Back to Archived Journals » Biosimilars » Volume 6
Considerations in biosimilar insulin device development
Authors Fry A, Krentz A, Hompesch M
Received 8 October 2015
Accepted for publication 28 October 2015
Published 16 February 2016 Volume 2016:6 Pages 9—15
DOI https://doi.org/10.2147/BS.S77034
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Shein-Chung Chow
Andy R Fry,1 Andrew J Krentz,2 Marcus Hompesch2
1Team Consulting Limited, Ickleton, Cambridge, UK; 2Profil Institute for Clinical Research, Chula Vista, CA, USA
Abstract: Biosimilar insulins, also known as follow-on biologics, are modifications of an originator insulin that are intended to be clinically equivalent to the licensed product. For injectable insulins, or other peptides used in the management of diabetes, regular use of an insulin pen injector or other device to administer therapy is part of the patient's self-management regimen. By definition, the biosimilar product should have comparable pharmacokinetic and pharmacodynamic properties to the reference product. However, the device is the initial interaction for the patient rather than the product contained within. We consider the regulatory aspects of insulin device development. The options for manufacturers bringing a biosimilar insulin to market, including whether to outsource development of delivery devices, are explored. The structure of a device development program is outlined and issues of accuracy, safety, ease of use, and attractiveness of modern insulin delivery devices are discussed.
Keywords: biosimilar insulin, delivery devices, diabetes mellitus
Introduction
Insulin is used in the treatment of all patients with type 1 diabetes and in a substantial proportion of patients with type 2 diabetes.1 Recombinant technology is the mainstay of modern insulin manufacture.2 When a licensed insulin product reaches the end of its patent life, the market opens for biosimilar products, also known as follow-on biologics. These are modifications of the originator insulin that are intended to be clinically equivalent to the licensed product.3
Devices are a major issue that any pharmaceutical manufacturer needs to acknowledge when considering the development of a drug product that relies upon a device for its delivery. For the patient, the delivery device is their interface with the drug. The drug concerned may or may not offer significant improvements compared to other therapies, but in the first instance, the patient’s experience is that of the delivery device itself. Perhaps this is especially the case for insulins or other peptides used in the management of diabetes, where regular use of the insulin pen or other device to administer therapy becomes part of the patient’s way of life (assuming the patient adheres to the prescribed regimen), and comparison with other products on the basis of pharmacokinetics and pharmacodynamics may not necessarily be immediately obvious to the patient. So the starting point for any consideration of the activities required for a successful product development, where a delivery device is an absolute necessity, will be broadly the same whether the drug concerned is an “originator product” or a “biosimilar”.
This paper focuses specifically on what are commonly referred to as “insulin pens”, or “needle-based injection systems (NISs) for medical use”, to use the general title as in ISO 11608-1:2014,4 the applicable international standard. The first insulin pen was the NovoPen (subsequently referred to as the NovoPen 1), introduced in October 1985 by Novo Nordisk A/S, Bagsvaerd, Denmark and offering a compact, convenient, and easy to use alternative to the vial and syringe. The NovoPen offered “one click per unit” dosing, with an on-board disposable insulin cartridge and a short needle. The original NovoPen was superseded by the NovoPen 2, which included dial-up dosing and a “dial-up, dial-down, press-to-inject” operating format that has since been followed in virtually all pen injectors wherever made. Pen injectors are the predominant delivery means for insulin and related injectable therapies for diabetes management both in well established Western markets and in those emerging markets that have experienced a very rapid growth in diabetes in recent years (Figure 1). Reported advantages of insulin pens over the vial and syringe method of insulin delivery include greater patient satisfaction and adherence, greater ease of use and social acceptability, and improved dosing accuracy.5
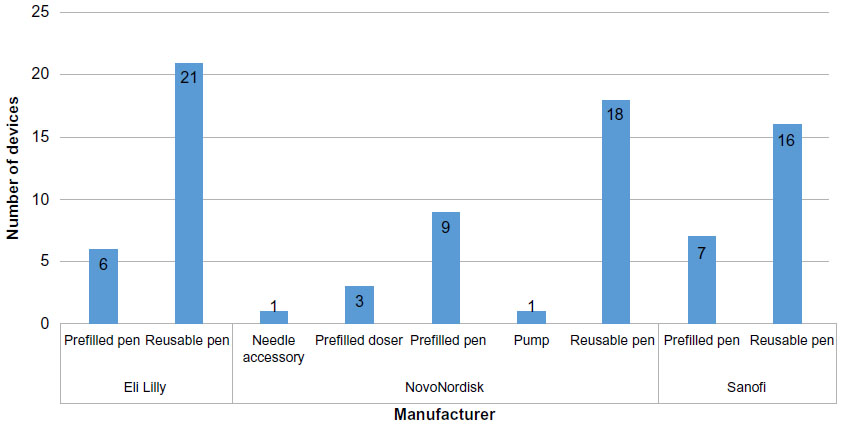 | Figure 1 Manufacturer versus number of device types marketed for market-leader manufacturers (till August 2015). |
When we consider biosimilar insulins, the precise product definition is important. There is currently only one insulin biosimilar approved in Western markets; Abasaglar/Basaglar (insulin glargine, a biosimilar of Sanofi’s Lantus) marketed by Eli Lilly and Company (Indianapolis, IN, USA) together with Boehringer Ingelheim GmbH (Ingelheim am Rhein, Germany). Lantus is a so-called basal insulin intended for the control of blood glucose, typically between meals and overnight, and has a duration of action extending that permits once-daily dosing in many patients with type 1 or type 2 diabetes. This product was given European Medicines Agency approval in September 2014 with the product name Abasaglar (formerly Abasria).6 The same product, under the name Basaglar, was given tentative approval by the US Food and Drug Administration (FDA) for the US, pending a patent infringement suit filed by Sanofi; an agreement between the companies was reached in September 2015. Abasaglar/Basaglar is offered in the latest version of the Eli Lilly KwikPen, a prefilled product originally launched in 2008 and also as cartridges for use with Lilly’s Savvio refillable pen. As a part of the legal settlement, Lilly and Boehringer Ingelheim will have the ability to launch Basaglar in the USA in December 2016. Under the terms of the agreement, Sanofi has granted Eli Lilly a royalty-bearing license permitting Eli Lilly to manufacture and sell Basaglar in the KwikPen device globally.7 Several other companies from developed and emerging markets are currently engaged in programs to develop biosimilar insulins. Some of these are in Phase III (Sanofi, Mylan & Biocon, and MSD & Samsung), whereas others still in Phase I (Wockhardt and Gan & Lee) (Table 1).
 | Table 1 Insulin biosimilars in development |
However, beyond the current, single example of Abasaglar/Basaglar, as a product which has gained approval in the EU and US, and the products mentioned above that are in development, there are a number of locally produced copies of different insulins that are already available in the emerging markets that have less rigorous regulatory requirements (Table 2). These products have not been approved via a biosimilar pathway, and there is a significant lack of clarity regarding their intellectual property status and regulatory position.
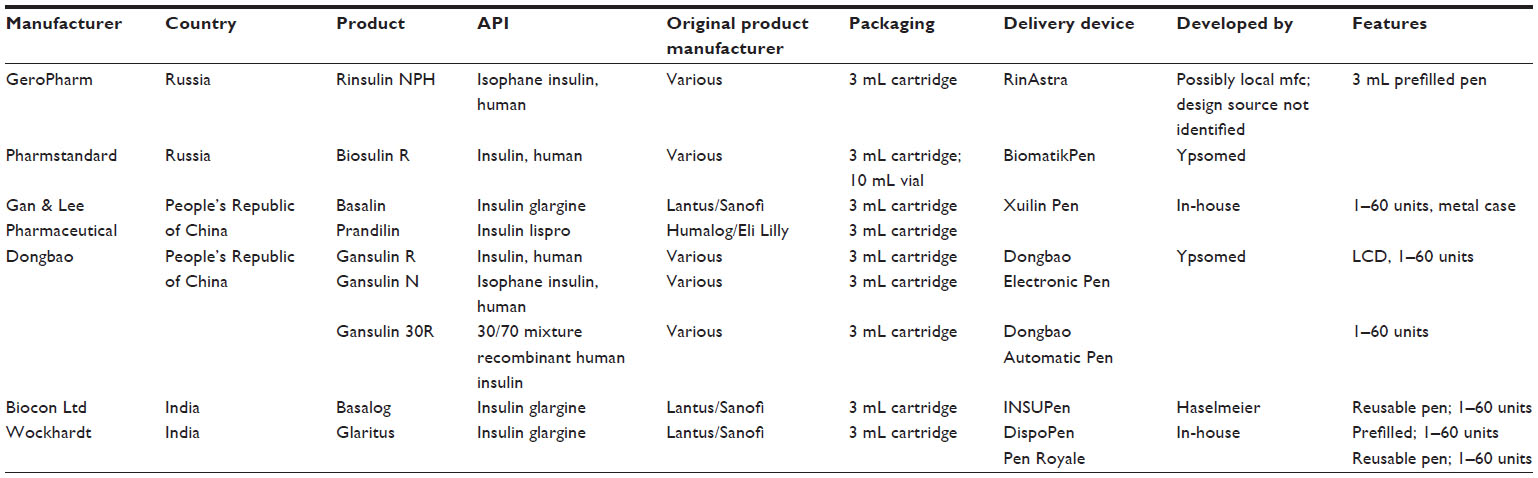 | Table 2 Table of emerging market copy-insulin manufacturers – drug-device product combinations |
Of these non-Western market-approved “copy insulins”, three manufacturers out of the six identified use pens from two established European manufacturers (Ypsomed and Haselmeier). The other three have pens developed in-house or developed by third-party suppliers in the countries of origin for the drug product. All pens mentioned however, irrespective of the market or manufacturer, follow the “dial-up, dial-down, press-to-inject format”; the pens sourced from Ypsomed and Haselmeier comply fully with ISO 11608-1. Locally produced pens appear to have adopted the general requirements of ISO 11608-1 in a somewhat looser fashion. Studies comparing pens from emerging market suppliers indicate that products from established European suppliers, such as Ypsomed and Haselmeier, which provide superior performance in key aspects such as lower operating force and better dose-to-dose variability,8 are preferred by users who were interviewed.9
One illustration of potential risks associated with a less than complete adoption of good practice as set out in ISO 11608-1 concerns dose setting and delivery when the cartridge (insulin primary container) is approaching “empty”. Section 5.5 General design requirements, section j of 11608-1 2012 states that the design of the variable multidose NISs must prevent the user from presetting, or attempting to deliver, a larger dose than that remaining in the container. Alternatively, the design must make the user aware of the amount of drug delivered, or the “shortfall”, ie, the amount of the preset dose not delivered.13
In compliance with the above requirement, pens offered in Western markets deploy a range of strategies to ensure that if a patient attempts to deliver a dose greater than is available within the device, the patient is either prevented from attempting the chosen dose, or enabled to deliver what is available but, as the wording from ISO 11608-1 emphasizes, the user is informed very clearly of the remaining dose needed for completion of the required amount. Thus, the patient can use a new pen or replacement cartridge to complete the required dose. However, some emerging market “home-grown” pens had no such safety feature. Indeed, in at least one design, the patient could dial up a dose of (say) 12 IU, when only 6 IU was available, deliver the injection, and be utterly unaware that he or she had only received half the dose intended.8 The implications of such a design shortcoming are potentially serious.
There is a whole spectrum of issues to consider and requirements to be satisfied, which may relate specifically to a biosimilar drug product and the specification of a suitable delivery device; there may be novel and imaginative features of design, presentation, or marketing approach, which may be of particular relevance to a new product, which may be a biosimilar. But a majority of the “best practice” aspects (eg, dose management, anti-tamper mechanisms) of device development are the same whether the label on the drug product is “biosimilar” or “originator”. A good insulin pen is just that, and all patients deserve a safe, effective, and reliable product.
From a marketing perspective, a biosimilar may require additional differentiation in the form of device features which may improve usability perhaps, or possibly make the pen preferable to competitor products (size or appearance being typical aspects which could be iterated to attract users). Such aspects might be given added emphasis compared to an originator product, though even this would depend on the market within which it is to be offered and, indeed, the features present in competing products (not only from other manufacturers but also from other products in the range from the manufacturer of the biosimilar itself).
What distinguishes a biosimilar device from an originator device?
There should be no fundamental difference between a device for delivering an originator versus a biosimilar product – after all, the device is the drug-to-patient interface, irrespective of whether the drug is an originator or biosimilar. That said, in a crowded market, there is an argument that the main differentiator between a biosimilar and an originator product may only be in the means of delivery. If the biosimilar really is that close to the originator product, then what else is there to tell the two apart from the means of delivery? Therefore, the opportunity exists to offer the biosimilar product in a pen with features particularly important to a specific subgroup. Senior citizens, often with type 2 diabetes, may well have other conditions resulting in limited dexterity or motor power (eg, rheumatoid arthritis), but also are more likely to have some level of visual impairment or aural impairment. Design variants may be developed which appeal to younger patient subgroups, for instance pediatric patients (for whom half-unit dosing may be particularly important), or adolescents (for whom a pen which is discreet or “cool” may be desirable). Considering the importance of the delivery device, when developing a biosimilar product, may also be a factor in helping to improve adherence within particular patient populations.10,11 Such feature enhancements may then give advantageous positioning to what would otherwise just be a “me too” biosimilar product. The key point which one always returns to is that the patient sees the device, not the molecule.
This has been the underlying philosophy of the “supergeneric” product in the small-molecule arena, whereby a more elegant or more feature-filled presentation for a relatively simple product such as an ibuprofen tablet can create customer/patient preference and hence command a premium price, even if it sits on the same supermarket or pharmacy shelf as a therapeutically identical “vanilla” generic of the same drug which is a fraction of the price.
Differentiation of the biosimilar product, via the design and features of the drug delivery device, is strategically more important in markets in which substitution of the originator product, by the pharmacist or physician, is permitted. Interchangeability, if allowed in a specific market, may mean a change in the associated delivery device. If a pharmacist substitutes a product with a user-friendly, familiar, device for one with a less user-friendly, or more complex device, what is the impact to the patient? How similar should the devices be to prevent use error? Should a patient be trained in the use of the switched device?
How should a development program for a “biosimilar device” be structured?
The question of an appropriate development program has been raised in some quarters, with the implication that there will be a fundamental difference of approach between development for an originator drug and a biosimilar one. Although the starting point for an originator drug requiring a delivery device will differ from that for a biosimilar one, the fundamental activities for a well-managed and successful development program should be fundamentally the same in both cases. Furthermore, there is a worthwhile opportunity to inform the development process for a biosimilar product by observation of the devices in use for originator products (it is, of course, also the case that this opportunity exists for those developing new originator products).
The FDA issued its “Design Control Guidance for Medical Device Manufacturers” (relating to FDA 21 CFR 820.30 and ISO 9001 subclause 4.4) in 1997.12 As stated in the foreword of this guidance document; “the regulation does not prescribe the practices that must be used. Instead, it establishes a framework that manufacturers must use when developing and implementing design controls”.
The third page of the introduction to the 53-page guidance document includes the “waterfall diagram”, which has become a familiar illustration for describing the structure of a design (and development) control process. Originally developed by Health Canada, this diagram has been adopted (and adapted) by many device development organizations and pharmaceutical companies worldwide.
It is perhaps most useful to regard the diagram as an illustration of a “closed-loop process”, wherein outputs are continually assessed against the specification set out at the appropriate earlier stage (sometimes referred to as “checks against specs”), as shown in Figure 2.
 | Figure 2 Device development process – “Waterfall” model. |
The key control documents and their functions include: a target product profile (TPP), which contributes to a thorough assessment of user needs. A TPP results in the generation of: a User Requirements Specification (URS); a Product Requirements Specification (PRS); and a Detailed Design Specification (DDS). The URS defines what the users will get from the product/device and will typically require validation through summative user trials and Phase III clinical trials. The PRS defines what the product/device will do (or not do!) and will typically require verification as part of the overall design verification process. The DDS defines what the product/device is and provides the reference against which the manufacturer is able to validate production.
What are relevant standards?
The ISO 11608 series of standards, in particular ISO 11608-1-201213 (and ISO 11608-1-2014,4 especially for EU markets), should be studied by any interested party. As the introduction in ISO 11608-1-2012 states: “[…] it [ie, ISO 11608-1] provides performance requirements regarding essential aspects so that variations of design are not unnecessarily restricted”.13
The rest of the ISO 11608 series of standards relating to other aspects of NISs for medical use, cover the requirements and test methods for: Needles (Part 2); Finished containers (Part 3); Requirements and test methods for electronic and electromechanical pen-injectors (Part 4); and Automated functions (Part 5).
In addition, ISO 11608-1 provides a list of normative references, recommended as “indispensable for the application of this document” (Table 3).13 While this may appear to be a long list, a full set of all these standards and guides is a very useful toolkit for any organization involved in device-dependent medication.
 | Table 3 ISO 11608-1 normative references |
Make or buy?
On the face of it, there is a fairly straightforward “make or buy” option for an insulin manufacturer when it comes to insulin pens. On the one hand, the decision to develop your “own” pen, or have a third-party develop one for you as a unique, new product should mean that the pharma company can have exactly what it wants. However, the crowded patent landscape and the cost and time involved are significant hurdles. Furthermore, for those who have not undertaken such a development before, there is a very steep learning curve!
Not surprisingly, a number of pharma companies elect to outsource pens, often from a handful of established organizations in Western Europe or the USA, or possibly from a growing number of suppliers in other regions. The obvious appeal is the opportunity to save time and development cost. However, time and cost savings will depend heavily on the degree of customization required. The initial stage to identify and define the product opportunity (in device terms) will be very similar irrespective of whether the “make” or “buy” option is taken. This stage is essentially bracketed by the generation of a TPP, assessment of user needs, and issue of the URS. The design process itself, from URS issue through generation and issue of the PRS and approaching the issue of a DDS (hence detail design stage, heading for pilot manufacture), holds the prospect for significant savings if a chosen “off the shelf” design is acceptable with little customization. Much the same will apply in the final stages of the process, from DDS issue through pilot manufacture and design verification testing, to industrialization. Here again, the cost and timescale will depend heavily on the extent of any customization. At one extreme, extensive customization can result in timescales and costs which approach those of a full-scale new development. At the other extreme, minor changes (eg, in external body color or labeling) can often be accommodated fairly quickly, at relatively modest cost, and at a lower risk. Whichever route is taken, the resulting product will still be subject to the same regulatory approval requirements and relevant clinical and user trials.
This spectrum of choice, and related increase in risk, cost and development time exists for almost any device, including pen injectors, and is illustrated in Figure 3.
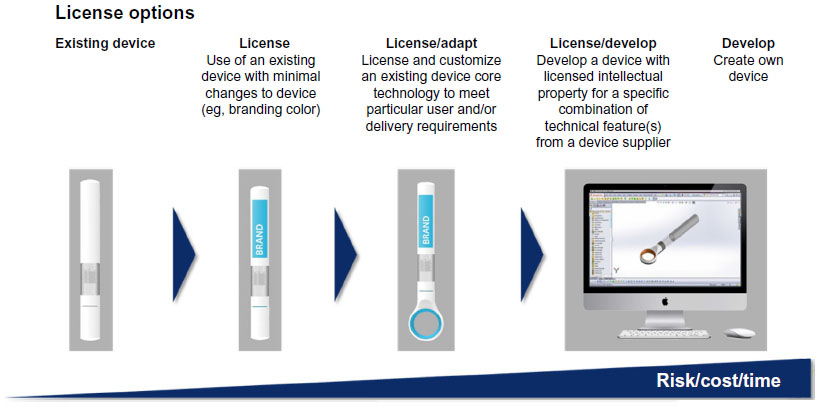 | Figure 3 Device development – a spectrum of choice and related risk/cost/time. |
Although the above comments relate principally to the pen “mechanism” and associated user interface, the primary container (or Container Closure System) and any related drug-contacting parts of the device are of major importance given the sensitive nature of insulin or similar peptide and protein drugs. Kuhlman and Schmidt14 refer to the “implications for patients, physicians, and health care systems” of silicone oil from syringe lubricant, silica nanoparticles, tungsten leachates, and stainless steel microparticles, any of which may interact with or adsorb therapeutic proteins. The same article concludes that, specific regulatory standards for the approval of biosimilar insulins are well defined in Europe, but there is significant variability in the extent of clinical and quality evidence required for approval outside the EU.
How does the future look?
As a postscript to the above comments and cautions regarding biosimilar insulins and in particular, the pens which are the “sine qua non” for their delivery, it is perhaps interesting to look at how at least one manufacturer has approached the delivery of a new diabetes management therapy. In September 2014, Trulicity (dulaglutide), from Eli Lilly and Company, was approved by the FDA for adults with type 2 diabetes. Trulicity is the first ready-to-use, once weekly glucagon-like receptor-1 (GLP-1) receptor agonist (competitor GLP-1 receptor agonist products require reconstitution). The drug is delivered using a single-use autoinjector with a hidden needle and a simple, four-step use sequence. It is unlikely that this type of drug product and approach to delivery device will replace the dial-a-dose pens in the short-term; they clearly have and will retain a very strong presence across markets. Nevertheless, the direction which future biosimilar products, perhaps with a similar dosing regimen and therapeutic effect to that of dulaglutide, might take will be of interest; as will the role of the associated delivery devices required to support these biosimilar products.
Acknowledgments
The authors acknowledge the support of the following colleagues in the preparation of this article: Dr Stella Wooder (Team Consulting Ltd), Dr Max Rubin (Cambridge Healthcare Research Ltd), Dr Matteo Perucchini (Cambridge Healthcare Research Ltd), and Mark DiCioccio (Team Consulting Ltd).
Disclosure
The contents of the article and the opinions expressed within are those of the authors. The authors report no conflicts of interest in this work.
References
Nathan DM. Diabetes: Advances in Diagnosis and Treatment. JAMA. 2015;314:1052–1062. | |
Gerich JE. Novel insulins: expanding options in diabetes management. Am J Med. 2002;113:308–316. | |
Heinemann L, Hompesch M. Biosimilar Insulins: Basic Considerations. J Diabetes Sci Technol. 2014;8:6–13. | |
International Organization for Standardization. ISO 11608-1:2014. Needle-based injection systems for medical use-Requirements and test methods – Part 1: Needle-based injection systems. 2014. | |
Pearson TL. Practical aspects of insulin pen devices. J Diabetes Sci Technol. 2010;4:522–531. | |
Krentz AJ, Hompesch M. Biosimilar insulins: Current and future perspectives. Diabetes Management. 2015;5:405–409. | |
Sanofi, Eli Lilly reach deal on insulin product. at http://www.reuters.com/article/2015/09/28/us-sanofi-patent-idUSKCN0RS1R220150928.) Accessed August 1, 2015. | |
Friedrichs A, Bohnet J, Korger V, Adler S, Schubert-Zsilavecz M, Abdel-Tawab M. Dose accuracy and injection force of different insulin glargine pens. J Diabetes Sci Technol. 2013;7:1346–1353. | |
Tschiedel B, Almeida O, Redfearn J, Flacke F. Initial experience and evaluation of reusable insulin pen devices among patients with diabetes in emerging countries. Diabetes Ther. 2014;5:545–555. | |
Bolge SC, Goren A, Tandon N. Reasons for discontinuation of subcutaneous biologic therapy in the treatment of rheumatoid arthritis: a patient perspective. Patient Prefer Adherence. 2015;9:121–131. | |
Class JN, Langis L. A patient-centred paradigm for the biosimilars market. GaBI J. 2012;1:17–21. | |
Food and Drug Administration. 1997. Design control guidance for medical device manufacturers. http://www.fda.gov/downloads/Medical Devices/…/ucm070642.pdf. Accessed August 1, 2015. | |
International Organization for Standardization. ISO 11608-1:2012. Needle-based injection systems for medical use – Requirements and test methods – Part 1: Needle-based injection systems: BSI Standards Limited. | |
Kuhlmann MK, Schmidt A. Production and manufacturing of biosimilar insulins: implications for patients, physicians, and health care systems. Biosimilars. 2014;4:44–58. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.