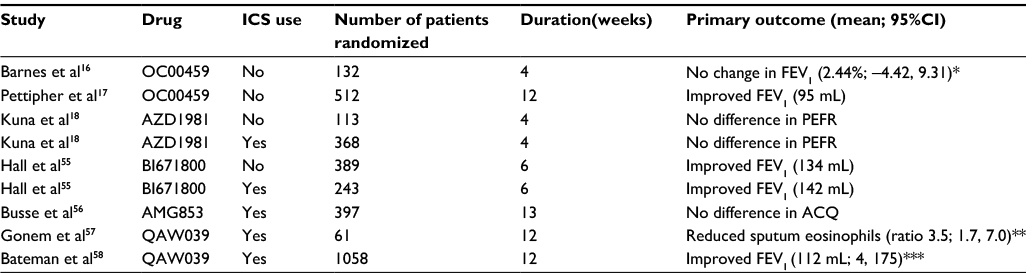 | Table 1 Clinical trials of CRTH2 antagonists in asthma Notes: Only placebo-controlled trials of ≥4 weeks duration have been included. Primary outcome shows the difference versus placebo; only statistically significant changes are shown. 95% CI are shown if presented in the publication.*A secondary analysis in the per protocol population was significant; mean difference 7.66% (0.49, 14.82). **Ratio of geometric mean sputum eosinophil changes for fevipiprant versus placebo. ***Model-averaged FEV1 for different fevipiprant doses. Abbreviations: ACQ, asthma control questionnaire; CRTH2, chemoattractant receptor-homologous molecule on T helper type 2 cells; FEV1, forced expiratory volume in one second; ICS, inhaled corticosteroids; PEFR, peak expiratory flow rate. |
OC000459 was subsequently assessed at lower doses (25 mg once daily, 200 mg once daily, and 100 mg twice daily) in a placebo-controlled study over 12 weeks in asthma patients not using ICS (n=512 randomized).17 There was a similar increase in FEV1 in each OC000459 treatment group, with the pooled active treatment dose groups showing a 95 mL difference compared to placebo at week 12 (p=0.024). There were also improvements in the asthma control questionnaire (ACQ) and asthma quality of life questionnaire (AQLQ) scores with OC000459 compared with placebo. The effect of OC000459 on lung function appeared to be greater in patients with atopy and those with higher blood eosinophil counts (>250 cells/µL).
OC000459 has been studied using the inhaled allergen challenge model.53 This drug attenuated the late asthmatic response, with a less pronounced fall in FEV1 after inhaled allergen, compared with placebo in steroid-naïve patients with asthma. OC000459 also reduced the magnitude of sputum eosinophilia caused by allergen exposure. There was a significant period effect in this placebo-controlled crossover design, with no therapeutic effect on FEV1 observed in patients who received active treatment first. At first glance, one would assume that this was a carryover effect in a crossover study. However, the screening allergen challenge data showed that this group actually had no therapeutic response with OC000459 compared to the screening challenge, suggesting that the drug had no effect in this group rather than a carryover effect. These patients with no drug response had a significantly lower FEV1 at screening, suggesting a variation between individuals in drug response, which is dependent on clinical characteristics. Nevertheless, the CRTH2 antagonist setipiprant also inhibited the allergen-induced late asthmatic response, providing further confirmation of the effects of this drug class on allergic inflammation.54
The effects of the CRTH2 antagonist BI671800 have been investigated in asthma patients not using ICS and in a population of asthma patients taking ICS.55 BI671800 (50, 200, and 400 mg), inhaled fluticasone propionate (220 µg), and placebo (all administered twice daily) were studied over 6 weeks using a parallel group design in asthma patients not using ICS (Trial 1), while a similar design was used in asthma patients taking ICS to evaluate BI671800 400 mg twice daily, montelukast 10 mg once daily, and placebo (Trial 2). In Trial 1, BI671800 caused FEV1 improvements of ~3%–4% compared with placebo (3.98% or 134 mL, p=0.0078 at 400 mg), while a greater improvement was observed with fluticasone propionate (8.62% or 293 mL, p<0.0001). In Trial 2, BI671800 significantly improved FEV1 compared with placebo (3.87%, 142 mL, p=0.005), while the change with montelukast was not statistically significant (2.37%, 80 mL). In both studies, a greater effect of BI671800 was observed in patients with higher blood eosinophil counts (using a threshold of 350 cells/mm3).
The CRTH2 antagonist AZD1981 was also investigated in one trial involving asthma patients where previous ICS treatment was withdrawn at randomization (Study 1), and in another trial in asthma patients where ICS treatment was continued (Study 2).18 Study 1 was a parallel group study comparing AZD1981 1000 mg twice daily with placebo, with low-dose ICS being withdrawn on the day of randomization. Study 2 was also a placebo-controlled, parallel group study, but patients continued medium- to high-dose ICS, with any long-acting beta-agonist use stopped. The active treatments were AZD1981 50, 400, or 1000 mg twice daily. The treatment duration in both studies was 4 weeks, and peak expiratory flow rate (PEFR) was the primary end point. In both studies, there were no differences in PEFR between active treatments and placebo. In Study 2, there was evidence of greater lung function improvements at 4 weeks in patients who were atopic, with significant improvements of ~170–180 mL compared with placebo at the two higher doses.
AMG853 is a dual antagonist of DP1 and CRTH2. AMG853 had no effect on lung function, ACQ, AQLQ, or symptoms compared to placebo after 12 weeks treatment in asthma patients using ICS. No subgroups could be identified that showed a significant response.56
Fevipiprant (QAW039) has been studied in patients with moderate to severe asthma and sputum eosinophil counts >2% (n=61).57 This patients selection logically follows from the previous studies already described where the effects of CRTH2 antagonists appeared to be greater in patients with higher blood eosinophil counts. This was a 12-week, placebo-controlled, parallel group study. There was a significant decrease in sputum eosinophil percentage (p=0.0014) with fevipiprant (geometric means at baseline and 12 weeks were 5.4% and 1.1%, respectively) compared with placebo (geometric means at baseline and 12 weeks were 4.6% and 3.9%, respectively). There was also a significant reduction in bronchial submucosal eosinophil counts. This study was not sufficiently powered to properly assess lung function and symptoms, but it confirmed that the principal mechanism of action of CRTH2 antagonists is through blocking eosinophil activation and chemotaxis.
A recent publication has described a dose-ranging, parallel-group Phase II study of fevipiprant in patients with uncontrolled, allergic asthma (n=1058 randomized).58 Asthma patients taking ICS with evidence of allergy (by skin prick or Immunoglobulin E test) were recruited and treated with inhaled budesonide 200 µg twice daily during the run-in and treatment periods. Patients with an ACQ score ≥1.5 at the end of the run-in period, indicating uncontrolled symptoms, were randomized. There were 13 fevipiprant-treatment arms, a placebo arm, and a montelukast arm. Fevipiprant doses from 1 to 450 mg were used, with once daily and twice daily regimes, and studied for 12 weeks. The primary end point was trough FEV1 at 12 weeks; fevipiprant caused a model-averaged FEV1 change of 112 mL compared with placebo (p=0.0035), while for montelukast, 134 mL was observed (p=0.0033). The total fevipiprant daily dose of 150 mg caused the greatest FEV1 change, with 179 and 164 mL observed after 75 mg twice daily (p=0.0059) and 150 mg once daily doses (p=0.0075), respectively. No changes in symptoms for fevipiprant compared to placebo were observed, but there was a numerical reduction in exacerbations with both fevipiprant and montelukast treatments compared with placebo. This study provides information on the optimal doses to be used in Phase III studies of fevipiprant.
In general, CRTH2 antagonists have been well tolerated in these clinical trials. The adverse events profile in these studies has been reported to be similar to placebo.
The effects of CRTH2 antagonists on nasal allergic inflammation
CRTH2 antagonism has been demonstrated to attenuate nasal allergic inflammation. A two-way, placebo-controlled cross-over study in patients with allergic rhinitis (n=36) using OC000459 (200 mg twice daily) administered for 8 days exposed subjects to grass pollen for 6 h in an environmental challenge chamber on days 2 and 8 of each treatment period.59 The primary end point was the total nasal symptom score (TNSS) averaged over 6 h of allergen challenge on day 8; although there was a significant reduction in TNSS with OC000459 (p=0.035), there was also evidence of a significant carryover effect between treatment periods.
Krug et al performed a randomized, double-blind, placebo-controlled, two-way (partial) cross-over study in patients with allergic rhinitis.60 Patients (n=146) were randomized to receive placebo and one of the following active treatments; BI671800 twice daily (50, 200, or 400 mg bid), montelukast 10 mg once daily, or fluticasone propionate 200 μg nasal spray once daily. Patients received treatment for 2 weeks, with a grass pollen challenge in an environmental challenge chamber performed at the end of each treatment period. The primary efficacy end point was the TNSS area under the curve from 0 to 6 hours (AUC)0–6h during the allergen challenge. Additional end points included measurement of nasal secretion weight, nasal flow (rhinomanometry), nasal secretion inflammatory mediators, and nasal secretion inflammatory cells. BI671800 200 mg caused a statistically significant difference in adjusted mean TNSS AUC0–6h values versus placebo (−17%; p=0.0026). BI671800 50 and 400 mg caused numerically lower TNSS AUC0–6h values compared with placebo, but these did not reach statistical significance. Statistically significant differences were observed in the montelukast and the fluticasone propionate (−15%; p=0.0115 and −33%; p<0.0001, respectively). BI671800 significantly reduced nasal eosinophil counts at all the doses studied. In general, fluticasone propionate had a greater effect than BI671800 on secondary end point measurements.
Nasal challenge studies are technically demanding, and the carryover effect observed in the study with OC000459 highlights the practical issues that may be encountered. Nevertheless, these nasal challenge studies show that CRTH2 antagonism inhibits allergic nasal inflammation, although the effect is lower in magnitude compared to ICS.
Future perspectives
Clinical studies of CRTH2 antagonists have shown a degree of efficacy in patients with mild to moderate asthma.16,17,53,55 However, the unmet medical need in these patients is low, with ICS or ICS/LABA combinations providing effective treatment. Furthermore, CRTH2 antagonists are unlikely to replace ICS treatment, as the clinical benefits of the former appear to be lower than the latter in mild to moderate asthma.55 There is a better way of using CRTH2 antagonists as an additional anti-inflammatory treatment in patients already using ICS, and early phase clinical trial evidence has demonstrated efficacy of this approach.55
There is an upregulation of the PGD2–CRTH2 signaling axis in severe asthma compared with mild to moderate asthma patients and healthy controls.15 Also, there is an unmet medical need for additional anti-inflammatory treatments in patients with severe asthma, and the future development of CRTH2 antagonists should be focused on this patient subset. Fevipiprant caused a reduction in airway eosinophil numbers in patients with severe asthma,57 while other CRTH2 antagonists have shown increased clinical efficacy in patients with higher blood eosinophil counts.55,60 Overall, these findings suggest that CRTH2 antagonists would be better targeted toward severe asthma patients with eosinophilic inflammation. This strategy is supported by the well-described inhibitory effect of CRTH2 antagonists on eosinophil activation and chemotaxis.52,61,62
Blood eosinophils have been used as a biomarker of eosinophilic airway inflammation, although the correlation between these measurements is often modest, leading to false-positive and false-negative results.63 Nevertheless, blood eosinophil counts remain a practical method for analyzing eosinophils in clinical practice, in contrast to sputum analysis or bronchoscopic sampling. The cut-off point that should be used to identify eosinophilic asthma is controversial, with 150, 300, and 400 cells/µL being used in various clinical trials of novel drugs.64–66 If one assumes that the effects of CRTH2 antagonists follow a (blood eosinophil) concentration–clinical response curve, then the drug effect sizes will show incremental increases at higher blood eosinophil concentrations, rather than being an “all or nothing” phenomenon. Exacerbations are the key clinical end points to be assessed in severe asthma long-term Phase III studies. The effect size of CRTH2 antagonists on exacerbations is unknown, but is likely to be greater at higher blood eosinophil concentrations.
Biological treatments targeting eosinophilic inflammation have been successfully developed and are now being used in clinical practice.66–69 There are also other biological treatments targeting T2 cytokines, such as IL-4 and IL-13, that are in the late-stage clinical development.70,71 The optimum patient selection for these different classes of biological treatments remains under debate, as individuals with T2 inflammation could be potential candidates for anti-eosinophil treatments (such as mepolizumab or benralizumab) or anti-IL4/IL-13 treatment with dupilumab. There are no head-to-head studies to compare such treatments, so clinicians may need to make decisions based on criteria such as higher blood eosinophil counts, which could favor anti-IL5 treatment, and the presence of atopic dermatitis, which may favor treatment with dupilumab. Although these biological treatments have shown good efficacy in severe asthma, there are practical issues to be addressed concerning the regular administration of these systemically administered drugs at intervals such as fortnightly and the high costs of biological treatments. Furthermore, there are some concerns with the long-term side effects of such treatments.72 Orally administered CRTH2 antagonists may offer an alternative to these biologics based on the good safety profile observed in clinical trials performed to date and the practically easier method of administration. The possibility for CRTH2 antagonists to carve a space in the marketplace based on practicality and safety is dependent on the clinical benefits on exacerbations to be proved in Phase III studies.
Conclusion
CRTH2 antagonists provide a potentially convenient oral treatment for severe asthma. The results of long-term studies in severe asthma focusing on exacerbations will dictate whether these drugs have sufficient efficacy to be approved by drug regulatory authorities for the treatment of asthma. The biology and clinical data indicate that CRTH2 antagonists should be targeted toward eosinophilic asthma. CRTH2 antagonists could provide a practical alternative to biological treatments for this subgroup of patients.
Disclosure
DS has received sponsorship to attend international meetings, honoraria for lecturing or attending advisory boards, and research grants from various pharmaceutical companies including Almirall, AstraZeneca, Boehringer Ingelheim, Chiesi, Genentech, GlaxoSmithKline, Glenmark, Johnson and Johnson, Merck, NAPP, Novartis, Pfizer, Skyepharma, Takeda, Teva, Theravance and Verona. AR and TS report no conflicts of interest in this work.
References
Schatz M, Rosenwasser L. The allergic asthma phenotype. J Allergy Clin Immunol Pract. 2014;2(6):645–648; quiz 649. | ||
Peters SP. Asthma phenotypes: nonallergic (intrinsic) asthma. J Allergy Clin Immunol Pract. 2014;2(6):650–652. | ||
Lotvall J, Akdis CA, Bacharier LB, et al. Asthma endotypes: a new approach to classification of disease entities within the asthma syndrome. J Allergy Clin Immunol. 2011;127(2):355–360. | ||
Medrek SK, Parulekar AD, Hanania NA. Predictive biomarkers for asthma therapy. Curr Allergy Asthma Rep. 2017;17(10):69. | ||
Agusti A, Bel E, Thomas M, et al. Treatable traits: toward precision medicine of chronic airway diseases. Eur Respir J. 2016;47(2):410–419. | ||
Woodruff PG, Modrek B, Choy DF, et al. T-helper type 2-driven inflammation defines major subphenotypes of asthma. Am J Respir Crit Care Med. 2009;180(5):388–395. | ||
Morita H, Moro K, Koyasu S. Innate lymphoid cells in allergic and nonallergic inflammation. J Allergy ClinImmunol. 2016;138(5):1253–1264. | ||
Fahy JV. Type 2 inflammation in asthma--present in most, absent in many. Nat Rev Immunol. 2015;15(1):57–65. | ||
Brusselle GG, Maes T, Bracke KR. Eosinophils in the spotlight: eosinophilic airway inflammation in nonallergic asthma. Nat Med. 2013;19(8):977–979. | ||
Cosmi L, Liotta F, Annunziato F. Th17 regulating lower airway disease. Curr Opin Allergy ClinImmunol. 2016;16(1):1–6. | ||
Aalbers R, Vogelmeier C, Kuna P. Achieving asthma control with ICS/LABA: a review of strategies for asthma management and prevention. RespirMed. 2016;111:1–7. | ||
Aalbers R, Park HS. Positioning of long-acting muscarinic antagonists in the management of asthma. Allergy Asthma Immunol Res. 2017;9(5):386–393. | ||
Walsh GM. Biologics targeting IL-5, IL-4 or IL-13 for the treatment of asthma - an update. Expert Rev Clin Immunol. 2017;13(2):143–149. | ||
Hirai H, Tanaka K, Yoshie O, et al. Prostaglandin D2 selectively induces chemotaxis in T helper type 2 cells, eosinophils, and basophils via seven-transmembrane receptor CRTH2. J Exp Med. 2001;193(2):255–261. | ||
Fajt ML, Gelhaus SL, Freeman B, et al. Prostaglandin D(2) pathway upregulation: relation to asthma severity, control, and TH2 inflammation. J Allergy Clin Immunol. 2013;131(6):1504–1512. | ||
Barnes N, Pavord I, Chuchalin A, et al. A randomized, double-blind, placebo-controlled study of the CRTH2 antagonist OC000459 in moderate persistent asthma. Clin Exp Allergy. 2012;42(1):38–48. | ||
Pettipher R, Hunter MG, Perkins CM, et al. Heightened response of eosinophilic asthmatic patients to the CRTH2 antagonist OC000459. Allergy. 2014;69(9):1223–1232. | ||
Kuna P, Bjermer L, Tornling G. Two Phase II randomized trials on the CRTh2 antagonist AZD1981 in adults with asthma. Drug Des Dev Ther. 2016;10:2759–2770. | ||
Murata T, Maehara T. Discovery of anti-inflammatory role of prostaglandin D2. J Vet Med Sci. 2016;78(11):1643–1647. | ||
Hamberg M, Fredholm BB. Isomerization of prostaglandin H2 into prostaglandin D2 in the presence of serum albumin. Biochim Biophys Acta. 1976;431(1):189–193. | ||
Beynon RC, Robinson C, Church MK. Differential release of histamine and eicosanoids from human skin mast cells activated by IgE dependent and non-immunogenic stimuli. Br J Clin Pharmacol. 1989;97(3):898–904. | ||
Hirai H, Tanaka K, Yoshie O, et al. Prostaglandin D2 selectively induces chemotaxis in T helper type 2 cells, eosinophils and basophils via seven-transmembrane receptor CRTH2. J Exp Med. 2001;193(2):255–261. | ||
Tanaka K, Ogawa K, Sugamura K, Nakamura M, Takano S, Nagata K. Cutting edge: differential production of prostaglandin D2 by human helper T CELL SUBSETS. J Immunol. 2000;164(5):2277–2280. | ||
Urade Y, Ujihara M, Horiguchi Y, Ikai K, Hayaishi O. The major source of endogenous prostaglandin D2 production is likely antigen-presenting cells. Localization of glutathione-requiring prostaglandin synthetase in histiocytes, dendritic and Kupffer cells in various tissues. J Immunol. 1989;143(9):2982–2989. | ||
Luna-Gomes T, Magalhães KG, Mesquita-Santos FP, et al. Eosinophils as a novel cell source of prostaglandin D2: autocrine role in allergic inflammation. J Immunol. 2011;187(12):6518–6526. | ||
Schuligoi R, Schmidt R, Geisslinger G, Kollroser M, Peskar BA, Heinemann A. PGD2 metabolism in plasma: kinetics and relationship with bioactivity on DP1 and CRTH2 receptors. Biochem Pharmacol. 2007;74(1):107–117. | ||
Heinemann A, Schuligoi R, Sabroe I, Hartnell A, Peskar BA. Δ12-prostaglandin J2 a plasma metabolite of prostaglandin D2, causes eosinophil mobilization and primes eosinophils for chemotaxis. J Immunol. 2003;170(9):4752–4758. | ||
Monneret G, Li H, Vasilescu J, Rokach J, Powell WS. 15-Deoxy-Δ12, 14-prostaglandin D2 and J2 are potent activators of human eosinophils. J Immunol. 2002;168(7):3563–3569. | ||
Garcia-Solaesa V, Sanz-Lozano C, Padron-Morales J, et al. The prostaglandin D2 receptor (PTGDR) gene in asthma and allergic diseases. Allergol Immunopathol. 2014;42(1):64–68. | ||
Stubbs VE, Schratl P, Hartnell A, et al. Indomethacin causes prostaglandin D(2)-like and eotaxin-like selective responses in eosinophils and basophils. J Biol Chem. 2002;277(29):26012–26020. | ||
Nagata K, Tanaka K, Ogawa K, et al. Selective expression of a novel surface molecule by human Th2 cells in vivo. J Immunol. 1999;162(3):1278–1286. | ||
Nagata K, Hirai H, Tanaka K, et al. CRTH2, an orphan receptor of T-helper-2-cells, is expressed on basophils and eosinophils and responds to mast cell-derived factor(s). FEBS Lett. 1999;459(2):195–199. | ||
Stinson SE, Amrani Y, Brightling CE. D prostanoid receptor 2 (chemoattractant receptor-homologous molecule expressed on TH2 cells) protein expression in asthmatic patients and its effects on bronchial epithelial cells. J Allergy ClinImmunol. 2015;135(2):395–406. | ||
Xue L, Salimi M, Panse I, et al. Prostaglandin D2 activates group 2 innate lymphoid cells through chemoattractant receptor-homologous molecule expressed on TH2 cells. J Allergy ClinImmunol. 2014;133(4):1184–1194. | ||
Moon TC, Campos-Alberto E, Yoshimura T, et al. Expression of DP2 (CRTh2), a prostaglandin D(2) receptor, in human mast cells. PLoS One. 2014;9(9):e108595. | ||
Honda K, Arima M, Cheng G, et al. Prostaglandin D2 reinforces Th2 type inflammatory responses of airways to low-dose antigen through bronchial expression of macrophage derived chemokine. J Exp Med. 2003;198(4):533–543. | ||
Böhm E, Sturm GJ, Weiglhofer I, et al. 11-Dehydro-thromboxane B2, a stable thromboxane metabolite, is a full agonist of chemoattractant receptor-homologous molecule expressed on TH2 cells (CRTH2) in human eosinophils and basophils. J Biol Chem. 2004;279(9):7663–7670. | ||
Nagata K, Hirai H, Tanaka K, et al. CRTH2, an orphan receptor of T-helper-2-cells, is expressed on basophils and eosinophils and responds to mast cell-derived factors. FEBS Lett. 1999;459(2):195–199. | ||
Wojno ED, Monticelli LA, Tran SV, et al. The prostaglandin D2 receptor CRTH2 regulates accumulation of group 2 innate lymphoid cells in the inflamed lung. Mucosal Immunol. 2015;8(6):1313–1323. | ||
Spik I, Brénuchon C, Angéli V, et al. Activation of the prostaglandin D2 receptor DP2/CRTH2 increases allergic inflammation in mouse. J Immunol. 2005;174(6):3703–3708. | ||
Crea AE, Nakhosteen JA, Lee TH. Mediator concentrations in bronchoalveolar lavage fluid of patients with mild asymptomatic bronchial asthma. Eur Respir J. 1992;5(2):190–195. | ||
Wenzel SE, Westcott JY, Smith HR, Larsen GL. Spectrum of prostanoid release after bronchoalveolar allergen challenge in atopic asthmatics and in control groups. An alteration in the ratio of bronchoconstrictive to bronchoprotective mediators. Am Rev Respir Dis. 1989;139(2):450–457. | ||
Liu MC, Bleecker ER, Lichtenstein LM, et al. Evidence for elevated levels of histamine, prostaglandin D2, and other bronchoconstricting prostaglandins in the airways of subjects with mild asthma. Am Rev Respir Dis. 1990;142(1):126–132. | ||
Balzar S, Fajt ML, Comhair SA, et al. Mast cell phenotype, location, and activation in severe asthma. Data from the Severe Asthma Research Program. Am J Respir Crit Care Med. 2011;183(3):299–309. | ||
Murray JJ, Tonnel AB, Brash AR, et al. Release of prostaglandin D2 into human airways during acute antigen challenge. N Engl J Med. 1986;315(13):800–804. | ||
Macfarlane AJ, Dworski R, Sheller JR, Pavord ID, Kay AB, Barnes NC. Sputum cysteinyl leukotrienes increase 24 hours after allergen inhalation in atopic asthmatics. Am J Respir Crit Care Med. 2000;161(5):1553–1558. | ||
Schulman ES, Newball HH, Demers LM, Fitzpatrick FA, Adkinson NF Jr. Anaphylactic release of thromboxane A2, prostaglandin D2, and prostacyclin from human lung parenchyma. AmRevRespirDis. 1981;124(4):402–406. | ||
Mutalithas K, Guillen C, Day C, Brightling CE, Pavord ID, Wardlaw AJ. CRTH2 expression on T cells in asthma. Clin Exp Immunol. 2010;161(1):34–40. | ||
Hirai H, Tanaka K, Takano S, Ichimasa M, Nakamura M, Nagata K. Cutting edge: agonistic effect of indomethacin on a prostaglandin D2 receptor, CRTH2. JImmunol. 2002;168(3):981–985. | ||
Nantel F, Fong C, Lamontagne S, et al. Expression of prostaglandin D synthase and the prostaglandin D2 receptors DP and CRTH2 in human nasal mucosa. Prostaglandins Other Lipid Mediat. 2004;73(1–2):87–101. | ||
Fujishima H, Fukagawa K, Okada N, et al. Chemotactic responses of peripheral blood eosinophils to prostaglandin D2 in atopic kerato conjunctivitis. Ann Allergy Asthma Immunol. 2013;111(2):126–131.e124. | ||
Pettipher R, Vinall SL, Xue L, et al. Pharmacologic profile of OC000459, a potent, selective, and orally active D prostanoid receptor 2 antagonist that inhibits mast cell-dependent activation of T helper 2 lymphocytes and eosinophils. J Pharmacol Exp Ther. 2012;340(2):473–482. | ||
Singh D, Cadden P, Hunter M, et al. Inhibition of the asthmatic allergen challenge response by the CRTH2 antagonist OC000459. Eur Respir J. 2013;41(1):46–52. | ||
Diamant Z, Sidharta PN, Singh D, et al. Setipiprant, a selective CRTH2 antagonist, reduces allergen-induced airway responses in allergic asthmatics. Clin Exp Allergy. 2014;44(8):1044–1052. | ||
Hall IP, Fowler AV, Gupta A, et al. Efficacy of BI 671800, an oral CRTH2 antagonist, in poorly controlled asthma as sole controller and in the presence of inhaled corticosteroid treatment. Pulm Pharmacol Ther. 2015;32:37–44. | ||
Busse WW, Wenzel SE, Meltzer EO, et al. Safety and efficacy of the prostaglandin D2 receptor antagonist AMG 853 in asthmatic patients. J Allergy Clin Immunol. 2013;131(2):339–345. | ||
Gonem S, Berair R, Singapuri A, et al. Fevipiprant, a prostaglandin D2 receptor 2 antagonist, in patients with persistent eosinophilic asthma: a single-centre, randomised, double-blind, parallel-group, placebo-controlled trial. Lancet Respir Med. 2016;4(9):699–707. | ||
Bateman ED, Guerreros AG, Brockhaus F, et al. Fevipiprant, an oral prostaglandin DP2 receptor (CRTh2) antagonist, in allergic asthma uncontrolled on low-dose inhaled corticosteroids. Eur Respir J.2017;50(2):1700670. | ||
Horak F, Zieglmayer P, Zieglmayer R, et al. The CRTH2 antagonist OC000459 reduces nasal and ocular symptoms in allergic subjects exposed to grass pollen, a randomised, placebo-controlled, double-blind trial. Allergy. 2012;67(12):1572–1579. | ||
Krug N, Gupta A, Badorrek P, et al. Efficacy of the oral chemoattractant receptor homologous molecule on TH2 cells antagonist BI 671800 in patients with seasonal allergic rhinitis. J Allergy Clin Immunol. 2014;133(2):414–419. | ||
Schmidt JA, Bell FM, Akam E, et al. Biochemical and pharmacological characterization of AZD1981, an orally available selective DP2 antagonist in clinical development for asthma. Br J Pharmacol. 2013;168(7):1626–1638. | ||
Royer JF, Schratl P, Lorenz S, et al. A novel antagonist of CRTH2 blocks eosinophil release from bone marrow, chemotaxis and respiratory burst. Allergy. 2007;62(12):1401–1409. | ||
Silkoff PE, Laviolette M, Singh D, et al. Identification of airway mucosal type 2 inflammation by using clinical biomarkers in asthmatic patients. J Allergy Clin Immunol. 2017;140(3):710–719. | ||
Casale TB, Chipps BE, Rosen K, et al. Response to omalizumab using patient enrichment criteria from trials of novel biologics in asthma. Allergy.Epub2017 Aug 31. | ||
Goldman M, Hirsch I, Zangrilli JG, Newbold P, Xu X. The association between blood eosinophil count and benralizumab efficacy for patients with severe, uncontrolled asthma: subanalyses of the Phase III SIROCCO and CALIMA studies. Curr Med Res Opin. 2017;33(9):1605–1613. | ||
Castro M, Zangrilli J, Wechsler ME, et al. Reslizumab for inadequately controlled asthma with elevated blood eosinophil counts: results from two multicentre, parallel, double-blind, randomised, placebo-controlled, phase 3 trials. Lancet RespirMed. 2015;3(5):355–366. | ||
Bermejo I, Stevenson M, Cooper K, et al. Mepolizumab for treating severe eosinophilic asthma: an evidence review group perspective of a NICE single technology appraisal. Pharmacoeconomics.Epub2017 Sep 20. | ||
Albers FC, Price RG, Smith SG, Yancey SW. Mepolizumab efficacy in patients with severe eosinophilic asthma receiving different controller therapies. J Allergy ClinImmunol. 2017;140(5):1464–1466.e4. | ||
Maspero J. Reslizumab in the treatment of inadequately controlled asthma in adults and adolescents with elevated blood eosinophils: clinical trial evidence and future prospects. Ther Adv Respir Dis. 2017;11(8):311–325. | ||
Bleecker ER, FitzGerald JM, Chanez P, et al. Efficacy and safety of benralizumab for patients with severe asthma uncontrolled with high-dosage inhaled corticosteroids and long-acting beta2-agonists (SIROCCO): a randomised, multicentre, placebo-controlled phase 3 trial. Lancet. 2016;388(10056):2115–2127. | ||
Wenzel S, Castro M, Corren J, et al. Dupilumab efficacy and safety in adults with uncontrolled persistent asthma despite use of medium-to-high-dose inhaled corticosteroids plus a long-acting beta2 agonist: a randomised double-blind placebo-controlled pivotal phase 2b dose-ranging trial. Lancet. 2016;388(10039):31–44. | ||
Hansel TT, Kropshofer H, Singer T, Mitchell JA, George AJ. The safety and side effects of monoclonal antibodies. Nat Rev Drug Discov. 2010;9(4):325–338. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.