Back to Journals » International Journal of Nanomedicine » Volume 11
Continuous synthesis of drug-loaded nanoparticles using microchannel emulsification and numerical modeling: effect of passive mixing
Authors Ortiz de Solorzano I, Uson L, Larrea A, Miana M, Sebastian V, Arruebo M
Received 19 March 2016
Accepted for publication 30 April 2016
Published 25 July 2016 Volume 2016:11 Pages 3397—3416
DOI https://doi.org/10.2147/IJN.S108812
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Thomas Webster
Video abstract presented by Isabel Ortiz de Solorzano.
Views: 5093
Isabel Ortiz de Solorzano,1,2,* Laura Uson,1,2,* Ane Larrea,1,2,* Mario Miana,3 Victor Sebastian,1,2 Manuel Arruebo1,2
1Department of Chemical Engineering and Environmental Technologies, Institute of Nanoscience of Aragon (INA), University of Zaragoza, 2CIBER de Bioingeniería, Biomateriales y Nanomedicina (CIBER-BBN), Centro de Investigación Biomédica en Red, Madrid, 3ITAINNOVA, Instituto Tecnológico de Aragón, Materials & Components, Zaragoza, Spain
*These authors contributed equally to this work
Abstract: By using interdigital microfluidic reactors, monodisperse poly(d,l lactic-co-glycolic acid) nanoparticles (NPs) can be produced in a continuous manner and at a large scale (~10 g/h). An optimized synthesis protocol was obtained by selecting the appropriated passive mixer and fluid flow conditions to produce monodisperse NPs. A reduced NP polydispersity was obtained when using the microfluidic platform compared with the one obtained with NPs produced in a conventional discontinuous batch reactor. Cyclosporin, an immunosuppressant drug, was used as a model to validate the efficiency of the microfluidic platform to produce drug-loaded monodisperse poly(d,l lactic-co-glycolic acid) NPs. The influence of the mixer geometries and temperatures were analyzed, and the experimental results were corroborated by using computational fluid dynamic three-dimensional simulations. Flow patterns, mixing times, and mixing efficiencies were calculated, and the model supported with experimental results. The progress of mixing in the interdigital mixer was quantified by using the volume fractions of the organic and aqueous phases used during the emulsification–evaporation process. The developed model and methods were applied to determine the required time for achieving a complete mixing in each microreactor at different fluid flow conditions, temperatures, and mixing rates.
Keywords: microchannel emulsification, high-throughput synthesis, drug-loaded polymer nanoparticles, passive mixing, numerical modeling
Introduction
Poly(D,L lactic-co-glycolic acid) (PLGA) is a commercially available biodegradable polymer widely used in drug delivery applications such as vaccinations,1–3 cancer treatment,4–6 and management of inflammatory diseases.7–9 Among the different polymers developed for biomedical applications, PLGA has attracted considerable attention due to its interesting properties:10 1) predictable biodegradability and biocompatibility, 2) US Food and Drug Administration and European Medicine Agency approvals for its drug delivery systems intended for parenteral administration, 3) well-described formulations and methods of production adapted to the encapsulation of various types of hydrophilic or hydrophobic active pharmaceutical ingredients, 4) drug protection from biochemical degradation, 5) possibility to target its based nanoparticles (NPs) to specific tissues or cells, 6) possibility of sustained release, and 7) possibility to easily modify its surface properties.
In liquid phase, PLGA NPs are generally prepared using single or double emulsion solvent evaporation techniques, nanoprecipitation, membrane emulsification, spontaneous emulsion solvent diffusion techniques, and microfluidic reactors. Their degradation rates can be tuned depending on the polymer molecular weight and the copolymer composition. Physical methods such as spray-drying, supercritical antisolvent precipitation, electrospining, and so on can also be used for their formulation. Despite the current advances in the design of PLGA-based NPs, their translation into the clinic has been slow because it remains challenging to produce NPs that are consistent “batch-to-batch”,11 easily reconstituted after lyophilization and in sufficient quantities for clinical production scale.12 In fact, the strict quality requirements on the characteristics of NPs pose serious challenges to their mass production, including limited batch-to-batch reproducibility and controllability, in terms of morphological and physicochemical properties. A wide variety of multistep batch laboratory procedures are not amenable to large-scale production,11 and there is often a balance between maintaining the ability to control the desired nanoscale features and achieving high throughput.11 Consequently, the development of new technologies tackling some of these challenges could significantly accelerate the clinical translation of nanomedicines.11
Microfluidic systems are a powerful tool to carry out a wide range of chemical reactions, and their use in the synthesis of NPs is attracting a remarkable interest on account of the inherent properties associated with microreaction technology:13 the small channel dimensions lead to a relatively large surface area-to-volume ratios and increased driving forces for heat and mass transport. Thus, compared to conventional batch synthesis, microfluidic systems allow a precise control of the reaction conditions (reaction time, temperature, reactant concentration, and stoichiometry). Their high surface-area-to-volume ratios and mixing characteristics help to reduce or avoid temperature and concentration heterogeneities, decreasing NP polydispersity, and guaranteeing a specific composition and structure. Because of the accurate control and reproducibility of physicochemical properties achieved by microfluidic systems, they are considered as the technology of choice for mass production of nanomaterials.11
In this regard, controlled synthesis of polymeric NPs by rapid mixing in microfluidic platforms has been shown to improve their homogeneity and physicochemical properties.14–16 Microchannel emulsification was introduced as a novel emulsification method less than 2 decades ago.17 This method produces monodisperse droplets with a polydispersity below 5%, with the distinguishing feature that no shear forces are needed compared to the traditional batch production where ultrasonic or mechanical (ie, homogenizer) forces are needed. This fact is highly remarkable if the payload in the NPs is subjected to thermal or shear-sensitive degradation. However, besides the continuous production mode of microfluidic reactors, the throughput required to fulfil the clinical translation is still a challenge, and high-throughput procedures are highly desired. For instance, in vitro drug delivery studies require low amounts of NPs (eg, ~50 μg/well), but the subsequent in vivo studies require significantly larger amounts (eg, ~5 mg/mouse).15 This means that single microfluidic reactors with a typically low productivity (<0.3 g/h)15 fall short in the production rates that are typically required for clinical studies and industry. However, parallelization of NP synthesis by 3D hydrodynamic flow focusing using PDMS-based microfluidic systems can improve the production rates up to 84 mg/h.15 However, those systems require sophisticated fabrication procedures, and the lack of robustness at high flow rates turns these designs unappropriated for long-term use. Recently, a glass flow focusing reactor16 and 3D hydrodynamic flow focusing microfluidic reactor fabricated in polyimide,18 a more durable material, enhanced the productivity of polymeric PLGA NPs achieving production rates of ~10 g/h. New millifluidic reactors based on coaxial turbulent jet mixers have also arisen to address the insufficient production of NPs, achieving a throughput as high as 126 g/h.19 However, this type of mixer is extremely sensitive to the hydrodynamic focusing of streamlines, and if the inner coaxial tubing is poorly aligned during fabrication, the resulting NPs would differ from the expected results.20 Consequently, a different type of microfluidic reactor exploiting the advantages of the reported ones should contribute to clinical translation.
Here, a simple interdigital micromixer is demonstrated, which is mechanically durable and easy to operate and still enables high production rates close to 10 g/h, which are suitable for in vivo studies and large-scale production of drug-loaded NPs. In addition, this microfluidic reactor retains the advantages of processing small volumes: homogeneity, reproducibility, and control over the NP properties. The synthesis conditions promoted zero reactant adsorption on the channel surfaces and no particle aggregation, thus rendering it appropriated for continuous production. In addition, we have studied both the importance of passive mixing and the influence of the geometry analyzing various mixing junctions by evaluating the physicochemical properties of the resulting NPs. The mixing process within the microfluidic junctions was further analyzed by computational fluid dynamics (CFD) modeling in order to understand the hydrodynamic process and its implication for the polymeric NPs formation and to validate our experimental data.
Materials and methods
Reagents
The following reactant materials were used in the experiments: polymer poly(D,L-lactic acid/glycolic acid) 50/50 (PLGA; molecular weight 38–54 kDa), under the commercial name Resomer® RG 504, which was purchased from Evonik Industries AG (Darmstadt, Germany). Drugs cyclosporine A and dexamethasone (used as HPLC internal standard), surfactant Pluronic® F-68, and the solvents ethyl acetate, acetonitrile, and methanol (HPLC grade) were supplied by Sigma-Aldrich Co. (St Louis, MO, USA) and were used as received without further purification. All references to water imply the use of Milli-Q water previously filtered through a 0.2-μm cellulose nitrate membrane.
Synthesis of PLGA NPs in batch reactors
PLGA NPs were prepared by the oil-in-water (O/W) emulsion solvent evaporation method. In this method, 50 mg of the polymer PLGA (50:50) and 150 mg of Pluronic F68 used as surfactant were dissolved in 5 mL of ethyl acetate. The organic phase was then emulsified with 10 mL of Milli-Q water used as aqueous phase. This mixture was sonicated in an ice bath for 25 seconds with a sonicator (Digital Sonifier 450; Branson Ultrasonics, North Olmsted, OH, USA) using a probe of 3 mm diameter and 40% amplitude. After the formation of a stable emulsion, the organic solvent was evaporated under continuous stirring (600 rpm) for 3 hours. The same protocol was followed for the scaled-up experiments.
Synthesis of PLGA NPs in microreactors
PLGA NPs were prepared by O/W microchannel emulsification process and solvent evaporation method. Approximately 200 mg of polymer PLGA (50:50) and 600 mg of Pluronic F68 used as surfactant were dissolved in 20 mL of ethyl acetate (organic solvent). The organic phase was then introduced in a plastic syringe (QO) and emulsified with Milli-Q water (QA) fed with another plastic syringe, using different flow ratios by the aid of a syringe pump (Harvard Apparatus, Holliston, MA, USA). Both the solutions were fed through a 1/16″ OD PTFE tubing and then mixed inside different Polyether ether ketone (PEEK) junctions or in the PEEK-based interdigital micromixer (Micro4 Industries GmbH, Mainz, Germany). The temperatures of QO and QA streams were monitored and modified accordingly. Emulsification temperature of 14°C was obtained by mixing the aqueous and the organic streams both at 14°C, and emulsion temperature of 17.5°C was reached by mixing the aqueous and the organic streams at 14°C and 24°C, respectively. Emulsion temperature of 22°C was reached by mixing both streams at room temperature. After the formation of a stable emulsion, the organic solvent was evaporated under continuous stirring (600 rpm) in an open flask during 3 hours.
Cyclosporine-loaded PLGA NPs were prepared by following the same protocol mentioned earlier, but by dissolving the drug (10% w/w) in the organic phase before the formation of the emulsion. See additional details in the Supplementary materials.
Characterization techniques
Determination of NP morphology
Electron microscopy observations were carried out at the LMA-INA-Universidad Zaragoza facilities. Scanning electron microscopy (SEM, Inspect F50; FEI, Eindhoven, the Netherlands) at an accelerating voltage of 10–15 kV was employed to determine the size (100 NPs were measured for performing the statistical analysis) and morphology of the synthesized NPs. The freshly prepared NPs were mixed during 1.5 hours with the same volume of phosphotungstic acid solution (7.5% w/v) used as contrast agent. The dispersion was centrifuged and washed three times with Milli-Q water and later resuspended. A drop of the resulting NP suspension was placed on a glass slide, dried in air, and coated with platinum under vacuum before SEM observation.
Transmission electron microscopy (TEM) observations were carried using a T20-FEI microscope with a LaB6 electron source fitted with a “SuperTwin®” (FEI Company, Hillsboro, OR, USA) objective lens allowing a point-to-point resolution of 2.4 Å. Approximately 2.5 μL suspension of stained PLGA NPs was pipetted onto a transmission electron microscopy copper grid with a continuous carbon film. Samples were let to evaporate completely and then were analyzed.
Size and size distribution measurement
NP size and size distribution were determined by Dynamic Light Scattering (Zeta Plus; Brookhaven Instruments Corporation, NY, USA) after appropriate dilution with Milli-Q water. At least five replicate measurements were recorded in each case.
Entrapment efficiency of cyclosporine A
The drug content in NPs was determined directly by measuring the encapsulated cyclosporine amount in PLGA NPs. A known quantity of NPs was dissolved in acetonitrile mixed for 1 hour with the internal standard (dexamethasone). Then, methanol was added into the mixture and mixed in an ultrasonic bath for 15 minutes to enhance PLGA precipitation. The dispersion was centrifuged at 12,000 rpm for 20 minutes to remove the polymeric residue, and the supernatant was filtered using 0.22-μm PTFE syringe filters and placed in a vial for HPLC analysis. Experiments were performed in triplicate. Cyclosporine A content was determined by HPLC (Waters Instrument 2690 Alliance, Milford, MA, USA). A Kinetex C18 column with a 2.6 μm of filler particle size and dimensions of 50×4.6 mm was used. The mobile phase consisted of a 80:20 (v/v) mixture of acetonitrile:water and a phosphoric acid concentration of 200 ppm. The detector wavelength, injection volume, flow rate, and column temperature were 210 nm, 5 μL, 0.5 mL/min, and 70°C, respectively. The HPLC method was validated with respect to linearity, repeatability, limit of quantitation, and limit of detection.
Drug encapsulation efficiency (EE) was calculated using the following equation:
|
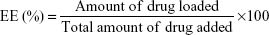
CFD study
Numerical models
CFD simulations were performed using the ANSYS FLUENT 16.0 pressure-based segregated double precision solver.21 The volume of fluid method was used to determine the position of the interface between the aqueous and organic phases. The flow inside the interdigital micromixer (inner volume =8 μL) was defined by the unsteady three-dimensional Navier–Stokes equations, assuming the hypothesis of continuous, Newtonian fluid and laminar flow.
The temporal term was discretized using a first-order implicit scheme. The Pressure Staggering Option scheme22 was applied for interpolation of pressure gradient and A quadratic upwind interpolation scheme23 for discretization of convective terms. Pressure and velocity fields were coupled by the Pressure Implicit with Splitting of Operator algorithm.24,25 The convergence criterion was set as 10−3 for all variables and 10−6 for energy. Approximately 100 iterations were calculated in each time step. The time step size was set to 10−5 seconds.
The volume fraction equation was solved in an explicit manner at the beginning of each time step. Parabolic velocity profiles were considered at the inlets to yield the calculated mass flow rates described in the “Results and discussion” section. The initial temperature of the water stream was set at 287.15 K and the temperature of the organic stream at 297.15 K. At the wall of the microreactor, nonslip boundary conditions were imposed on the velocity components, the contact angle was set at 70°, while the thermal boundary condition imposed a conductive resistance through the PEEK wall (having a thickness of 0.794 mm and a thermal conductivity of 0.25 W/m·K) and a constant outside temperature of 273.15 K, since the micromixer was submerged into an ice bath. Fluid properties were linearly interpolated with temperature among the values compiled in Table S1. It is important that the modeled simulation is a simplified model of the real scenario where a polymer with different nature (a hydrophobic lactic acid part and a hydrophilic glycolic acid part) is dissolved in an organic solvent under the presence of an amphiphilic molecule (surfactant). Hence, in our model only two phases, an aqueous and an oily phase, were considered to simplify the conditions.
The surface tension was set to 0.0239 N/m. At the outlet, variables were extrapolated from the values at the adjacent cell centroids, assuming a zero diffusion flux. The mesh was composed of 1,889,480 hexahedral cells for the case of micromixer, 159,156 quadrilateral cells for T junction, 159,156 mixed cells for Y junction, and 161,757 quadrilateral cells for the cross junction.
Results and discussion
Continuous production of PLGA NPs by emulsification
The most widely used method to produce PLGA NPs is based on the O/W emulsification–evaporation process of a mixture of an organic dissolved polymer and a surfactant using water as antisolvent.26 The dispersion method used to prepare the emulsion is directly related to the particle size and morphology. Highly energetic homogenization techniques, such as probe sonication, have been used to effectively reduce the particle sizes <1 μm. In this study, a well-known batch-modified synthesis method27 was reproduced, which renders good quality PLGA NPs with sizes (measured by DLS) approximately 128.1±4.7 nm after only 25 seconds of energetic sonication using a sonotrode in a reaction volume of 15 mL. We then attempted to scale up the production to different reaction volumes, up to a factor of 20 (150 and 300 mL), but heterogeneous particle distributions with medium particle sizes of 464±100 nm and 1,119±300 nm (measured by SEM) with reactor volumes of 150 and 300 mL, respectively, were obtained (Figure S1). These results clearly show that the emulsification process works well for small batches, but scale-up is difficult. As the intensity of the ultrasound in a liquid decreases rapidly with the distance to the sonotrode, larger volumes may not be well homogenized, promoting a heterogeneous distribution of acoustic waves. Besides the limited production obtained by ultrasonic emulsification, this approach is mainly driven by cavitation. Consequently, ions or particles can be emitted into the product by cavitational abrasion of the sonotrode since they consist of metallic alloys, promoting a feasible product contamination.28 In addition, high-energy inputs involved during the ultrasonic emulsion could also induce the loss of the functional properties of thermal or shear-sensitive encapsulated molecules. Consequently, alternative production approaches are demanded to enable a scaled-up production.
The shear stress produced at the interphase of immiscible liquid microflows is energetic enough to promote the emulsion formation without the need of external mechanical or ultrasonic forces.14 Passive mixing by the reduction of the diffusion distance allows a rapid diffusive mixing with time scales ranging from tens to hundreds of milliseconds.29 Nevertheless, the grade of mixing and shear stress not only depend on the fluid properties, the diffusion distance or linear velocity, but also on the stream layout.30 Figure 1 shows a comparative study of PLGA production when the emulsion precursor streams were mixed at different contact angles using three different micromixers. Two separate phases were obtained at low flow rates (<95 mL/min) after mixing at the assessed junctions. The formation of a milky white dispersion, characteristic of emulsion formation, was obtained at a mean velocity >8 m/s (Figure S2). This confirms that the shear stress generated at the mixing unit is an essential variable to generate emulsions. In those mixers, the initial emulsion is broken down sequentially forming a fine emulsion in which the polymer hardens after organic solvent diffusion toward the aqueous media (Figure 2). In fact, the particle size distribution of emulsified PLGA NPs decreased as the total flow rate increased (Figure S3). For instance, the PLGA NPs produced at a residence time of 0.5 millisecond, when the aqueous and the organic streams were mixed in a Y junction, were highly polydisperse (1,499±743 nm, measured by SEM; Figure 1A). The mixing, and consequently the PLGA particle size distribution, was improved when the inlet configuration was changed to a T junction. However, this particle-size distribution was also sensitive to the inlet configuration. The system where the two inlets (QO and QA) were introduced counter current along the top arm of the T-micromixing unit enabled to narrow down the PLGA size distribution (972±469 nm, measured by SEM; Figure S3) in comparison with the configuration where the disperse phase (QO) entered the micromixing unit via the side arm, with the continuous phase introduced through the top arm (1,024±448 nm, measured by SEM; Figure 1B).
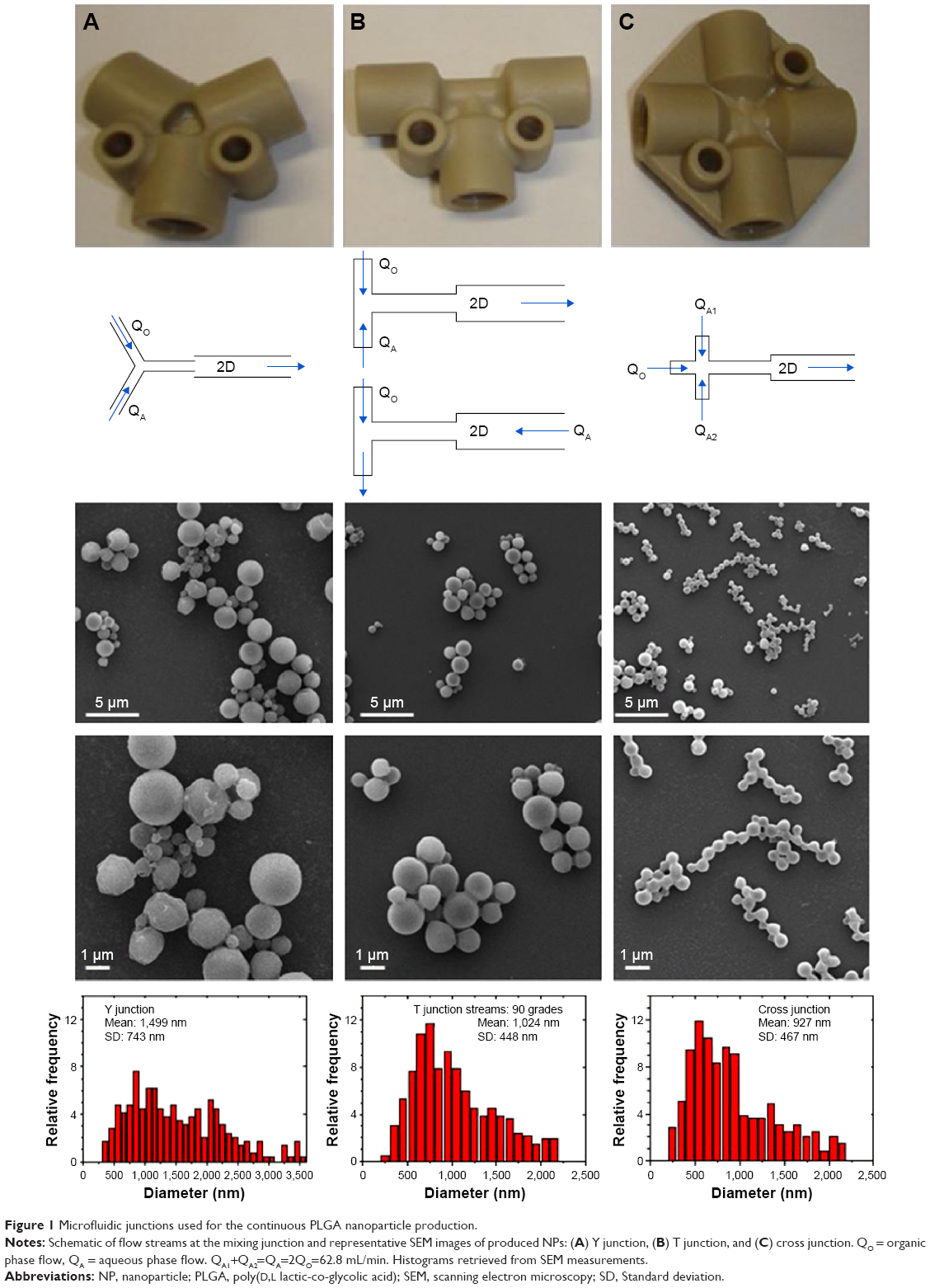 | Figure 1 Microfluidic junctions used for the continuous PLGA nanoparticle production. |
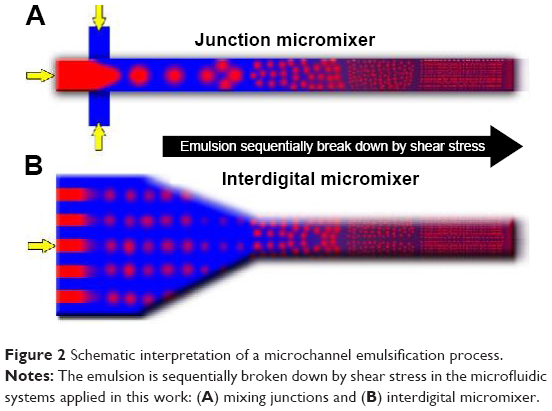 | Figure 2 Schematic interpretation of a microchannel emulsification process. |
Finally, the best results were obtained at the cross-micromixing unit (Figure 1C). The tangential injection of the continuous phase enables the confinement of the disperse phase and maximizes the interface mixing, which enables to decrease the particle size distribution (measured by SEM) up to 927±467 nm. Then, the lamination of the aqueous and organic phases reduces the polydispersity of PLGA emulsified NPs.
Considering the previous results, the use of a multilamination (interdigital) micromixer, where the microchannel dimensions (45 μm width) were thinner that the cross junction unit, was proposed. These dimensions should promote the production of monodisperse NPs because of an improved mixing and the higher shear stress produced at the microchannel junctions (Figure 2). Figure 3 shows the slit interdigital micromixer selected to fulfil the aforementioned requirements. Each input flow (QA and QO) diverges into 16 slits where QO and QA laminate following a 90° trajectory to accelerate the mixing of reagents and promoting high shear stress (Figure 3A–D). Finally, the streams flowed out and converged into an outlet.
 | Figure 3 The slit interdigital micromixer. |
In agreement with the results obtained at Y, T, and cross-micromixing junctions, by simply varying the mixing time from 20 to 10 milliseconds, the size of the produced NPs can be reproducibly controlled in a range of 700–300 nm, respectively (Figures 3A and B and S4). The results were in agreement with the previous analysis carried out for PLGA microparticles where Freitas et al31 showed that larger mixer diameters produced larger particles when using passive mixers. The mixing time can be estimated in the range of 6 milliseconds, using a two-dimensional flow focusing hydrodynamic equation14 (Equation 1).
|
|
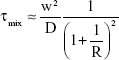
where D is the diffusivity of the solvent (10−9 m2/s), w is the microchannel width, and R is the ratio of flow rate of the organic stream to the total flow rate of water.
Then, it seems reasonable that the best control on particle size is achieved at 10 milliseconds of residence time because higher flow rates increase local mixing and thus smaller particles can be produced. A fast flow induced a fast convective mixing, avoiding the generation of larger particles because of the much faster mixing time relative to the aggregation time as compared to a slow diffusive mixing.18
For intravenous or topic delivery of soft biodegradable NPs, particle sizes of <300 nm are a requirement.32 However, under those conditions and using the interdigital micromixer, the NP size could not be decreased under 300 nm since a mixing time <10 milliseconds increased the set-up pressure drop and modified the flow regime from laminar to turbulent. The size of the droplet is determined by a competition between the pressure due to viscous shear stresses and the capillary pressure resisting deformation, both related in the dimensionless capillary number (Ca) (Equation 2).
|
|
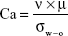
where ν is the mean velocity at the channel section, μ is the dynamic viscosity and σw–o is the surface tension at the water–oil interphase.
For Ca >0.01, the shear action by the continuous phase on the emerging dispersed phase is the dominating mechanism,33 which promotes the formation of homogenous polymer NPs. It is generally accepted that if the mixing temperature is conveniently decreased, the viscous shear stress can be increased over the capillary pressure, promoting a decrease in the particle size.33,34 Figure 4 shows that the mean particle size can be considerably decreased when the mixing unit was placed in an ice bath at 4°C, decreasing the emulsification temperature from 24°C to 22°C. This continuous emulsification at 22°C enables the production of polymeric NPs with mean sizes below 300 nm (215±67 nm, measured by SEM) (Figure S5). Besides the well-known heat transfer of microfluidics, the fluid temperature was vaguely decreased by 2°C (inlet temperature was set at 24°C) through the residence time of 10 milliseconds, due to the fast flow of precursors through the micromixer. Then, the temperature of the inlet streams was modified before the mixing in order to credit the key role of shear stresses and capillary forces on the resulting sizes of the PLGA NPs. The temperature of QO stream was set at 22°C to prevent the potential precipitation of the polymer and surfactant dissolved and the consequent blockage of the microreactor. The temperature of the QA was set at 24°C, 14°C, and 8°C, obtaining an emulsion stream at 22°C, 17.5°C, and 14°C, respectively. Figure 5 depicts the PLGA NPs produced at QA temperature of 24°C, 14°C, and 8°C. The particle size distribution analysis and transmission electron microscopy images confirm that decreasing the emulsion temperature from 22°C to 17.5°C enables to produce the smallest PLGA NPs (220±54 nm, measured by SEM) as well as the narrowest size distribution (Figure 5). If the emulsification temperature was further decreased to 14°C, the NP average size and distribution were not well controlled (535±296 nm). This trend can be justified by the lower solubility of reagents in the reaction media and the notable increase in the calculated surface tension (~30%) (Table 1). These results show that there is an optimum temperature to decrease the size of PLGA NPs. The optimum temperature is the result of a balance between the viscous shear stress and the surface tension, both related in the Ca number.
 | Figure 4 SEM images of PLGA nanoparticles produced by microchannel emulsification in a interdigital micromixer at different conditions. |
 | Figure 5 SEM and TEM images of PLGA nanoparticles produced by microchannel emulsification in a interdigital micromixer at different conditions. |
 | Table 1 Summary of the physical properties of the organic stream and the emulsion formed at QA/QO =2 |
Regarding the residence time contribution in the continuous emulsification process, the interdigital microreactor corroborated the same findings observed in Y, T, and cross junctions. The independent injection of QO and QA phases at the microfluidic reactor enabled a fast screening of the proper synthesis conditions. The ratio of QA/QO was varied to study the effect of modifying the proportion of the dispersed phase during the emulsification process. Figures 4B and D and S5 show that the best results were obtained when the QA/QO was 2. This circumstance could be attributed to the fact that the viscous force becomes less important as QA increases.
On the other hand, it was also observed that the procedure followed to evaporate the solvent from the PLGA NPs was directly related with the final NP morphology. The vacuum rotative evaporation is usually more efficient in the polymer precipitation process than other evaporation methods since the rate of diffusion of the organic solvent through the solvent interphase is maximized.35 However, an increase in the particle size by a coalescence phenomenon as well as the presence of collapsed NPs was observed when using the rotavapor (Figure 5D).
On the contrary, the use of magnetic stirring at high speed, 600 rpm, still enabled a fast solvent displacement, evaporation, and polymer precipitation and hardening in a short time, but without altering the NPs morphology.
As mentioned earlier, biodegradable PLGA polymeric NPs have received considerable attention in nanomedicine as carriers for targeted and controlled delivery of therapeutic and diagnostic agents.36 As proof of concept, the interdigital micromixer was considered to easily encapsulate a therapeutic drug, cyclosporine, normally used to prevent transplant rejection. Cyclosporine was encapsulated in PLGA NPs in a continuous manner by using the interdigital micromixer by premixing the therapeutic drug in the polymeric NP precursor (Figure 6). No significant differences were observed while comparing the sizes of empty PLGA NPs with the ones loaded with cyclosporine, obtaining a mean particle of 211±62 nm (measured by SEM). The drug EE, defined as the percentage of initial drug which is encapsulated size in the NPs, was considerably high (91±5%) out of three runs. Consequently, the EE was highly reproducible which is of great importance since discontinuous production in batch-type reactors suffer from a considerable irreproducibility.11 This results are similar to the ones recently reported by Liu et al16 using a nanoprecipitation process instead of emulsification to encapsulate drugs in PLGA.
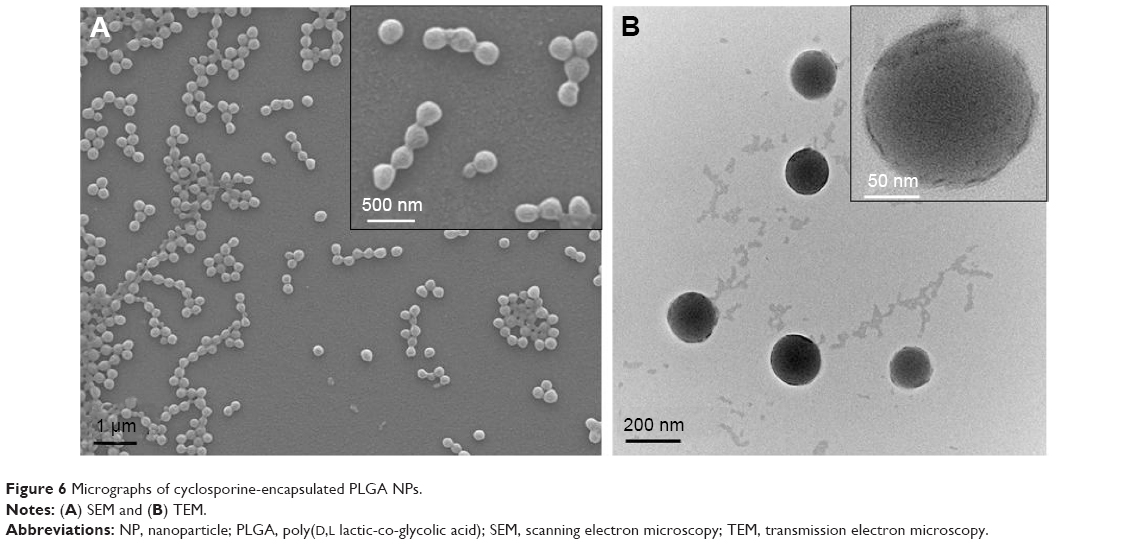 | Figure 6 Micrographs of cyclosporine-encapsulated PLGA NPs. |
CFD simulation
A CFD simulation was carried out to elucidate the flow pattern under the different synthesis conditions and stream arrangements to corroborate our experimental observations. The study proved to be useful for visualizing the flow patterns in opaque microchannels allowing us to make a correlation among the different operating conditions and the size of the NPs obtained.
Two inlet streams, organic (QO) and aqueous (QA), were considered. The properties and flow rates used were those experimentally determined to be optimum (QA =32 mL/min, QO=16 mL/min) to produce the NPs with a low polydispersity.
As an indicator to correlate particle size with simulation data, we used the degree of mixing, as we consider that improving the mixing enables a great uniformity in the sizes of the NPs due to the interphase shear stress that has been generated (Figure 2). According to inlet streams composition, the optimum mixing point at the microchannel emulsification should be considered when the outlet stream composition was approximately 66% of aqueous phase (volume fraction) and 33% of organic phase (volume fraction), which also correlates with the PLGA cloud point.37 The validation of the experimental results obtained with micromixing junctions confirms that mixing criteria is a key parameter to understand the PLGA emulsification process. The simulation shows that the most viscous fluid (QO) was detached from the walls and was unsheathed by the less viscous fluid (QA), causing a progressive increase in the surface-to-volume ratio of the contact interface. This behavior results in a fast mixing, high shear stress, and thus smaller and less polydisperse PLGA NPs. Figure 7A–C depicts the CFD contours of volume fraction of each micromixing junction used, confirming that the mixture was enhanced in the cross junction in comparison with the Y and T junctions. However, the presence of areas where the organic stream was not unsheathed by water was still observed. This lack of mixing control can consequently support high polydispersity and large particle size of the NPs produced with those micromixing junctions. On the other hand, the CFD study reveals that the mixing grade of QO and QA was considerably improved in the interdigital micromixer (Figure 7D), achieving a fast mixing that promotes the controlled production of PLGA NPs. Consequently, the experimental results previously stated credit the computational fluid dynamic analysis.
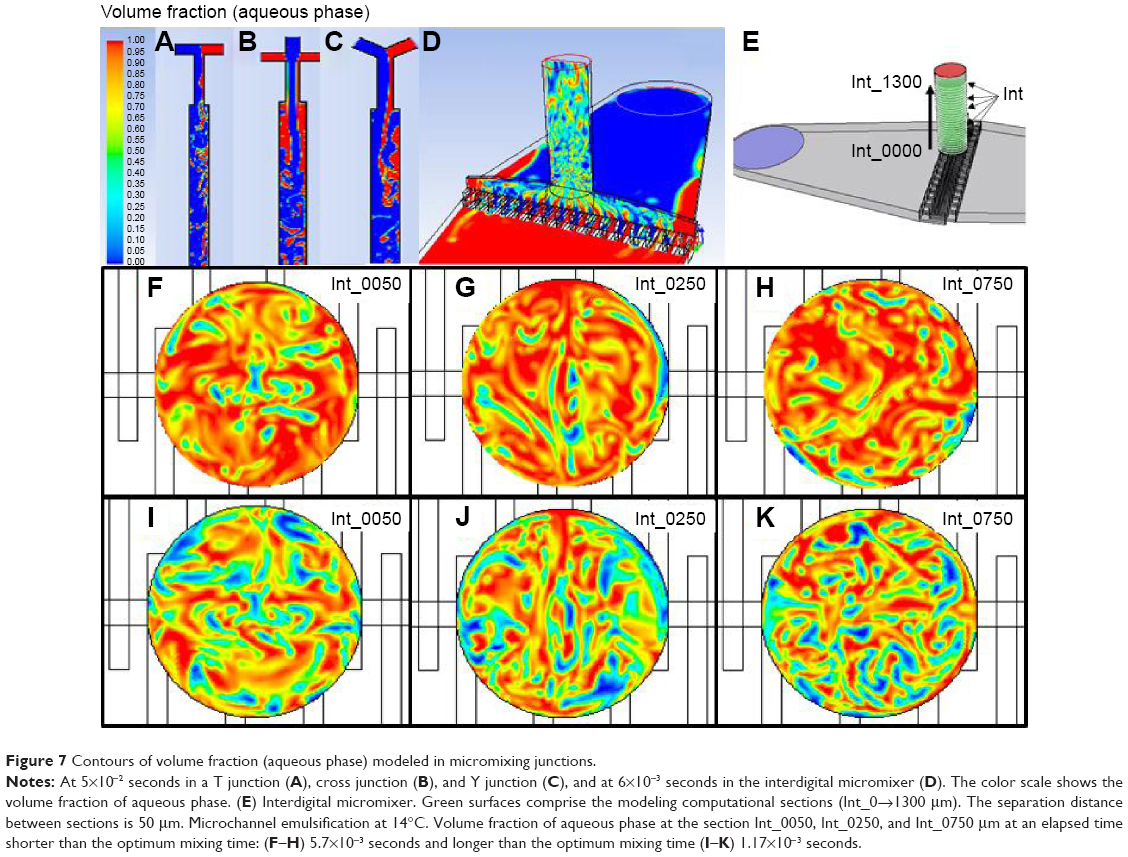 | Figure 7 Contours of volume fraction (aqueous phase) modeled in micromixing junctions. |
To further demonstrate the correlation between shear stress/mixing process and resulting monodispersity in the NPs produced as a function of the emulsification temperature, the flow focusing outlet stream of the interdigital micromixer was divided into 26 sections (from z=0 μm (Int_0000) to z=1,300 μm (Int_1300). The distance between the sections was 50 μm (Figure 7E and Supplementary materials for more details on the numerical model). Simulations of the aqueous volumetric fraction in the different flow focusing sections show that a uniform mixing was achieved at the downstream section Int_0500. The variation in mixing performance regarding the emulsification temperature is evident from Figure 8A; the elapsed time required to achieve a steady state was shorter at 24°C (5×10−3 seconds) than at 14°C (7×10−3 seconds), which is reasonable in terms of diffusive mixing enhancement.38 On the other hand, the mixing performance along the different sections was studied by two-dimensional concentration color maps (Figures 7 and S6). The map analysis evidences that the section area percentage with a volumetric fraction close to the optimum mixing point is higher at 14°C (15%) than at 24°C (10%; Figure 8A). Figures 7F–K and S7 compare the two-dimensional volumetric fraction maps at 14°C and 24°C in sections Int_0050, Int_0250, and Int_0750 at a transient and steady elapsed state, respectively. It can be observed that the variation in the mixing performance is evident at 5.7×10−3 seconds and 1.17×10−2 seconds.
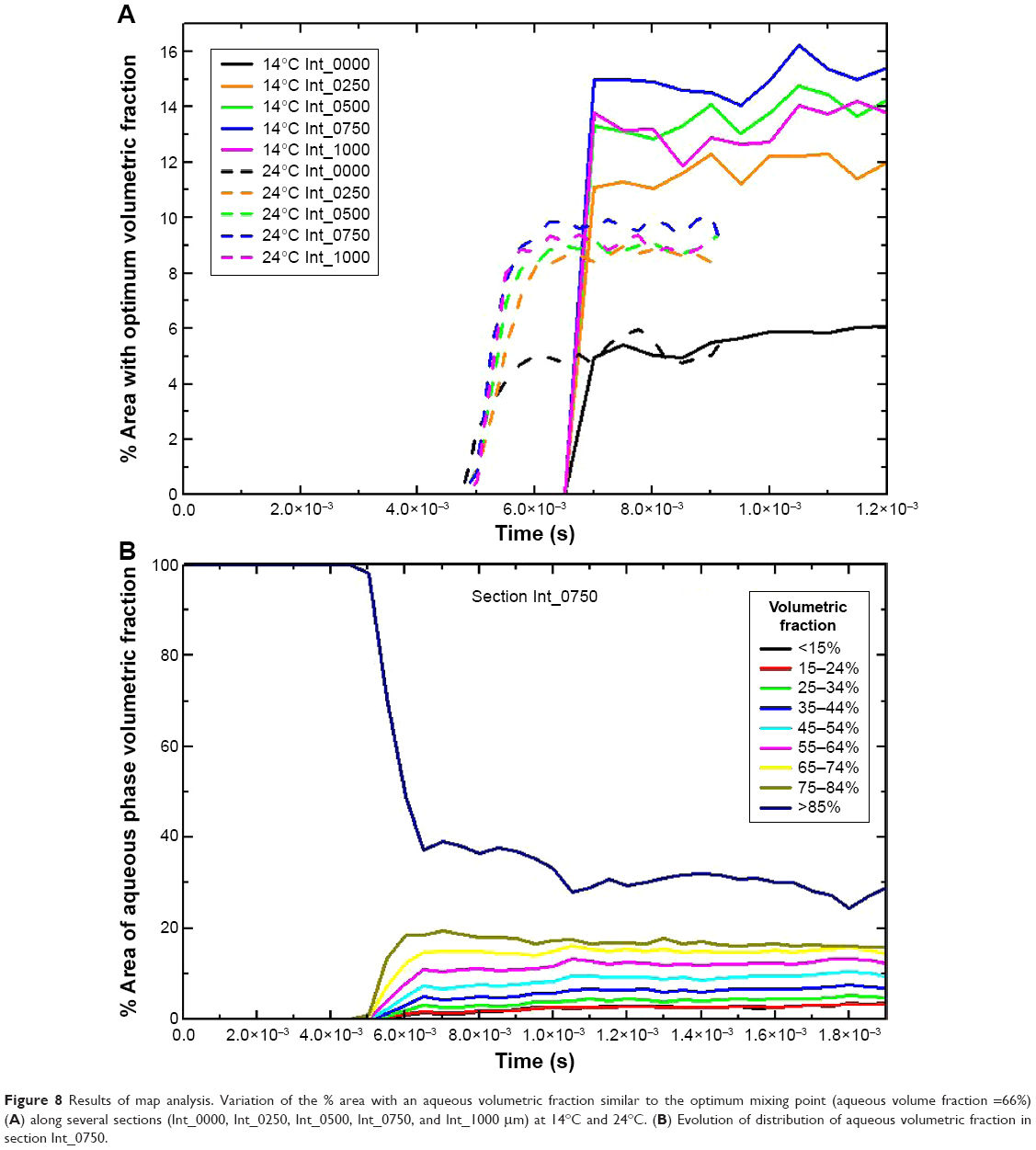 | Figure 8 Results of map analysis. Variation of the % area with an aqueous volumetric fraction similar to the optimum mixing point (aqueous volume fraction =66%) (A) along several sections (Int_0000, Int_0250, Int_0500, Int_0750, and Int_1000 μm) at 14°C and 24°C. (B) Evolution of distribution of aqueous volumetric fraction in section Int_0750. |
The most viscous fluid (QO) was unsheathed, achieving a surface area interface at the section Int_0750. It should be highlighted that a high surface area interface is required to promote a high shear stress between the aqueous and oily phases, as well as to induce the sequential break down of the emulsion (Figure 2). The aqueous volumetric fraction in terms of % area with similar composition at the section Int_0750 μm is presented in Figure 8B, achieving both a homogenous and fast mixing in a short time. It can be observed that initially the flow-focusing channel contains only the aqueous phase, which is mixed with the organic phase over time. The area related with 100% of aqueous volume fraction is decreased with elapsed time, arising the profiles related to the unsheathed organic phase. It can also be inferred from Figure 8B that the optimum conditions to generate a stable emulsion (%H2O v/v <60) are highly dependent on the elapsed time. Consequently, the simulations carried out in the flow-focusing section of the interdigital micromixer remark the influence of the emulsification temperature as well as the elapsed time in the mixing performance. Both the findings are in full agreements with the experimental results obtained, which implies that the proposed computational model is powerful enough to predict the evolution of the mixing during the microchannel emulsification.
Conclusion
In summary, using the O/W emulsification–evaporation technique, monodisperse PLGA NPs can be prepared by controlling the fluid flow conditions and temperatures of the mixing streams interfaced in interdigital micromixers. The shear stress between the mixing streams in the microchannels is enough to generate stable emulsions without the need of mechanical or sonic disruption. Low temperatures and laminar flow regimes (ReH2O =1,193) rendered highly monodisperse NPs with a high throughput production. In microfluidic channels, the NP size can be modified depending on the Re number used. The optimum synthesis temperature is a result of the balance between the viscous shear stress and the surface tension in the microchannels. Three-dimensional CFD simulations represent a valuable tool to gain insight into the understanding of the mixing phenomena during emulsification–evaporation processes. Both simulations and experimental results supported the production of drug-loaded monodisperse PLGA NPs.
Acknowledgments
The EU CIG–Marie Curie under the REA grant agreement no 321642 and the ERC Consolidator Grant program (ERC-2013-CoG-614715, NANOHEDONISM) are gratefully acknowledged.
Disclosure
The authors reports no conflicts of interest in this work.
References
Clawson C, Huang CT, Futalan D, et al. Delivery of a peptide via poly(D,L-lactic-co-glycolic) acid nanoparticles enhances its dendritic cell-stimulatory capacity. Nanomed-Nanotechnol. 2010;6(5):651–661. | ||
Diwan M, Elamanchili P, Lane H, Gainer A, Samuel J. Biodegradable nanoparticle mediated antigen delivery to human cord blood derived dendritic cells for induction of primary T cell responses. J Drug Target. 2004;11(8–10):495–507. | ||
Thomas C, Rawat A, Hope-Weeks L, Ahsan F. Aerosolized PLA and PLGA nanoparticles enhance humoral, mucosal and cytokine responses to hepatitis B vaccine. Mol Pharmaceut. 2011;8(2):405–415. | ||
Derakhshandeh K, Erfan M, Dadashzadeh S. Encapsulation of 9-nitrocamptothecin, a novel anticancer drug, in biodegradable nanoparticles: factorial design, characterization and release kinetics. Eur J Pharm Biopharm. 2007;66(1):34–41. | ||
Danhier F, Lecouturier N, Vroman B, et al. Paclitaxel-loaded PEGylated PLGA-based nanoparticles: in vitro and in vivo evaluation. J Control Release. 2009;133(1):11–17. | ||
Choi SH, Park TG. G-CSF loaded biodegradable PLGA nanoparticles prepared by a single oil-in-water emulsion method. Int J Pharm. 2006;311(1–2):223–228. | ||
Horisawa E, Kubota K, Tuboi I, et al. Size-dependency of DL-lactide/glycolide copolymer particulates for intra-articular delivery system on phagocytosis in rat synovium. Pharmaceut Res. 2002;19(2):132–139. | ||
Higaki M, Ishihara T, Izumo N, Takatsu M, Mizushima Y. Treatment of experimental arthritis with poly(D, L-lactic/glycolic acid) nanoparticles encapsulating betamethasone sodium phosphate. Ann Rheum Dis. 2005;64(8):1132–1136. | ||
Horisawa E, Hirota T, Kawazoe S, et al. Prolonged anti-inflammatory action of DL-lactide/glycolide copolymer nanospheres containing betamethasone sodium phosphate for an intra-articular delivery system in antigen-induced arthritic rabbit. Pharmaceut Res. 2002;19(4):403–410. | ||
Danhier F, Ansorena E, Silva JM, Coco R, Le Breton A, Preat V. PLGA-based nanoparticles: an overview of biomedical applications. J Control Release. 2012;161(2):505–522. | ||
Sebastian V, Arruebo M, Santamaria J. Reaction engineering strategies for the production of inorganic nanomaterials. Small. 2014;10(5):835–853. | ||
Valencia PM, Farokhzad OC, Karnik R, Langer R. Microfluidic technologies for accelerating the clinical translation of nanoparticles. Nat Nanotechnol. 2012;7(10):623–629. | ||
Marre S, Jensen KF. Synthesis of micro and nanostructures in microfluidic systems. Chem Soc Rev. 2010;39(3):1183–1202. | ||
Karnik R, Gu F, Basto P, et al. Microfluidic platform for controlled synthesis of polymeric nanoparticles. Nano Lett. 2008;8(9):2906–2912. | ||
Lim JM, Bertrand N, Valencia PM, et al. Parallel microfluidic synthesis of size-tunable polymeric nanoparticles using 3D flow focusing towards in vivo study. Nanomedicine. 2014;10(2):401–409. | ||
Liu DF, Cito S, Zhang YZ, Wang CF, Sikanen TM, Santos HA. A versatile and robust microfluidic platform toward high throughput synthesis of homogeneous nanoparticles with tunable properties. Adv Mater. 2015;27(14):2298–2304. | ||
Kawakatsu T, Kikuchi Y, Nakajima M. Regular-sized cell creation in microchannel emulsification by visual microprocessing method. J Am Oil Chem Soc. 1997;74(3):317–321. | ||
Min KI, Im DJ, Lee HJ, Kim DP. Three-dimensional flash flow microreactor for scale-up production of monodisperse PEG-PLGA nanoparticles. Lab Chip. 2014;14(20):3987–3992. | ||
Lim JM, Swami A, Gilson LM, et al. Ultra-high throughput synthesis of nanoparticles with homogeneous size distribution using a coaxial turbulent jet mixer. Acs Nano. 2014;8(6):6056–6065. | ||
Dziubinski M. Hydrodynamic focusing in microfluidic devices. In: Kelly DR, editor. Advances in Microfluidics. Rijeka: Intech; 2012. | ||
ANSYS Fluent theory guide [computer program]; 2015. | ||
Bender E. Numerical heat transfer and fluid flow. Von S. V. Patankar. Hemisphere Publishing Corporation, Washington – New York – London. McGraw Hill Book Company, New York 1980. 1. Aufl., 197 S., 76 Abb., geb., DM 71,90. Chemie Ingenieur Technik. 1981;53(3):225. | ||
Leonard BP. ULTRA-SHARP nonoscillatory convection schemes for high-speed steady multidimensional flow [microform]/Leonard BP, Mokhtari S. [Washington, DC]: [Cleveland, OH]: [Springfield, VA]: NASA; Institute for Computational Mechanics in Propulsion, NASA Lewis Research Center, Case Western Reserve University; For sale by the National Technical Information Service; 1990. | ||
Issa RI. Solution of the implicitly discretised fluid flow equations by operator-splitting. J Comput Phys. 1986;62(1):40–65. | ||
Issa RI, Gosman AD, Watkins AP. The computation of compressible and incompressible recirculating flows by a non-iterative implicit scheme. J Comput Phys. 1986;62(1):66–82. | ||
Cohen S, Yoshioka T, Lucarelli M, Hwang LH, Langer R. Controlled delivery systems for proteins based on poly(lactic glycolic acid) microspheres. Pharmaceut Res. 1991;8(6):713–720. | ||
Manchanda R, Fernandez-Fernandez A, Nagesetti A, McGoron AJ. Preparation and characterization of a polymeric (PLGA) nanoparticulate drug delivery system with simultaneous incorporation of chemotherapeutic and thermo-optical agents. Colloid Surface B. 2010;75(1):260–267. | ||
Freitas S, Hielscher G, Merkle HP, Gander B. Continuous contact- and contamination-free ultrasonic emulsification – a useful tool for pharmaceutical development and production. Ultrason Sonochem. 2006;13(1):76–85. | ||
Gavi E, Marchisio DL, Barresi AA. CFD modelling and scale-up of confined impinging jet reactors. Chem Eng Sci. 2007;62(8):2228–2241. | ||
Lester E, Blood PJ, Denyer JP, Azzopardi BJ, Li J, Poliakoff M. Impact of reactor geometry on continuous hydrothermal synthesis mixing. Mater Res Innov. 2010;14(1):19–26. | ||
Freitas S, Merkle HP, Gander B. Microencapsulation by solvent extraction/evaporation: reviewing the state of the art of microsphere preparation process technology. J Control Release. 2005;102(2): 313–332. | ||
Shekunov B. Nanoparticle technology for drug delivery – from nanoparticles to cutting-edge delivery strategies – Part II – 21–22 March 2005, Philadelphia, PA, USA. Idrugs. 2005;8(5):402–403. | ||
Li J, Renardy YY. Shear-induced rupturing of a viscous drop in a Bingham liquid. J Non-Newton Fluid. 2000;95(2–3):235–251. | ||
Raj R, Mathur N, Buwa VV. Numerical simulations of liquid-liquid flows in microchannels. Ind Eng Chem Res. 2010;49(21):10606–10614. | ||
Mainardes RM, Evangelista RC. PLGA nanoparticles containing praziquantel: effect of formulation variables on size distribution. Int J Pharm. 2005;290(1–2):137–144. | ||
Ansary RH, Awang MB, Rahman MM. Biodegradable poly(D,L-lactic-co-glycolic acid)-based micro/nanoparticles for sustained release of protein drugs – a review. Trop J Pharm Res. 2014;13(7):1179–1190. | ||
Murakami H, Kobayashi M, Takeuchi H, Kawashima Y. Preparation of poly(DL-lactide-co-glycolide) nanoparticles by modified spontaneous emulsification solvent diffusion method. Int J Pharm. 1999;187(2):143–152. | ||
Tamam L, Pontoni D, Sapir Z, et al. Modification of deeply buried hydrophobic interfaces by ionic surfactants. Proc Natl Acad Sci. 2011;108(14):5522–5525. |
Supplementary materials
Synthesis of poly(D,L lactic-co-glycolic acid) nanoparticles loaded with cyclosporine
Cyclosporine–poly(D,L lactic-co-glycolic acid) (PLGA) nanoparticles (NPs) were prepared using an Interdigital micromixer by oil-in-water microchannel emulsification process and solvent evaporation method. Briefly, 1% (w/v) of polymer PLGA (50:50), 0.1% (w/v) of cyclosporine and 3% (w/v) of Pluronic F68 used as surfactant were dissolved in 30 mL of ethyl acetate (organic solvent). The organic phase was then introduced in a plastic syringe (QO) and emulsified with MilliQ water (QA) fed with another plastic syringe, using 16 and 32 mL/min flow ratios, respectively, with the aid of a syringe pump (Harvard Apparatus). Both the solutions were fed through a 1/16″ OD PTFE tubing and then mixed inside different PEEK junctions or in the PEEK-based interdigital micromixer. The micromixer is placed in an ice bath to control the reaction temperature. After the formation of a stable emulsion, the organic solvent was evaporated under continuous stirring (600 rpm) in an open flask during 3 hours.
Entrapment efficiency of cyclosporine A
Drug content in NPs was determined directly by measuring the encapsulated cyclosporine amount in PLGA NPs. Approximately 2.5 mg of PLGA–cyclosporine nanoparticles were dissolved in 1.15 mL of acetonitrile mixed for 1 hour with 100 μL of the internal standard (dexamethasone 1,500 ppm). Then, 250 μL of methanol was added into the mixture and mixed in a sonifier bath for 15 minutes to enhance the PLGA precipitation. The dispersion was centrifuged at 12,000 rpm for 20 minutes to remove the polymeric residue, and the supernatant was filtered using 0.22-μm PTFE syringe filters and placed in a vial for HPLC analysis. Experiments were performed in triplicate.
Cyclosporine A content of the samples was determined by HPLC (Waters Instrument 2,690 Alliance, USA). A Kinetex C18 column with a filler particle of size 2.6 μm and dimensions of 50×4.6 mm was used. The mobile phase consisted of a 80:20 (v/v) mixture of acetonitrile:water and a phosphoric acid concentration of 200 ppm. The detector wavelength, injection volume, flow rate, and column temperature were 210 nm, 5 μL, 0.5 mL/min, and 70°C, respectively. The HPLC method was validated with respect to linearity, repeatability, limit of quantitation (LOQ), and limit of detection (LOD). The linearity range was 30–200 μg/mL (r2 =0.9995) and the LOD and LOQ were 8.23 and 24.70 μg/mL, respectively.
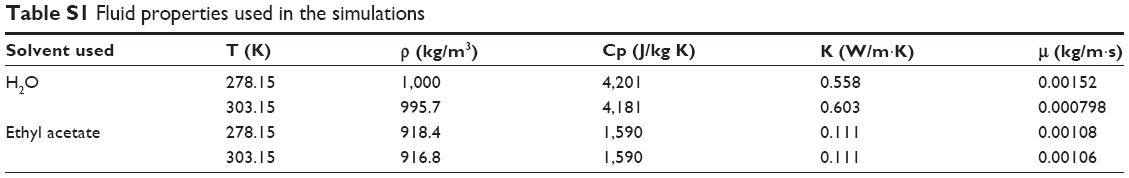 | Table S1 Fluid properties used in the simulations |
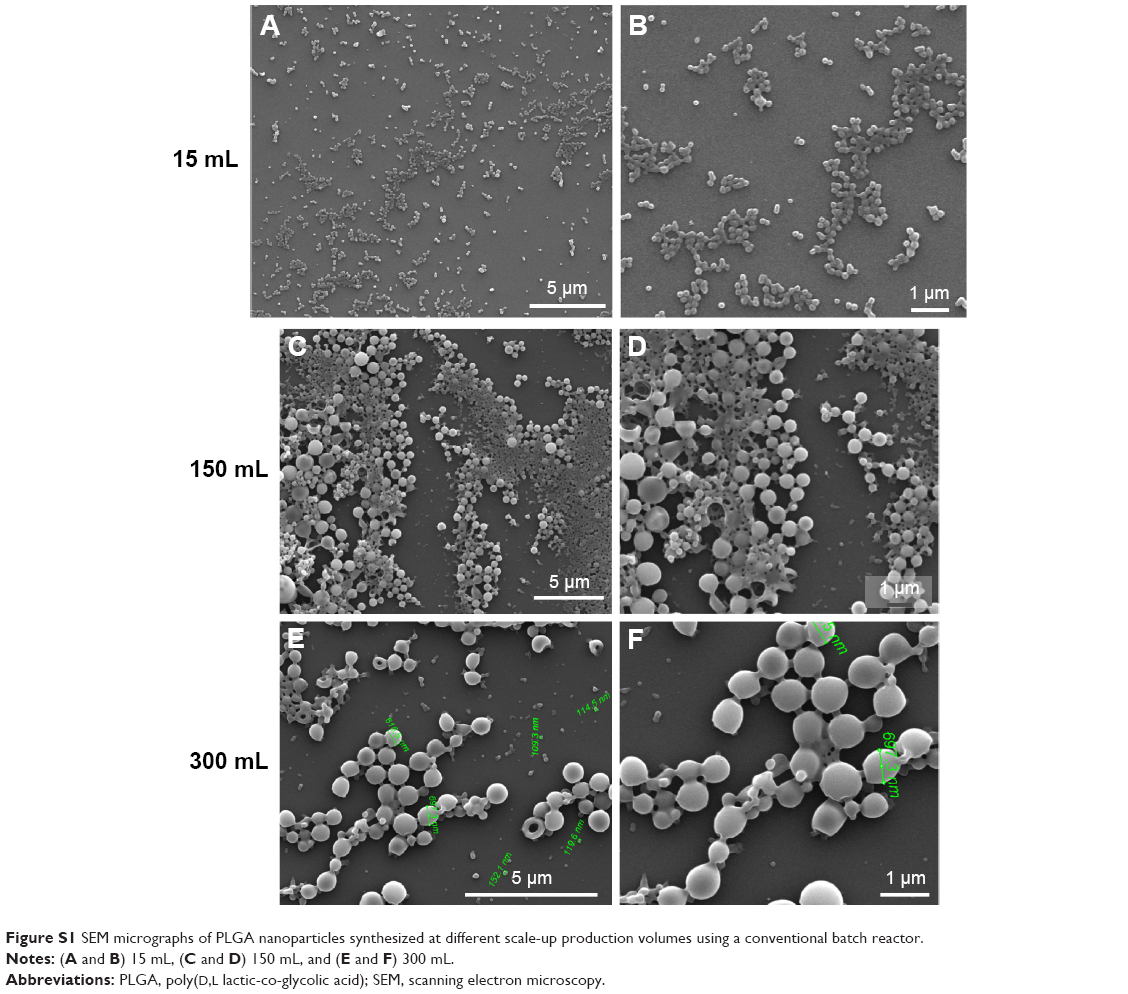 | Figure S1 SEM micrographs of PLGA nanoparticles synthesized at different scale-up production volumes using a conventional batch reactor. |
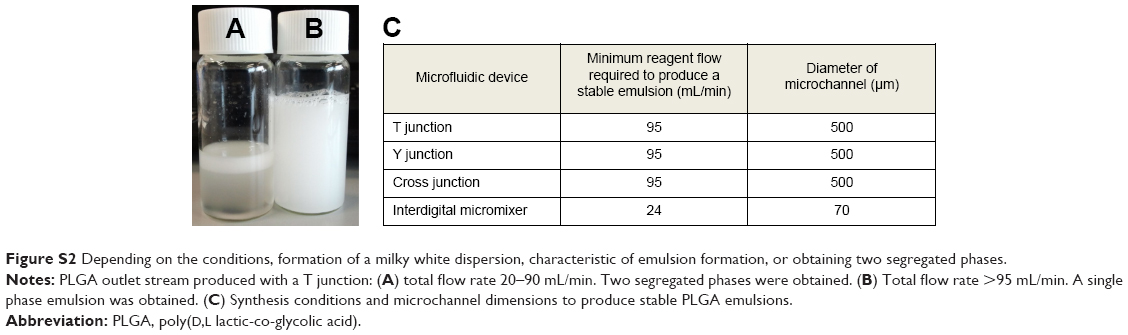 | Figure S2 Depending on the conditions, formation of a milky white dispersion, characteristic of emulsion formation, or obtaining two segregated phases. |
 | Figure S3 SEM micrographs and particle size distribution of PLGA nanoparticles synthesized with the T junction at different residence times. |
 | Figure S4 SEM, TEM micrographs and particle size distribution of PLGA nanoparticles synthesized with an interdigital micromixer at different conditions. |
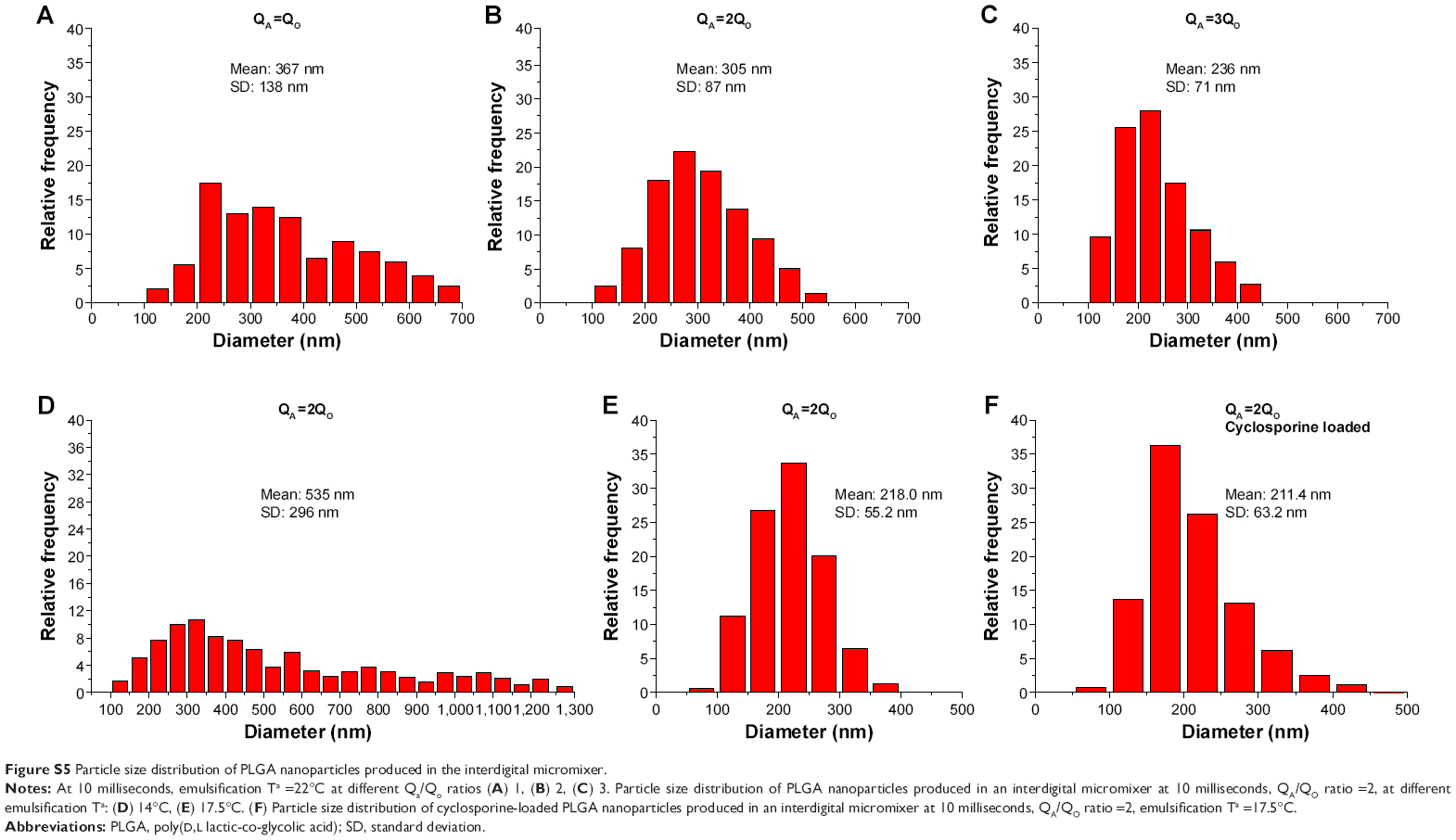 | Figure S5 Particle size distribution of PLGA nanoparticles produced in the interdigital micromixer. |
 | Figure S6 SEM micrographs and particle size distribution of PLGA nanoparticles synthesized with an interdigital micromixer at 10 milliseconds. |
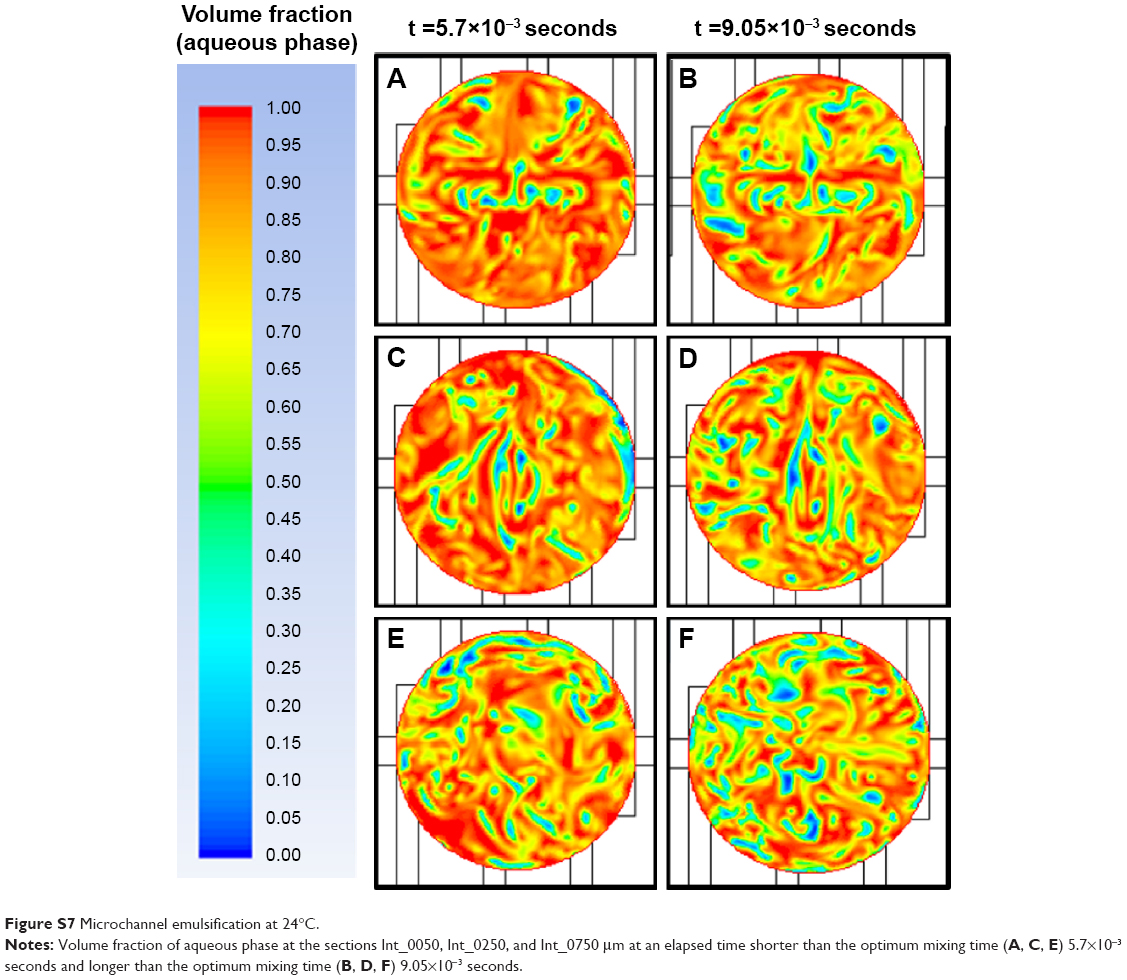 | Figure S7 Microchannel emulsification at 24°C. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.