Back to Journals » Risk Management and Healthcare Policy » Volume 15
Critical Factors and Economic Methods for Regulatory Impact Assessment in the Medical Device Industry
Authors Maci J ![]() , Marešová P
, Marešová P ![]()
Received 29 October 2021
Accepted for publication 30 December 2021
Published 19 January 2022 Volume 2022:15 Pages 71—91
DOI https://doi.org/10.2147/RMHP.S346928
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Kyriakos Souliotis
Jan Maci, Petra Marešová
Department of Economics, Faculty of Informatics and Management, University of Hradec Králové, Hradec Králové, Czech Republic
Correspondence: Petra Marešová
Department of Economics, Faculty of Informatics and Management, University of Hradec Králové, Rokitanskeho 62, Hradec Králové, 50003, Czech Republic
Tel +420 737928745
Email [email protected]
Introduction: The regulatory area is one that restricts human behaviour and opportunities, but it also allows the prevention of loss of property, health, or even life in various fields. Regulations provide the market with public confidence, which is extremely important in the field of innovative medical devices. The aim of this article is to analyse critical factors and economic methods for regulatory impact assessment in the medical device industry, to focus on the finances, processes, or innovation activity of organisations operating in the medical device sector.
Methods: The paper consists of a scoping review according to the PRISMA methodology of the available literature in Web of Science and Scopus database, whereby combing the keywords “regulation” AND “innovation” AND “medical device” AND “economic impact,” we obtained a set of 156 results in the form of English-written articles. The output was then limited to the period between 2011 and 2020. Finally, 23 papers were used based on the exclusion and inclusion criteria.
Results: The resulting challenges of the identified problems in particular are the amount of high-quality data available at an appropriate cost and the availability of a flexible notified body. There are also challenges specific to the situation, such as demands on the safety of medical devices for children. From a public expectations point of view, there is a continuing need to maintain the urgency of the balance between available innovation and safety.
Discussion: As for the methods of economic assessment in general, or methods for assessing the economic impact of regulations in particular, cost-effectiveness analysis is the most commonly used method for research and development, while internal rate of return is frequently used for the producers, and budget impact analysis is typically used for healthcare service providers. A non-financial indicator that is often discussed is the time demands associated with meeting compliance requirements. The time-to-market indicator is also often mentioned. Economic and financial topics are not discussed in depth, as the reviewed articles simply mention the generally high costs attendant on complying with regulations and obtaining certificates.
Keywords: medical device, regulatory assessment, critical factors, economic impact, innovation
Introduction
Regulation can be seen as a basic tool of government to pursue economic growth and, especially in the field of healthcare, society’s well-being. The challenges of the contemporary world, such as the complexity of the system supported by foreign trade, new technologies, and other influences, make regulation an increasingly difficult discipline.1 Regulation limits or, on the contrary, stimulates and pushes for some behaviour. This is one of the reasons why the topic of regulating economic activity is a very sensitive one, with overlaps into various scientific disciplines and human activities, such as political economy, law, and environmental sciences – including engineering, urban planning, health policy services, etc. Moreover, regulation is also a significant sociopolitical force.2 In addition, regulation is such a complex issue that it meets the full extent of the term “regulatory science,” defined by the Food and Drug Administration (FDA) “the science of developing new tools, standards, and approaches to assess the safety, efficacy, quality, and performance of all FDA-regulated products”.3 Due to the afore-mentioned complexity, the approach of regulatory impact analysis (RIA) has prevailed in the field of regulation for about 40 years. The concept of RIA, which is defined by International Telecommunication Union4 as “a systematic, structured, evidence-based analysis of the prospective impacts of a proposed policy measure against possible alternatives,” is promoted not only in the USA, where it was established, but also, owing to the OECD and the World Bank, it has been implemented across both developed and developing economies.
While some regulations limit activity, at the same time they provide opportunities for pursuing new activities – in other words, opportunities for innovation.5 Some innovations are ahead of regulations (ie technology-push innovation), while others only appear after regulations are introduced (ie technology-pull innovation). As stated by Faulkner,2 technological innovation is commonly considered to be ahead of regulation, which means that regulation lags behind innovation. Examples of recent innovations that are ahead of the regulatory framework are platforms offering taxi services (Uber) or accommodation (Airbnb). In contrast, regulations are often ahead of innovations in the automotive industry, such as the requirements for maximum CO2 emissions. Examples of push and pull schemes or models are shown in Rothwell’s diagram.6 Regulation also plays an important role in the innovative field of medical devices, and as Bonnin Roca and O’Sullivan7 observe, regulatory agencies in this sector focus on ensuring patient safety rather than promoting innovation. According to Johnson,8 this affects variables such as costs, quality, and availability in the healthcare system, but also financing options as shown by his observation about the venture capital outflow due to a regulation favouring “sure bets.”9
At present, there is a change in regulatory legislation on the medical devices market in EU countries. The main objective of the newly introduced legislative changes is to increase patient safety by tightening the requirements for a medical device to be approved for market.10 Among other measures, even more rigorous quality assurance is required to enable fast tracking of individual devices in case of emergency. Furthermore, the new regulations prescribe setting up a specialist position open only to candidates with a proven track record of experience in the field of medical device regulations. This role of this specialist will be to manage and oversee the fulfilment of all regulatory requirements.11
The medical device industry is particularly heterogeneous and overlaps with other fields of healthcare and manufacturing, as it covers a wide range of devices and tools intended for patient diagnostics, surgery, therapy, and monitoring.12 The industry is attractive for developers, producers, and investors alike because of a consistently high demand on the part of patients and healthcare providers, as well as the high profit margins for companies.13,14 Considering the increasing life expectancy and decreasing fertility rates, there is a clear trend of population ageing, which further increases the demand for healthcare in general and medical devices in particular. As confirmed by macroeconomic data, this situation presents challenges especially to countries with lower standards of health and education, where it is difficult to achieve sustained growth. At the same time, the medical device industry is among the most heavily regulated ones, and any company on the market must comply with stringent laws and procedures to ensure the highest standards of quality and safety. There are specific regulations affecting all aspects of medical device development, production, and marketing, including device design, manufacturing, testing, storage, advertising, and distribution. These government regulations effectively restrict competition in the area of medical devices.
In order to establish a regulatory framework and assess the impact of the regulation, it is necessary to have a detailed knowledge of the methods that can be used to evaluate the effect of the regulation and be aware of the indicators that will be affected by regulation change. We did not find any comprehensive source of information for the medical device industry specifically, and thus decided to fill this research gap.
Therefore, the aim of this article is to analyse the impact of regulations on the finances, processes, or innovation activity of organisations operating in the medical device sector. At the same time, research questions concerning regulation were formulated to aid in the fulfilment of the main aim:
- RQ1: What methods are used to assess the economic impact of medical device regulation?
- RQ2: Which specific indicators are used to assess the impact on stakeholders in this sector?
- RQ3: What are the critical moments and processes in the manufacture, development, and distribution of medical devices?
To fulfil the set goal and answer the research questions, the following sections first introduce the method of data collection, followed by an evaluation of the outputs in terms of bibliometrics and content, and a summary of the findings and contributions of the reviewed studies. Finally, there is a discussion section of our results.
Theoretical Background
The European trade association for the medical technology industry (MedTech) states that “medical technologies can save lives, improve health, and contribute to sustainable healthcare”.15 The medical health literature states,
Medical devices are used for the diagnosis, monitoring, and treatment of virtually every disease or condition, and include familiar objects such as simple bandages to high-end MRI scanners
regulated by Council Directive.16
Within the framework of economic evaluation, a wide range of methods is used to cover the above-mentioned aspects. The purpose of behind using the method determines which is the best choice.
The basic method of medical devices evaluation, which also contains guidelines or recommendations for economic evaluation, is the Health Technology Assessment (HTA) method. HTA evaluates the social, economic and, organisational issues of a health intervention in the following areas: 1. Health problem and current use of technology; 2. Description and technical characteristics; 3. Safety; 4. Clinical effectiveness; 5. Costs and economic evaluation; 6. Ethical analysis; 7. Organisational aspects; 8. Patient and social aspects; 9. Legal aspects.17 The outcome measure of the health economic benefit assessment is the composite measure of Quality-adjusted life years (QALYs). QALY weights are usually estimated using the generic multi-attribute instrument EQ- 5D3 (EuroQol). A comparison of the costs of the current and the new solution are performed so that the calculation of the incremental cost-effectiveness ratio (ICER) and cost-effectiveness can be ensured. Then cost-effectiveness analysis (CEA) with regard to the primary clinical outcome measure and a cost-utility analysis (CUA) are performed as part of HTA. For a complete HTA it is also necessary to evaluate under what financial and time possibilities the given treatment or device can get to the given market, and whether it is possible/necessary to try to set/change guidelines defining treatment procedures. It is also important to realise that one of the necessary regulatory requirements is to set up a quality management system. This system is reflected in all individual activities related to the management of design and development, production, management of clinical trials, and communication with users and physicians.
Furthermore, for some types of medical devices which are related to data transfer and processing on new platforms, the metric of key performance indicators (KPI) is used. In healthcare and elsewhere, KPIs serve to monitor performance, collect data, analyse it, and use the resulting insights to optimise processes. Healthcare providers use KPIs to compare their performance with other providers. This practice enables healthcare facilities to make data-based decisions leading to minimising the waste of resources and maximising the value provided. There are five parameters that are considered, including a) operational metrics, b) financial metrics, c) healthcare facility, d) emergency department healthcare KPIs, and f) care quality metrics.18
Finally, the Balanced scorecard company’s performance evaluation method is also modified for use in the area of healthcare. This method is used to monitor, implement, and evaluate company strategies, particularly in mergers and acquisitions. In the healthcare sector, this method must be applied carefully, since there is a number of stakeholders to consider.
In health care, however, the three customer functions are performed by three different groups. Physicians, especially primary care physicians, select (or strongly influence) the hospital or clinical practice where their patients will receive care; private health plans or the government pay the provider for the care; and patients receive the care. All three groups—referring physicians, payers, and patients—are customers of the provider organisation, and the strategy of the provider organisation should explicitly recognise and measure how it creates value for all three customer types. An academic medical centre has additional customer types: interns and residents for its educational function, and academic scholars and practicing physicians for its research function. If five customer groups were not complicated enough, all hospitals also have responsibility for the health and well-being of residents in the communities where they operate. Suitable performance metrics for the diverse customers of health care providers are from the patient’s point of view: Index of Patient Outcomes: • Incidence of complications or readmissions, • Patient Net Promoter Score. From the perspective of payers: • Price index relative to regional competitors, • Cost of invoice processing; from community’s health care: Rating of hospital by community advocacy groups and local government health agencies and • Number of collaborative community health care initiatives.
Finally, according to Brockis et al19 there are several programmes under which medical devices can be evaluated, including the Technology Appraisal Programme (TAP), Medical Technologies Guidance (MTG), the Diagnostics Assessment Programme (DAP), and Clinical Guidelines (CG). The choice of programme is based on the type of device and its value proposition. Non-diagnostic devices fall under the TAP programme when a cost-effectiveness analysis is required, but under the MTG programme the devices have a potential to save cost. While potentially cost-saving diagnostic devices may be evaluated under the same programme, other diagnostic devices fall under the DAP programme. The different programmes utilise different evaluation methods: TAP, DAP, and CG conduct a cost-utility analysis, while MTG performs cost-consequence analysis. In the former, benefits are measured in quality-adjusted life years, while in the latter, the unit of measure is the clinical outcome.
These are examples of methods used by companies or medical entities (hospitals) in order to express the effectiveness of new solutions, medical devices or procedures. The aim of the article is to address not only these approaches, but also an external view of the economic situation of companies, the effects of decisions of external entities establishing regulations, and also with economic method showing the impact on the company itself.
Methodology
For a systematic literature review, a number of available databases can be utilised. Scopus and Web of Science (WoS) are among the most respected and widely used international databases, which is why these two services were selected to research the available literature and process the relevant analyses. For a more detailed analysis of the outputs, specifically for building keyword clusters, the VOSviewer application was used. This posed a challenge in terms of combining the outputs from WoS and Scopus—for an example of how to merge outputs from WoS and Scopus, see Echchakoui.20 Further details on the individual steps of preparing the bibliometric analysis are listed below. The contribution of possible relevant publications outside of WoS or Scopus is covered in chapter 4, which is devoted to discussion.
When searching in the WoS and Scopus databases, the following procedure was followed with respect to the aim of the article and in keeping with the basic elements of the PRISMA guidelines:21
- Step 1. In the first step, the following search parameters were set: results in the category “article, book, review, book chapter, conference paper,” written in English, without time limitations, with the keywords “regulation” AND “innovation” AND “medical device” AND “economic impact.” The filter for where to look for these keywords was set to “All fields.” The results obtained in this step were used for a bibliometric analysis, including a cluster analysis performed using the VOSviewer program.
- Step 2. In the second step, the selection parameters were narrowed down to a period of the last 10 calendar years, ie, between the years 2011 and 2020. The purpose of this time limitation was to consider the most recent and current approaches in the field. Included in the results were also outputs other than just literature reviews (eg analyses of government reports, professional reports, and guidelines) and outputs primarily focused on a medical problem, that is, outputs where the economic aspect was not the key finding obtained through the review. In this step, a PRISMA-compliant procedure was utilised, and based on article abstract screening, the least thematically relevant articles were excluded. The exclusion criteria were as follows: no or insufficient economic background (no economic variables or no method of assessing economic impacts of regulation), purely medical focus, and purely technical or procedural focus, including a simple description of the regulatory environment. Finally, non-medical sources investigating a topic unrelated to medical device regulation were excluded as well. The remaining results were subjected to a qualitative analysis, where articles were excluded due to their unavailability, or a detailed review eventually revealed they should be excluded for the same reasons as those stated at the beginning of the PRISMA procedure. The decision to include or exclude articles in the identification and screening steps was made by two researchers independently according to predefined inclusion or exclusion criteria. Articles that were not in the joint intersection were discussed, and then either finally included or excluded.
In the Scopus and Web of Science (WoS) databases, entering the parameters from the first step returned 156 matching results in the Scopus database and, perhaps a little surprisingly, no outputs in WoS. Not wishing to neglect the added value of WoS, we tried deleting the keyword “economic impact” and repeated the first step. In this case, a reasonable output of 67 results in the WoS database was obtained for subsequent analysis, but the output in Scopus increased to about 4400 items. Such a high number can be hardly analysed in a systematic literature review. Yet, combining the results from Scopus with results from WoS based on a different number of keywords did not appear methodologically correct. Therefore, we decided to perform a bibliometric analysis (see step 1 above), including a procedure according to the PRISMA guidelines (see step 2), using the Scopus output only. The WoS output was analysed according to the second step only, and the results are described separately in sections 3.2 and 3.3, respectively. The final date on which the database outputs were retrieved was May 2021.
As mentioned in the previous paragraph, a total of 156 outputs were found in the first step. The time constraint of step 2 (that is, the narrowing to the last 10 calendar years) reduced 156 outputs to 129. Following the individual steps according to PRISMA, these 129 outputs were reduced to 45 and then to 36 outputs to be used for a qualitative analysis, out of which 23 outputs were eventually included in this review (see Figure 1, depicting the PRISMA diagram).
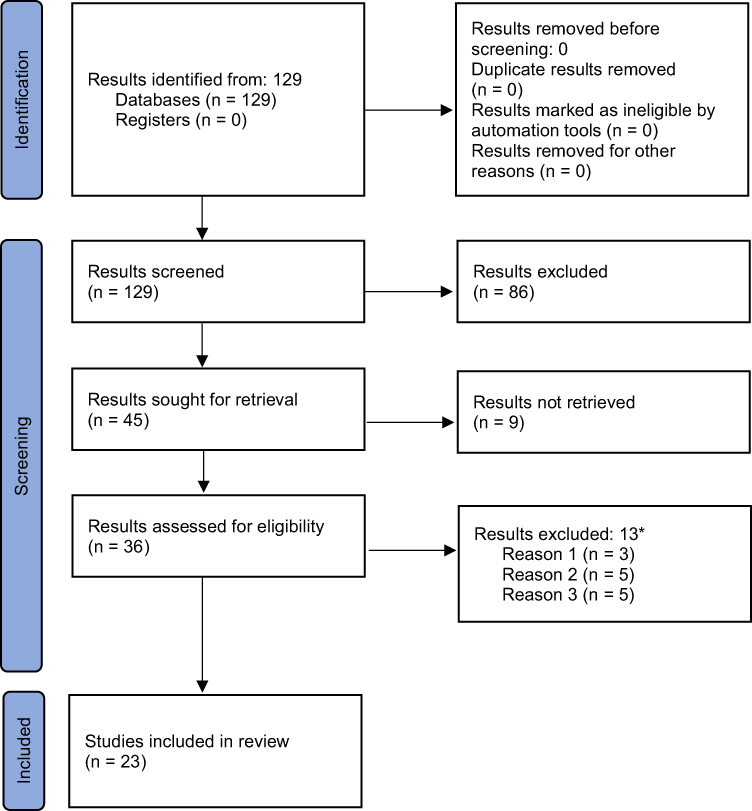 |
Figure 1 PRISMA 2020 flow diagram of outputs obtained in step 2. Notes: PRISMA figure adapted from Page MJ, McKenzie JE, Bossuyt PM et al. The PRISMA 2020 statement: an updated guideline for reporting systematic reviews. BMJ. 2021;372:n71. Creative Commons.22 *Results excluded following screening (reasons): 1) Focused on processes and their relation to regulation (influence of non-financial, non-medical variables). 2) Focused on regulation as such; methods for evaluating regulation are lacking. 3) Economic circumstances described only very superficially (eg financial data are missing); focused on the medical aspect. Necessary economic context in the form of costs, benefits, and financial impact as well as methods for an economic evaluation of the research problem are missing (missing numbers, data, methods). Regarding the work with articles classified as review, such articles were included, provided that the area of review concerned a medical problem, ie, the economic issue was not the primary topic but economic issues were desirably represented in the review. |
The following chapter 3 presents the results of the work based on steps 1 and 2.
Search Results on Regulation, Innovation, Economic Impact, and Medical Device
Chapter 3 presents the main results of this article. The first part, Chapter 3.1, presents bibliometric results derived from the output described in the methodology as Step 1. Chapter 3.2 contains qualitative results focused on market regulation, innovation, economic impact assessment, and variables in the field of medical devices. It is based on a sample that was obtained according to the criteria described in the methodology as Step 2. Chapter 3.3 complements Chapter 3.2 with resources focused on critical moments and processes in the manufacture, development, and distribution of medical devices. These results are collected from the Web of Science database (WoS), which have not been included in previous chapters. Although WoS documents did not fall into the filter according to steps 1 and 2 described in the methodology, we included them to minimise the error in the form of missing information. Finally, Chapter 3.4 provides a synthesis of the findings contained in Chapters 3.2 and 3.3.
Bibliometric Analysis
In the first step, a total of 156 outputs were found, falling into the categories of articles (57), books (41), reviews (41), book chapters (12), and conference papers (5). After narrowing the search parameters to the last 10 complete calendar years (ie 2011–2020) in the second step, the number of outputs for a more detailed analysis dropped to 129. This shows that a significant shortening of the time period (a period between 1981 and 2021 in step 1 vs a period between 2010 and 2020 in step 2) led to only about a one-fifth decrease in outputs (20 outputs for 1981–2010 and 7 outputs for 2021).
Figure 2 illustrates a trend that becomes apparent when narrowing the search criteria to a shorter period—20 years in this case, since the years before 2002 are largely underrepresented. The figure makes it apparent that a significant accumulation of outputs occurs in the most recent years. Apart from objective facts and trends in academic publishing as such, an initial look at the list of results does not reveal any apparent explanation for the fluctuation. The only exception is that in 2020, the issue of coronavirus plays a minor role.
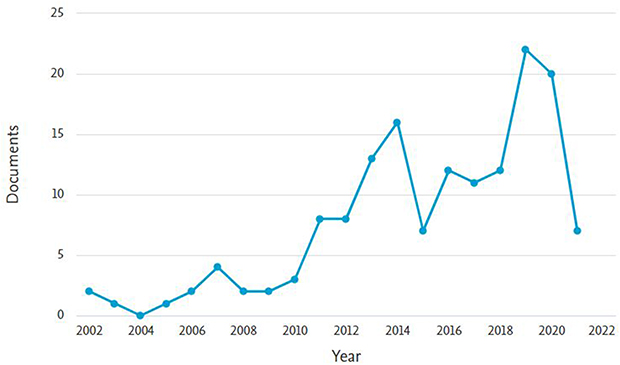 |
Figure 2 Number of publications according to the filter from step 1 for the last 20 years. |
As for the field of study, most of the outputs from step 1 were categorised in the field of “medicine” (23.3%), followed by “social sciences” (14.3%), and “business, management, and accounting” (10.2%). Next, with a representation ranging between 7.9% and 6.4%, were the fields of “engineering,” “pharmacology, toxicology, and pharmacies,” “biochemistry, genetics, and molecular biology,” and “economics, econometrics, and finance.”
The last task involving the outputs from step 1 is a cluster analysis of keywords performed in the VOSviewer program. The value of the minimum co-occurrence of keywords was set to 6, which resulted in a map of 38 words (see the upper left corner in Figure 3). Quite surprisingly, the words “innovation” and “economic impact” did not appear among the keywords with the strongest links, although they were part of the search filter. For the keywords “regulation” and “medical device,” the most frequent co-occurrences are presented in Figure 3 in the upper right corner and in the lower right corner, respectively. Instead of “economic impact,” the keyword “economics” is highlighted to represent economic contexts (see the lower left corner in Figure 3). The most frequent co-occurrences were found with the keywords human or humans (55 and 35 occurrences, respectively), article (26), review (26), priority journal (23), economics (15), medical devices (12), Europe (11), healthcare policy (11), healthcare cost (11), and healthcare delivery (10). The word regulation occurs in two variations, as “regulation” (6 occurrences) and “government regulation” (7 occurrences). The keyword innovation does not appear in Figure 3 at all because its occurrence value was only 3. The topic of innovation is probably so obvious for this area of publications that it is rarely explicitly included in the list of keywords.
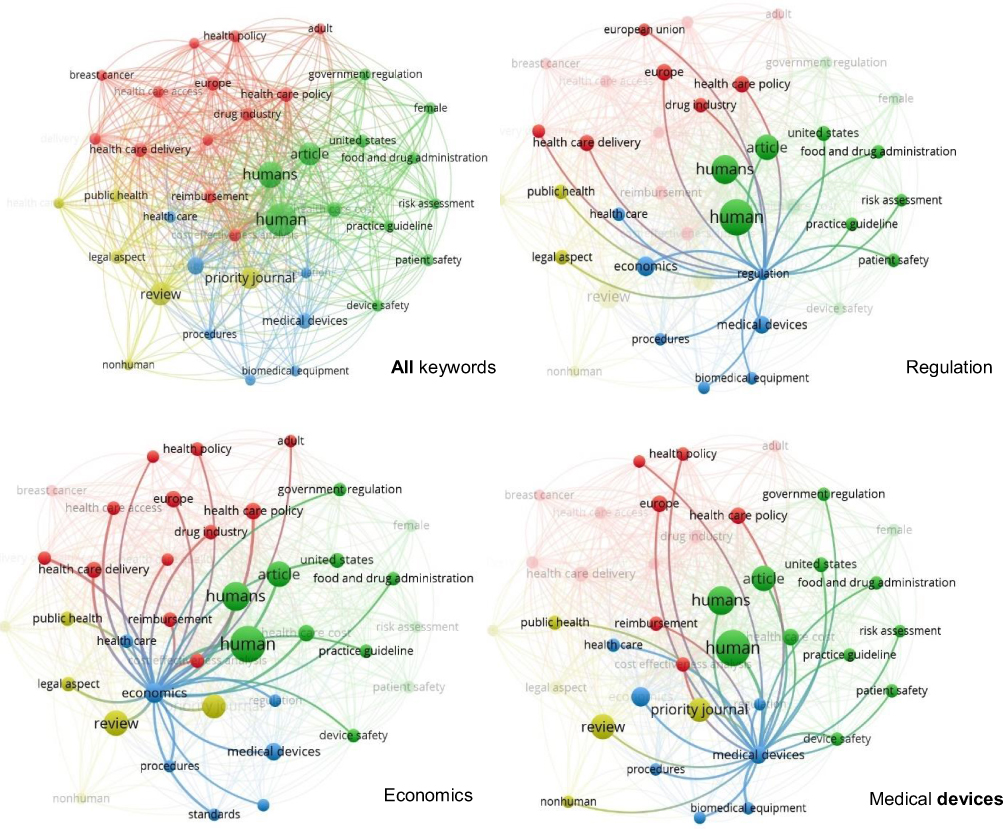 |
Figure 3 Cluster analysis of keywords in step 1 (all, regulation, cost-effectiveness analysis, economy). |
In terms of clusters, the VOSviewer analytical tool categorised the keywords into 4 non-overlapping clusters, each including between 13 and 6 items (see Table 1; clusters are also recognisable by colour coding in Figure 3). The words “regulation” or “economics,” together with “medical device” as the main subject of research, can be found in Cluster 3, which has a total of 7 items. The same cluster also includes biomedical equipment, healthcare, procedures, and standards (the blue colour in Figure 3).
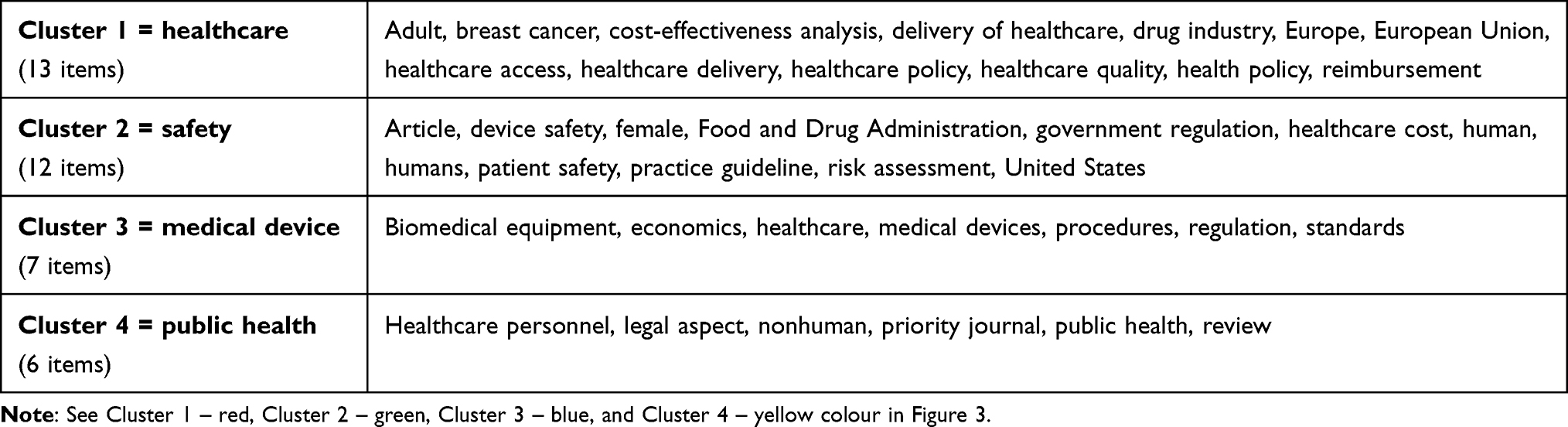 |
Table 1 Clusters of Keywords in WoS |
Having described the bibliometric analysis of results obtained in step 1, the following sections describe the procedures involved in step 2, followed by conclusions derived from a qualitative analysis of selected articles in keeping with the PRISMA procedure.
Market Regulation, Innovation, Economic Impact Assessment, and Variables in the Field of Medical Devices
In this section, a qualitative analysis is performed on selected outputs obtained using the PRISMA methodology, which best fulfils the aim of this article. Let us remember that excluded from the selection were purely medical sources or, on the contrary, non-medical sources covering a topic unrelated to medical device regulation. Following the individual steps according to PRISMA, the initial 129 outputs were reduced to 45 and then to 36 outputs to be used for a qualitative analysis, out of which 23 outputs were eventually included in this review (see Figure 1 above in methodology, depicting the PRISMA diagram).
The following table (Table 2) presents the remaining 23 publications that bear the highest relevance to the aim of this article (ie they are closely related to market regulation, innovation, medical devices, and economic impact, and at the same time they provide answers to the above-formulated research questions). The column in Table 2 titled “Article context” characterises the focus of the reviewed article. With respect to our research questions, the most relevant columns are “Economic evaluation method mentioned or used” and “Economic variable considered.”
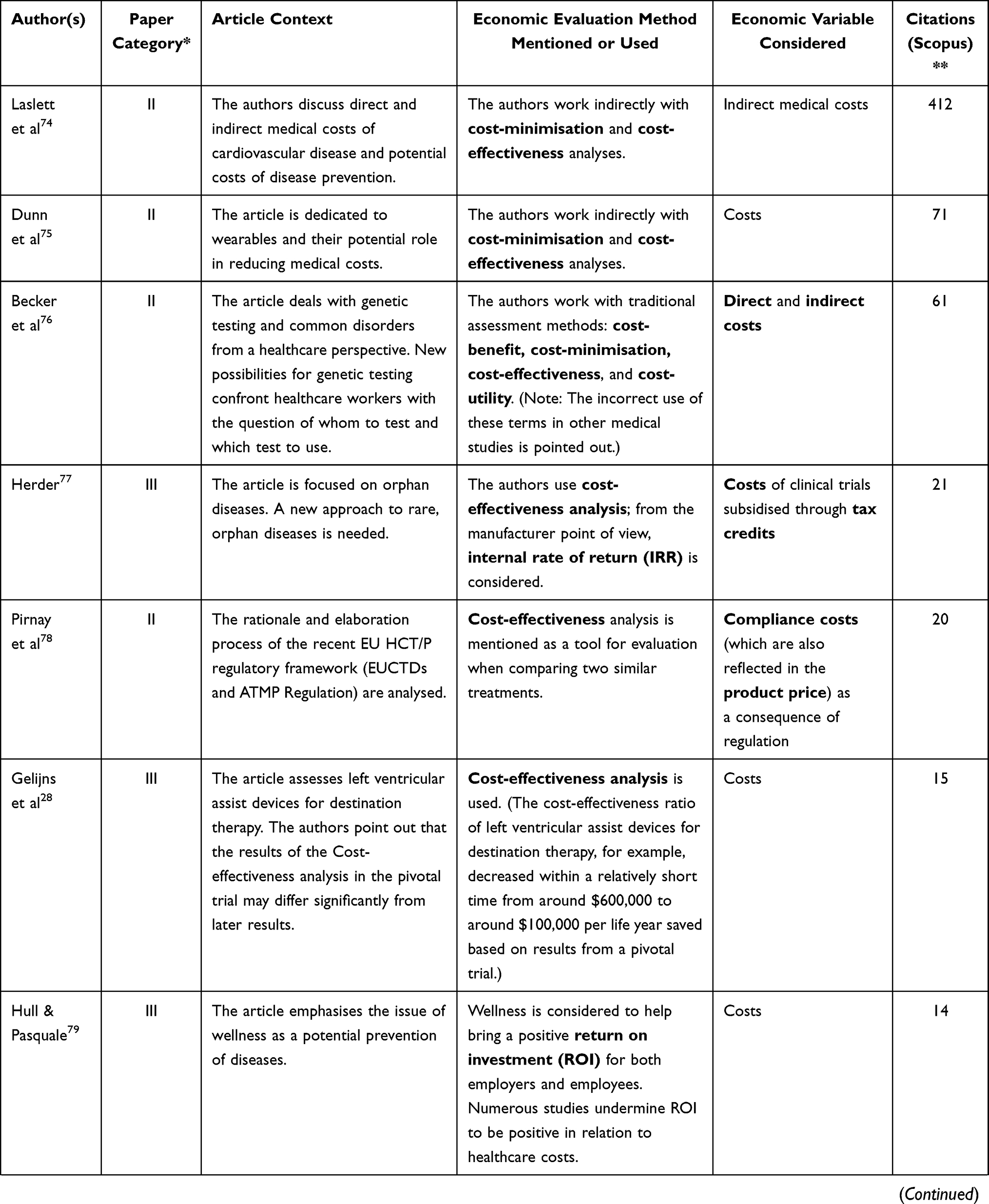 | 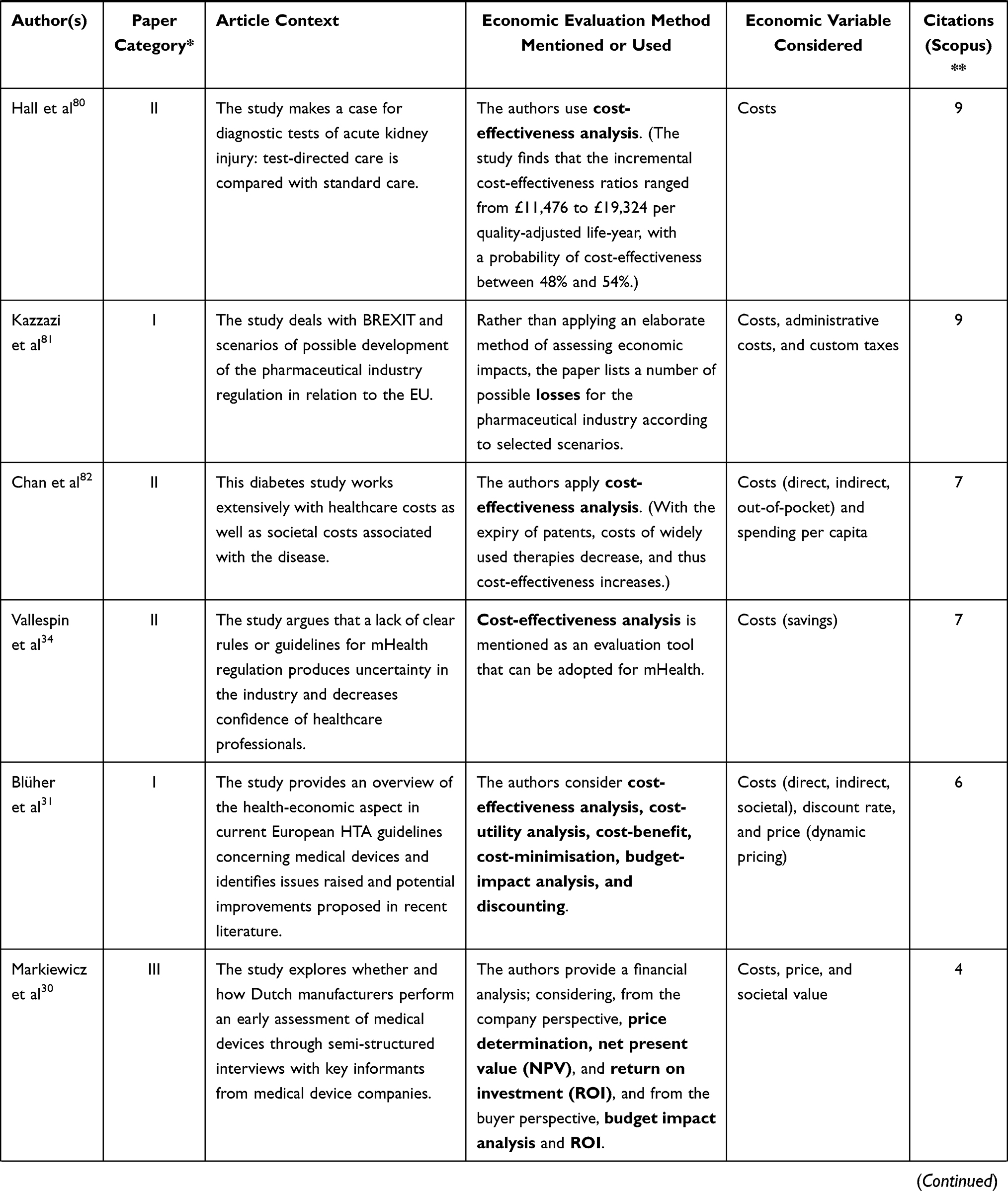 | 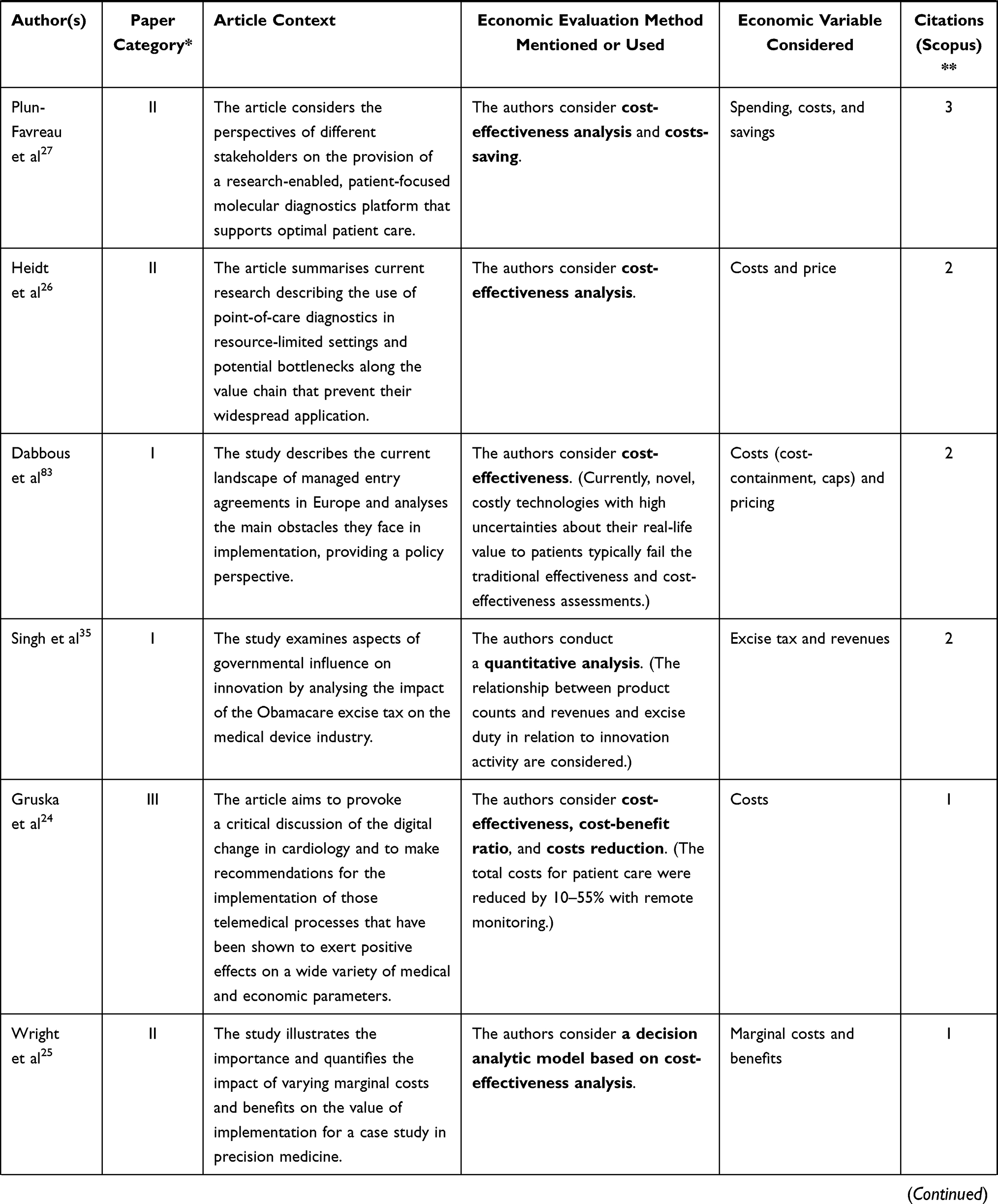 | 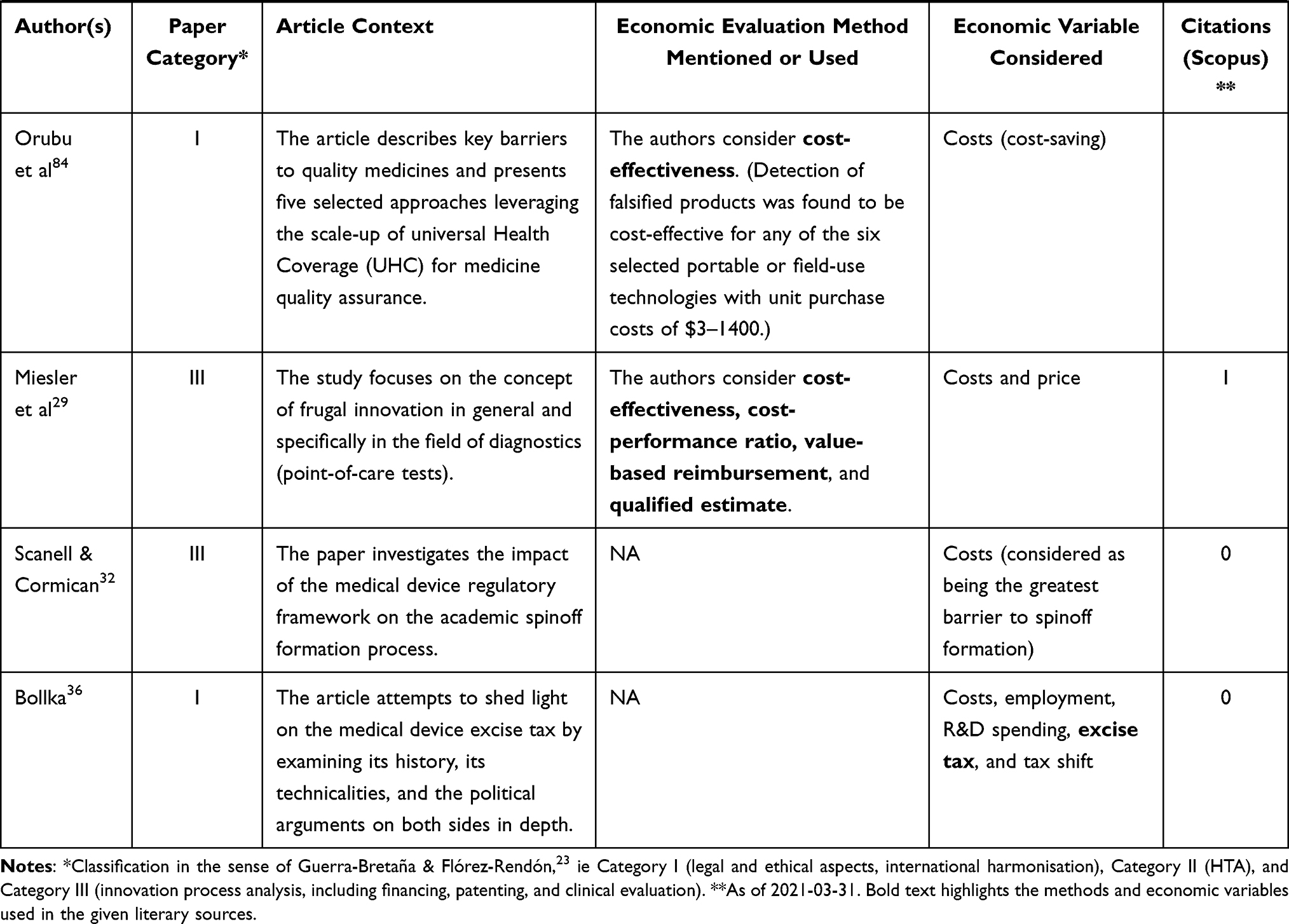 |
Table 2 Step 2 – Medical Device, Regulation, and Innovation – Economic Context |
Table 2 offers several major findings. According to a classification given by Guerra-Bretaña and Flórez-Rendón,23 most of the 23 outputs under consideration would fall into Category II, ie health technology assessment (HTA). This group includes 10 outputs. Category III (ie innovation, process analysis, including financing, patenting, and clinical evaluation) includes 7 outputs. Finally, Category I (ie legal and ethical aspects, international harmonisation) includes 6 outputs.
Among the economic methods of assessing regulation impact and overcoming regulatory barriers, cost-effectiveness analysis is clearly the most common one. As for business indicators, it is worth mentioning return on investment (ROI), net present value (NPV), and internal rate of return (IRR) on the part of the manufacturer and budget-impact analysis or cost-minimisation on the part of the purchaser. By far the most frequently considered variable when evaluating the impacts of regulations or overcoming obstacles posed by them, “costs” are mentioned in various forms (compliance, direct, indirect, etc.). Other variables include, for example, “price” or “tax,” specifically excise duty.
A more specific in-depth, but still synthesised view of the individual sources listed in Table 2 is provided in the following paragraphs.
Where the reviewed articles employ any methods of assessing the impact of regulations or meeting the conditions of regulations, especially in the case of HTA, it is usually the method of cost-effectiveness, the approach of cost per patient, or methods of cost-saving—eg Gruska et al.24 Wright et al.25 Heidt et al26 and Plun-Favreau et al.27 On the other hand, Gelijns et al28 emphasise that it is important to consider the specificity of pharmaceuticals and medical devices when using the cost-effectiveness method. It is not uncommon that shortly after the introduction of a medical devices, additional adjustments are made due to the “learning-by-using” effect. This means that the cost-effectiveness value subsequently changes and may differ significantly from initial assumptions.
As for the methods of assessing regulation impact, it is not uncommon for authors to work with a mere qualified estimate. To name but one, Miesler et al29 consider the introduction of EU Regulation 2017/746 on the example of point-of-care tests (POTC), assuming that on one hand, stringent regulation may lead to higher performance and safety outcomes and thus increased rate of reputation and scope of application; on the other hand, such regulation might be challenging for some especially small companies.
Markiewicz, Van Til, and IJzerman30 offer a detailed insight into the thought process of manufacturers when developing a medical device, specifically in the stage of early assessment, through results derived from semi-structured interviews. The interviewed companies performed extensive economic evaluations from the perspectives of both the producer and the customer. On the part of the producer, the methods utilised are price determination, net present value, and return on investment; on the part of the purchaser, the common approaches are budget impact analysis and return on investment.
An important variable for assessing economic impacts is the discount rate. As to its amount when converting costs to the same point in time (ie using net present value), Blüher et al31 suggest a number between 3% and 5% in their critical literature review.
Considering the process of overcoming regulatory limitations, Scannell and Cormican32 use the example of academic spinoffs in the field of medical devices and conclude that, unlike other commercialisation activities, these spinoffs lag behind, which is why they recommend adding an expert on the issue of regulations to the team more promptly. On the other hand, these authors also present costs as the main identified barrier, which may be one of the reasons why the regulatory expert is not invited to the team earlier (see other results presented by the same paper).
From the point of view of basic economic variables (eg price and costs), Jogova, Shaw, and Jamieson33 highlight the disparity between the willingness to pay for an mHealth medical device and the cost of overcoming regulatory constraints. The willingness to pay for an mHealth application is relatively low, while the cost of complying with regulations is high. At the same time, these authors point out the significant differences between traditional medical devices and software applications. In conclusion, the authors propose that a clear regulatory framework may reduce market entry barriers, foster innovation, and facilitate exportation. On the same topic, Vallespin, Cormet, and Kotzeva34 discuss that mHealth has a significant cost-saving potential for healthcare (especially wellness and prevention, and treatment and monitoring), but the potential cannot yet be realised due to a lack of regulations, or rather, the existing conventional regulations are costly and incompatible with the nature of mHealth. According to the same authors, regulation through certification (proof of the benefits of mHealth), standardisation, and interoperability will help increase the confidence of patients as well as healthcare professionals, thereby opening new market opportunities.
As for other economic variables, Singh, Russ, and Terzidis35 use quantitative analysis to examine the influence of excise tax applicable to medical devices (ie a tax levied in 2013 in the USA as part of Obamacare) on the innovation performance. Bolka36 works with the same variable and makes a general estimate of the economic impacts of this tax; for example, in the need to reduce producer costs through job cuts, moving jobs abroad, or downsizing research and development activities. The possibility of the excise tax being carried by the purchaser rather than the manufacturer is also mentioned.
An important element in reducing regulatory costs is assessment by equivalence, ie assessing a new medical device based on data of an already existing equivalent device. White and Walters37 point out possible problems caused by the equivalence approach and conclude that the US FDA seems to systematically promote innovation rather than adhering to maximum safety. The authors believe that this support for innovation should be counterbalanced by post-market mechanisms to safeguard against residual and developmental risks.
Critical Moments and Processes in the Manufacture, Development, and Distribution of Medical Devices
This section discusses thematically related outputs from the WoS database, which complement the conclusions made on the basis of the systematic literature review above. Unlike when searching the Scopus database, the keyword “economic impact” was omitted in WoS, while the other search parameters remained identical. As stated above in the methodology section, due to the omission of one keyword, WoS outputs are presented separately. Using the same steps as when searching the Scopus database, 67 outputs were identified, out of which 24 of the most relevant ones were selected. The findings of these articles concerning regulation, innovation, medical devices, and selected economic manifestations are described below.
One thing the WoS articles reveal is the time period of product launch versus regulatory issues and their economic manifestations. When it comes to new medical devices, Howard38 states that although stricter regulatory requirements may cause delays in market introduction, ultimately, avoiding malfunctioning devices will eliminate the risk of eroding public confidence and the consequent financial and reputational problems for the companies of medical device developers. In addition, Richards and Hudson39 point out not only the extensive time demands on new product development, but also the increasing pressure faced by the regulatory bodies and government in recent years – the public demands a fast and easy implementation of product innovations, but it does not consider the risk of the product not working correctly. The public also controls and criticises more; patients are more informed, for example, owing to online searches and patient group forums. The solution to shortening time-to-market, and subsequently reducing development costs, could be a dedicated public institution to provide consultations on a timely overcoming of regulatory barriers.
Furthermore, it can be observed that outputs focused on regulation associated with the development and implementation of innovative medical devices tend to consider the impact on all stakeholders (most often patients, manufacturers, practitioners, and others). In the case of developers, however, specific economic impacts and regulatory aspects are mentioned only in passing and are discussed in general terms as costs and implementation barriers (such as long device development time, high development costs, and regulatory considerations – see Bowsher et al.40 De Maria et al41). Some articles, such as Faulkner2, simply communicate the finding of a European Commission survey that “Regulatory costs were prohibitive for some companies.”
As in the Scopus articles, cost-effectiveness analysis is a commonly used method of economic assessment in the WoS articles. Specifically, Boudard et al42 deal with the quality of clinical studies, including clinical trials and cost-effectiveness studies. The authors screened and classified 217 studies according to the Sackett 5-level evidence scale. Only a few among the reviewed studies provided high-quality clinical evidence. Hence, methodological quality did not correlate with the risk level of the medical device.
Dr. Michael Dunbar, quoted in Howard,38 considers the shift of decision-making from the surgeon to the administrator to be beneficial. Sauerland et al43 note the issue of a “pioneering” medical device and its followers on the market. Derivative devices tend to be cheaper for the purchaser as their manufacturers do not expend as high development costs as the developer of the pioneering device, nor do they need to submit the same amount of clinical data; however, subsequent devices tend to have difficulties with demonstrating product safety (p. 281). In financial terms, Lennox44 uses an example from the 2005 US market, where the assessment of a new device through a pre-market approval submission cost the Center for Devices and Radiological Health at the FDA approximately $870,000, while the assessment of a new device through a 510(k) submission based on equivalency with an already approved device cost only about $18,200. Lennox44 also adds that while the time required for the pre-market approval review process in 2011 was around 15 months, with the 510(k) submission, it was around 150 days (ie 5 months). This approach therefore saves both money and time.
Furthermore, based on Gardner45 and Altenstetter,46 geographical differences can be observed between the USA and Europe in the definition of the term “efficacy.” Geographical differences are also considered in a comparative study by De Maria et al41 who compare Africa and the EU. The authors note that the European regulatory framework is stricter, but they also consider the benefits of common regulatory requirements shared across regions, such as reducing the device development costs associated with regulatory compliance. Kale47 analyses the situation in India and demonstrates the disadvantages of a poorly regulated medical device market. Due to the lack of collective action (ie insufficient regulation), there is a high risk of a poor choice when purchasing locally manufactured medical devices, and therefore foreign devices are imported. Imported devices are, however, up to 50% more expensive than their domestic equivalents. Kale47 argues that foreign-owned multinational corporations thus effectively operate under a monopoly rent. In addition, in some segments of medicine, India is about 90% dependent on imports. Insufficient regulation not only makes India dependent on expensive imports but also has a negative impact on the development of local technological capacities.
The thematic focus of the reviewed articles makes it obvious that there has been an appearance in the last few years of smart technology devices (eg wearables) in research publications relating to the economic impacts of regulation – see the terms “mHealth”, standing for mobile health, in Munos et al48 and in Onodera and Sengoku,49 or simply “wearable sensor” in Ravizza et al.50 As a specific impact on the healthcare sector, Munos et al48 state that the benefits of involving wearables in drug development is expected to save about 80% of the cost of data collection. Ravizza et al50 make a general statement that wearable sensors are expected to reduce healthcare costs. On the other hand, as Munos et al48 state, mobile health developers have to deal not only with the FDA (Food and Drug Administration, specifically its Center for Devices and Radiological Health) but also with the FCC (Federal Communications Commission). It follows that mHealth devices can expect higher compliance costs. Onodera and Sengoku49 observe that there is a significant mismatch between the number of mHealth applications and the evidence of their benefits. This can reduce the credibility of applications and hinder the overall market development (p. 69).
Drukker, Noble, and Papageorghiou51 focus on the involvement of artificial intelligence (AI) in medicine, specifically in medical image analysis for repetitive ultrasound tasks. The authors mention the increase in AI solutions submitted to regulators, but they also observe that there is a lack of publicly available data verifying the results, and thus a lack of data on the potential economic impacts. Kesavan and Dy52 summarise progress in healthcare reforms that address price transparency or device approval through market approval regulations, post-market surveillance, or comparative effectiveness research. Challenges faced by the industry, physicians, and patients as a result of the reforms are also discussed. Ibrahim et al53 discuss the difficulties of developing medical devices for children, including considerations of the return on investment indicator. The authors believe that the market of medical devices for children does not allow for returns to scale, which is a shortcoming that undermines potential innovation efforts. In conclusion, the authors identify an opportunity for reducing R&D costs, and thus reaching a more favourable ROI and decreasing the need for returns to scale in the use of computational modeling and simulation. Onur and Söderberg54 analyse changes in regulatory review time (often considered as synonymous to higher compliance costs) in relation to the choice of companies to opt for a radical or an incremental innovation. Based on FDA data on US high-risk medical device industry, these authors demonstrate a greater sensitivity of companies to changes in their application review time in the case of incremental innovations when compared to radical innovations. Grennan and Town55 refer to the example of coronary stents when comparing the US and EU regulatory frameworks in terms of pre-market testing and post-market observational studies, and the effects of this legislature on public welfare. The authors work with an economic model that seeks to measure the total surplus. They55 conclude that, for coronary stents, the USA come close to the optimal policy in terms of achieving a balance between testing and access to innovation, while the EU is too lax. The authors also believe that their approach is applicable to other Class III medical devices.
Finally, Maresova et al5 in relation to the new medical device regulation EU 2017/745 (MDR), point out that innovations in the field of medical devices are mostly driven by small and medium-sized enterprises (SMEs). At the same time, it is SMEs rather than large companies that are vulnerable to changes in device development regulation due to the limited possibilities of co-financing additional administrative costs. Letourneur et al56 and Vasiljeva et al57 also comment on the effectiveness of the new EU regulation on medical devices. Vasiljeva et al57 draw attention to the increasing strictness of the regulatory framework, represented by the new MDR legislation, in the field of medical devices on the common EU market. The authors point out that while the new regulatory measures further increase patient safety, on the other hand, they are becoming a considerable barrier to innovation and may ultimately reduce patient benefits. In terms of economic impact, Vasiljeva et al57 note that there is no certainty as to the change in compliance costs resulting from the MDR legislation. Letourner et al56 perform a SWOT analysis and conclude, among other findings, that the tightening regulatory demands are resource-intensive (eg the increased time-to-market for the originator). However, the authors believe that the new legislation brings new opportunities for spin-off and start-up companies collaborating with academic institutions. What is more, academic research should provide a guarantee of return on investment and cost-effectiveness in health policy. Wilkinson and van Boxtel58 also see new opportunities associated with the new regulation:
The MDR’s requirement to include ‘a specification of the clinical benefits to be expected’ in a device’s IFU represents an opportunity for manufacturers to highlight these benefits to potential users.
On the other hand, as the authors acknowledge, the new regulatory requirements mean additional workload and costs for producers. While the authors work with the concept of cost-effectiveness, they do not attempt to quantify either the benefits or the additional costs that they discuss.
Dai, Ma, and Li59 offer a specific view on the regulation and economic development of the medical device market. Using the example of the Chinese market, the authors consider the compliance costs for local medical device developers, but they also point out the existence of a regulation relevant for purchasers, ie, Chinese top hospitals, which are required to cover 50% of their demand for medical devices with locally manufactured products. The authors conclude that this step presents an opportunity to develop domestic production and stimulate innovation.
Findings Synthesis
This chapter, which synthesises the content of sections 3.2 and 3.3, answers the research questions RQ1, “What methods are used to assess the economic impact of medical device regulation?” and RQ2, “Which specific indicators are used to assess the impact on stakeholders in this sector?” posed in the introduction. First, as to the methods of economic assessment in general, or methods for assessing the economic impact of regulations in particular, cost-effectiveness analysis is the most commonly used method for research and development, while internal rate of return is frequently used by the producers, and budget impact analysis is typically used by healthcare service providers. Second, specific financial indicators are either absolute, such as costs and price, or relative, such as return on investment. Non-financial indicators that are often discussed are the time demands associated with meeting compliance requirements. The time-to-market indicator is also often mentioned.
Figure 4 schematically illustrates the basic economic variables and impact assessment methods in the medical environment discussed in sections 3.2 and 3.3.
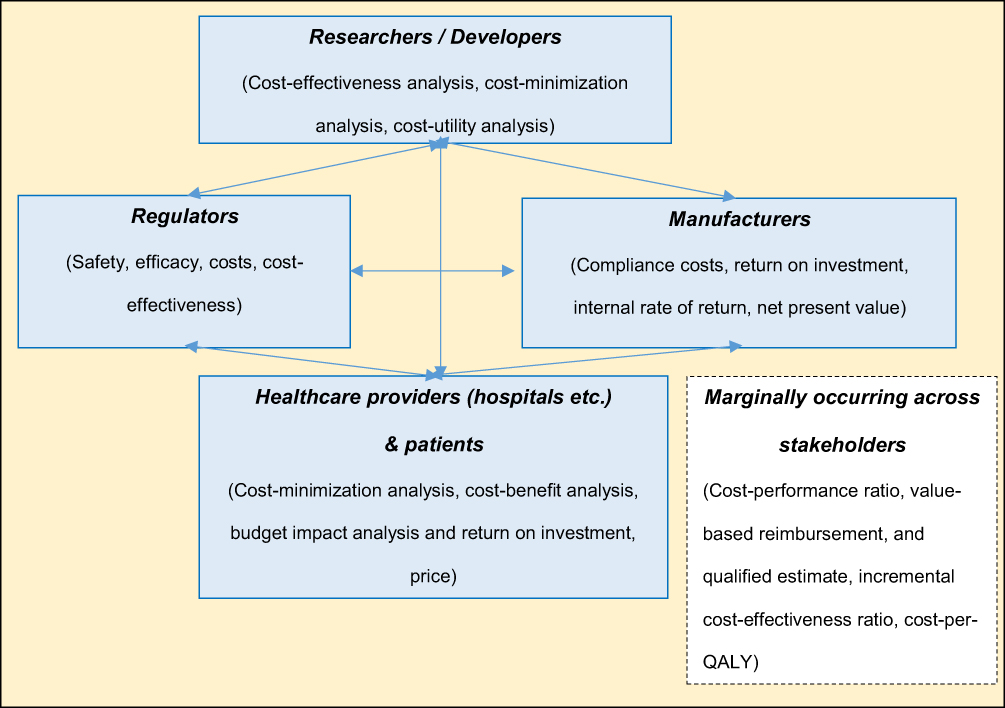 |
Figure 4 Basic economic variables and frequently used methods of economic impact assessment – key stakeholders. |
In addition, common problems with close economic ties can be observed. The diagram shown in Figure 5 answers the third research question, “What are the critical moments and processes in the manufacture, development, and distribution of medical devices?”. The scheme is divided into three parts called “problem”, “critical factor”, and “resulting challenges”. In particular, problems related to the use of ICT (whether AI solutions or mHealth), a narrow market problem, or public pressure for rapid innovation in conflict with security issues were identified.
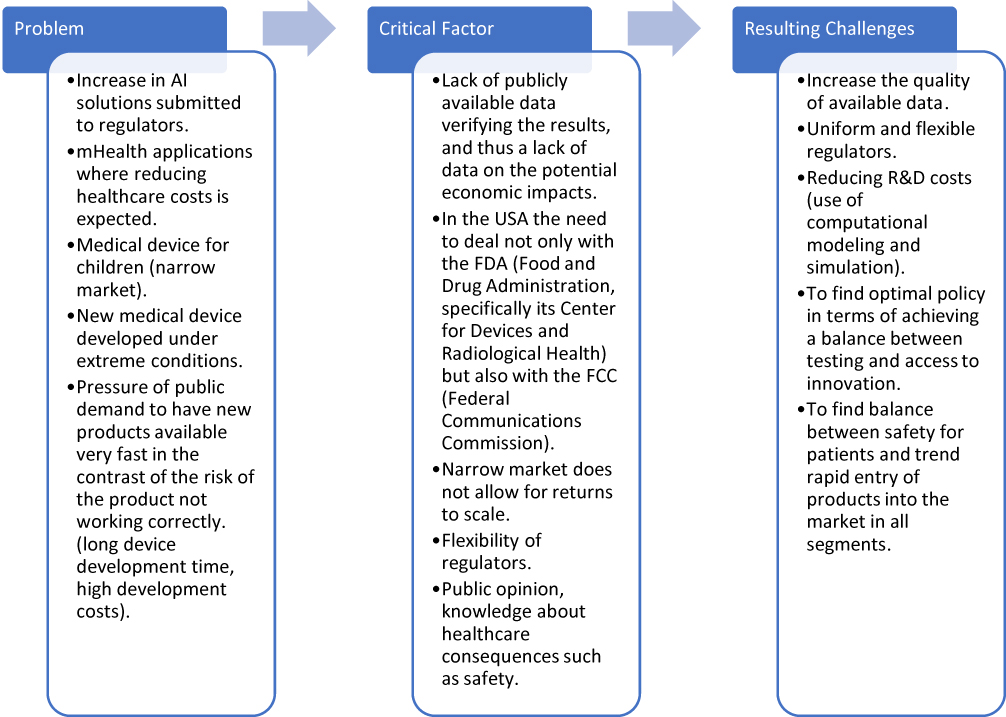 |
Figure 5 Critical moments and processes. |
As shown in Figure 5, the resulting challenges of the identified problems are in particular high-quality data available at an appropriate cost and the availability of a flexible notified body. There are also challenges specific to certain situations, such as demands on the safety of medical devices for children. From a public expectations point of view, there is a continuing need to maintain the urgency of the balance between available innovation and safety.
Discussion
The discussion held here is composed in the spirit of the main answers to the research questions asked. The research questions were “What methods are used to assess the economic impact of medical device regulation?”, “Which specific indicators are used to assess the impact on stakeholders in this sector?”, and “What are the critical moments and processes in the manufacture, development, and distribution of medical devices?”.
The question of why cost-effectiveness is the most frequently utilised method of economic evaluation in the above-discussed systematic literature review can be answered simply. Cost-effectiveness is a commonly used standard method in HTA. Its use is guided, where applicable, by guidelines for the economic evaluation of health technologies—see, for example, the Irish Guidelines for the Economic Evaluation of Health Technologies in Ireland.60 Local HTAs may also contain local specifics for economic evaluation, such as discount rate. A review of economic guidelines across Europe was recently compiled by Blüher.31
This also corresponds to the fact that if the area of medical device development and economic view are combined, most of the attention is focused on proving the effectiveness of the new solution with regard to how it will reduce costs. What impacts it has on the company, its processes, or how everything is related to the issue of regulation, which may affect the performance and output of the company, is less addressed. Overall, the view of the company is less discussed at the professional level.
Considering the goal of this article, the following remarks should be added. Regulation in the field of medical devices is considered so essential by some that it can be found on university curriculum in Biomedical Engineering. For example, Di Pietro et al61 in their paper present the incorporation of regulatory issues in the Medical Device Design course taught at the University of Pisa, Italy. Introducing a university course focused on medical device regulation, on one hand, can be seen as an increase in the indirect costs of the entire regulatory system. On the other hand, in the last analysis, it may lead to a better management of compliance with regulatory requirements in private companies and public institutions alike, thus reducing both costs and time-to-market.
Finally, types of regulation and methods of evaluating regulations, including their impact in the medical device sector, can be compared with methods used in other sectors and economic activities. Examples include data envelopment analysis (DEA),62 big data for simulations and modeling,63 stochastic frontier analysis,64 network planning model,65 and others, such as stochastic simulation, simulation study, general equilibrium model, regression or multiple regression analysis, panel factor-augmented VAR model, or business simulation games (eg Joskow & Rose66; Musshoff & Hirschauer;67 Parker & Kirkpatrick68).
In general, methods for regulating economic activity can be divided into financial and process/technological ones. Specific financial methods of regulation that can and do find their use in the healthcare sector are price regulation (see, eg69), setting ratios such as rate of return (RoR)70 or ROI,53 and taxation.35,36 Process/technological methods include system throughput,71 maximum product output,72 or the certification system, on which the medical device sector is in fact based. The process associated with the certification of (not only) medical devices before their introduction on the market is often time-consuming and therefore expensive. This is one of the reasons why stakeholders strive to shorten this time by deriving and optimising various methods—see, for example, Vidal, Beuscart, and Chevallier,73 who conducted a review of adaptive methods applied in the phase of clinical trials.
Last but not least, our present study is best compared with that of Guerra-Bretaña and Flórez-Rendón,23 who use a similar methodology but have a different goal. While our study seeks to emphasise economic evaluation, Guerra-Bretaña and Flórez-Rendón are focused on innovation activity, using the PubMed database for their research. There are also the following intersections and differences between our research and that of Guerra-Bretaña and Flórez-Rendón.23 The first difference is that compared with the article of Guerra-Bretaña and Flórez-Rendón, we slightly modified the basic filter. We added the keyword “economic impact” and searched for the keywords in “all fields,” rather than only in the title and abstract fields. Second, the analysis of Scopus articles presented here contains a much larger ratio of outputs focused on HTA, ie, category II (43.5% vs 12.1%), while almost the same number of papers is in category III (30.4% vs 31.0%), and there is again a significant difference in category I (26.1% vs 56.9%). Despite the best efforts of the authors, it is possible that this search may have limits because the review articles are subject to potential bias, including the influence of the authors’ personal viewpoints, as well as gaps in literature searching practices that may lead to the omission of relevant research. Therefore, we state that the difference in the ratios of categories I and II may be due to the subjective perception of the content of the reviewed articles.
Conclusion
Regulation is a complex process that in many ways determine the environment where economic processes, among others, take place. In some cases, regulation shapes the environment beforehand; in others, regulation catches up with the shortcomings of the environment created by the participants’ innovative practices. Regulation is often a continuous process that moves between the extremes of insufficient regulation and over-regulation, while attempting to reach the optimal state of equilibrium that does not place unnecessary burden on the affected entities, including their efforts to innovate.
This article focused on illustrating the economic aspects of medical device regulation and highlighting new and current trends in this area (see mHealth or digital health). With regard to research questions concerning methods and indicators which are used to assess the economic impact of medical device regulation and impact on stakeholders, we can state that these methods are commonly used: cost-effectiveness analysis, cost-minimisation, analysis, cost-utility analysis, safety, efficacy, costs, cost effectiveness, compliance costs, return on investment, internal, rate of return, net present value, cost-minimisation analysis, cost-benefit analysis, budget impact analysis and return on investment, price, cost-performance ratio, value-based, reimbursement, and qualified estimate, incremental cost-effectiveness ratio, cost-per-QALY. With their use, the availability or cost of securing data is discussed in many cases, which is considered time consuming. Data availability is at the same time one of the critical factors in the development and distribution of medical devices. Other important critical factors are pressure of public demand to have new products available very fast, medical device for children, increase in AI solutions submitted to regulators and other mentioned in Chapter 3.
The mentioned critical points draw attention to problems that are currently being partially solved by methods such as new legislative conditions. For example, the implementation of the MDR (EU) 2017/745 in the countries of the European Union brings new challenges to changing processes, financial flows, increased costs. Sometimes there are also concerns about the possibility of ensuring these conditions, due to concerns about the unavailability of the regulatory body. However, the demand for greater patient safety is the challenge for innovation. Many of the above points still remain challenges that require a continuous solution.
Acknowledgments
The paper was supported by the project Excellence 2021 at the Faculty of Informatics and Management of the University of Hradec Kralove, Czech Republic.
Disclosure
The authors report no conflicts of interest in this work.
References
1. OECD. Measuring regulatory performance. OECD; September 21, 2020. Available from: http://www.oecd.org/gov/regulatory-policy/measuring-regulatory-performance.htm.
2. Faulkner A. Tissue engineered technologies: regulatory pharmaceuticalization in the European Union. Innov Eur J Soc Sci Res. 2012;25(4):389–408. doi:10.1080/13511610.2012.723333
3. FDA. Advancing regulatory science. U.S. Food & Drug Administration; October 4, 2019. Available from: https://www.fda.gov/science-research/science-and-research-special-topics/advancing-regulatory-science.
4. ITU. Using regulatory impact analysis to improve decision making in the ICT sector. International Telecommunication Union; Union internationale des télécommunications; 2014. Available from: http://handle.itu.int/11.1002/pub/80cc9798-en.
5. Maresova P, Hajek L, Krejcar O, Storek M, Kuca K. New regulations on medical devices in Europe: are they an opportunity for growth? Adm Sci. 2020;10(1):16. doi:10.3390/admsci10010016
6. Rothwell R, Zegveld WC. Reindustrialization and Technology. Sharpe; 1985.
7. Bonnin Roca J, O’Sullivan E. The role of regulators in mitigating uncertainty within the valley of death. Technovation. 2020:102157. doi:10.1016/j.technovation.2020.102157
8. Johnson JA. FDA regulation of medical devices. In: Medical Devices & the FDA: Regulation, User Fees & Tort Claims. Nova Science; Gazelle [distributor]; 2013:1–38.
9. Ganz RA. The impact of health care reform on innovation and new technology. Gastrointest Endosc Clin N Am. 2012;22(1):109–120. doi:10.1016/j.giec.2011.08.006
10. Pane J, Coloma PM, Verhamme KMC, Sturkenboom MCJM, Rebollo I. Evaluating the safety profile of non-active implantable medical devices compared with medicines. Drug Saf. 2017;40(1):37–47. doi:10.1007/s40264-016-0474-1
11. Maresova P, Rezny L, Peter L, Hajek L, Lefley F. Do regulatory changes seriously affect the medical devices industry? Evidence from the Czech Republic. Front Public Health. 2021;9:666453. doi:10.3389/fpubh.2021.666453
12. Medical devices in the EU: a global leader in safety, availability and innovation; November, 2015. Available from: https://www.medtecheurope.org/news-and-events/default/medical-devices-in-the-eu-a-global-leader-in-safety-availability-and-innovation/.
13. Wisła R, Sierotowicz T. Innovation in the pharmaceutical and medical technologies industries of Poland. WIPO World Intellectual Property Organization; 2018. Available from: https://www.wipo.int/edocs/pubdocs/en/wipo_pub_econstat_wp_45.pdf.
14. Yamaue H. Innovation of Diagnosis and Treatment for Pancreatic Cancer. SPRINGER Verlag; 2017.
15. MedTech Europe. MedTech Europe, from diagnosis to cure – homepage; 2020. Available from: https://www.medtecheurope.org/.
16. Council of the European Union. Regulation (EU) 2017/745 of the European Parliament and of the council of 5 April 2017 on medical devices, Amending Directive 2001/83/EC, Regulation (EC) No 178/2002 and Regulation (EC) No 1223/2009 and Repealing council directives 90/385/EEC and 93/42/EEC; 2017. Available from: https://eur-lex.europa.eu/legal-content/EN/ALL/?uri=CELEX%3A32017R0745.
17. EUnetHTA. HTA Core Model®. EUnetHTA; 2021. https://www.eunethta.eu/hta-core-model/.
18. Burlea-Schiopoiu A, Ferhati K. The managerial implications of the key performance indicators in healthcare sector: a cluster analysis. Healthcare. 2020;9(1):19. doi:10.3390/healthcare9010019
19. Brockis E, Marsden G, Cole A, Devlin N. A review of NICE methods across health technology assessment programmes: differences, justifications and implications. Office of Health Economics Research; 2016. Available from: https://www.ohe.org/system/files/private/publications/NICE%20HTA%20methods%20RP%20FINAL_0.pdf.
20. Echchakoui S. Why and how to merge Scopus and Web of Science during bibliometric analysis: the case of sales force literature from 1912 to 2019. J Mark Anal. 2020;8(3):165–184. doi:10.1057/s41270-020-00081-9
21. Moher D, Liberati A, Tetzlaff J, Altman DG; The PRISMA Group. Preferred reporting items for systematic reviews and meta-analyses: the PRISMA statement. PLoS Med. 2009;6(7):e1000097. doi:10.1371/journal.pmed.1000097
22. PRISMA. PRISMA flow diagram; 2021. Available from: http://prisma-statement.org/prismastatement/flowdiagram.aspx.
23. Guerra-Bretaña RM, Flórez-Rendón AL. Impact of regulations on innovation in the field of medical devices. Res Biomed Eng. 2018;34(4):356–367. doi:10.1590/2446-4740.180054
24. Gruska M, Aigner G, Altenberger J, et al. Recommendations on the utilization of telemedicine in cardiology. Wien Klin Wochenschr. 2020;132(23–24):782–800. doi:10.1007/s00508-020-01762-2
25. Wright SJ, Paulden M, Payne K. Implementing interventions with varying marginal cost-effectiveness: an application in precision medicine. Med Decis Making. 2020;40(7):924–938. doi:10.1177/0272989X20954391
26. Heidt B, Siqueira W, Eersels K, et al. Point of care diagnostics in resource-limited settings: a review of the present and future of PoC in its most needed environment. Biosensors. 2020;10(10):133. doi:10.3390/bios10100133
27. Plun-Favreau J, Immonen-Charalambous K, Steuten L, et al. Enabling equal access to molecular diagnostics: What are the implications for policy and health technology assessment? Public Health Genomics. 2016;19(3):144–152. doi:10.1159/000446532
28. Gelijns AC, Russo MJ, Hong KN, Brown LD, Ascheim DD, Moskowitz AJ. Dynamics of device innovation: implications for assessing value. Int J Technol Assess Health Care. 2013;29(4):365–373. doi:10.1017/S0266462313000561
29. Miesler T, Wimschneider C, Brem A, Meinel L. Frugal innovation for point-of-care diagnostics controlling outbreaks and epidemics. ACS Biomater Sci Eng. 2020;6(5):2709–2725. doi:10.1021/acsbiomaterials.9b01712
30. Markiewicz K, Van Til J, IJzerman M. Early assessment of medical devices in development for company decision making: an exploration of best practices. J Commer Biotechnol. 2017;23(2). doi:10.5912/jcb780
31. Blüher M, Saunders SJ, Mittard V, Torrejon Torres R, Davis JA, Saunders R. Critical review of European health-economic guidelines for the health technology assessment of medical devices. Front Med. 2019;6:278. doi:10.3389/fmed.2019.00278
32. Scannell P, Cormican K. Spinning out of control? How academic spinoff formation overlooks medical device regulations. J Technol Manag Innov. 2019;14(3):82–92. doi:10.4067/S0718-27242019000300082
33. Jogova M, Shaw J, Jamieson T. The regulatory challenge of mobile health: lessons for Canada. Healthc Policy Polit Santé. 2019;14(3):19–28. doi:10.12927/hcpol.2019.25795
34. Vallespin B, Cornet J, Kotzeva A. Ensuring evidence-based safe and effective mHealth applications. In: Evidence-Based Health Informatics. Studies in Health Technology and Informatics. IOS Press; 2016:248–261. doi:10.3233/978-1-61499-635-4-248
35. Singh MD, Russ J, Terzidis O. The impact of the Obamacare excise tax on innovation and entrepreneurship early empirical findings. Int J Innov Sci. 2015;7(2):75–90. doi:10.1260/1757-2223.7.2.75
36. Bolka EM. The medical device excise tax: an unfair burden. Indiana Law J. 2014;89(4):1691–1722.
37. White SK, Walters AN. Assessing risk by analogy: a case study of us medical device risk management policy. Health Risk Soc. 2018;20(7–8):358–378. doi:10.1080/13698575.2018.1554802
38. Howard J. Balancing innovation and medical device regulation: the case of modern metal-on-metal hip replacements. Med Devices Evid Res. 2016;9:267–275. doi:10.2147/MDER.S113067
39. Richards N, Hudson I. UK medicines regulation: responding to current challenges: UK medicines regulation: responding to current challenges. Br J Clin Pharmacol. 2016;82(6):1471–1476. doi:10.1111/bcp.13077
40. Bowsher K, Civillico EF, Coburn J, et al. Brain–computer interface devices for patients with paralysis and amputation: a meeting report. J Neural Eng. 2016;13(2):023001. doi:10.1088/1741-2560/13/2/023001
41. De Maria C, Di Pietro L, Díaz Lantada A, et al. Safe innovation: on medical device legislation in Europe and Africa. Health Policy Technol. 2018;7(2):156–165. doi:10.1016/j.hlpt.2018.01.012
42. Boudard A, Martelli N, Prognon P, Pineau J. Clinical studies of innovative medical devices: what level of evidence for hospital-based health technology assessment?: Innovative medical devices and level of evidence. J Eval Clin Pract. 2013;19(4):697–702. doi:10.1111/jep.12024
43. Sauerland S, Brockhaus AC, Fujita-Rohwerder N, Saad S. Approaches to assessing the benefits and harms of medical devices for application in surgery. Langenbecks Arch Surg. 2014;399(3):279–285. doi:10.1007/s00423-014-1173-y
44. Lennox K. Substantially unequivalent: reforming FDA regulation of medical devices. Univ Ill LAW Rev. 2014;2014(4):1363–1400.
45. Gardner J. A history of deep brain stimulation: technological innovation and the role of clinical assessment tools. Soc Stud Sci. 2013;43(5):707–728. doi:10.1177/0306312713483678
46. Altenstetter C. Medical device regulation and nanotechnologies: determining the role of patient safety concerns in policymaking: medical device regulation. Law Policy. 2011;33(2):227–255. doi:10.1111/j.1467-9930.2010.00337.x
47. Kale D. Mind the gap: investigating the role of collective action in the evolution of Indian medical device regulation. Technol Soc. 2019;59:101121. doi:10.1016/j.techsoc.2019.03.004
48. Munos B, Baker PC, Bot BM, et al. Mobile health: the power of wearables, sensors, and apps to transform clinical trials: mobile technology and clinical trials. Ann N Y Acad Sci. 2016;1375(1):3–18. doi:10.1111/nyas.13117
49. Onodera R, Sengoku S. Innovation process of mHealth: an overview of FDA-approved mobile medical applications. Int J Med Inf. 2018;118:65–71. doi:10.1016/j.ijmedinf.2018.07.004
50. Ravizza A, De Maria C, Di Pietro L, Sternini F, Audenino AL, Bignardi C. Comprehensive review on current and future regulatory requirements on wearable sensors in preclinical and clinical testing. Front Bioeng Biotechnol. 2019;7:313. doi:10.3389/fbioe.2019.00313
51. Drukker L, Noble JA, Papageorghiou AT. Introduction to artificial intelligence in ultrasound imaging in obstetrics and gynecology. Ultrasound Obstet Gynecol. 2020;56(4):498–505. doi:10.1002/uog.22122
52. Kesavan P, Dy CJ. Impact of health care reform on technology and innovation. Hand Clin. 2020;36(2):255–262. doi:10.1016/j.hcl.2020.01.008
53. Ibrahim N, Gillette N, Patel H, Peiris V. Regulatory science, and how device regulation will shape our future. Pediatr Cardiol. 2020;41(3):469–474. doi:10.1007/s00246-020-02296-0
54. Onur I, Söderberg M. The impact of regulatory review time on incremental and radical innovation: evidence from the high-risk medical device market. J Regul Econ. 2020;57(2):134–158. doi:10.1007/s11149-020-09401-4
55. Grennan M, Town RJ. Regulating innovation with uncertain quality: information, risk, and access in medical devices. Am Econ Rev. 2020;110(1):120–161. doi:10.1257/aer.20180946
56. Letourneur D, Joyce K, Chauvierre C, Bayon Y, Pandit A. Enabling MedTech translation in academia: redefining value proposition with updated regulations. Adv Healthc Mater. 2021;10(1):2001237. doi:10.1002/adhm.202001237
57. Vasiljeva K, Van Duren BH, Pandit H. Changing device regulations in the European Union: impact on research, innovation and clinical practice. Indian J Orthop. 2020;54(2):123–129. doi:10.1007/s43465-019-00013-5
58. Wilkinson B, van Boxtel R. The medical device regulation of the European Union intensifies focus on clinical benefits of devices. Ther Innov Regul Sci. 2020;54(3):613–617. doi:10.1007/s43441-019-00094-2
59. Dai Z, Ma Y, Li Q. China’s particle therapy equipment market: opportunities outweigh challenges. Front Public Health. 2020;8:602776. doi:10.3389/fpubh.2020.602776
60. Health Information and Quality Authority. Guidelines for the economic evaluation of health technologies in Ireland; September 28, 2020. Available from: https://www.hiqa.ie/reports-and-publications/health-technology-assessment/guidelines-economic-evaluation-health.
61. Di Pietro L, Botte E, Granati R, et al. Teaching design standards and regulations on medical devices through a collaborative project-based learning approach. Int J Eng Educ. 2019;35(6A):1803–1815.
62. Adler N, Liebert V. Joint impact of competition, ownership form and economic regulation on airport performance and pricing. Transp Res Part Policy Pract. 2014;64:92–109. doi:10.1016/j.tra.2014.03.008
63. Erdman AG, Keefe DF, Schiestl R. Grand challenge: applying regulatory science and big data to improve medical device innovation. IEEE Trans Biomed Eng. 2013;60(3):700–706. doi:10.1109/TBME.2013.2244600
64. Franco C, Pieri F, Venturini F. Product market regulation and innovation efficiency. J Product Anal. 2016;45(3):299–315. doi:10.1007/s11123-015-0441-3
65. Huang Y, Söder L. Evaluation of economic regulation in distribution systems with distributed generation. Energy. 2017;126:192–201. doi:10.1016/j.energy.2017.03.019
66. Joskow PL, Rose NL. The effects of economic regulation. In: Schmalensee R, Willig R, editors. Handbook of Industrial Organization.
67. Musshoff O, Hirschauer N. Using business simulation games in regulatory impact analysis – the case of policies aimed at reducing nitrogen leaching. Appl Econ. 2014;46(25):3049–3060. doi:10.1080/00036846.2014.920482
68. Parker D, Kirkpatrick C. Measuring Regulatory Performance. The Economic Impact of Regulatory Policy: A Literature Review of Quantitative Evidence. OECD; 2012.
69. Radcliffe JC. Water recycling in Australia – during and after the drought. Environ Sci Water Res Technol. 2015;1(5):554–562. doi:10.1039/C5EW00048C
70. Simões P, Marques RC. Influence of regulation on the productivity of waste utilities. What can we learn with the Portuguese experience? Waste Manag. 2012;32(6):1266–1275. doi:10.1016/j.wasman.2012.02.004
71. Keyaerts N, Delarue E, Rombauts Y, D’haeseleer W. Impact of unpredictable renewables on gas-balancing design in Europe. Appl Energy. 2014;119:266–277. doi:10.1016/j.apenergy.2014.01.011
72. Cossent R, Gómez T, Olmos L. Large-scale integration of renewable and distributed generation of electricity in Spain: current situation and future needs. Energy Policy. 2011;39(12):8078–8087. doi:10.1016/j.enpol.2011.09.069
73. Vidal C, Beuscart R, Chevallier T Contribution of methodologies adapted to clinical trials focusing on high risk medical devices: In:
74. Laslett LJ, Alagona P, Clark BA, et al. The worldwide environment of cardiovascular disease: prevalence, diagnosis, therapy, and policy issues. J Am Coll Cardiol. 2012;60(25):S1–S49. doi:10.1016/j.jacc.2012.11.002
75. Dunn J, Runge R, Snyder M. Wearables and the medical revolution. Pers Med. 2018;15(5):429–448. doi:10.2217/pme-2018-0044
76. Becker F, van El CG, Ibarreta D, et al. Genetic testing and common disorders in a public health framework: how to assess relevance and possibilities. Eur J Hum Genet. 2011;19(S1):S6–S44. doi:10.1038/ejhg.2010.249
77. Herder M. When everyone is an orphan: against adopting a U.S.-styled orphan drug policy in Canada. Account Res. 2013;20(4):227–269. doi:10.1080/08989621.2013.793120
78. Pirnay JP, Vanderkelen A, De Vos D, et al. Business oriented EU human cell and tissue product legislation will adversely impact Member States’ health care systems. Cell Tissue Bank. 2013;14(4):525–560. doi:10.1007/s10561-013-9397-6
79. Hull G, Pasquale F. Toward a critical theory of corporate wellness. BioSocieties. 2018;13(1):190–212. doi:10.1057/s41292-017-0064-1
80. Hall PS, Mitchell ED, Smith AF, et al. The future for diagnostic tests of acute kidney injury in critical care: evidence synthesis, care pathway analysis and research prioritisation. Health Technol Assess. 2018;22(32):1–274. doi:10.3310/hta22320
81. Kazzazi F, Pollard C, Tern P, Ayuso-Garcia A, Gillespie J, Thomsen I. Evaluating the impact of Brexit on the pharmaceutical industry. J Pharm Policy Pract. 2017;10(1):32. doi:10.1186/s40545-017-0120-z
82. Chan JCN, Lim LL, Wareham NJ, et al. The Lancet Commission on diabetes: using data to transform diabetes care and patient lives. Lancet. 2020;396(10267):2019–2082. doi:10.1016/S0140-6736(20)32374-6
83. Dabbous M, Chachoua L, Caban A, Toumi M. Managed entry agreements: policy analysis from the European Perspective. Value Health. 2020;23(4):425–433. doi:10.1016/j.jval.2019.12.008
84. Orubu ESF, Ching C, Zaman MH, Wirtz VJ. Tackling the blind spot of poor-quality medicines in Universal Health Coverage. J Pharm Policy Pract. 2020;13(1):40. doi:10.1186/s40545-020-00208-4
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.