Back to Archived Journals » Oncolytic Virotherapy » Volume 3
Critical analysis of an oncolytic herpesvirus encoding granulocyte-macrophage colony stimulating factor for the treatment of malignant melanoma
Authors Hughes T, Coffin R, Lilley C, Ponce R, Kaufman H
Received 6 August 2013
Accepted for publication 11 November 2013
Published 15 January 2014 Volume 2014:3 Pages 11—20
DOI https://doi.org/10.2147/OV.S36701
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 5
Tasha Hughes,1 Robert S Coffin,2 Caroline E Lilley,2 Rafael Ponce,2 Howard L Kaufman1
1Departments of General Surgery and Immunology and Microbiology, Rush University Medical Center, Chicago IL, 2BioVex, Inc, a subsidiary of Amgen, Inc, Sherman Oaks, CA, USA
Abstract: Oncolytic viruses that selectively lyse tumor cells with minimal damage to normal cells are a new area of therapeutic development in oncology. An attenuated herpesvirus encoding the granulocyte-macrophage colony stimulating factor (GM-CSF), known as talimogene laherparepvec (T-VEC), has been identified as an attractive oncolytic virus for cancer therapy based on preclinical tumor studies and results from early-phase clinical trials and a large randomized Phase III study in melanoma. In this review, we discuss the basic biology of T-VEC, describe the role of GM-CSF as an immune adjuvant, summarize the preclinical data, and report the outcomes of published clinical trials using T-VEC. The emerging data suggest that T-VEC is a safe and potentially effective antitumor therapy in malignant melanoma and represents the first oncolytic virus to demonstrate therapeutic activity against human cancer in a randomized, controlled Phase III study.
Keywords: granulocyte-macrophage colony stimulating factor, herpesvirus, melanoma, oncolytic virus, treatment
Introduction
Oncolytic viruses represent a novel form of cancer therapy that are uniquely designed to treat established tumors and are usually injected directly into tumors, obviating the need to have defined tumor-associated antigens. Oncolytic virotherapy has three unique features relating to mechanism and immunity that make it a promising frontier in the treatment of cancer. First, oncolytic viruses provide a means for highly selective targeting to tumor cells. This occurs in some cases through innate properties of native viral particles that may be trophic for tumor cells or able to selectively replicate in tumor cells where there is an abundance of nucleic acids or where the endogenous antiviral response is impaired. In other cases, oncolytic viruses can be engineered through deletion of nonessential viral genes or insertion of tumor targeting sequences to provide the virus with the property of either tumor-selective infection or tumor-selective replication. The second critical feature of oncolytic viruses is their ability to induce tumor cell lysis. Viruses with lytic potential that selectively replicate in tumor cells readily induce cell death, and progeny viral particles may then be able to enter nearby cells, resulting in a cascade of infection and cell lysis within an established tumor mass. The third mechanism that oncolytic viruses utilize to treat cancer is induction of local and systemic antitumor immunity. Local lysis of tumor cells will initiate a local immune response wherein antigen-presenting cells will take up dying tumor cells, tumor antigens, and viral particles to activate a local immune response. These responses can initiate antigen-specific tumor rejection and may also promote a bystander effect in which local release of perforins and granzyme B by activated T-cells and natural killer cells can kill uninfected tumor cells. In some cases, the infection can lead to generation of systemic tumor-specific immunity, which can then mediate tumor rejection at sites distant from the oncolytic virus injection. Thus, oncolytic viruses provide a highly selective method for targeting tumor cells and promote direct lytic destruction of the tumor and initiation of patient-specific, tumor-specific, and antigen-specific antitumor immunity.
To date, several native and genetically modified viruses have been utilized as oncolytic agents for cancer therapy. These include adenoviruses, poxviruses, herpesviruses, reoviruses, coxsackieviruses, Newcastle disease virus, and others.1 These oncolytic viruses have demonstrated therapeutic activity against cancer in murine tumor models and many have entered into early-phase clinical trials. An attenuated herpesvirus encoding granulocyte-macrophage colony stimulating factor (GM-CSF), known as talimogene laherparepvec (T-VEC), has been extensively evaluated in clinical trials, including a prospective, multi-institutional, randomized Phase III study. T-VEC is optimized for tumor-selective replication, minimal pathogenicity, and induction of immune responses through local production of GM-CSF. This review describes the basic biology of T-VEC, characterizes the preclinical and clinical studies, discusses the role of GM-CSF expressed from T-VEC, provides a critical analysis of the role of T-VEC in the treatment of melanoma, and suggests potential future directions for research and development.
Basic biology of T-VEC
T-VEC is based on a herpes simplex type 1 virus (HSV-1). HSV-1 is a member of the alphaherpesvirus family and contains a large 152 kb, double-stranded DNA genome.1 The genome consists of two covalently linked components, designated as long (L) and short (S). Each component consists of unique sequences bracketed by inverted repeats (Figure 1A). The DNA exists in a central capsid that is surrounded by a glycoprotein-rich envelope. Between the capsid and the envelope is the tegument, which carries certain viral proteins into infected cells to aid the priming of target cells for productive infection.1 The virus infects mucosal epithelial cells and utilizes a variety of receptors for cell entry, including herpesvirus entry mediator and various nectins. Herpesvirus entry mediator is a tumor necrosis factor superfamily member with expression on natural killer and cluster of differentiation (CD)8+ T-cells. Herpesvirus entry mediator is also expressed at lower levels on CD4+ T-cells, dendritic cells, B-cells, fibroblasts, and epithelial cells. Nectins are part of the immunoglobulin gene superfamily and are expressed on a variety of cell types, including epithelial cells, neurons, and fibroblasts.2 The ability to use a number of different receptors, at least one of which is expressed on most cell types, means that HSV can enter a very wide variety of cell types, including most cancers. Following infection of a permissive cell type, the virus replicates in the host cell nucleus but does not integrate into the genome, so there is no risk of insertional mutagenesis.3
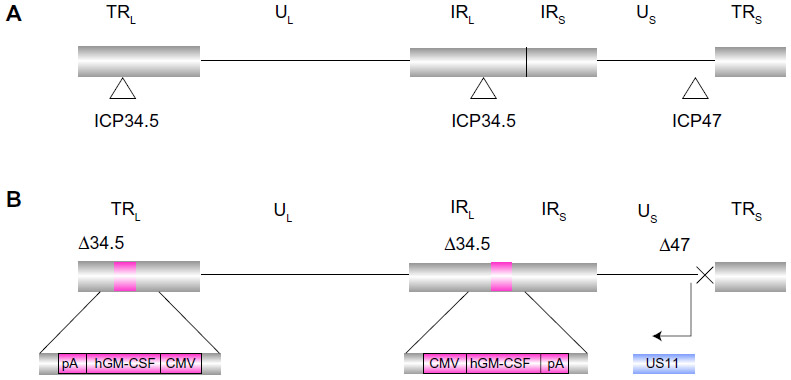 | Figure 1 Schematic diagram showing the herpes simplex type 1 virus from which T-VEC was developed (A) and the modified vector known as talimogene laherparepvec (T-VEC) (B). Viral ICP34.5 neurovirulence gene and the ICP47 immunogenicity gene have been deleted in T-VEC. The human GM-CSF gene driven by a CMV promoter has been inserted into two deleted ICP34.5 loci and the deleted ICP47 results in early activation of the US11 promoter. |
The lytic cycle consists of a cascade of gene expression typically resulting in replication, lysis of the infected cell, and release of viral progeny within 15 hours. In the in vivo human situation, herpes simplex infection begins with exposure to mucosal surfaces followed by infection of the axonal termini of sensory neurons. This results in transport to the cell body in the spinal ganglia where latency is established, with reactivation occurring usually infrequently thereafter, months to years later.4 Approximately 60%–80% of the human population has antibodies to HSV-1, and therefore probably harbor latent HSV, although only a small proportion regularly if ever reactivate the virus to show symptoms of the disease.5
HSV-1 causes the typical “cold sore” or fever blister. Rarely, infection with HSV can cause more serious complications such as retinitis or encephalitis.5 During latency, the virus does not replicate, but remains in a dormant state during which it only expresses RNA species, named latency-associated transcripts, from a small portion of the genome. The functions of these latency-associated transcripts still remain largely unknown, although roles in neuronal survival and transcriptional regulation through micro (mi)RNA expression seem likely.6,7 While the mechanisms resulting in HSV reactivation are not well understood, recurrence is often associated with various kinds of stress, such as exposure to ultraviolet light, fever, or hormonal changes.4 Following viral reactivation, viral particles travel in an antegrade fashion to epithelial cells at the site of the initial infection (usually the oral mucosa for HSV-1 or the genital mucosa for HSV-2), where the characteristic herpetic lesions occur. HSV-1 is immunogenic and induces neutralizing antibodies and a T-cell response, and thus entry into a latent state where no viral proteins are expressed enables the virus to hide from the immune system and persist in the long-term, assumed to usually be for the lifetime of the host.8
T-VEC (formerly known as OncoVEXGM-CSF) is an attenuated strain of HSV-1 encoding human GM-CSF and has been extensively evaluated as an oncolytic cancer therapeutic in preclinical models and in clinical trials. T-VEC is derived from the new HSV strain, JS1, and is attenuated by deletion of nonessential genes encoding the HSV proteins ICP (infected cell protein) 34.5 and ICP47, modified to express GM-CSF, and expresses US (unique short) 11 as an immediate early gene (Figure 1B). As discussed below, the genetic modifications in T-VEC limit viral pathogenesis in normal tissues, while allowing selective replication in tumor tissues and enhancing the immunogenicity of infected cells.
Activation of the type 1 interferon (IFN-α/β) signaling cascade is central to cellular defense following HSV-1 infection.9 One of the many activities regulated by IFNs is induction of protein kinase R synthesis. Upon detection of viral RNA, protein kinase R is activated via dimerization and autophosphorylation.10 Activated protein kinase R regulates cellular RNA translation via phosphorylation of the alpha subunit of eukaryotic initiation factor 2 (eIF2), and also induces apoptosis and autophagy. Collectively, these processes protect the infected cell by blocking viral protein synthesis prior to completion of the viral life cycle, inducing death of the infected cell, sequestering viral DNA, and presenting viral antigens to initiate an adaptive immune response.9,11–15 Thus, the response of normal cells to HSV-1 infection drives both intracellular and systemic responses designed to limit viral replication and damage to normal cells, and to clear infection both locally and systemically.
The HSV-1 protein ICP34.5 has a number of identified activities, including viral evasion of the host antiviral defense mediated by protein kinase R. Specifically, the C-terminal domain of ICP34.5 recruits protein phosphatase 1 and dephosphorylates the alpha subunit of eIF2, thereby restoring protein translation and thus viral replication. Through a separate binding region, ICP34.5 can also bind Beclin-1 to interfere with autophagy and immune-mediated clearance of virally infected cells.16–19
Because T-VEC has been engineered to be functionally deficient in ICP34.5, normal cells infected with this virus should be fully competent to mount an effective antiviral defense. In contrast with normal cells, tumors are often impaired in various pathways affecting host immunity or translational control, including the IFN/protein kinase R response, so may be impaired in their ability to mount an effective antiviral defense even in the absence of ICP34.5.20–25 Other functions of ICP34.5 that may contribute to HSV-1 replication and virulence include binding to proliferating cell nuclear antigen and blocking viral egress.26–28 Whether and how these functional characteristics contribute to preferential viral replication in and destruction of cancer cells is not fully established; however, available nonclinical pharmacology models and toxicology studies confirm tumor-selective destruction in animals and humans exposed to ICP34.5-deficient HSV-1.29–31
In addition to conferring tumor-selective replication, ICP34.5 underlies the neurovirulence of HSV-1, and HSV-1 strains lacking ICP34.5 are 10,000-fold to 1,000,000-fold less neurovirulent than wild-type HSV-1.32–35 Although the precise mechanisms for ICP34.5-mediated neurovirulence have not been established, ICP34.5 can bind Beclin-1, a key regulator of autophagy, and Beclin binding domain-deficient HSV-1 demonstrates enhanced autophagy activity and adaptive immune responses, and reduced neurovirulence as compared with wild-type HSV-1.17 These results indicate that, by binding Beclin-1, ICP34.5 may drive neurovirulence by reducing autophagy and impairing systemic antiviral immunity.19
In wild-type HSV-1, ICP47 impairs transporter associated with antigen presentation (TAP) and blocks the binding of antigenic peptides, and subsequent CD8+ T cell immunity.36–39 Removal of ICP47 thus permits proper antigen processing (for both viral and tumor antigens) and is intended to aid the generation of a productive T-cell adaptive immune response. Additionally, deletion of ICP47 increases viral growth in tumor cells through modulation of the herpes US11 promoter, which results in immediate-early, rather than late, US11 gene expression.40 US11 expression appears to restore efficient replication of ICP34.5-deficient HSV-1 in tumors, without restoring neurovirulence or loss of tumor selectivity.41 T-VEC also encodes human GM-CSF, which is thought to enhance immunogenicity through activation of local macrophages and dendritic cells, resulting in an enhanced cellular immune response both locally and systemically.31
Role of GM-CSF in T-VEC immunotherapy
While T-VEC incorporates local GM-CSF production in the treatment strategy, the general role of GM-CSF as monotherapy or adjuvant therapy for melanoma is controversial, and merits brief review. More complete reviews of GM-CSF as a cancer therapy are available elsewhere.42–44 GM-CSF is an immune-modulating cytokine that stimulates macrophage activation, recruits peripheral blood monocytes, and activates dendritic cells, promoting antigen presentation and development of a T-cell-mediated immune response. The theoretical rationale for including GM-CSF as part of the oncolytic virus therapy strategy is that virus-induced lysis of tumor cells will attract host macrophages and dendritic cells to the tumor microenvironment, where these professional antigen-presenting cells will engulf dying tumor cells and initiate antigen presentation of viral antigens and potentially tumor-associated antigens. This process can be enhanced by high levels of local GM-CSF, which can promote loco-regional, and possibly systemic, adaptive immune responses against the injected tumor cells.
Although GM-CSF is controversial for the immunotherapy of cancer, the cytokine plays an important role in the recruitment and regulation of antigen-presenting cells, such as dendritic cells.45 Thus, addition of GM-CSF as an adjuvant to a vaccine or oncolytic immunotherapy might be expected to have a beneficial impact on induction of tumor-specific immunity. In the case of T-VEC, the virus infects tumor cells, resulting in cell death and release of potential tumor-associated antigens. Dying cells and soluble antigen could be taken up by local dendritic cells and other antigen-presenting cells for presentation to T-cells in draining lymph nodes. This could then prime a T-cell response capable of tumor cell recognition at both injected and noninjected sites. This was the rationale for including GM-CSF in the viral backbone, and the improved therapeutic response of herpesviruses containing GM-CSF has been confirmed in preclinical tumor models, as detailed below.
Preclinical data supporting T-VEC in immunotherapy for cancer
A series of preclinical studies contributed to the development of T-VEC. The JS1 strain of HSV-1 was selected for vector development based on in vitro studies suggesting superior tumor cell killing and at lower doses than observed with other strains, such as the laboratory strain HSV-1 17 syn+.29 Additional in vitro studies comparing HSV-1 17 syn+ and JS1 strains, both deficient in the ICP34.5 neurovirulence factor (ICP34.5−), demonstrated that JS1/ICP34.5− was superior at killing a number of tumor cell lines compared with HSV-1 17 syn+/ICP34.5−. The cell lines in which JS1/ICP34.5− induced superior killing included colorectal adenocarcinoma, prostate adenocarcinoma, malignant melanoma, and glioblastoma.29 T-VEC is less virulent than wild-type HSV-1, which was confirmed in vitro and in vivo by demonstrating limited growth in cell cultures known to support growth of wild-type HSV-1, and by the absence of pathogenicity when injected into mice via intravenous or subcutaneous injection (Coffin, unpublished observations, 2010). The absence of productive replication in normal eukaryotic cells confers a significant advantage to this strain as a therapy for patients with cancer.29 T-VEC demonstrated approximately 10,000-fold less neurovirulence at the estimated LD50 (lethal dose to 50% of patients) following intracerebral injection as compared with that reported for wild-type HSV-1 (MacLean et al, unpublished observations, 1991).
Deletion of ICP47 resulted in enhanced major histocompatibility complex class I expression in infected human breast cancer cell lines.29 The JS1/ICP34.5−/ICP47− attenuated strain also resulted in enhanced therapeutic activity against established tumors, including HT-29 colon adenocarcinoma, Fadu (hypopharyngeal) carcinoma, and U87MG glioma, in nude Balb/c mice.29 Recent pharmacologic data using in vitro tumor cell killing assays indicate broad antitumor activity across a range of solid tumor types, but not in hematologic malignancies (Amgen, unpublished data, 2013). Deletion of ICP47 alters the expression of US11 from late to immediate early, as described previously. Based on these murine models, it is thought that this early expression of US11 contributes to the enhanced tumor lysis seen with the ICP47− strain, given that ICP47 does not function in mice.29 Using a strain of JS1/ICP34.5−/ICP47− encoding murine GM-CSF, superior therapeutic activity was observed in a bilateral flank tumor model using the A20 cell line. In this model, tumors established in the right flank were injected with JS1/ICP34.5−/ICP47− or JS1/ICP34.5−/ICP47−/murine (m) GM-CSF, and the lesions on the left flank were not injected. While the injected tumors showed comparable regression, the contralateral noninjected tumors in the left flank showed regression only in mice treated with JS1/ICP34.5−/ICP47−/mGM-CSF virus. Splenocytes cultured from JS1/ICP34.5−/ICP47−/mGM-CSF virus-treated animals had higher IFN-γ expression in vitro, suggesting an augmentation of cellular immunity.29 Based on these studies, the JS1/ICP34.5−/ICP47− virus encoding human GM-CSF was generated for clinical trials.
Clinical data for oncolytic herpesvirus encoding GM-CSF
In a Phase I clinical trial, T-VEC was administered to 30 patients with melanoma, breast, gastrointestinal, or head and neck malignancies with accessible subcutaneous or cutaneous metastases.46 The study was designed to evaluate the safety profile, dosing regimen, and objective response rate of the vaccine. Thirteen patients received single doses of 106, 107, or 108 plaque-forming units (pfu)/mL and 17 patients received various multidose regimens. Local erythema, inflammation, and pyrexia were the most common side effects across all doses. HSV-seronegative patients developed more severe local injection site reactions. The major dose-limiting toxicity was local injection site reactions and, based on these, 107 pfu/mL was determined to be the maximum tolerated dose in seronegative patients. HSV-naive patients who received a single dose of 106 pfu/mL seroconverted and were then able to tolerate subsequent doses of 108 pfu/mL, establishing this as a safe and tolerable dosing regimen for future studies. Viral clearance was also evaluated by injection site swabs. Virus was detected at the injection site up to 2 weeks post-injection in patients receiving a single dose, supporting the 2–3-week dosing schedule that was implemented in the multidose cohorts.
Post-treatment biopsies of injected lesions suggested that T-VEC could induce significant tumor necrosis. In the above Phase I trial, a subset of 19 post-treatment biopsies contained residual tumor. Of these, 14/19 had evidence of tumor necrosis or apoptosis. These same biopsies were then stained for the presence of HSV. All 14 specimens containing significant tumor necrosis also stained positive for HSV, while HSV was rarely detected in non-necrotic or non-tumor tissue. Additionally, non-tumor tissue evaluated in these biopsies did not show any evidence of necrosis. These post-treatment biopsies support both the biological effectiveness and tumor-specific effects of the injections. In this Phase I trial, stable disease was noted in 3/26 patients for whom follow-up data were available, with no patients having a complete or partial response during the study period.
A Phase II clinical trial of T-VEC evaluated 50 patients with unresectable stage IIIc or IV melanoma receiving 106 pfu/mL T-VEC injections followed 3 weeks later by 108 pfu/mL T-VEC given by intratumoral administration every 2 weeks for up to 24 treatments. Safety parameters, clinical activity, and survival were reported based on a median follow-up of 18 months. In this trial, the vaccine was again well tolerated, with toxicity limited to low-grade fever and local injection site reactions, noted in 85% of patients. Although not a primary outcome in this study, injection sites were also swabbed for evidence of viral shedding. Consistent with the findings of the Phase I trial, only one swab from one patient (from a total of 28 patients who provided swab samples) was positive at any time point tested.
In this Phase II study, eight patients (16%) had progressive disease before receiving four injections and were subsequently removed from the study. The overall objective response rate among the remaining patients was 26%, with eight patients having a complete response and an additional five patients having a partial response by standard Response Evaluation Criteria In Solid Tumors criteria. Two additional patients were rendered disease-free by surgical resection (ie, a surgical complete response). One-year survival was 58% for all patients, and 40% in a subgroup analysis of stage IVc patients, with a median survival of >16 months in all the patients. Among patients who had a partial response, complete response, or surgical complete response, the 1-year survival was 93%.
A dose-escalation, open-label Phase I/II trial of 17 patients with stage III or IV squamous cell carcinoma of the head and neck was conducted to evaluate the combined effect of T-VEC and chemoradiation on locoregional disease control. All patients were treated with an initial dose of 106 pfu/mL T-VEC followed by three doses of either 106 pfu/mL, 107 pfu/mL, or 108 pfu/mL T-VEC by intratumoral injection.46 The primary endpoint was safety and secondary endpoints included response rate, relapse rate, and survival. In this study, all patients completed the dosing schedule without dose-limiting toxicity, supporting the safety and tolerability of the injections in patients with squamous cell carcinoma of the head and neck. All patients experienced at least one adverse event that was considered treatment-related, although 86% of these events were grade I or II. More severe adverse events (grade III or IV) were noted during the study period, but were determined by the investigators to be unrelated to treatment. Across all treatment cohorts, 4/17 (23.5%) patients had a complete response and 10/17 (58.8%) had a partial response. The locoregional control rate was 100% at all doses. Disease-specific survival was 82.4% and overall survival was 70.6% at a median follow-up of 29 months. The results of this small trial support the safety and tolerability of T-VEC in patients with squamous cell carcinoma of the head and neck, and suggest a potential therapeutic benefit of this agent in combination with chemotherapy and radiation treatment.
Based on the safety profile and encouraging clinical responses seen in these early trials, a multi-institutional, prospective, randomized Phase III clinical trial has been conducted in 439 patients enrolled with unresectable stage IIIb, IIIc, or IV melanoma.47 Melanoma is considered to be a highly immunogenic cancer based on the identification of defined melanoma-associated antigens, the infrequent regression of both primary lesions and metastatic disease, and the therapeutic response to immunotherapy agents, such as high-dose interleukin-(IL)-2 and ipilimumab (an anti-cytotoxic T lymphocyte-associated antigen 4 [CTLA] monoclonal antibody) for stage IV melanoma.48,49 The inherent immunogenicity of melanoma has made vaccine development a major goal, and recently a prospective, randomized clinical trial did demonstrate an improvement in progression-free survival for patients treated with a human leukocyte antigen (HLA)-A2-restricted gp (glycoprotein)100 peptide vaccine administered with high-dose IL-2 compared with IL-2 alone.50 Eligible patients with accessible lesions measuring at least 1 cm were randomized in a 2:1 fashion to T-VEC, administered at an initial dose of 106 pfu/mL followed by 108 pfu/mL every 2 weeks for up to 24 doses by intratumoral injection, or to recombinant GM-CSF, administered at 125 μg/kg by subcutaneous injection every 14 days in each 28-day cycle. In the treatment arm, all accessible lesions could be injected (up to 4 cc of virus per visit) and any new lesions that developed while patients were enrolled could also be injected. Patients were removed from the study if they had a complete response, developed predefined high-grade toxicity, or had significant disease progression. Patients were monitored with complete body imaging, and the primary endpoint was a durable disease response lasting at least 6 months. Secondary endpoints were disease-free and overall survival. Duration of response and quality of life parameters were also collected as part of the protocol.47
The study completed accrual in June 2011 and follow-up is ongoing. There were 436 patients in the intent-to-treat population. Based on the 2:1 randomization scheme, 295 (68%) patients received T-VEC and 141 (32%) received GM-CSF. The median age of the patients was 63 years and 57% were male. Of the 436 patients, 30% had stage III, 37% had stage IV M1a, 21% had stage IV M1b, and 22% had stage IV M1c disease. T-VEC was well tolerated, with fatigue, chills, and fever being the most commonly reported adverse events. Serious adverse events were reported in 26% of T-VEC patients and 13% of GM-CSF patients. Individual grade III or greater events accounted for less than 3% of the adverse events in either arm. The primary endpoint was durable response rate, defined as partial or complete response measured within 12 months of starting treatment and duration of this response for at least 6 months once achieved. The durable response rate was 16% in the T-VEC arm compared with 2% in the GM-CSF arm (P<0.0001). Improvement in durable response rate was seen for T-VEC-treated patients at all stages, but was most pronounced for stage III and IV M1a patients. In this subset of patients, there was a 33% and 16% durable response rate, respectively, for T-VEC patients, compared with 0% and 2% responders amongst patients treated with GM-CSF. The reason why T-VEC may be better in stage III/IV M1a patients is not known. T-VEC induces antitumor activity through a direct lytic effect and induction of antitumor immunity. It is possible that the lytic effect may be more prominent or potent and, thus, earlier-stage lesions, which are generally more accessible for direct injection, might respond better. The immune response may also require intact host immunity, which becomes weakened as the disease progresses. Thus, induction of tumor immunity may also be more challenging in advanced disease. Further work is needed to clarify why these earlier tumors respond better, but this may also be very promising as it may prevent patients with earlier-stage disease from progressing to advanced disease or delay disease progression.
The best overall response rate was 26% for patients treated with T-VEC compared with 6% for subjects receiving GM-CSF. The interim overall survival demonstrates a trend favoring T-VEC (hazards ratio 0.79; 95% confidence interval 0.61–1.02), but final survival data are not mature at the time of this report.51 Results of clinical data currently available for T-VEC are summarized in Table 1.
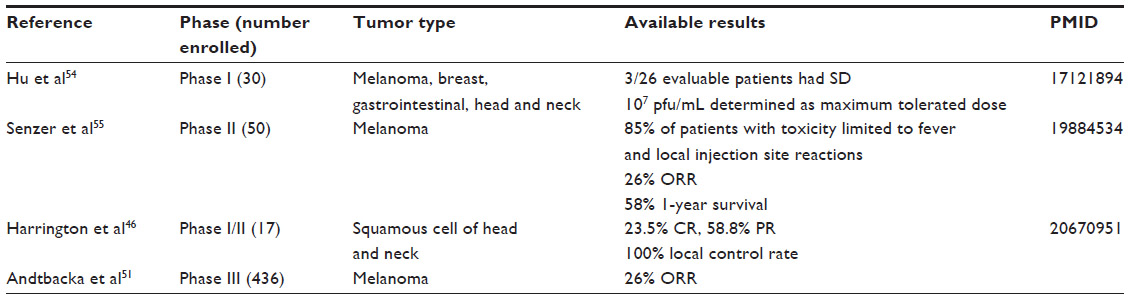 | Table 1 Phase I, II, and III clinical trials of talimogene laherparepvec |
Induction of antitumor immunity by T-VEC
Analysis of a subset of patients enrolled in the Phase II clinical trial in melanoma confirmed the presence of local and distant antitumor immune responses following administration of T-VEC. Eleven patients underwent biopsy after their sixth intratumoral injection, and tissue was compared with that of uninjected patients with metastatic melanoma.52 Additionally, peripheral blood mononuclear cells were collected from study patients, non-study melanoma patients, and healthy controls. Following treatment, tumors demonstrated extensive necrosis and lymphocyte infiltration compared with melanomas that were not injected with T-VEC. T-cells present among tumor-infiltrating lymphocytes revealed a higher proportion of activated (CD45RO+) and antigen-specific (melanoma antigen recognized by T-cells 1 [MART 1]) T-cells compared with peripheral blood mononuclear cell-derived T-cells. Further analysis found that, among tumor-infiltrating lymphocytes, there was an increased number of CD8+ T-cells expressing CD25, HLA-DR, and PD (programmed death)-1 compared with peripheral blood mononuclear cells in the same patient. Tumor-infiltrating lymphocytes also contained an increase in T-cells expressing perforin and granzyme B. In addition, injected lesions demonstrated a decrease in regulatory CD4+FoxP3+ T-cells, suppressor CD8+FoxP3+ T-cells, and myeloid-derived suppressor cells. These findings suggest that there is induction of local activated T-cell responses and inhibition of immune suppressive factors following T-VEC administration.52
Evaluation of tumor cells from injected and noninjected lesions in the same patient revealed a qualitatively similar response at both sites, with a predominant MART-1-specific CD8+ T-cell response. There was, however, a quantitative difference, with fewer activated MART-1-specific T-cells and more regulatory CD4+FoxP3+ T-cells in noninjected tumor sites compared with injected lesions. While more research is needed to better understand the mechanisms through which antitumor immunity is induced with T-VEC, these data do support the presence of a cell-mediated, antigen-specific antitumor immune response in patients treated with T-VEC.
Critical analysis and future directions
The Phase III clinical trial is the first prospective, randomized, clinical trial to demonstrate a clinical benefit of an oncolytic virus in the treatment of human cancer. Further work is needed to better understand this vector’s antitumor effects through investigation of how the virus both mediates cell death and induces antitumor immunity. There is some evidence supporting a role for immune induction and so a natural next step would be to consider combining T-VEC with other immunotherapy strategies, especially those designed to promote expansion of CD8+ T-cell responses, such as the anti-CTLA-4 (ipilimumab), anti-PD-1, or anti-PD ligand 1 (L1) monoclonal antibodies. In fact, a Phase I/II clinical trial of T-VEC and ipilimumab is already in progress.53
Another potential approach is to evaluate the role of T-VEC as neoadjuvant therapy, particularly in patients with in-transit metastases. These patients are often difficult to treat, and given the particularly strong effects observed in the unresectable stage III melanoma patients in the Phase III clinical trial,51 a study of T-VEC in patients with in-transit melanoma metastasis would be warranted. In addition, T-VEC could be tested in other tumor types and with other standard cancer therapeutics, as supported by the high response rate observed in the previously discussed Phase I/II trial of head and neck cancer patients.46 In addition to head and neck cancer, T-VEC could potentially be utilized for other tumors, provided the tumors are accessible for direct injection. The use of interventional radiology or surgical approaches to identify tumor masses for injection could potentially allow any tumor to be evaluated for treatment with the oncolytic virus. Intravenous injection is also possible but, to date, no clinical trials have been conducted to determine the safety and feasibility of an intravenous approach. Different tumors may have different lytic responses to virus delivery based on the innate antiviral response of the parent tumor, changes within the transformed cells, and impact of other oncogene and tumor suppressor gene activity within specific tumor types. The mechanism of T-VEC-induced cytotoxicity and the status of the tumor antiviral cell response should be a high priority for future investigation.
One of the most notable and desirable features of T-VEC is the relatively mild toxicity profile of the treatment, especially when compared with cytotoxic chemotherapy, radiation therapy, and other forms of systemic immunotherapy. Long-term follow-up of patients is needed to ensure that there are no signs of latent infection or late development of side effects. A registry trial for patients treated with T-VEC is available and should provide this information in the future. The combination of safety, patient tolerability, and potential therapeutic benefit makes this a promising therapy in patients with advanced cancer.
To date, the virus has generally been given only to clinically accessible cutaneous, subcutaneous, and nodal tumors. Given the acceptable safety profile, it may be possible to consider injection of visceral lesions by utilizing ultrasound or computed tomography guidance for tumor access. In fact, a Phase I clinical trial was conducted in patients with pancreatic cancer where T-VEC was given by endoscopic ultrasound-guided injection.56 Although results from this trial have not been published, the treatment was well tolerated and clinical activity was seen at the highest dose given (Coffin, personal communication, 2013). There will also be a need to educate clinicians on the proper preparation and administration of this agent, as this is fundamentally different from other forms of treatment that utilize standard infusions or oral delivery methods. The lack of viral shedding, however, suggests that the virus can be safely given in the ambulatory setting by trained personnel.
The Phase III clinical trial51 results provide evidence of a clinical benefit for T-VEC in the treatment of patients with advanced melanoma. These represent the first successful clinical results for an oncolytic virus in the treatment of cancer, and should inspire new research with other oncolytic viruses and enable further research into understanding the molecular and cellular mechanisms through which these novel agents mediate antitumor activity.
Disclosure
The authors report no conflicts of interest in this work.
References
Goldufsky J, Sivendran S, Pan M, et al. Oncolytic virus therapy for cancer. Oncolytic Virotherapy. 2013;2:31–46. | |
Eisenberg RJ, Atanasiu D, Cairns TM, Gallagher JR, Krummenacher C, Cohen GH. Herpes virus fusion and entry: a story with many characters. Viruses. 2012;4:800–832. | |
Mellerick DM, Fraser NW. Physical state of the latent herpes simplex virus genome in a mouse model system: evidence suggesting an episomal state. Virology. 1987;158:265–275. | |
Preston CM, Efstathiou S. Human Herpesviruses: Biology, Therapy, and Immunoprophylaxis. 7th ed. Arvin A, Campadelli-Fiume G, Mocarski E, Moore PS, Roizman B, Whitley R, Yamanishi K, editors. Cambridge, UK: Cambridge University Press; 2007. | |
Fatahzadeh M, Schwartz RA. Human herpes simplex virus infections: epidemiology, pathogenesis, symptomatology, diagnosis, and management. J Am Acad Dermatol. 2007;57:764–766. | |
Bloom DC. HSV LAT and neuronal survival. Int Rev Immunol. 2004; 23:187–198. | |
Umbach JL, Kramer MF, Jurak I, Karnowski HW, Coen DM, Cullen BR. MicroRNAs expressed by herpes simplex virus 1 during latent infection regulate viral mRNAs. Nature. 2008;454:780–783. | |
Knickelbein JE, Khanna KM, Yee MB, Baty CJ, Kinchington PR, Hendricks RL. Noncytotoxic lytic granule-mediated CD8+ T cell inhibition of HSV-1 reactivation from neuronal latency. Science. 2008;322:268–271. | |
Gale M, Katze MG. Molecular mechanisms of interferon resistance mediated by viral-directed inhibition of PKR, the interferon-induced protein kinase. Pharmacol Ther. 1998;78:29–46. | |
Clemens MJ, Elia A. The double-stranded RNA-dependent protein kinase PKR: structure and function. J Interferon Cytokine Res. 1997;17:503–524. | |
He B, Gross M, Roizman B. The gamma134.5 protein of herpes simplex virus 1 has the structural and functional attributes of a protein phosphatase 1 regulatory subunit and is present in a high molecular weight complex with the enzyme in infected cells. J Biol Chem. 1998;273:20737–20743. | |
Dey M, Cao C, Dar AC, et al. Mechanistic link between PKR dimerization, autophosphorylation, and eIF2alpha substrate recognition. Cell. 2005;122:901–913. | |
Li Y, Zhang C, Chex X, et al. ICP34.5 protein of herpes simplex virus facilitates the initiation of protein translation by bridging eukaryotic initiation factor 2alpha (eIF2alpha) and protein phosphatase 1. J Biol Chem. 2011;286:24785–24792. | |
Talloczy Z, Jiang W, Virgin HW 4th, et al. Regulation of starvation- and virus-induced autophagy by the eIF2alpha kinase signaling pathway. Proc Natl Acad Sci U S A. 2002;99:190–195. | |
Esclatine A, Chaumorcel M, Codogno P. Macroautophagy signaling and regulation. Curr Top Microbiol Immunol. 2009;335:33–70. | |
Talloczy Z, Virgin HW 4th, Levine B. PKR-dependent autophagic degradation of herpes simplex virus type 1. Autophagy. 2006;2:24–29. | |
Orvedahl A, Alexander D, Talloczy Z, et al. HSV-1 ICP34.5 confers neurovirulence by targeting the Beclin 1 autophagy protein. Cell Host Microbe. 2007;1:23–35. | |
Alexander DE, Leib DA. Xenophagy in herpes simplex virus replication and pathogenesis. Autophagy. 2008;4:101–103. | |
Leib DA, Alexander DE, Cox D, Yin J, Ferguson TA. Interaction of ICP34.5 with Beclin 1 modulates herpes simplex virus type 1 pathogenesis through control of CD4+ T-cell responses. J Virol. 2009;83:12164–12171. | |
Meurs EF, Galabru J, Barber GN, Katze MG, Hovanessian AG. Tumor suppressor function of the interferon-induced double-stranded RNA-activated protein kinase. Proc Natl Acad Sci U S A. 1993;90:232–236. | |
Haus O. The genes of interferons and interferon-related factors: localization and relationships with chromosome aberrations in cancer. Arch Immunol Ther Exp. 2000;48:95–100. | |
Liang XH, Jackson S, Seaman M, et al. Induction of autophagy and inhibition of tumorigenesis by Beclin 1. Nature. 1999;402:672–676. | |
Sarinella F, Calistri A, Sette P, Palù G, Parolin C. Oncolysis of pancreatic tumour cells by a gamma34.5-deleted HSV-1 does not rely upon Ras-activation, but on the PI 3-kinase pathway. Gene Ther. 2006;13:1080–1087. | |
Smith KD, Mezhir JJ, Bickenbach K, et al. Activated MEK suppresses activation of PKR and enables efficient replication and in vivo oncolysis by deltagamma(1)34.5 mutants of herpes simplex virus 1. J Virol. 2006;80:1110–1120. | |
Farassati F, Yang AD, Lee PW. Oncogenes in Ras signalling pathway dictate host-cell permissiveness to herpes simplex virus 1. Nat Cell Biol. 2001;3:745–750. | |
Brown SM, MacLean AR, Aitken JD, Harland J. ICP34.5 influences herpes simplex virus type 1 maturation and egress from infected cells in vitro. J Gen Virol. 1994;75 Pt 12:3679–3686. | |
Brown SM, MacLean AR, McKie EA, Harland J. The herpes simplex virus virulence factor ICP34.5 and the cellular protein MyD116 complex with proliferating cell nuclear antigen through the 63-amino-acid domain conserved in ICP34.5, MyD116, and GADD34. J Virol. 1997;71:9442–9449. | |
Harland J, Dunn P, Cameron E, Conner J, Brown SM. The herpes simplex virus (HSV) protein ICP34.5 is a virion component that forms a DNA-binding complex with proliferating cell nuclear antigen and HSV replication proteins. J Neurovirol. 2003;9:477–488. | |
Liu BL, Robinson M, Han ZQ, et al. ICP34.5 deleted herpes simplex virus with enhanced oncolytic, immune stimulating, and anti-tumour properties. Gene Ther. 2003;10:292–303. | |
Campadelli-Fiume G, De Giovanni C, Gatta V, Nanni P, Lollini PL, Menotti L. Rethinking herpes simplex virus: the way to oncolytic agents. Rev Med Virol. 2011;21:213–226. | |
Russell SJ, Peng KW, Bell JC. Oncolytic virotherapy. Nat Biotechnol. 2012;30:658–670. | |
Chou J, Kern ER, Whitley RJ, Roizman B. Mapping of herpes simplex virus-1 neurovirulence to gamma 134.5, a gene nonessential for growth in culture. Science. 1990;250:1262–1266. | |
Bolovan CA, Sawtell NM, Thompson RL. ICP34.5 mutants of herpes simplex virus type 1 strain 17 syn+ are attenuated for neurovirulence in mice and for replication in confluent primary mouse embryo cell cultures. J Virol. 1994;68:48–55. | |
MacLean AR, ul-Fareed M, Robertson L, Harland J, Brown SM. Herpes simplex virus type 1 deletion variants 1714 and 1716 pinpoint neurovirulence-related sequences in Glasgow strain 17+ between immediate early gene 1 and the ‘a’ sequence. J Gen Virol. 1991;72 Pt 3:631–639. | |
Whitley RJ, Kern ER, Chatterjee S, Chou J, Roizman B. Replication, establishment of latency, and induced reactivation of herpes simplex virus gamma 1 34.5 deletion mutants in rodent models. J Clin Invest. 1993;91:2837–2843. | |
Hill A, Jugovic P, York I, et al. Herpes simplex virus turns off the TAP to evade host immunity. Nature. 1995;375:411–415. | |
Ahn K, Meyer TH, Uebel S, et al. Molecular mechanism and species specificity of TAP inhibition by herpes simplex virus ICP47. EMBO J. 1996;15:3247–3255. | |
Galocha B, Hill A, Barnett BC, et al. The active site of ICP47, a herpes simplex virus-encoded inhibitor of the major histocompatibility complex (MHC)-encoded peptide transporter associated with antigen processing (TAP), maps to the NH2-terminal 35 residues. J Exp Med. 1997;185:1565–1572. | |
Goldsmith K, Chen W, Johnson DC, Hendricks RL. Infected cell protein (ICP)47 enhances herpes simplex virus neurovirulence by blocking the CD8+ T cell response. J Exp Med. 1998;187:341–348. | |
Mohr I, Gluzman Y. A herpesvirus genetic element which affects translation in the absence of the viral GADD34 function. EMBO J. 1996;15:4759–4766. | |
Mohr I, Sternberg D, Ward S, et al. A herpes simplex virus type 1 gamma34.5 second-site suppressor mutant that exhibits enhanced growth in cultured glioblastoma cells is severely attenuated in animals. J Virol. 2001;75:5189–5196. | |
Grotz TE, Kottschade L, Pavey ES, Markovic SN, Jakub JW. Adjuvant GM-CSF improves survival in high-risk stage IIIc melanoma a single-center study. Am J Clin Oncol. February 20, 2013. [Epub ahead of print.] | |
Spitler LE, Grossbard ML, Ernstoff MS, et al. Adjuvant therapy of stage III and IV malignant melanoma using granulocyte-macrophage colony-stimulating factor. J Clin Oncol. 2000;18:1614–1621. | |
Spitler LE, Weber RW, Allen RE, et al. Recombinant human granulocyte-macrophage colony-stimulating factor (GM-CSF, Sargramostim) administered for 3 years as adjuvant therapy of stages II (T4), III, and IV melanoma. J Immunother. 2009;32(6):632–637. | |
Steinman RM. The dendritic cell system and its role in immunogenicity. Annu Rev Immunol. 1991;9:271–296. | |
Harrington KJ, Hingorani M, Tanay M, et al. Phase I/II study of oncolytic HSVGM-CSF in combination with radiotherapy and cisplatin in untreated stage III/IV squamous cell cancer of the head and neck. Clin Cancer Res. 2010;16:4005–4015. | |
Kaufman HL, Bines SD. The OPTiM trial: a Phase III prospective randomized clinical trial of an oncolytic herpesvirus encoding GM-CSF in patients with unresectable stage III or IV melanoma. Future Oncol. 2010;6:941–949. | |
Atkins MB, Lotze MT, Dutcher JP, et al. High-dose recombinant interleukin 2 therapy for patients with metastatic melanoma: analysis of 270 patients treated between 1985 and 1993. J Clin Oncol. 1999;17(7):2105–2116. | |
Hodi FS, O’Day SJ, McDermott DF, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363(8):711–723. | |
Schwartzentruber DJ, Lawson DH, Richards JM, et al. Gp100 peptide vaccine and interleukin-2 in patients with advanced melanoma. N Engl J Med. 2011;364:2119–2127. | |
Andtbacka RHI, Collichio F, Amatruda T, et al. OPTIM: a randomized phase 3 trial of talimogene laherparepvec (T-VEC) vs subcutaneous (SC) granulocyte-macrophage colony-stimulating factor (GM-CSF) for the treatment of unresectable stage IIIB/C and IV melanoma (abstract). J Clin Oncol. 2013;31 Suppl:Abstr LBA9008. | |
Kaufman HL, Kim DW, DeRaffele G, Mitcham J, Coffin RS, Kim-Schulze S. Local and distant immunity induced by intralesional vaccination with an oncolytic herpes virus encoding GM-CSF in patients with stage IIIc and IV melanoma. Ann Surg Oncol. 2010;17:718–730. | |
Amgen. Ipilimumab With or Without Talimogene Laherparepvec in Unresected Melanoma. Available from: http://clinicaltrials.gov/show/NCT01740297. NLM identifier: NCT01740297. Accessed November 28, 2013. | |
Hu JC, Coffin RS, Davis CJ, et al. A phase I study of OncoVEXGM-CSF, a second-generation oncolytic herpes simplex virus expressing granulocyte macrophage colony-stimulating factor. Clin Cancer Res. 2006;12:6737–6747. | |
Senzer NN, Kaufman HL, Amatruda T, et al. Phase II clinical trial of a granulocyte-macrophage colony-stimulating factor-encoding, second-generation oncolytic herpesvirus in patients with unresectable metastatic melanoma. J Clin Oncol. 2009;27:5763–5771. | |
BioVex Limited. OncoVEX GM-CSF in Patients With Unresectable Pancreatic Cancer. Available from: http://clinicaltrials.gov/show/NCT00402025. NLM identifier: NCT00402025. Accessed December 12, 2013. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.