Back to Journals » Clinical Interventions in Aging » Volume 11
Correlation between vitamin D levels and apoptosis in geriatric patients infected with hepatitis C virus genotype 4
Authors Gabr S ![]() , Alghadir A, Allam A, Ajarem J, Al-Basher G, Abdel-Maksoud M, Ghfar A, Aboud A
, Alghadir A, Allam A, Ajarem J, Al-Basher G, Abdel-Maksoud M, Ghfar A, Aboud A
Received 19 January 2016
Accepted for publication 2 March 2016
Published 4 May 2016 Volume 2016:11 Pages 523—533
DOI https://doi.org/10.2147/CIA.S104599
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Richard Walker
Sami A Gabr,1,2 Ahmad H Alghadir,1 Ahmed A Allam,3,4 Jamaan Ajarem,3 Ghada Al-Basher,3 Mostafa A Abdel-Maksoud,3 Ayman A Ghfar,5 Alaa Aboud6
1Rehabilitation Research Chair, College of Applied Medical Sciences, King Saud University, Riyadh, Kingdom of Saudi Arabia; 2Department of Anatomy, Faculty of Medicine, Mansoura University, Mansoura, Egypt; 3Zoology Department, College of Science, King Saud University, Riyadh, Saudi Arabia; 4Zoology Department, Faculty of Science, Beni-Suef University, Beni Suef, Egypt; 5Department of Chemistry, College of Science, King Saud University, Riyadh, Saudi Arabia; 6Internal Endemic Medicine Department, College of Medicine, Beni-Suef University, Beni Suef, Egypt
Background: Vitamin D levels play a pivotal role in most biological processes and differ according to age. A deficiency of vitamin D in chronic hepatitis C (CHC) patients has been shown to be linked with the severity of liver fibrosis, but little is known about the mechanism of this association.
Objective: In this study, we evaluate the potential interrelation between vitamin D levels, oxidative stress, and apoptosis, based on liver fibrosis in geriatric patients infected with hepatitis C virus (HCV) genotype 4.
Subjects and methods: A total of 120 adult individuals aged 30–68 years were recruited in this study. Of these, 20 healthy subjects (15 men and five women) with a mean age of 48.3±6.1 years were selected as controls, and 100 patients with a mean age of 47.8±4.9 years with chronic HCV (CHC) who had undergone liver biopsy (80 men and 20 women) were included in this study. Based on liver radiographic (computed tomography, magnetic resonance imaging) and histological Metavir system analyses, the CHC patients were classified into three groups: asymptomatic CHC carriers (n=30), fibrosis (n=25), and cirrhosis (n=45). HCV RNA, HCV genotypes, inflammatory cytokines AFP and TNFα, 25-hydroxyvitamin D (25[OH]D) levels, apoptotic markers single-stranded DNA (ssDNA) and soluble Fas (sFas), and oxidative stress markers nitric oxide (NO) and total antioxidant capacity (TAC) were estimated by using molecular, immunoassay, and colorimetric techniques.
Results: Approximately 30% of the study population (n=30) were diagnosed as asymptomatic CHC carriers, and 70% of the study population (n=70) had severe fibrosis; these were classified into fibrosis and cirrhosis. There was a significant reduction in 25(OH)D levels and TAC activity, along with an increase in levels of NO, AFP, TNFα, ssDNA, and sFas in fibrosis and cirrhosis subjects compared with those of asymptomatic CHC carriers and health controls. The deficiency in 25(OH)D levels correlated positively with sFas, ssDNA, AFP, TNFα, NO, and TAC, and negatively with age, sex, liver function, body mass index, homeostatic model assessment – insulin resistance, HCV RNA, and viral load. Significant intercorrelation was reported between serum 25(OH)D concentrations and apoptotic and oxidative markers, which suggested progression of liver pathogenesis and fibrogenesis via oxidative and apoptotic mechanisms.
Conclusion: The data showed that vitamin D status was significantly correlated with pathogenesis and fibrogenesis of the liver in geriatric patients infected with HCV genotype 4. The deficiency in 25(OH)D levels was shown to have a pivotal role in the pathogenesis of liver via apoptotic, oxidative stress, and inflammatory mechanistic pathways. The data point to adequate vitamin D levels being recommended for a good response to treatment strategies, especially in older CHC patients.
Keywords: 25(OH)D, HCV, apoptosis, Fas antigen, liver fibrosis, oxidative stress, geriatrics
Introduction
Hepatitis C virus (HCV) is considered one of the major causes of liver injury worldwide.1 Globally, >170 million individuals suffer from different HCV health problems, ranging from liver injury and cirrhosis to hepatocellular carcinoma (HCC).2–4 Acute and chronic stages are the most reported stages of HCV infection, with significant appearance of HCV RNA, elevation of serum ALT levels, jaundice, and consequently appearance of HCV antibodies (anti-HCV). In the chronic hepatitis stage, progressive expression of hepatic fibrosis has been reported, ultimately leading to the progression of cirrhosis and HCC.5
In HCV-infected patients, oxidant–antioxidant imbalance status has been reported, with a significant increase in reactive oxygen species (ROS).6 These oxidative free radicals result in more free radical initiation via liver cell-damage mechanisms.7,8 Most studies have reported that the HCV core protein and nonstructural proteins are responsible for the biological changes in human liver cells, such as an increase in ROS production, inhibition of the electron-transport chain, and altering apoptosis, the transcription process, and cell signaling.9 Also, these viral proteins provide substantial alterations in endogenous antioxidants and antioxidant enzymes within the human body:10 an excessive amount of ROS was produced from mitochondria, inflammatory cells, and peroxisomes of the infected liver cells. Besides their role in the promotion of HCV RNA replication and suppression of gene regulation of liver cells, these oxidative free radicals produce more free radicals from the fat and protein constituents of the cell wall and the cell’s genetic materials via lipid peroxidation.11,12 These biologically active oxidative radicals promote the progression of liver-cell fibrosis,13 and the excessive generation of ROS by HCV infection leads to activation of oncogenic transcription factors and mutagenesis that ultimately ends in a state of HCC.14
It has been reported that the integrity of liver tissues depends on homeostatic balance between liver-cell proliferation and apoptosis, and that any disruption of this balance by core proteins of HCV may lead to hepatic carcinogenesis, whereas viral proteins, along with activating transcription of viral and cellular genes, coordinate the balance between proliferation and death processes of liver cells by inducing or blocking apoptosis.15 In patients with acute and chronic hepatitis (CH), the expression of apoptosis-related proteins was shown to be linked with intrahepatic bile-duct development.16 The change in 25-hydroxyvitamin D (25[OH]D) levels in the serum of patients with CH is significantly associated with the expression of apoptosis-related proteins, especially in HCC subjects.17
Besides its role in the regulation of bones and calcium homeostasis, 25(OH)D is involved in many biological processes.18 25(OH)D deficiencies have been reported in hepatitis B/C patients.19,20 Recent studies suggest a relation of vitamin D status with fibrosis progression and response to the IFN-based therapy.21 It was reported that during the treatment of CHC with polyethylene glycol–IFNα, considerable amounts of vitamin D administration were shown to increase sustained viral response.22–24
Most studies have reported that the response to therapy depends mainly on HCV genotypes,25 targeting of apoptosis,26 and 25(OH)D levels in CH patients.17,27,28 Considerable benefits of therapeutic treatments have been achieved in patients with HCV genotype 2 and 3 infections than genotype 1 or 4, which had significantly lower response rates.29 Also, vitamin D deficits were significantly reported in HCV genotype 4-infected subjects compared with healthy controls.30,31
It has been shown that the hydroxylated form of 25(OH)D controls a variety of genes that regulate more biological processes, including apoptosis and angiogenesis.32 There was a significant correlation among HCV genotypes, hepatic fibrosis scores, and 25(OH)D deficiency in treated and untreated CHC patients.33,34
With regard to age, elderly patients with CHC showed significant decreases in serum vitamin D concentration compared with young patients, especially in elderly women,21 and young patients were shown to be at lower risk of developing HCC than elderly patients.35,36 However, little is known about the link between severity of fibrosis, higher 25(OH)D deficiency, and apoptosis as a target for therapeutic treatments in genotype 4 CHC patients. Therefore, in this study, we evaluate the potential interrelation between vitamin D levels, oxidative stress, and apoptosis based upon liver fibrosis in geriatric patients infected with HCV genotype 4.
Subjects and methods
Patients
A total of 120 adult individuals aged 30–68 admitted to the outpatient department of the Gastroenterology Surgical Centre, Faculty of Medicine, Mansoura University, Mansoura, Egypt, were recruited in this retrospective study. Of these, 20 healthy subjects (15 men and five women) with a mean age of 48.3±6.1 years were selected as controls from a population undergoing routine medical investigations for medical insurance, and 100 patients with CHC who had undergone liver biopsy (80 men and 20 women) with a mean age of 47.8±4.9 years were included in the study. Full reported informed consent was obtained from all participants prior to liver biopsy.
Patient selection
Patients with proven HCV viremia, HCV RNA positivity, and genotype determinations were selected. Liver biopsy was taken from patients prior to antiviral therapy or any other antifibrotic therapy. Serum marker levels (such as AST, ALT, and AFP) were performed on the day of biopsy or within 5 days after liver biopsy.
Exclusion criteria were presence of HIV and/or HBV coinfection, other causes of chronic liver diseases, HCC, and prior liver transplantation. Also, subjects taking iron supplementation, the overweight and obese (body mass index [BMI] ≥25 and ≥30 kg/m2, respectively), with previous IFN therapy, and insufficient liver biopsy were excluded from this study.
The study protocol conformed with the ethical guidelines of the 1975 Declaration of Helsinki and was reviewed and approved by the ethical committee of the Gastroenterology Surgical Centre, Faculty of Medicine, Mansoura University. Also, the study was conducted after obtaining clearance from the institutional ethics committee and written informed consent from each participant. All subjects completed a structured questionnaire with questions regarding demographics and daily medication use. Venous blood samples from each patient were collected either before the administration of preoperative drugs on the day of biopsy or within 5 days after biopsy. Samples were given a coded study identification number and were shipped frozen at −80°C for analysis.
Virology
Patients with CHC were diagnosed by elevated levels of ALT and higher titers of anti-HCV, established by third-generation enzyme immunoassay (AxSym HCV 3.0; Abbott Laboratories, Abbott Park, IL, USA). Also, HCV RNA as a measure of diagnosis was reported qualitatively using a nested polymerase chain-reaction Qiagen RNA-extraction kit (Thermo Fisher Scientific, Waltham, MA, USA) and quantitatively using Smart Cycler II real-time polymerase chain reaction (Cepheid, Sunnyvale, CA, USA) with HCV RNA-quantification kits (Sacace Biotechnologies, Como, Italy) for estimation of HCV RNA-positive subjects, as previously described.37,38 In addition, reverse hybridization was performed to identify HCV genotypes using a line-probe assay (Inno-LiPA HCV II kit; Innogenetics, Ghent, Belgium).39
Laboratory determinations
A previously validated questionnaire40 was used to collect demographic and medical information. Laboratory test results used in this study were serum ALT, AST, and blood platelet counts and fasting blood glucose, using standard methods. Serum AFP and 25(OH)D levels were measured by sandwich enzyme-linked immunosorbent assay (ELISA; R&D Systems, Minneapolis, MN, USA) for and immunoassay kits (IDS, Boldon, UK), respectively. Also, TNFα (Orgenium Laboratories, Helsinki, Finland) and insulin (Monobind Inc, Lake Forest, CA, USA) were estimated for all participants using ELISA kits. Insulin resistance was determined by homeostatic model assessment (HOMA)41 using the formula: fasting insulin (μIU/mL) × fasting blood glucose (mg/dL)/405. Serum total antioxidant capacity (TAC) was measured using a colorimetric assay kit (K274-100; BioVision, Milpitas, CA, USA). The data were measured and calculated as previously reported.42 Plasma NO concentration as measure of oxidative stress parameter was estimated as nitrate and nitrite using a Griess reagent. The concentration of nitrite was measured at 540 nm using high-performance liquid chromatography technology as previously reported.43 Plasma soluble Fas (sFas; ng/mL) and single-stranded DNA (ssDNA) concentrations were determined as early markers for liver-cell apoptosis using a commercial quantitative enzyme immunoassay kit (Quantikine®; R&D Systems) for Fas antigens and an ssDNA ELISA kit (EMD Millipore, Billerica, MA, USA) for ssDNA.
Histologic examination
After informed consent documents had been submitted, hepatic biopsies were obtained from all cases by a surgeon after computed tomography or magnetic resonance imaging scans. A preoperative clinical diagnosis of primary liver cancer was made on the basis of an elevated serum AFP level (≥400 ng/mL) and characteristic features of the disease that were visible in the scans. Histological diagnosis of cirrhosis and HCC was based on international criteria. For histological examination, liver biopsies were obtained using an automatic 16-gauge Trucut needle (biopsy gun), which provides adequate specimens for evaluation and fewer cases with tissue fragmentations. Liver biopsy specimens analyzed were at least 15–25 mm long with complete portal tracts (>10). Formalin-fixed, paraffin-embedded sections were stained with hematoxylin and eosin and Masson’s trichrome. Slides were labeled with patient-identification numbers and then reviewed and graded blindly by a senior pathologist. The mean length of liver biopsies and the number of portal tracts were assessed (including only the complete, intact portal tracts). The degree of fibrosis was scored according to the Metavir system,44 and no fibrosis was defined as F0, mild fibrosis as F1, moderate fibrosis as F2, severe fibrosis as F3, and cirrhosis as F4. Significant fibrosis was also defined as F2–F4. Hepatic inflammatory activity was also scored.
Statistical analysis
SPSS version 16 was used for statistical analysis. Data are expressed as mean ± standard deviation or number (percentage) of patients with a condition. Correlation analysis between the studied variables was done by Student’s t-test, one-way analysis of variance, and Mann–Whitney U-test for continuous variables.
Results
A total of 120 subjects aged 18–68 years with a mean age of 48.3±6.1 years were recruited in this study. Based on clinical investigations, Anti-HCV titers, HCV RNA, and pathological examination, the subjects were classified into control healthy subjects (n=20) and CHC patients (n=100), as shown in Table 1. Approximately 70% of CHC patients showed severe pathological symptoms: they were classified according to fibrosis scores into 25% with fibrosis (n=25) and 45% with cirrhosis (n=45), and only 30% (n=30) of patients were diagnosed as asymptomatic CHC (ASC) carriers. There were significant changes in BMI, increases in serum levels of AST, ALT, and bilirubin, and decreases in albumin in CH patients compared with the control group.
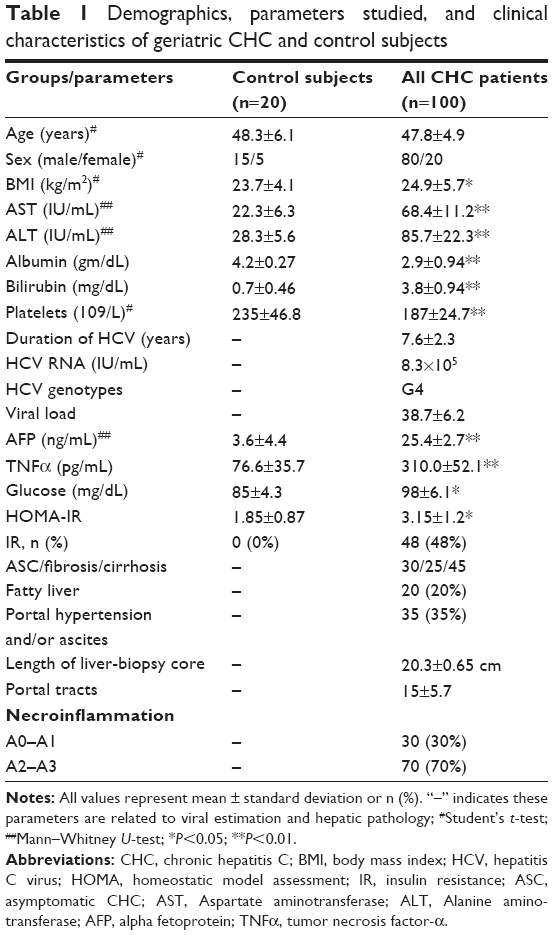 | Table 1 Demographics, parameters studied, and clinical characteristics of geriatric CHC and control subjects |
Also, significant changes in serum inflammatory markers, such as AFP and TNFα, along with glucose and HOMA – insulin resistance (IR) were reported in CHC patients compared to control subjects (Table 1). Also, in HCV patients with cirrhosis, there were significant increases in serum levels of AST, ALT, bilirubin, AFP, TNFα, glucose, and HOMA-IR, along with decreases in albumin compared with ASC patients (Table 2). However, significant reductions in levels of AST, ALT, albumin, and HOMA-IR were reported in cirrhotic patients compared with those with fibrosis (Table 2).
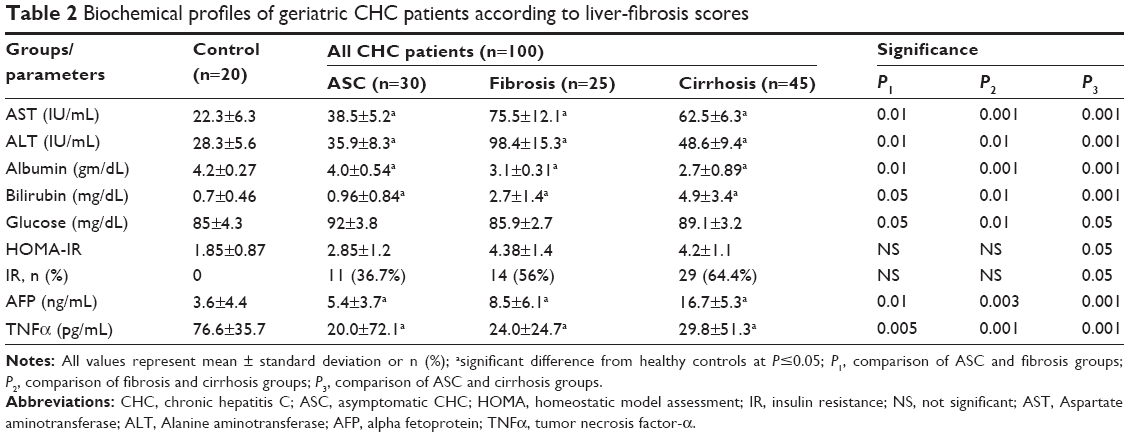 | Table 2 Biochemical profiles of geriatric CHC patients according to liver-fibrosis scores |
With regard to the association between 25(OH)D levels and severity of liver fibrosis, this was significantly lower in CHC patients compared to the control group (P≤0.05). Also, in cirrhotic patients, the reduction in 25(OH)D levels was shown to be significantly correlated (P=0.001) with ASC and fibrosis cases. HCV patients with fibrosis showed lower 25(OH)D levels when compared with either controls or ASC cases. There was significant correlation between lower levels of 25(OH)D and severity of liver fibrosis. HCV-related disease progression was associated with lower median values of 25(OH)D levels compared to nondiseased control patients, while those with cirrhosis had the lowest 25(OH)D concentrations among those with liver disease (Table 3). Also, there was significant association between severity of HCV infection and the deficiency in 25(OH)D levels. In CHC patients with fibrosis and cirrhosis, there were significant increases in viral load, HCV RNA-expression rates, and depletion in 25(OH)D levels compared with the ASC group (Figure 1).
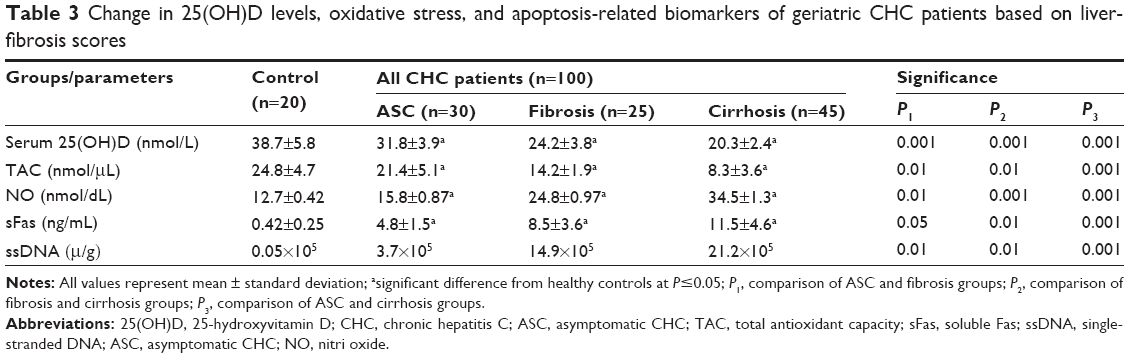 | Table 3 Change in 25(OH)D levels, oxidative stress, and apoptosis-related biomarkers of geriatric CHC patients based on liver-fibrosis scores |
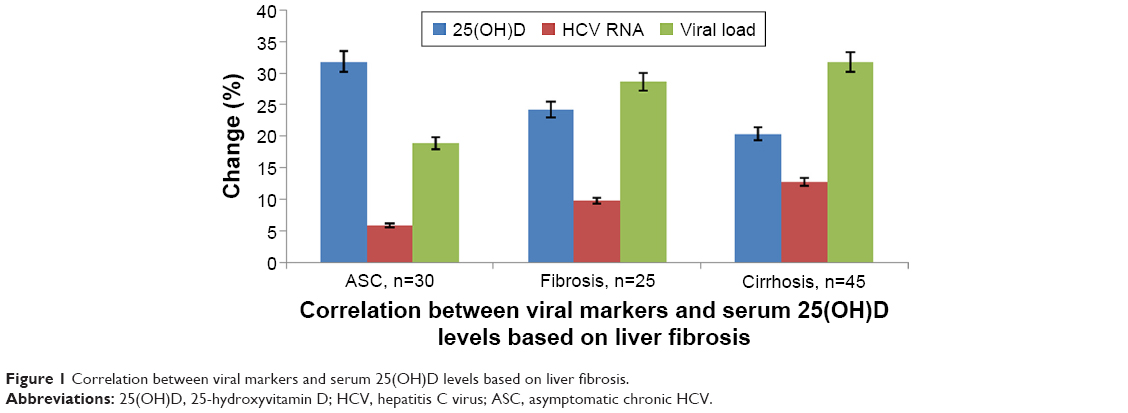 | Figure 1 Correlation between viral markers and serum 25(OH)D levels based on liver fibrosis. |
To study the role of inflammation in the pathogenesis of CHC, the cytokines/inflammatory markers AFP and TNFα were estimated in CHC patients. There were significant increases in AFP and TNFα levels among CHC patients with cirrhosis compared with ASC and control subjects, as shown in Table 2.
To study the relationship between oxidative stress status and severity of liver fibrosis, NO and TAC as biomarkers of oxidative free radicals were measured in all participants. All CH patients showed significant increases in NO levels and depletion in TAC activity when compared with control subjects. However, patients with cirrhosis showed significantly higher increases in NO free radical levels and decreases in TAC activity when compared with patients of ASC and fibrosis groups, as shown in Table 3.
In order to estimate the possible association between apoptosis and liver fibrosis, sFas and ssDNA were estimated as apoptosis-related biomarkers in the plasma of all participants. HCV patients with fibrosis and cirrhosis showed significant increases in plasma levels of sFas and ssDNA when compared with ASC patients and healthy controls, as shown in Table 3; however, significant increases in sFas and ssDNA as apoptosis markers were reported in cirrhotic patients compared with ASC and fibrosis (P=0.001) patients. Expression of the apoptosis-related markers ssDNA and sFas showed positive significant correlations with the increase in HCV RNA, viral load, AFP, TNFα, and NO, and decreases in levels of TAC activity (Table 4).
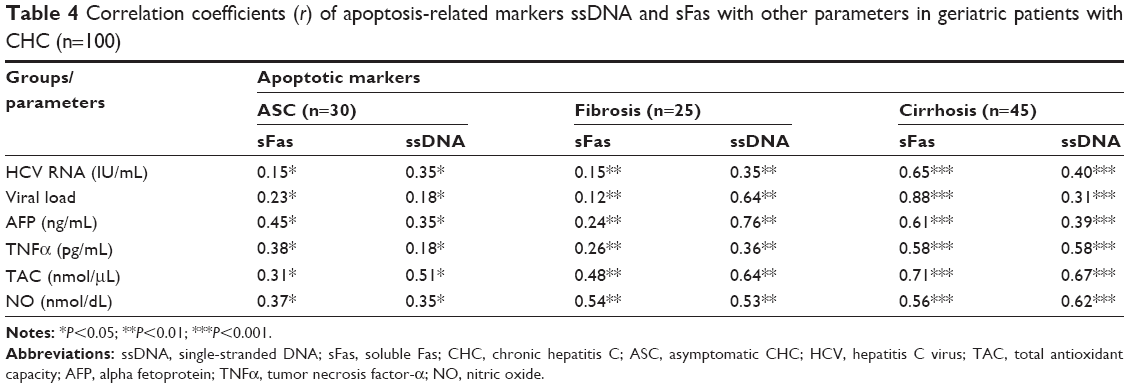 | Table 4 Correlation coefficients (r) of apoptosis-related markers ssDNA and sFas with other parameters in geriatric patients with CHC (n=100) |
In all CHC patients, deficient 25(OH)D levels showed significant correlations with the parameters studied in all groups. Lower 25(OH)D levels correlated positively with increases in sFas, ssDNA, AFP, TNFα, NO, and decreases in TAC activity, and negatively with age, sex, liver function, BMI, HOMA-IR, HCV RNA, and viral load in all patients with varying liver fibrosis, as shown in Table 5. Such a relation appeared to have higher significance in cirrhosis patients (P=0.001) compared to ASC or fibrosis patients (P=0.01).
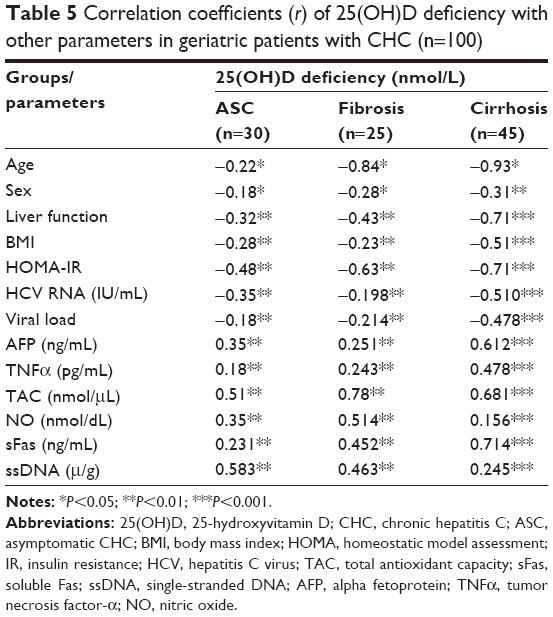 | Table 5 Correlation coefficients (r) of 25(OH)D deficiency with other parameters in geriatric patients with CHC (n=100) |
Discussion
CH is one of the most pathological disorders caused by HCV worldwide. During progression of the disease, there is a significant increase in HCV RNA expression, elevated ratios of viral loads, and significant fibrotic protein deposits.1,4,5 Vitamin D deficits have been shown to be commonly linked with chronic liver diseases. While 93% of these patients reported insufficient vitamin D levels,45 almost a third of these showed severe deficiency.46 The concentration of 25(OH)D in plasma is the most reliable indicator of vitamin D status. It reflects the total amount of vitamin provided from all natural sources, including conversion from fatty deposits of the liver.47 Therefore, we evaluate the association between 25(OH)D levels, apoptosis, and liver fibrosis in genotype 4 CHC patients.
In the current study, based up on Metavir fibrosis-scoring system, only 70% of CHC patients were diagnosed with severe progression of fibrosis: they are classified into 25% with fibrosis (n=25) and 45% with cirrhosis (n=45), and only 30% (n=30) of patients were diagnosed as ASC carriers. There were significant changes in BMI, increases in serum levels of AST, ALT, and bilirubin, and decreases in albumin in CH patients compared with the control group. Our results are in line with other research, where the progression of hepatic fibrosis is significantly associated with higher rates of obesity-related factors, such as BMI.48 Abnormal liver functions are expected to be associated with the progression of hepatic fibrosis. The obtained elevated serum levels of AST, ALT, and bilirubin and decreased albumin in CHC patients in our study were in accordance with most of the published data.21,49,50 Other important inflammatory biomarkers released during CHC infection are AFP and TNFα levels in plasma. The elevated levels of AFP and TNFα in plasma of CHC patients compared to the controls were in agreement with other reports.51,52
The clinical data of our patients were correlative and supportive of previous reports that showed significant decreases in platelet counts and increases in glucose and HOMA-IR compared to controls.53,54 Also, the results of our study showed that activity and progression of chronic liver disease measured by Metavir fibrosis stages (F0–F4), necroinflammatory activity grades (A0–A3), and viral load (38.7±6.2) were similar to those previously reported.55,56
Significant 25(OH)D deficits have been reported in almost half of the healthy population of developed countries.51 Besides its effects in bone health, 25(OH)D deficiency has been reported in the progression of many diseases, such as several types of cancers and cardiovascular and infection-related diseases, including CHC.57–60
Correlations between 25(OH)D levels and the progression of liver fibrosis were estimated in CHC patients under this study. Lower levels of 25(OH)D were significantly reported in CHC patients with fibrosis, cirrhosis, and ASC compared with control subjects. The data showed that 25(OH)D deficits correlated negatively with abnormal liver function, viral load, and HCV RNA expression. The deficiency in 25(OH)D levels among our patients was shown to be associated with liver severity, as reported in most of the literature, whereas vitamin D deficiency has been estimated among CHC cases with higher rates of mortality,61–63 portal hypertension,64 and fibrosis severity.65 25(OH)D deficits were proposed to be linked with poor hepatic hydroxylation of vitamin D to 25(OH)D or calcidiol,47 lower albumin, and decreased expression of DBP, which is synthesized mainly in the liver, and in normal liver cells ~88% of serum 25(OH)D is attached to a DBP.66 It was reported that only 5% of DBP-binding sites were required for the binding of vitamin D metabolites,67 and this confirms that liver dysfunction should be severe enough to decrease DBP levels and ultimately produce vitamin D deficiency.68 Therefore, these previous reports explained the variability in deficiency rates of vitamin D levels among our CHC patients, especially those with cirrhosis.
Also, other studies also showed a close association between 25(OH)D deficiencies, degree of fibrosis and necrosis in CHC patients, and decrease in sustained viral response factor toward HCV viral treatments, especially IFN/ribavirin (RBV)-based therapies.5,21,50,69
Vitamin D defects in CHC patients with fibrosis were shown to be inversely correlated with age, sex, HOMA-IR, and obesity (BMI). Negative correlations between 25(OH)D levels and obesity, glucose intolerance, IR, and BMI have been reported in many diseases, especially liver disorders.70,71 In addition, previous studies have shown an increase in the accumulation of vitamin D in adipose tissue of obese patients, which might relate to vitamin D deficiency.72,73 In contrary, although vitamin D levels are inversely correlated with fibrosis and necroinflammation of liver tissues, this correlation is independent of age, sex, BMI, HOMA-IR score, and presence of metabolic syndrome,74–76 whereas normal vitamin D levels have been shown to enhance HCV response to IFN and RBV therapy77 via improving sensitivity to insulin78 and prevent the development and progression of fatty liver by modulation of lipid metabolism.79
In the present study, the deficiency in vitamin D levels also showed significant associations with inflammation and fibrosis among CHC patients. There were positive correlations among vitamin D defects and the release of both AFP and TNFα as inflammatory markers in patients with severe fibrosis scores. Previous evidence that matches our results suggests that during inflammatory diseases of the liver, the production of active vitamin D is affected due to significant decreases in the expression of vitamin D receptors. These receptors were significantly expressed in tissue macrophages, whereas it comprises of ~90% in the liver, non-parenchymal cells, and biliary epithelial cells.80,81 Vitamin D in its active form is involved in the decrease of inflammation.21,76,82 Also, it has been reported that excessive inflammatory response was regulated by proinflammatory signals present in monocytes and macrophages. These signals may regulate the local metabolism of vitamin D through autoexpression of CYP27B1 and the local production of 1α, 25(OH)2D. It has been proposed that antifibrotic and anti-inflammatory activity of vitamin D decreases the persistence of HCV against IFN/RBV therapy. This occurs via minimizing the induction of proinflammatory cytokines.20,83
In various liver diseases, pathogenesis and progression attributed to initiation of higher amounts of oxidative stress free radicals, such as ROS. Mitochondria, cytochrome P450 enzymes, Kupffer cells, and neutrophils were shown to be the main sources of ROS.9,84
The excessive production of ROS produces significant damage to cellular proteins, the cell wall, and DNA. These harmful effects can be nullified with both enzymatic (superoxide dismutase, catalase, glutathione peroxidase), and nonenzymatic antioxidants, such as vitamins (A, C, E, D), and reduced glutathione.85 Therefore, CHC patients included in this study were subjected to estimation of the nitric oxide free radical (NO) and TAC as markers of oxidative stress.
The consequence increase in the levels of NO and decrease in the levels of TAC activity among patients with cirrhosis and fibrosis were reported as Oxidative stress parameters compared with ASC subjects. These data were in line with other studies that reported the role of oxidative stress in potentiating the pathogenesis and progression of liver diseases.9,20,78–84,86,87 Also, the decrease in vitamin D levels of the studied subjects was positively correlated with changes in both NO and TAC in CHC patients with varying pathological lesions.
Previous studies have reported that low vitamin D levels were estimated in >92% of HCV patients, with 25% of these patients suffering from severe vitamin D deficiency.21,88 It was proposed that HCV infection may affect direct or indirect 25-hydroxylation of vitamin D through cytokine induction or oxidative stress,89 or suppression of 25(OH)D levels via a disruption in lipid metabolism.90
On a molecular basis, little is known about events leading to cellular damage and progression of liver diseases among CHC patients; however, host cells and viral and environmental factors may have a significant role in the pathogenesis of liver. Apoptosis is one of the molecular processes that occurs in both normal and pathological organs to maintain tissue development and homeostasis.91 Previous research has reported that hepatocyte apoptosis plays a role in liver fibrogenesis, persistent liver inflammation, and subsequent severe pathogenesis of the liver among patients with CHC.92–94
In the present study, higher rates of liver cell apoptosis were reported in CHC patients. There was a significant increase in the expression levels of ssDNA and sFas as apoptotic markers in patients with cirrhosis and fibrosis compared with ASC and healthy control subjects. The data were in line with many previous studies that reported significant increases in levels of ssDNA and sFas and their association with fibrosis.95–97 Similarly, a significant positive correlation was reported between increased ssDNA, sFas, and fibrosis levels in CHC patients,95,98 which suggests that increased levels of these markers may reflect the degree of liver fibrosis.
The overexpression of apoptotic markers was shown to be positively correlated with oxidative stress markers: NO, TAC, inflammatory cytokines, TNFα, AFP, viral load, and HCV RNA. The data were matched with previous reports that supported the association of apoptosis with the expression of oxidative stress,87 inflammatory cytokines,93 and HCV-infection parameters.99–101 Also, deficits in vitamin D levels showed positive significant correlation with apoptosis among our CHC patients under investigation. Our data are in agreement with previous studies supporting the association of vitamin D-deficient status and excessive expression of Fas and ssDNA apoptotic markers,17,95,102 whereas long-term vitamin D deficiency can activate chronic inflammation and subsequently higher oxidative stress that can induce apoptosis.
Finally, HCV genotype 4 is the most prevalent genotype in North Africa, the Middle East, and central and east sub-Saharan Africa, and prevalence has been increasing in Europe.103–106 Our study is considered the first to evaluate the potential role of 25(OH)D status in the progression of liver fibrosis via apoptotic and oxidative mechanisms in geriatric patients with HCV genotype 4.
Conclusion
The data showed that vitamin D status was significantly correlated with the pathogenesis and fibrogenesis of the liver in geriatric patients infected with HCV genotype 4. The deficiency in vitamin D levels was shown to have severe pathological effects on the liver via apoptosis, oxidative stress, and inflammatory mechanistic pathways. The data suggest that adequate vitamin D levels be recommended for good response to treatment strategies, especially in older CHC patients.
Acknowledgment
The authors would like to extend their sincere appreciation to the Deanship of Scientific Research at King Saud University for its funding of this research through the Research Group Project RGP-VPP-240.
Disclosure
The authors report no conflicts of interest in this work.
References
Ali I, Siddique L, Rehman LU, et al. Prevalence of HCV among the high risk groups in Khyber Pakhtunkhwa. Virol J. 2011;8:296. | ||
Ashfaq UA, Javed T, Rehman S, Nawaz Z, Riazuddin S. An overview of HCV molecular biology, replication and immune responses. Virol J. 2011;8:161. | ||
Attaullah S, Khan S, Ali I. Hepatitis C virus genotype in Pakistan: a systemic review. Virol J. 2011;8:433. | ||
Averhoff FM, Glass N, Holtzman D. Global burden of hepatitis C: considerations for healthcare providers in the United States. Clin Infect Dis. 2012;55 Suppl 1:S101–S105. | ||
Hoofnagle JH. Course and outcome of hepatitis C. Hepatology. 2002;36:S21–S29. | ||
Robinson WS. Hepatitis B virus and hepatitis D virus. In: Mandel GL, Bennett JE, Dolin R, editors. Principles and Practice of Infectious Diseases. 5th ed. New York: Churchill Livingston; 2000;1652–1684. | ||
Mottola G, Cardinali A, Ceccacci C, et al. Hepatitis C virus nonstructural proteins are localized in a modified endoplasmic reticulum of cells expressing viral subgenomic replicons. Virology. 2002;293:31–43. | ||
Zhang CH, Xu GL, Jia WD, Li JS, Ma JL, Ge YS. Effects of interferon treatment on development and progression of hepatocellular carcinoma in patients with chronic virus infection: a meta-analysis of randomized controlled trials. Int J Cancer. 2011;129:1254–1264. | ||
Loguercio C, Federico A. Oxidative stress in viral and alcoholic hepatitis. Free Radic Biol Med. 2003;34:1–10. | ||
Abd Ellah MR. The role of liver biopsy in detection of hepatic oxidative stress. Vet Med Int. 2011;14:1–7. | ||
Okuda M, Li K, Beard MR, et al. Mitochondrial injury, oxidative stress, and antioxidant gene expression are induced by hepatitis C virus core protein. Gastroenterology. 2002;122:366–375. | ||
Halliwell B. The wanderings of free radicals. Free Radic Biol Med. 2009;46:531–542. | ||
Takagi H, Kakizaki S, Sohara N, et al. Pilot clinical trial of the use of alpha-tocopherol for the prevention of hepatocellular carcinoma in patients with liver cirrhosis. Int J Vitam Nutr Res. 2003;73:411–415. | ||
Waris G, Ahsan H. Reactive oxygen species: role in the development of cancer and various chronic conditions. J Carcinog. 2006;5:14. | ||
Patel T. Apoptosis in hepatic pathophysiology. Clin Liver Dis. 2000;4:295–317. | ||
Tsamandas AC, Thomopoulos K, Gogos C, et al. Expression of bcl-2 oncoprotein in cases of acute and chronic viral hepatitis type B and type C: a clinicopathologic study. Dig Dis Sci. 2002;47:1618–1624. | ||
Fingas CD, Altinbas A, Schlattjan M, et al. Expression of apoptosis- and vitamin D pathway-related genes in hepatocellular carcinoma. Digestion. 2013;87:176–181. | ||
Refaat B, Ashour TH, El-Shemi AG. Ribavirin induced anaemia: the effect of vitamin D supplementation on erythropoietin and erythrocyte indices in normal Wistar rat. Int J Clin Exp Med. 2014;7:2667–2676. | ||
Huang YW, Liao YT, Chen W, et al. Vitamin D receptor gene polymorphisms and distinct clinical phenotypes of hepatitis B carriers in Taiwan. Genes Immun. 2010;11:87–93. | ||
Abu-Mouch S, Fireman Z, Jarchovsky J, Zeina AR, Assy N. Vitamin D supplement improves sustained virologic response in chronic hepatitis C (genotype 1)-naïve patients. World J Gastroenterol. 2011;17:5184–5190. | ||
Petta C, Cammà C, Scazzone C. Low vitamin D serum level is related to severe fibrosis and low responsiveness to interferon-based therapy in genotype 1 chronic hepatitis C. Hepatology. 2010;51:1158–1167. | ||
Atsukawa M, Tsubota A, Shimada N, et al. Efficacy of alfacalcidol on PEG-IFN/ribavirin combination therapy for elderly patients with chronic hepatitis C: a pilot study. Hepat Mon. 2013;13:e14872. | ||
Atsukawa M, Tsubota A, Shimada N, et al. Serum 25(OH)D3 levels affect treatment outcomes for telaprevir/PEG-interferon/ribavirin combination therapy in genotype 1B chronic hepatitis C. Dig Liver Dis. 2014;46:738–743. | ||
Atsukawa M, Tsubota A, Shimada N, et al. Serum 25-hydroxyvitamin D levels affect treatment outcome in PEGylated interferon/ribavirin combination therapy for compensated cirrhotic patients with hepatitis C virus genotype 1B and high viral load. Hepatol Res. 2014;44:1277–1285. | ||
Lee SS, Ferenci P. Optimizing outcomes in patients with hepatitis C virus genotype 1 or 4. Antivir Ther. 2008;13 Suppl 1:9–16. | ||
Muntané J. Targeting cell death and survival receptors in hepatocellular carcinoma. Anticancer Agents Med Chem. 2011;11:576–584. | ||
Blutt SE, McDonnell TJ, Polek TC, Weigel NL. Calcitriol-induced apoptosis in LNCaP cells is blocked by overexpression of Bcl-2. Endocrinology. 2000;141:10–17. | ||
Kwon HJ, Won YS, Suh HW, et al. Vitamin D3 upregulated protein 1 suppresses TNF-α-induced NF-κB activation in hepatocarcinogenesis. J Immunol. 2010;185:3980–3989. | ||
Dahlan Y, Ather HM, Al-Ahmadi M, Batwa F, Al-Hamoudi W. Sustained virological response in a predominantly hepatitis C virus genotype 4 infected population. World J Gastroenterol. 2009;15:4429–4433. | ||
El Husseiny NM, Fahmy HM, Mohamed WA, Amin HH. Relationship between vitamin D and IL-23, IL-17 and macrophage chemoattractant protein-1 as markers of fibrosis in hepatitis C virus Egyptians. World J Hepatol. 2012;4:242–247. | ||
Schaalan MF, Mohamed WA, Amin HH. Vitamin D deficiency: correlation to interleukin-17, interleukin-23 and PIIINP in hepatitis C virus genotype 4. World J Gastroenterol. 2012;18:3738–3744. | ||
Nagpal S, Na S, Rathnachalam R. Noncalcemic actions of vitamin D receptor ligands. Endocr Rev. 2005;26:662–687. | ||
Petta S, Grimaudo S, Marco VD. Association of vitamin D serum levels and its common genetic determinants, with severity of liver fibrosis in genotype 1 chronic hepatitis C patients. J Viral Hepat. 2013;20:486–493. | ||
Mohamed AA, Sabry NA, Abbassi MM, Ibrahim WA, Ali-Eldin ZA. Vitamin D levels in Egyptian HCV patients (genotype 4) treated with PEGylated interferon. Acta Gastroenterol Belg. 2013;76:38–44. | ||
Asahina Y, Tsuchiya K, Tamaki N, et al. Effect of aging on risk for hepatocellular carcinoma in chronic hepatitis C virus infection. Hepatology. 2010;52:518–527. | ||
Ikeda K, Saitoh S, Arase Y, et al. Effect of interferon therapy on hepatocellular carcinogenesis in patients with chronic hepatitis type C: a long-term observation study of 1,643 patients using statistical bias correction with proportional hazard analysis. Hepatology. 1999;29:1124–1130. | ||
Zhang Q, Cao LY, Cheng SJ, Zhang AM, Jin XS, Li Y. P53-induced microRNA-1246 inhibits the cell growth of human hepatocellular carcinoma cells by targeting NFIB. Oncol Rep. 2015;33:1335–1341. | ||
Abdel-Hamid M, El-Daly M, Molnegren V, et al. Genetic diversity in hepatitis C virus in Egypt and possible association with hepatocellular carcinoma. J Gen Virol. 2007;88:1526–1531. | ||
Allam A, Gabr S, Ajarem J, Abdel-Maksoud M. Bcl-2 and p53 expression in hepatic tissues of Egyptian patients with chronic hepatitis C. J Pak Med Assoc. 2015;65:1186–1192. | ||
Kazi AM, Khalid W. Questionnaire designing and validation. J Pak Med Assoc. 2012;62:514–516. | ||
Matthews DR, Hosker JP, Rudenski AS, Naylor BA, Treacher DF, Turner RC. Homeostasis model assessment: insulin resistance and β-cell function from fasting plasma glucose and insulin concentrations in man. Diabetologia. 1985;28:412–419. | ||
Alghadir AH, Gabr SA, Anwer S, Al-Eisa E. Fatigue and oxidative stress response to physical activity in type 2 diabetic patients. Int J Diabetes Dev Ctries. 2016;36:59–64. | ||
Tsuchiya M, Asada A, Kasahara E, et al. Smoking a single cigarette rapidly reduces combined concentrations of nitrate and nitrite and concentrations of antioxidants in plasma. Circulation. 2002;105:1155–1157. | ||
Bedossa P. Intraobserver and interobserver variations in liver biopsy interpretation in patients with chronic hepatitis C. Hepatology. 1994;20:15–20. | ||
Holick MF. Vitamin D: evolutionary, physiological and health perspectives. Curr Drug Targets. 2011;12:4–18. | ||
Fisher L, Fisher A. Vitamin D and parathyroid hormone in outpatients with noncholestatic chronic liver disease. Clin Gastroenterol Hepatol. 2007;5:513–520. | ||
Heaney RP. The vitamin D requirement in health and disease. J Steroid Biochem Mol Biol. 2005;97:13–19. | ||
Thomopoulos KC, Arvaniti V, Tsamantas AC, et al. Prevalence of liver steatosis in patients with chronic hepatitis B: a study of associated factors and of relationship with fibrosis. Eur J Gastroenterol Hepatol. 2006;18:233–237. | ||
Poortahmasebi V, Alavian SM, Keyvani H, Norouzi M, Mahmoodi M, Jazayeri SM. Hepatic steatosis: prevalence and host/viral risk factors in Iranian patients with chronic hepatitis B infection. Asian Pac J Cancer Prev. 2014;15:3879–3884. | ||
Baur K, Mertens JC, Schmitt J, et al. Combined effect of 25-OH vitamin D plasma levels and genetic vitamin D receptor (NR 1I1) variants on fibrosis progression rate in HCV patients. Liver Int. 2012;32:635–643. | ||
Aroucha DC, do Carmo RF, Moura P, et al. High tumor necrosis factor-α/interleukin-10 ratio is associated with hepatocellular carcinoma in patients with chronic hepatitis C. Cytokine. 2013;62:421–425. | ||
Neuman MG, Schmilovitz-Weiss H, Hilzenrat N, et al. Markers of inflammation and fibrosis in alcoholic hepatitis and viral hepatitis C. Int J Hepatol. 2012;2012:231210. | ||
El Raziky M, Fathalah WF, El-Akel WA, et al. The effect of peginterferon alpha-2a vs. peginterferon alpha-2b in treatment of naive chronic HCV genotype-4 patients: a single centre Egyptian study. Hepat Mon. 2013;13:e10069. | ||
Knobler H, Schattner A. TNF-α, chronic hepatitis C and diabetes: a novel triad. QJM. 2005;98:1–6. | ||
Evans JL, Maddux BA, Goldfine ID. The molecular basis for oxidative stress-induced insulin resistance. Antioxid Redox Signal. 2005;7:1040–1052. | ||
Awad MD, Shiha GE, Sallam FA, Mohamed A, El Tawab A. Evaluation of liver stiffness measurement by fibroscan as compared to liver biopsy for assessment of hepatic fibrosis in children with chronic hepatitis C. J Egypt Soc Parasitol. 2013;43:805–819. | ||
Holick MF. Vitamin D deficiency. N Engl J Med. 2007;357:266–281. | ||
Pérez-López FR. Vitamin D and its implications for musculoskeletal health in women: an update. Maturitas. 2007;58:117–137. | ||
Looker AC, Mussolino ME. Serum 25-hydroxyvitamin D and hip fracture risk in older U.S. white adults. J Bone Miner Res. 2008;23:143–150. | ||
Peterlik M, Cross HS. Vitamin D and calcium deficits predispose for multiple chronic diseases. Eur J Clin Invest. 2005;35:290–304. | ||
Trépo E, Ouziel R, Pradat P, et al. Marked 25-hydroxyvitamin D deficiency is associated with poor prognosis in patients with alcoholic liver disease. J Hepatol. 2013;59:344–350. | ||
Putz-Bankuti C, Pilz S, Stojakovic T, et al. Association of 25-hydroxyvitamin D levels with liver dysfunction and mortality in chronic liver disease. Liver Int. 2012;32:845–851. | ||
Gerova DI, Galunska BT, Ivanova II, et al. Prevalence of vitamin D deficiency and insufficiency in Bulgarian patients with chronic hepatitis C viral infection. Scand J Clin Lab Invest. 2014;74:665–672. | ||
Malham M, Jørgensen SP, Ott P, et al. Vitamin D deficiency in cirrhosis relates to liver dysfunction rather than aetiology. World J Gastroenterol. 2011;17:922–925. | ||
Barchetta I, Carotti S, Labbadia G, et al. Liver vitamin D receptor, CYP2R1, and CYP27A1 expression: relationship with liver histology and vitamin D3 levels in patients with nonalcoholic steatohepatitis or hepatitis C virus. Hepatology. 2012;56:2180–2187. | ||
Yamamoto N, Homma S. Vitamin D3 binding protein (group-specific component) is a precursor for the macrophage-activating signal factor from lysophosphatidylcholine-treated lymphocytes. Proc Natl Acad Sci U S A. 1991;88:8539–8543. | ||
White P, Cooke N. The multifunctional properties and characteristics of vitamin D-binding protein. Trends Endocrinol Metab. 2000;11:320–327. | ||
Masuda S, Okano T, Osawa K, Shinjo M, Suematsu T, Kobayashi T. Concentrations of vitamin D-binding protein and vitamin D metabolites in plasma of patients with liver cirrhosis. J Nutr Sci Vitaminol (Tokyo). 1989;35:225–234. | ||
García-Álvarez M, Pineda-Tenor D, Jiménez-Sousa MA, Fernández-Rodríguez A, Guzmán-Fulgencio M, Resino S. Relationship of vitamin D status with advanced liver fibrosis and response to hepatitis C virus therapy: a meta-analysis. Hepatology. 2014;60:1541–1550. | ||
Afzal S, Brøndum-Jacobsen P, Bojesen SE, Nordestgaard BG. Vitamin D concentration, obesity, and risk of diabetes: a Mendelian randomisation study. Lancet Diabetes Endocrinol. 2014;2:298–306. | ||
Liu E, Meigs JB, Pittas AG, et al. Plasma 25 hydroxyvitamin D is associated with markers of the insulin resistant phenotype in nondiabetic adults. J Nutr. 2009;139:329–334. | ||
Lim JS, Mietus-Snyder M, Valente A, Schwarz JM, Lustig RH. The role of fructose in the pathogenesis of NAFLD and the metabolic syndrome. Nat Rev Gastroenterol Hepatol. 2010;7:251–264. | ||
Goldner WS, Stoner JA, Thompson J, et al. Prevalence of vitamin D insufficiency and deficiency in morbidly obese patients: a comparison with non-obese controls. Obes Surg. 2008;18:145–150. | ||
Weintraub SJ, Fleckenstein JF, Marion TN, Madey MA, Mahmoudi TM, Schechtman KB. Vitamin D and the racial difference in the genotype 1 chronic hepatitis C treatment response. Am J Clin Nutr. 2012;96:1025–1031. | ||
Dasarathy J, Periyalwar P, Allampati S, et al. Hypovitaminosis D is associated with increased whole body fat mass and greater severity of non-alcoholic fatty liver disease. Liver Int. 2014;34:e118–e127. | ||
Targher G, Bertolini L, Scala L, et al. Associations between serum 25-hydroxyvitamin D3 concentrations and liver histology in patients with non-alcoholic fatty liver disease. Nutr Metab Cardiovasc Dis. 2007;17:517–524. | ||
Grasso A, Malfatti F, De Leo P, et al. Insulin resistance predicts rapid virological response in non-diabetic, non-cirrhotic genotype 1 HCV patients treated with peginterferon alpha-2b plus ribavirin. J Hepatol. 2009;51:984–990. | ||
Alvarez JA, Ashraf A. Role of vitamin D in insulin secretion and insulin sensitivity for glucose homeostasis. Int J Endocrinol. 2010;2010:351385. | ||
Yin Y, Yu Z, Xia M, Luo X, Lu X, Ling W. Vitamin D attenuates high fat diet-induced hepatic steatosis in rats by modulating lipid metabolism. Eur J Clin Invest. 2012;42:1189–1196. | ||
Bilzer M, Roggel F, Gerbes AL. Role of Kupffer cells in host defense and liver disease. Liver Int. 2006;26:1175–1186. | ||
Gascon-Barré M, Demers C, Mirshahi A, Néron S, Zalzal S, Nanci A. The normal liver harbors the vitamin D nuclear receptor in nonparenchymal and biliary epithelial cells. Hepatology. 2003;37:1034–1042. | ||
Adams JS, Hewison M. Unexpected actions of vitamin D: new perspectives on the regulation of innate and adaptive immunity. Nat Clin Pract Endocrinol Metab. 2008;4:80–90. | ||
Ng TI, Mo H, Pilot-Matias T, et al. Identification of host genes involved in hepatitis C virus replication by small interfering RNA technology. Hepatology. 2007;45:1413–1421. | ||
Moustafa AH, Ali EM, Mohamed TM, Abdou HI. Oxidative stress and thyroid hormones in patients with liver diseases. Eur J Intern Med. 2009;20:703–708. | ||
Parola M, Robino G. Oxidative stress related molecules and liver fibrosis. J Hepatol. 2001;35:297–306. | ||
Gabr SA, Alghadir AH. Prediction of fibrosis in hepatitis C patients: assessment using hydroxyproline and oxidative stress biomarkers. Indian J Virol. 2014;25:91–100. | ||
Osman HG, Gabr OM, Lotfy S, Gabr S. Serum levels of bcl-2 and cellular oxidative stress in patients with viral hepatitis. Indian J Med Microbiol. 2007;25:323–329. | ||
Lange CM, Bibert S, Kutalik Z, et al. A genetic validation study reveals a role of vitamin D metabolism in the response to interferon-alfa-based therapy of chronic hepatitis C. PLoS One. 2012;7:e40159. | ||
Bellecave P, Sarasin-Filipowicz M, Donzé O, et al. Cleavage of mitochondrial antiviral signaling protein in the liver of patients with chronic hepatitis C correlates with a reduced activation of the endogenous interferon system. Hepatology. 2010;51:1127–1136. | ||
Clark PJ, Thompson AJ, Vock DM, et al. Hepatitis C virus selectively perturbs the distal cholesterol synthesis pathway in a genotype-specific manner. Hepatology. 2012;56:49–56. | ||
Thompson CB. Apoptosis in the pathogenesis and treatment of disease. Science. 1995;267:1456–1462. | ||
Fischer R, Baumert T, Blum H. Hepatitis C virus infection and apoptosis. World J Gastroenterol. 2007;13:4865–4872. | ||
Valva P, De Matteo E, Galoppo MC, Gismondi MI, Preciado MV. Apoptosis markers related to pathogenesis of pediatric chronic hepatitis C virus infection: M30 mirrors the severity of steatosis. J Med Virol. 2010;82:949–957. | ||
Zuckerman E, Zuckerman T, Sahar D, et al. Bcl-2 and immunoglobulin gene rearrangement in patients with hepatitis C virus infection. Br J Haematol. 2001;112:364–369. | ||
Lapinski TW, Panasiuk A, Jaroszewicz J, Kowalczuk O, Flisiak R, Rogalska M. Specific ssDNA concentration in liver tissue as an index of apoptosis in hepatitis C virus-infected patients. World J Gastroenterol. 2005;11:6130–6133. | ||
Hahn CS, Cho YG, Kang BS, Lester IM, Hahn YS. The HCV core protein acts as a positive regulator of Fas-mediated apoptosis in a human lymphoblastoid T cell line. Virology. 2000;276:127–137. | ||
Zhu N, Ware CF, Lai MM. Hepatitis C virus core protein enhances FADD-mediated apoptosis and suppresses TRADD signaling of tumor necrosis factor receptor. Virology. 2001;283:178–187. | ||
Toyoda M, Kakizaki S, Horiguchi N, et al. Role of serum soluble Fas/soluble Fas ligand and TNF-α on response to interferon-α therapy in chronic hepatitis C. Liver. 2000;20:305–311. | ||
Capone F, Costantini S, Guerriero E, et al. Serum cytokine levels in patients with hepatocellular carcinoma. Eur Cytokine Netw. 2010;21:99–104. | ||
Pianko S, Patella S, Ostapowicz G, Desmond P, Sievert W. Fas-mediated hepatocyte apoptosis is increased by hepatitis C virus infection and alcohol consumption, and may be associated with hepatic fibrosis: mechanisms of liver cell injury in chronic hepatitis C virus infection. J Viral Hepat. 2001;8:406–413. | ||
Di Martino V, Brenot C, Samuel D, et al. Influence of liver hepatitis C virus RNA and hepatitis C virus genotype on Fas-mediated apoptosis after liver transplantation for hepatitis C. Transplantation. 2000;70:1390–1396. | ||
Zhu L, Kong M, Han YP, et al. Spontaneous liver fibrosis induced by long term dietary vitamin D deficiency in adult mice is related to chronic inflammation and enhanced apoptosis. Can J Physiol Pharmacol. 2015;93:385–394. | ||
Schnell G, Tripathi R, Beyer J, et al. Hepatitis C virus genotype 4 resistance and subtype demographic characterization of patients treated with ombitasvir plus paritaprevir/ritonavir. Antimicrob Agents Chemother. 2015;59:6807–6815. | ||
Gower E, Estes C, Blach S, Razavi-Shearer K, Razavi H. Global epidemiology and genotype distribution of the hepatitis C virus infection. J Hepatol. 2014;61:S45–S57. | ||
Kamal SM. Improving outcome in patients with hepatitis C virus genotype 4. Am J Gastroenterol. 2007;102:2582–2588. | ||
Kamal SM, Nasser IA. Hepatitis C genotype 4: what we know and what we don’t yet know. Hepatology. 2008;47:1371–1383. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.