Back to Journals » Drug Design, Development and Therapy » Volume 20
Computational Discovery and Optimization of a Potent, Selective, and Drug-Like Scaffold for p38α Inhibition
Authors Nelen J ![]() , Goettert MI, Forster M, Breuils L, Villalgordo-Soto JM, Laufer S, Pérez-Sánchez H
, Goettert MI, Forster M, Breuils L, Villalgordo-Soto JM, Laufer S, Pérez-Sánchez H
Received 29 November 2025
Accepted for publication 26 March 2026
Published 3 June 2026 Volume 2026:20 585325
DOI https://doi.org/10.2147/DDDT.S585325
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Anastasios Lymperopoulos
Jochem Nelen,1,2 Márcia Inês Goettert,3 Michael Forster,3 Laure Breuils,4 José Manuel Villalgordo-Soto,5 Stefan Laufer,3 Horacio Pérez-Sánchez1
1Structural Bioinformatics and High Performance Computing Research Group (BIO-HPC), UCAM HiTech, Universidad Católica de Murcia UCAM, Guadalupe, Murcia, 30107, Spain; 2Health Sciences PhD Program, Universidad Católica de Murcia UCAM, Guadalupe, Murcia, 30107, Spain; 3Department of Pharmaceutical/Medicinal Chemistry, Eberhard Karls University Tübingen, Tübingen, 72076, Germany; 4Eurofins-Cerep S.A., Celle-Lévescault, 86600, France; 5Eurofins-Villapharma Research, Murcia, 30320, Spain
Correspondence: Stefan Laufer, Department of Pharmaceutical/Medicinal Chemistry, Eberhard Karls University Tübingen, Auf der Morgenstelle 8, Tübingen, 72076, Germany, Email [email protected] Horacio Pérez-Sánchez, Structural Bioinformatics and High Performance Computing Research Group (BIO-HPC), UCAM HiTech, Universidad Católica de Murcia UCAM, Campus de los Jerónimos n° 135, Guadalupe, Murcia, 30107, Spain, Email [email protected]
Purpose: The aim of this study was to discover and optimize a novel chemical scaffold capable of selectively inhibiting p38α, a kinase involved in inflammatory and neurodegenerative diseases. Despite decades of work, most p38α inhibitors have failed clinically due to limited selectivity, compensatory signaling, and safety issues. We sought to combine computational and experimental approaches to identify potent, drug-like, and selective inhibitors suitable for further development.
Methods: A consensus virtual screening workflow (ESSENCE-Dock), integrating DiffDock, LeadFinder, and GNINA, was applied to the Eurofins–Villapharma compound library. The top hit guided similarity searching and clustering to identify related analogues for structure–activity relationship studies. Binding modes and substituent contributions were analyzed using molecular modeling and molecular dynamics simulations. Biochemical HTRF assays, ADME profiling, NanoBRET intracellular target engagement, and kinome-wide screening were used to evaluate potency, cellular activity, and selectivity.
Results: Virtual screening identified a previously unreported 3,5-disubstituted dihydropyrazolo[1,5-a]pyrazinone scaffold as a potent p38α inhibitor (IC50 = 26 nM). Evaluation of related analogues yielded several compounds with sub-10 nM activity. Molecular dynamics simulations supported stable binding through interactions with MET109, ASP168, and LYS53. Selected compounds demonstrated high plasma stability, moderate solubility and permeability, strong intracellular target engagement (IC50 < 10 nM), and excellent selectivity across a 468-kinase panel.
Conclusion: This study identifies and characterizes a novel and drug-like p38α inhibitor scaffold with potent biochemical activity, high kinome selectivity, and confirmed intracellular target engagement. The combined computational–experimental workflow provides a strong foundation for further optimization toward therapeutic candidates for inflammatory and neurodegenerative diseases. Graphical abstract showing discovery of p38α inhibitor compound 26 through ESSENCE-Dock virtual screening and SAR optimization. Initial hit compound 24 (IC₅₀ = 26.2 nM) was optimized to compound 26 (IC₅₀ = 13.3 nM) with improved selectivity and favorable ADME properties.Diagram showing virtual screening, initial hit and optimized compound with properties.
Keywords: p38α MAP kinase, virtual screening, structure-based drug design, computational drug discovery, kinase inhibitor
Introduction
Mitogen-activated protein kinase 14 (MAPK14), also known as p38α, is a stress-activated serine/threonine-specific kinase that plays a crucial role in the larger p38 MAPK family, which also comprises p38β, p38γ, and p38δ isoforms.1 As the most studied member of the MAP kinase family, p38α has been reported to be involved in the integration of multiple biochemical signals, influencing a wide range of cellular processes, including cell proliferation, differentiation, transcription regulation, and development.2
Due to its role in various cellular processes, p38α has been implicated in various physiological and pathological processes, including inflammation, cancer, and neurodegenerative disorders.3–5 Consequently, p38α has been a target of interest for drug development for many years, with numerous clinical trials aiming to safely and effectively modulate its activity. Although these trials have not yet yielded successful outcomes, ongoing research continues to explore the therapeutic potential of p38α inhibition or modulation.6
One of the earliest breakthroughs in p38α inhibitor research came with the discovery of SB203580 in 1994, marking a pivotal step in understanding the MAPK pathway.7 This compound, a competitive ATP-binding site type 1 inhibitor, provided foundational insights into p38α’s role in inflammatory processes, laying the groundwork for future research. In 2002, the first type 2 inhibitor, BIRB 796, was introduced.8 It employed an innovative mechanism, binding p38α by targeting a deep buried hydrophobic pocket which is only accessible in the inactive DFG-out conformation of the kinase. This approach enhanced potency and pioneered a novel class of MAPK inhibitors. In 2011, Pfizer introduced PF-03715455, a p38α/β inhibitor formulated for inhalation, aimed at treating chronic obstructive pulmonary disease (COPD).9 While its localized delivery strategy was promising, Phase 2 of clinical trials were discontinued for business reasons.10 In the same year, Skepinone-L emerged as a novel scaffold, and was reported as one of the most specific and potent inhibitors at the time. Its excellent in vivo efficacy and selectivity made it a standout in p38α research.11 Subsequent optimization of Skepinone-L focused on improving its target residence time (TRT), culminating in the development of LN2015, which further enhanced its therapeutic potential.12 In 2015, MW150 was reported as a selective, brain-penetrant p38α inhibitor, representing a significant advancement in CNS-targeted therapeutics.13 Proposed as a clinical candidate for Alzheimer’s disease, MW150 demonstrated promise in addressing neuroinflammatory processes, making it a compelling prospect for neurodegenerative conditions. It is currently in clinical trials and has recently been shown to improve systems including reduced behavioral impairment, reduction in tau phosphorylation in mice, and a partial normalization of electrophysiological parameters.14 Together, these examples illustrate the broader clinical and medicinal chemistry landscape of p38α inhibitor development, in which promising compounds have repeatedly emerged but have often been limited by challenges related to selectivity, safety, or clinical efficacy; recent reviews and patent analyses provide comprehensive overviews of these earlier efforts.15,16 Structures of the p38α inhibitors discussed in this section are shown in Figure 1.
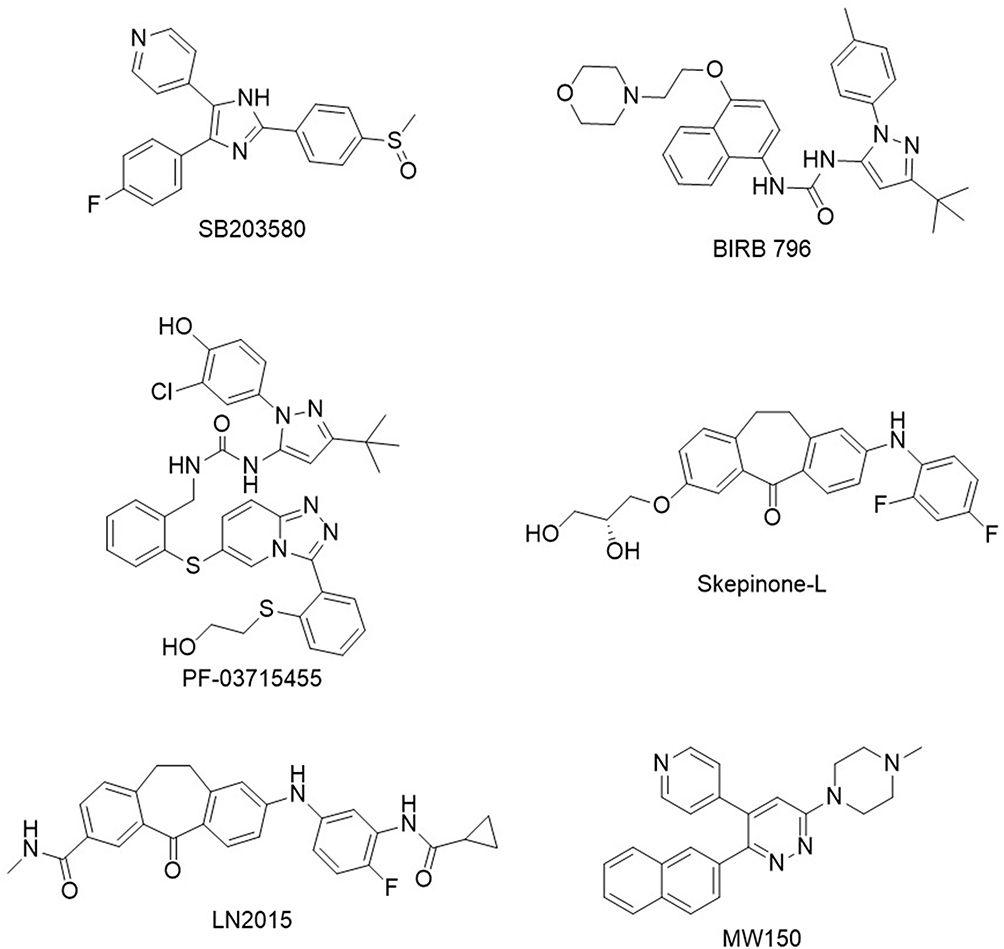 |
Figure 1 Structures of key p38α inhibitors. Shown are SB203580, BIRB 796, PF-03715455, Skepinone-L, LN2015, and MW150, representing major advances in p38α inhibitor development across different structural classes and mechanisms. |
From these examples, it is evident that despite encouraging preclinical data, p38α inhibitors have yet to achieve consistent clinical efficacy. Several factors may contribute to this challenge. One significant obstacle is the cellular adaptability to p38α inhibition, whereby compensatory activation of other p38 MAPK family members or alternative pathways may diminish the therapeutic impact of selective inhibitors.17 Additionally, the intricate interplay between the p38α MAPK pathway and other signaling networks often leads to pleiotropic effects, complicating the prediction of treatment outcomes. Off-target interactions, including those with other kinases or non-kinase proteins, may further undermine the specificity and safety of these compounds.18 Moreover, the therapeutic window for p38α inhibitors is frequently constrained by dose-dependent toxicities, limiting their clinical utility. These complexities underscore the inherent challenges of targeting the p38 MAPK pathway and emphasize the need for innovative and more sophisticated strategies to surmount these barriers.
In this study, we report and comprehensively characterize a novel p38α inhibitor scaffold that exhibits robust biochemical activity, favorable ADME properties, high kinome selectivity, and potent intracellular target engagement. The scaffold demonstrates favorable synthetic accessibility, enabling efficient chemical synthesis and high yields, making it a practical candidate for further exploration. These attributes position it as a compelling starting point for optimization efforts, paving the way for the development of more potent and selective p38α inhibitors with potential therapeutic applications.
Materials and Methods
Chemical Synthesis
Unless otherwise noted, all reagents were purchased from commercial suppliers and used without purification. Thin Layer Chromatography was carried out on Merck precoated silica gel 60 F254 plates. MPLC purifications were performed using Biotage isolera One using pre-packed SiO2 cartridges 60Å (40–60 µm). NMR spectra were recorded on a Bruker Ascend AVNEO400 MHz (400 MHz for 1H) with a Bruker 5mm PA BBO 400S1 BBF-H-D-05-Z SP probe, using a SampleXpress sample changer. For the 1H spectra, all chemical shifts are reported in part per million (δ) units, and are relative to the residual signal at 7.26 and 2.50 ppm for CDCl3 and DMSO, respectively, at 25 °C. High-resolution mass spectra (HRMS) were obtained on an Agilent 6230 LC/TOF instrument.
All compounds were characterized by LC–TOF MS and 1H NMR to confirm identity. Purity was assessed by HPLC with a PDA detector. All key compounds advanced to ADME profiling, kinome selectivity studies, and cellular assays (Compounds 26, 28, 47, 48, 50, and 53) had purities ≥95%. The general synthetic strategy is described below, while detailed experimental procedures, including a representative synthesis of Compound 26, are provided in the Supporting Information (Experimental Details, Chemical Synthesis). In addition, full characterization data for the key compounds, including full HPLC traces, LC–TOF MS and 1H NMR spectra are provided in Figures S1–S6 in the Supporting Information.
General Synthesis
The synthesis of the corresponding 3,5-disubstituted-4,5-dihydropyrazolo[1,5-a]pyrazinones was efficiently achieved starting from acetylenic ketone 1 (Scheme 1).
 |
Scheme 1 Synthesis of key intermediate 3-iodopyrazolopyrazinone 4. Reagents and conditions: (i) ethyl hydrazinoacetate hydrochloride, DIPEA, EtOH, 0°C-r.t. 5 h., (78%); (ii) HCl (4N in Dioxane), CH2Cl2, 0°C-r.t. overnight, then NaHCO3, Toluene, 110°C, 12 h. (92%, two steps). (iii) NIS, MeCN, 60°C, 12h. (92%). |
Thus, reaction of 1 with ethyl hydrazinoacetate hydrochloride in the presence of DIPEA in EtOH at 0°C, afforded almost exclusively the corresponding regioisomeric pyrazole derivative 2 in high yields. Subsequent cleavage of the Boc-protecting group by reacting 2 with 4N HCl in Dioxane at 0°C, followed by neutralization with a solution of saturated NaHCO3 of the resulting hydrochloride and heating the resulting free primary amine in toluene at 110°C yielded the corresponding 2-(4-fluorophenyl)-4,5-dihydropyrazolo[1,5-a]pyrazin-6-(7H)-one 3 in high yield. Next, reaction of 3 with NIS in MeCN at 60°C produced key intermediate iodinated fused pyrazole 4 also in excellent yield.
Then, from key intermediate 4, Suzuki and Stille coupling reactions catalyzed by Palladium afforded the corresponding pyridinyl- 5 and pyrimidinyl derivatives 7 in good yields (Scheme 2). From fluoropyridine derivative 5, nucleophilic displacement with cyclohexylamine afforded target derivative 6 also in good yields. From methylthiopyrimidinyl intermediate 7, oxidation of the thioether moiety yielded sulfone derivative 8 in quantitative yields. Intermediate 8 was then subjected to nucleophilic displacement with different primary amines in DMA at 140°C, affording targets 9a-c in good to moderate yields.
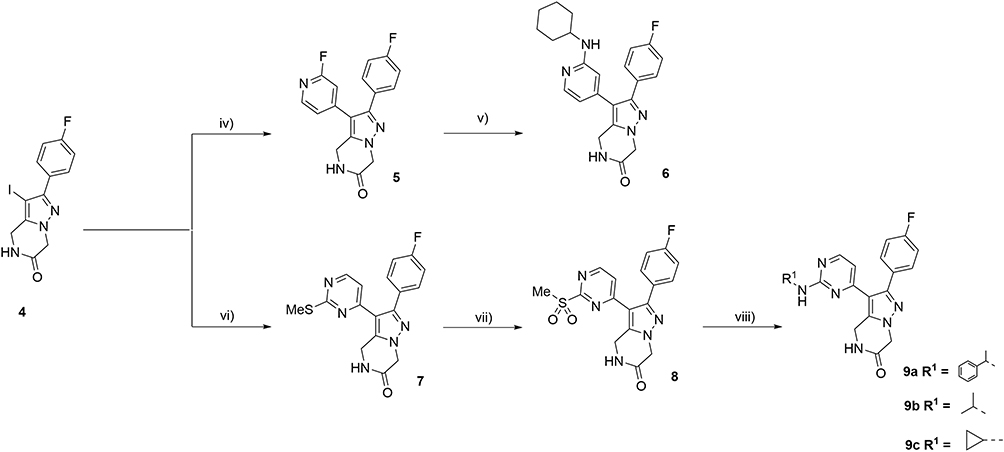 |
Scheme 2 (iv) (2-fluoropyridin-4-yl)boronic acid, Pd(dppf)Cl2, Cs2CO3, Dioxane/H2O (4:1), 115°C, 6 h. (87%); (v) cyclohexyl amine (4 eq).; DMA, 140°C, (68%). (vi) 2-(methylthio)-4-(tributylstannyl)pyrimidine, CuI, PPh3 (0.1 eq)., Pd(PPh3)4, Toluene, 110°C, 12 h. (62%). (vii) m-CPBA, CH2Cl2, 0°C-r.t. 12 h., (69%). (viii) Primary amine (4 eq)., DIPEA, DMA, 140°C, (56–62%). |
Alternatively, key intermediate 3 when subjected to N-alkylation in the presence of NaH, the corresponding N-alkylated products 10a,b were obtained in good yields (Scheme 3). Upon iodination with NIS in MeCN to 11a,b, and an analogous sequence as described above through Stille coupling, oxidation and final nucleophilic displacement with (S)-2-aminopropanol using analogous reaction conditions afforded the corresponding targeted 14a and 14b respectively in moderate to good overall yields.
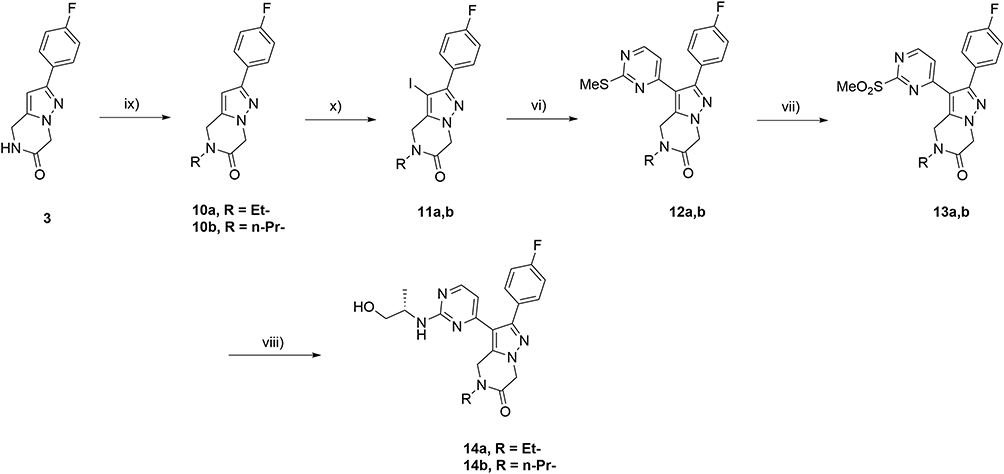 |
Scheme 3 (ix) NaH, DMF, R-I, 0°C-r.t., 5 h. (75–79%); (x) NIS, MeCN, 50°C, 12h., (90%); (vi) 2-(methylthio)-4-(tributylstannyl)pyrimidine, CuI, PPh3 (0.1 eq.), Pd(PPh3)4, Toluene, 110°C, 12 h. (58%); (vii) m-CPBA, CH2Cl2, 0°C-r.t. 12 h. (74%); (viii) (S)-2-amino-1-propanol (4 eq.), DIPEA, DMA, 140°C, (68–72%). |
ESSENCE-Dock Calculations
For the ESSENCE-Dock protocol,19 three different docking programs were employed as described in the original manuscript: LeadFinder,20 GNINA,21 and DiffDock.22 These programs require ligands in mol2, pdbt, and SDF formats respectively. The protein formats are of the same format, except for DiffDock, which uses pdb format for the target protein. To generate the correct ligand formats, the Eurofins-Villapharma library was converted from smiles to SDF format using RDKit’s ETKDG method.23 The resulting SDF file was then converted to mol2 format using ChemAxon’s Molconvert,24 and subsequently to pdbqt format using the “prepare_ligand4.py” script included with AutoDockTools.25
The p38α protein structure was prepared by importing the pdb code 3S4Q into Schrödinger’s Maestro tool.26,27 The Protein Preparation Wizard was used to remove crystallographic water molecules, add hydrogens and assign bond orders, after which the system was parameterized using the OPLS4 force field.28 The prepared protein was saved in mol2 and pdb formats for use with LeadFinder and DiffDock, respectively. Additionally, AutoDockTools was used to convert the pdb file to pdbqt format, adding hydrogens, assigning AD4 atom types, and computing Gasteiger charges.29
Docking calculations for GNINA and LeadFinder were conducted using Metascreener V1.2.30 GNINA docking was performed with GNINA v1.0.3 using pdbqt-formatted protein and ligand files. LeadFinder docking was carried out using version 2104 with mol2-formatted inputs. The protein was treated as rigid during docking, and no explicit water molecules were included. Default docking parameters were used unless otherwise specified. A cubic docking box (25 Å × 25 Å × 25 Å) centered on the binding site was used for both docking protocols to ensure consistent sampling of the active site. DiffDock calculations were performed with DiffDockHPC v1.0,31 a parallelized version of DiffDock optimized for high-throughput virtual screening. Standard settings were used, with the protein provided in pdb format and ligands in SDF format. As a blind docking method, DiffDock did not require predefined docking coordinates, identifying the binding site automatically.
Finally, after the prepared Eurofins-Villapharma library was screened using the three docking algorithms, the docking runs were integrated using ESSENCE-Dock as described in the original manuscript.
Consensus Based Fingerprint Similarity and Clustering
To explore the chemical space around Compound 24, a similarity search was conducted using ConFiLiS,32 which leverages a consensus of molecular fingerprints. The Eurofins-Villapharma library was encoded into three fingerprint types—ECFP6,33 PubChem,34 and Avalon35—using the PyFingerprint package.36 For each compound, the Euclidean distance to Compound 24 was calculated across all three fingerprints. These distances were normalized and averaged to obtain a single similarity score per compound. A similarity cutoff of −3 was chosen as it provided a good balance between structural similarity and the availability of a sufficient number of compounds for subsequent clustering. An open-source implementation of this methodology can be found on GitHub: https://github.com/Jnelen/ConFiLiS.
To ensure diversity in the final selection and reduce potential selection bias, a clustering approach was applied to the resulting 107 compounds. Butina clustering,37 implemented via RDKit and adapted from the TeachOpenCADD project,38 was performed using ECFP6 fingerprints with a clustering distance threshold of 0.25. Clusters with fewer than three compounds were excluded, yielding 11 clusters containing 3 to 8 compounds each. From each cluster, three compounds were randomly selected, resulting in a final set of 33 compounds for biological testing.
Homogeneous Time-Resolved Fluorescence (HTRF) Kinase Assay
Compounds were diluted with a Kinase buffer (with p38α enzyme) arising out of a stock solution of 10 mM in DMSO, giving a final concentration of 10 µM. Subsequently the compound dilution is added to the enzyme solution in a 96-well non-binding plate and pre incubated for 10 minutes at 37 °C. Afterwards, an ATP/ATF2 solution is added to the previous solution and incubated again for 30 minutes at 37 °C. The assay was performed using the HTRF detection kit (Cisbio, Bedford, MA) by adding 10 µL of the HTRF detection solution (2.5 µL of PAb Anti-phospho-ATF2-Eu cryptate; 5 µL of MAb Anti GST-d2; and 992.5 µL of HTRF detection buffer). The plate was incubated for 30 minutes at room temperature in the dark. HTRF signal was read out in the Victor Nivo®, and calculated as the ratio of signal from the 665 nm (acceptor) and 615 nm (donor) channels and multiplied by 10,000.
Internal assay controls for maximum activity (STIM; enzyme with vehicle only) and minimum activity (NSB; background signal in the absence of enzyme activity) were included in each experiment. The kinase activity was calculated by normalizing each the HTRF signal from each sample well to the mean HTRF signal from the DMSO only control wells, using the following equation:
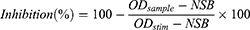
Where ODsample is the HTRF signal measured in the presence of the test compound, ODstim is the signal from stimulated control wells containing enzyme and vehicle only, and NSB is the background signal obtained when enzyme activity is absent.
All compounds were tested in triplicate in each experiment to ensure measurement reproducibility. Skepinone-L was included as a positive control in every assay to validate assay performance. IC50 values were determined by nonlinear regression analysis of the dose–response curves using GraphPad Prism 7.0 (GraphPad Software, Inc., San Diego, CA, USA) with default nonlinear regression settings.
QSAR Analysis Using MolSHAP
MolSHAP calculations were carried out using tools and scripts provided in the official GitHub repository.39 Experimental IC50 values were converted into pIC50 values and compiled into a CSV file, which included canonical SMILES strings alongside their corresponding pIC50 values. This dataset served as the primary input for automated model training and predictions as part of the MolSHAP workflow.
Automated R-group decomposition was the first step in the MolSHAP workflow. This process identified four distinct R-groups: R1 with 2 variants, R2 with 3 variants, R4 with 6 variants, and R3 with 20 variants. Using this information, a machine learning model was trained automatically which achieved a Pearson correlation coefficient of 0.74, indicating robust predictive performance. For compounds within the training dataset, the model exhibited good results, with both a MacroF1 score and accuracy of 0.97. When tested on previously unseen compounds, the model’s performance declined slightly, achieving a MacroF1 score of 0.74 and an accuracy of 0.78.
Molecular Dynamics Simulations
The initial complexes for the molecular dynamics (MD) simulations were generated using DynamicBind, a diffusion-based docking tool that incorporates protein flexibility into its calculations.40 This capability is particularly effective for proteins with highly dynamic regions, such as the DFG-loop found in kinases, including p38α. For the target protein, the full-length p38α protein model from the AlphaFold Protein Structure Database (UniProt ID: Q16539) was used to ensure no residues or loops were missing.41 The protein was further prepared using Schrödinger’s Maestro software suite, utilizing the Protein Preparation Wizard and the System Builder to correctly assign bond orders and charge the system.26,42 The DynamicBind calculations were performed using DynamicBindHPC v1.0, a custom fork simplifying the screening of multiple ligands by running calculations in parallel.43 Default settings were used, including automatic relaxation of the complexes after docking. All tested compounds exhibited favorable confidence metrics, with predicted local distance difference test (lDDT) scores ranging from 0.68 to 0.71, indicating their suitability as robust starting points for the MD simulations.
The resulting protein-ligand complexes were subsequently imported into Maestro, where system parameterization was performed using the OPLS4 force field.28 Each complex was placed in a simple point charge (SPC) water box with a 10 Å buffer zone to ensure full solvation. Physiological ionic conditions were simulated by introducing 0.15 M NaCl, with the appropriate number of Na⁺ and Cl− ions automatically calculated. MD simulations were carried out using Maestro-Desmond under constant temperature (300 K) and pressure (1 atm) conditions using the NPT ensemble to maintain stable system parameters.44 Each system underwent 100 nanoseconds of simulation, with trajectory data recorded at 4.8 picosecond intervals for detailed downstream analyses. Trajectory evaluations, including protein-ligand interaction profiling, were initially conducted using the Desmond simulation interaction diagram tool within Maestro. To enhance visualization and interpretability, protein-ligand interaction data were visualized using a custom script to process the data and generate the protein-ligand histogram figures.
NanoBRET Intracellular Target Engagement Assay for MAPK14
Intracellular binding to p38α (MAPK14) was assessed using the p38alpha (SAPK2A) (MAPK14) Human CMGC Kinase Cellular TE NanoBRET LeadHunter Assay (Eurofins-Cerep S.A., Celle-Lévescault France). HEK293-derived 293H cells were transfected with a construct encoding full-length human MAPK14 fused at the C-terminus to NanoLuc® luciferase. Cells were seeded in 384-well plates and incubated overnight.
Tested compounds were added in serial dilutions in the presence of 0.027 µM NanoBRET tracer K-4. After a 120-minute incubation at 37 °C, NanoBRET substrate and extracellular NanoLuc inhibitor were added, and BRET signals were measured using donor (450 nm) and acceptor (600 nm) filters.
The assay readout reflects competitive displacement of the tracer by test compounds. IC50 values were determined by nonlinear regression using the Hill equation. Dasatinib was included as a reference compound (IC50 = 130 nM), confirming expected assay performance.
Kinase Selectivity Assay
Kinase selectivity profiling was performed using the KINOMEscan® binding assay platform (DiscoverX Corporation). This assay measures compound binding across a large kinase panel using an active-site-directed competition binding format. DMSO was used as the negative control (100% control signal), while an internal reference inhibitor provided within the assay platform was used as the positive control (0% control signal). Selectivity profiles were visualized using TREEspot™ kinase dendrograms. Detailed experimental procedures are provided in the Supporting Information (Experimental Section, Kinase Selectivity Assay).
ADME Characterization
Physicochemical and ADME properties of the compounds were characterized by the Eurofins–Villapharma ADME team using validated and standardized experimental protocols. All assays were performed under controlled laboratory conditions to ensure data reliability and consistency. The evaluated parameters included plasma protein binding, aqueous solubility, lipophilicity (logD), Caco-2 permeability (C2BBe1 clone, ATCC CRL-2102), plasma stability, intrinsic clearance in human liver S9 fractions, and CYP3A inhibition. Detailed descriptions of the experimental procedures for each assay are provided in the Supporting Information (Experimental Details, ADMET Characterization).
Results and Discussion
Virtual Screening with ESSENCE-Dock
In order to efficiently screen the highly diverse and proprietary library of Eurofins-Villapharma, we employed ESSENCE-Dock,19 a consensus virtual screening approach. This method leverages the strengths of three distinct molecular docking algorithms: LeadFinder,20 GNINA,21 and DiffDock.22 By combining the output data from these calculations, ESSENCE-Dock rescores all compounds based on a comprehensive set of parameters, including docking scores, predicted binding poses, and ligand flexibility. This consensus approach enables the identification of compounds that exhibit consistent and similar predicted binding modes across the different docking methods. As a result, ESSENCE-Dock has been shown to reduce false positive hits and increasing the enrichment of active compounds, thereby enhancing the overall efficiency of the virtual screening process.19
Building on this capability, we leveraged ESSENCE-Dock to screen the entire proprietary Eurofins-Villapharma library against p38α. The highest-scoring compounds from the ESSENCE-Dock virtual screening campaign were selected by applying a stringent cutoff of −20 to the ESSENCE-Dock scores, which the original publication suggested is a reasonable cutoff. The measured IC50 values of the selected compounds, along with their individual docking scores, ESSENCE-Dock scores, and other relevant metrics, are presented in Table 1. The structures of the compounds showing any activity against p38α are shown in Figure 2.
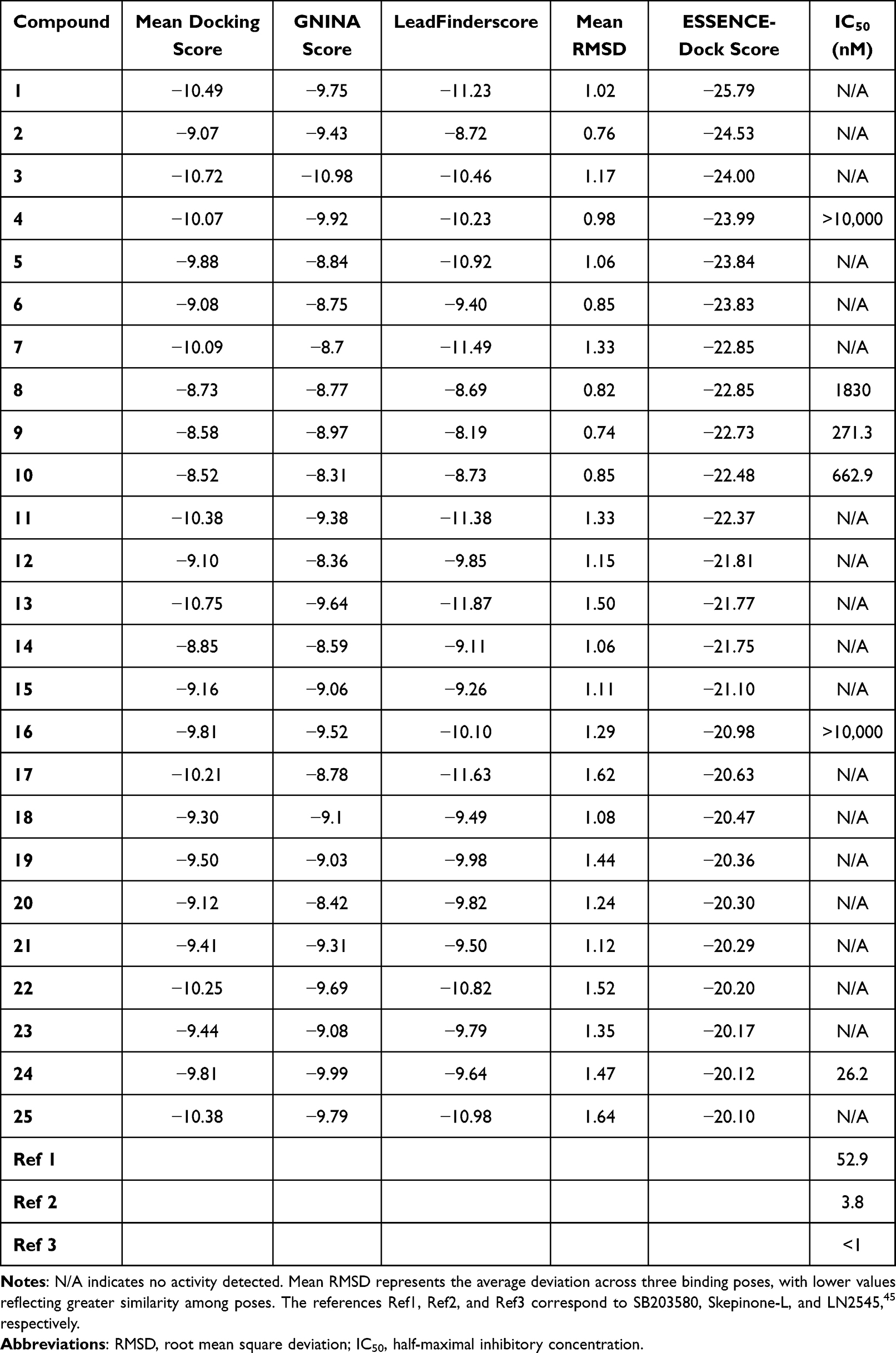 |
Table 1 Top ESSENCE-Dock Results with an ESSENCE-Dock Score < −20 |
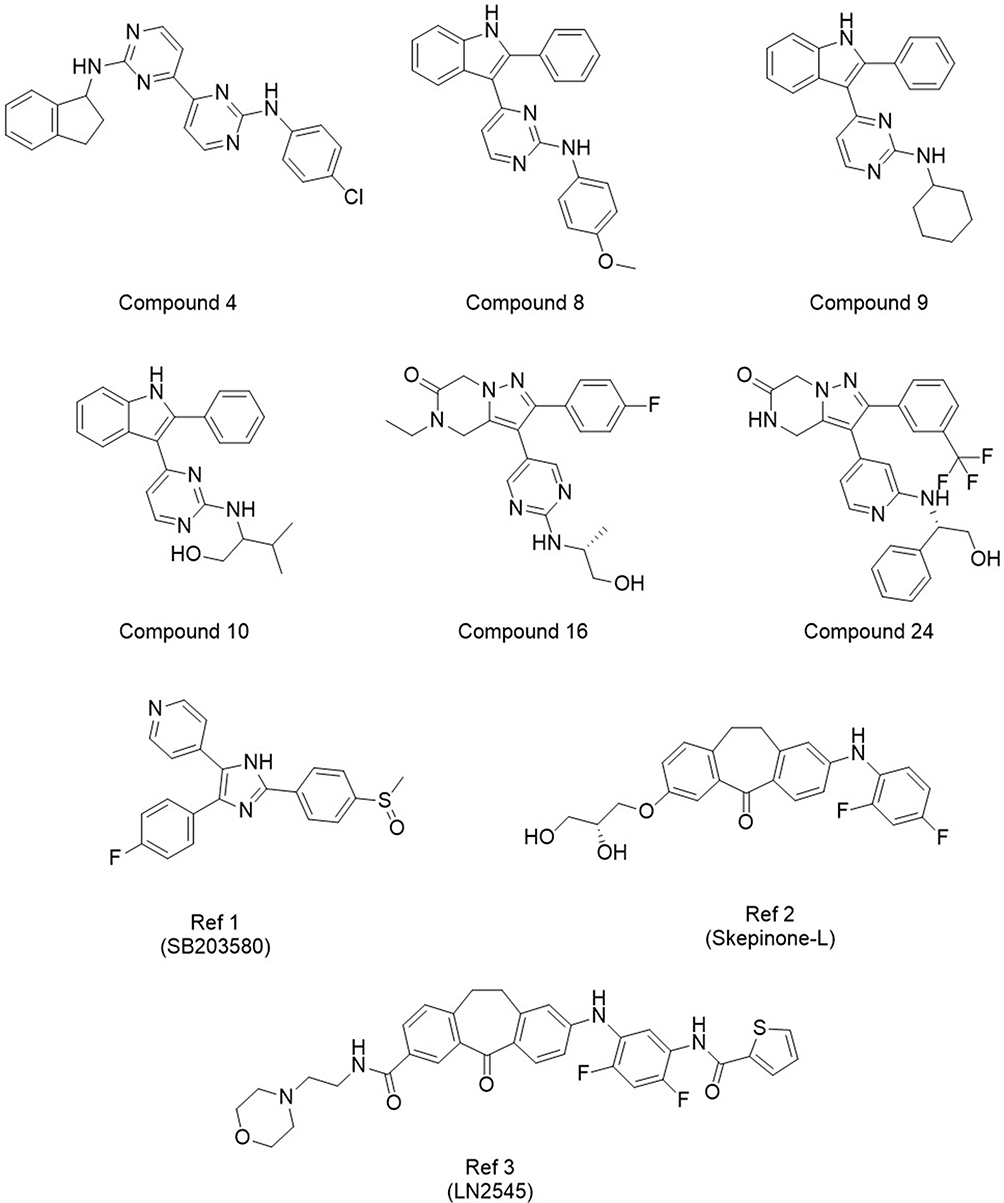 |
Figure 2 Villapharma and reference structures showing measurable activity from Round 1. The corresponding docking scores and measured activities can be found in Table 1. |
As reported in Table 1, several active compounds were identified in the initial round of virtual screening using ESSENCE-Dock. Of the 25 compounds tested, six demonstrated varying levels of activity, with IC50 values ranging from >10 µM to 26 nM. Notably, none of these active compounds would have been selected for testing based solely on the individual or average docking scores from LeadFinder and GNINA. Conversely, some compounds that were highly ranked by individual docking methods did not exhibit any measurable activity upon experimental validation. A concrete example is Compound 3, which ranked 7th based on GNINA’s docking score but failed to show any detectable activity in the experimental assays. This highlights the limitations of relying exclusively on traditional docking scores, which can often fail to predict experimental outcomes reliably. By integrating additional metrics such as binding pose similarity, ESSENCE-Dock effectively prioritizes compounds that demonstrate high consensus between docking methods.
Among the identified active compounds, Compound 4 demonstrated minimal activity, with a measured IC50 above 10 µM. Additionally, its structure was notably distinct from the other active compounds that were identified during this screening run. Due to its low potency and structural divergence from the other hits, the scaffold of Compound 4 was not prioritized for further investigation.
The remaining hits were grouped into two distinct series, each characterized by shared scaffolds. The first series includes Compounds 8, 9, and 10, which all share a phenyl-pyrimidine-indole scaffold. These compounds displayed moderate activity, with IC50 values ranging from 271.3 to 1830 nM. Despite the variation in activity, their ESSENCE-Dock scores were remarkably similar, ranging from −22.85 to −22.48, likely due to comparable docking scores and binding pose similarities. Interestingly, had the compounds been ranked using only individual docking methods or even average docking scores, none would have been selected for testing. Concretely, the highest-ranked compound in this series, Compound 9, was positioned outside the top 2500 by GNINA, while LeadFinder ranked this series even lower, placing it in the bottom 25%. While the moderate activity of the first series of compounds was encouraging, a more potent compound was discovered in the second series, offering a more promising lead for further optimization.
The second series of hits comprises Compounds 16 and 24, which share a novel and common 3,5-disubstituted dihydropyrazolo[1,5-a]pyrazinone scaffold. To the best of our knowledge, this scaffold has not previously been reported as a p38α inhibitor. Despite their structural similarity, their measured activities differed by several orders of magnitude. Compound 16 only showed minimal activity, with an IC50 greater than 10 µM. In contrast, Compound 24, despite their structural similarity, demonstrated very high activity and emerged as the most potent compound, measuring an IC50 of 26.15 nM. This significant difference in activity could be explained by small but important changes in the regioselectivity of the putative hinge-binding motif (aminopyrimidine and aminopyridine, respectively). We hypothesized that the 4-substitution in Compound 16 is not able to establish hydrogen bonds to the hinge region effectively, whereas the 3-substitution in Compound 24 could enable a crucial hinge interaction, leading to a substantial increase in binding affinity and potency. Notably, Compound 24 was also an order of magnitude more potent than the second-best hit, Compound 9 (271.3 nM). Its combination of high potency and a previously unexplored scaffold for p38α inhibition made it a promising candidate for further optimization.
Systematic Exploration and Optimization of Compound 24
Building on its high potency (IC50 = 26.15 nM) and novelty of the scaffold, Compound 24 served as the foundation for subsequent optimization efforts. Notably, its substantial activity advantage over the structurally related but much less potent Compound 16 (IC50 > 10 µM) underscored the need for a deeper exploration of the chemical space surrounding this lead compound.
To guide this exploration, we employed ConFiLiS,32 a consensus fingerprint similarity approach to identify a subset of around 100 compounds from the Eurofins-Villapharma library that shared structural features with Compound 24. To ensure a diverse and representative final selection, we applied Butina clustering,37 as described in the TeachOpenCADD project from the Volkamer lab.38 After clustering, we only kept clusters containing three or more compounds, which resulted in eleven clusters of varying sizes (3 to 8 compounds). From each of these clusters, we randomly selected three compounds for biological testing, yielding a total of 33 compounds. This sampling strategy helped minimize selection bias while maintaining structural diversity across the explored chemical space.
This selection strategy balanced the aim for diversity in compound structures with the goal of generating meaningful structure-activity relationship (SAR) insights. Key information about the selected compounds, including their cluster assignments, their Quantitative Estimate of Druglikeness (QED),46 and IC50 values, are detailed in Table 2. Their chemical structures are also presented in Figure 3, organized row-wise according to their respective clusters for clarity.
 |
Table 2 IC50 Results and QED Data for Clustered Compounds |
![Chemical structures of compounds 26–37, a clustered subset of Round 2 hits sharing a common 3,5-disubstituted dihydropyrazolo[1,5-a]pyrazinone scaffold. Compounds are grouped row-wise according to structural similarity identified by Butina clustering of the Eurofins–VillaPharma library.](article/fulltext_file/585325/aW1n/DDDT_A_585325_O_F0003ag.jpg) |
Figure 3 Clustered Structures of Selected Compounds for SAR Analysis in Round 2. Compounds are organized row-wise by clusters identified through Butina clustering of structurally similar compounds from the Eurofins-Villapharma library. Cluster assignments, QED values, and IC50 measurements are detailed in Table 2. |
![Chemical structures of compounds 38–52, a clustered subset of Round 2 hits sharing a common 3,5-disubstituted dihydropyrazolo[1,5-a]pyrazinone scaffold. Compounds are grouped row-wise according to structural similarity identified by Butina clustering of the Eurofins–VillaPharma library.](article/fulltext_file/585325/aW1n/DDDT_A_585325_O_F0003bg.jpg) |
Figure 3 Continued. |
![Chemical structures of compounds 53–58, a clustered subset of Round 2 hits sharing a common 3,5-disubstituted dihydropyrazolo[1,5-a]pyrazinone scaffold. Compounds are grouped row-wise according to structural similarity identified by Butina clustering of the Eurofins–VillaPharma library.](article/fulltext_file/585325/aW1n/DDDT_A_585325_O_F0003cg.jpg) |
Figure 3 Continued. |
As reported in Table 2, the second round of screening yielded promising results, with all tested compounds showing activity, ranging from low micromolar to low nanomolar range. Overall during this second round, six compounds had measured IC50 values below 10 nM, with three of them exhibiting values under 5 nM. However, two compounds, 40 and 44, could not be evaluated: compound 40 was unavailable, while compound 44 was insoluble and therefore could not be tested reliably.
When analyzing the results in more detail, it was interesting to note that the three compounds belonging to cluster 11 (Compounds 56, 57, and 58), although exhibiting low potency, did have some of the highest QED scores, ranging from 0.728 to 0.736. This suggests that these compounds possess favorable drug-like properties, despite their lower activity. This highlights the well-known problem of finding a good balance between activity and druglikeness: while potency is crucial, favorable ADME properties play a key role in determining a compound’s potential as a viable drug candidate.
Conversely, the two most potent compounds were found in Cluster 8, with the third and fourth most potent compounds being a part of Cluster 10. These compounds displayed high potency with IC50 values ranging from 1.2 nM to 5.6 nM. Notably, compound 47, the most potent in terms of activity (IC50 = 1.194 nM), had the lowest QED score among the top four compounds (QED = 0.503), indicating slightly less favorable drug-likeness. In contrast, the remaining three top compounds were still very potent but also had much higher QED scores, between 0.713 and 0.742, making them more attractive as potential lead candidates. For further assessment, ADME profiling was conducted on a selection of the available top compounds.
These findings highlight the value of combining ConFiLiS consensus fingerprint similarity with Butina clustering to explore the accessible chemical space around the initial scaffold. By selecting and testing compounds that balance structural diversity with relevance to the original hit, we generated meaningful structure–activity relationship insights and identified promising candidates for further optimization.
QSAR Analysis Using MolSHAP
Since all compounds in the second round revolved around a shared scaffold, the data was used to perform a QSAR analysis in order to gain more insights into the structure-activity relationship. More specifically, MolSHAP was used to analyze the results from round 2.39 This tool uses SHapley Additive exPlanations (SHAP),47 which is an interpretative ML technique in combination with R-group decomposition to automatically analyze the impact of different combinations of R-groups for a given dataset. Before analyzing the dataset, the IC50 values were converted to pIC50, as MolSHAP only works with pIC50.
A schematic overview with some of the most substantial results is shown in Table 3. The full data can be found in Table S1 in the Supporting Information (SI). MolSHAP identified four different substituent positions using automatic R-group decomposition. R1 only had two variations, R2 had three, while R4 had six structures. R3 was the largest and most diverse of all and contained 20 variations. The underlying model was trained with the data and exhibited fairly good performance, achieving a Pearson Correlation Coefficient of 0.74.
 |
Table 3 MolSHAP-Predicted Substituent Contributions to Activity (ΔpIC50) |
For R1, it appears that a trifluoromethyl group is most beneficial for activity, providing an average pIC50 uplift of around 0.3, with a hydrogen on this position being considered to be the baseline. For R-group 2, there were three variants: hydrogen (being the baseline), and two halogen substitutions, being chlorine or fluorine. Chlorine provides a very small increase in pIC50 0.018, while fluorine in this position is predicted to be more effective, increasing the pIC50 by 0.136. When combining the information from R-groups 1 and 2, one could assume that a combination of a trifluoromethyl group on position 1 and a fluorine on position 2 would be the best combination. However, we did not verify if this would actually be the case, since in all examples with trifluoromethyl on position 1, a hydrogen was present as R-group 2.
For position 3, the scaffolds are more numerous and diverse, making it difficult to establish clear SAR trends. However, some general patterns were still observed. The best-performing R-groups feature an amino-pyrimidine ring as the scaffold, connected to hydrophobic groups. The top performer is an amino-pyrimidine attached to an ethyl benzene substituent, achieving a predicted pIC50 uplift of +3.14. Other effective hydrophobic groups include isopropyl (+2.84), cyclohexane (+2.66), and cyclopropane (+2.47), highlighting the strong influence of hydrophobicity when paired with the amino-pyrimidine scaffold. Interestingly, when a cyclopropane group is attached to an amino-pyridine instead of an amino-pyrimidine, the pIC50 score drops substantially to +1.88—nearly 0.6 lower than its amino-pyrimidine counterpart.
This appeared to be a broader trend, as amino-pyridines consistently underperformed compared to R-groups containing amino-pyrimidine scaffolds, in agreement with MolSHAP-predicted ΔpIC50 values. This difference may reflect the electronic properties of the heterocycles, with the lower electron density of pyrimidine favoring more effective hinge binding. MolSHAP also highlighted that halogen-substituted pyridines showed reduced activity, likely due to the loss of the NH hydrogen bond donor, which is critical for maintaining interactions at the hinge region.
A fluorine-substituted pyridine was automatically determined as the baseline, having a neutral effect to the predicted pIC50 in this model. Substituting the fluorine with a chlorine atom provides only a minimal improvement (+0.03), demonstrating that minimal functionalization and simpler scaffolds offer little to no benefit. In summary, complex amino-pyrimidine scaffolds paired with hydrophobic groups yield the most significant improvements in pIC50, while amino-pyridines and halogen-substituted pyridines are far less effective.
For position 4, bulky substituents tend to have a negative effect on predicted pIC50. Most substituents reduce activity compared to the baseline (a single hydrogen). The exception is the propyl group, which was predicted to improve the pIC50 by approximately 0.12. Interestingly, even smaller alkyl groups like ethyl and methyl decrease activity. Ethyl has a minimal average effect (−0.012), while the methyl group shows a more pronounced negative SHAP contribution of nearly −0.5. Bulkier groups have an even greater negative impact, with phenethyl and cyclopropylethyl substitutions contributing −0.6 and −0.85, respectively, to the predicted pIC50.
In summary, MolSHAP analysis highlighted the importance of specific scaffold and substituent combinations in driving pIC50 improvements. Notably, amino-pyrimidine scaffolds paired with hydrophobic groups emerged as the most effective for R-group 3, while bulky groups in R-group 4 had consistently negative impacts on activity. These insights can provide a foundation for optimizing future compound designs.
Binding Mode Investigation Using Molecular Dynamics Simulations
To gain a deeper understanding of the binding mode and stability of our compounds, molecular dynamics (MD) simulations were conducted for the most potent candidates: Compounds 26, 28, 47, 48, 50, and 53. Compound 54 was omitted since it was also not available for the ADME testing. Initial protein-ligand complexes were generated using DynamicBind,40 a diffusion-based docking tool that incorporates protein flexibility into its calculations. The original publication demonstrated DynamicBind’s accuracy in predicting binding poses, particularly for proteins involving the DFG-loop, making it an excellent choice for generating initial protein-ligand complex poses. The method yielded favorable confidence metrics for all tested compounds, with predicted lDDT scores ranging from 0.68 to 0.71, indicating reliable protein–ligand complex predictions. An example for Compound 26 is shown in Figure 4.
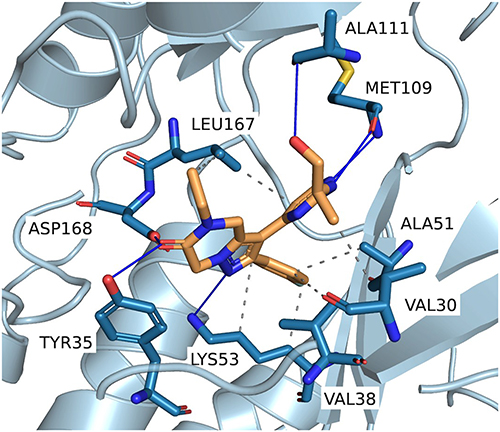 |
Figure 4 Representative binding pose of Compound 26 generated by DynamicBind. Predicted protein–ligand interactions were identified using PLIP,48 highlighting key contacts within the p38α MAP kinase binding site. |
The generated poses showed decent similarities to the binding mode of SB203580 and other pyridinylimidazole-based p38 inhibitors with the aminopyrimidine moiety forming a bidentate hydrogen bond pattern to the backbone of MET109 while the 4-fluorophenyl residue is positioned in the hydrophobic region I via circumvention of the gatekeeper amino acid THR106. The central dihydropyrazolopyrazine core is positioned in the sugar pocket and phosphate binding region with its N-substituent expanding towards the solvent-exposed area at the entrance of the ATP binding pocket. The unsubstituted nitrogen atom of the pyrazole core is directed into the back of the binding pocket enabling direct or water-mediated interactions with LYS53 and ASP168, while the kinase is arranged in a DFG-in conformation. In short, the complexes generated by DynamicBind provided structurally plausible binding poses and were used as starting structures for 100 ns molecular dynamics (MD) simulations to evaluate the dynamic stability of the predicted complexes and analyze ligand–protein interactions within the binding site.
The results of these simulations consistently indicated stable binding across all investigated compounds, as reflected in their RMSD profiles (Figure 5). Ligand RMSD values provided insight into the degree of positional fluctuations during the simulations, with lower values corresponding to higher stability. Compounds 26 and 28 (Figure 5A and B) emerged as particularly stable, exhibiting fluctuations predominantly between 1.0 and 1.5 Å, with only brief deviations reaching 2 Å. Compounds 47 and 48 (Figure 5C and D) also demonstrated sustained stability throughout the simulations. Compound 47 maintained RMSD values mostly within 1.0–1.5 Å, aside from occasional brief spikes approaching up to 3 Å. Compound 48 displayed a similar pattern, albeit slightly more stable. Similarly, Compounds 50 and 53 (Figure 5E and F) exhibited relatively consistent RMSD profiles. Compound 50 fluctuated primarily between 1.0 and 1.5 Å, but did have brief periods of peaking up to 3.5 Å, while Compound 53 showed a more stable progression with RMSD values staying below 2.5 Å. Importantly, none of the compounds exhibited signs of destabilization or unbinding during the simulations.
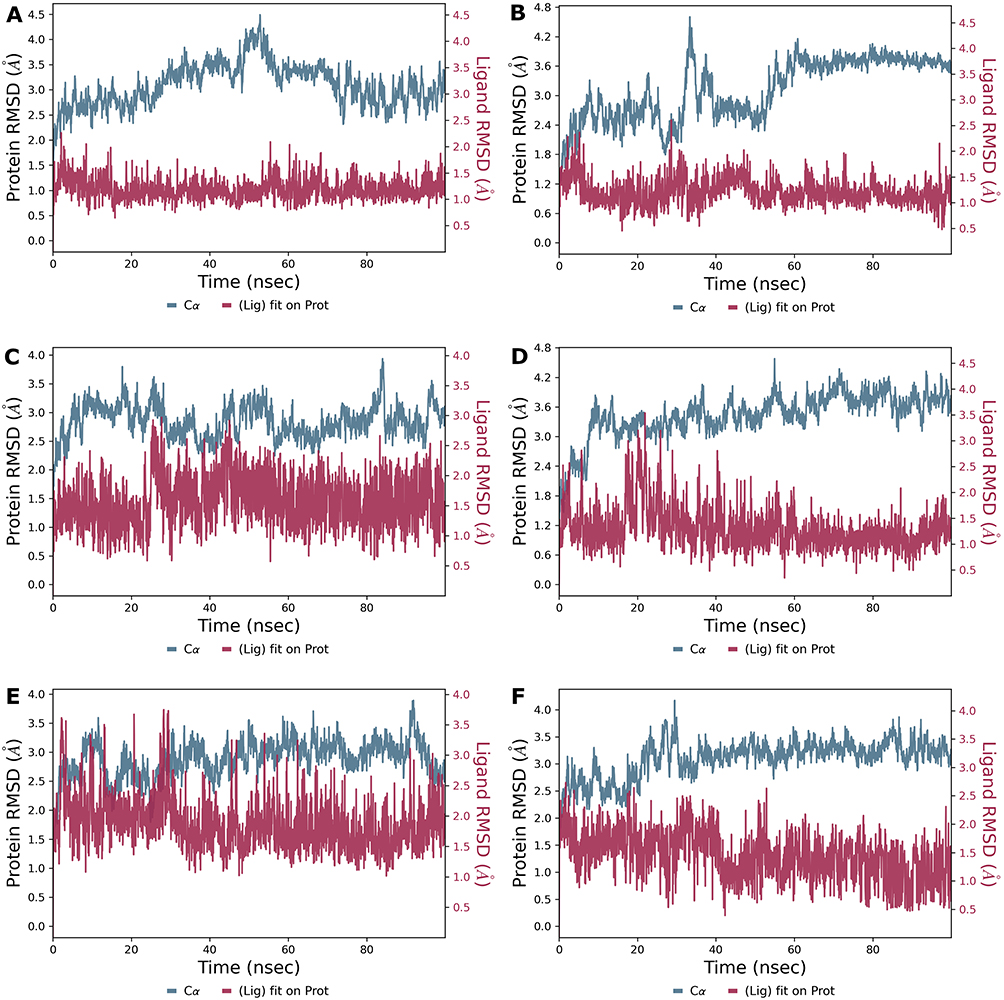 |
Figure 5 Ligand Stability Profiles from 100 ns Molecular Dynamics Simulations. This figure shows the RMSD data from 100 ns molecular dynamics simulations, performed using Maestro Desmond, to evaluate the structural stability of six ligand-protein complexes. Subplots (A–F) correspond to Compounds 26, 28, 47, 48, 50, and 53, respectively. In each plot, red lines represent ligand RMSD and blue lines represent protein RMSD, plotted on separate scales to highlight differences in fluctuations. Y-axis scales may vary across subplots to accommodate differences in RMSD values. |
The stability of ligand binding prompted a more in-depth investigation into protein-ligand interactions to identify general patterns and specific interactions contributing to binding affinity and stability. The interaction profiles for events occurring more than 10% of the time are summarized in Figure 6.
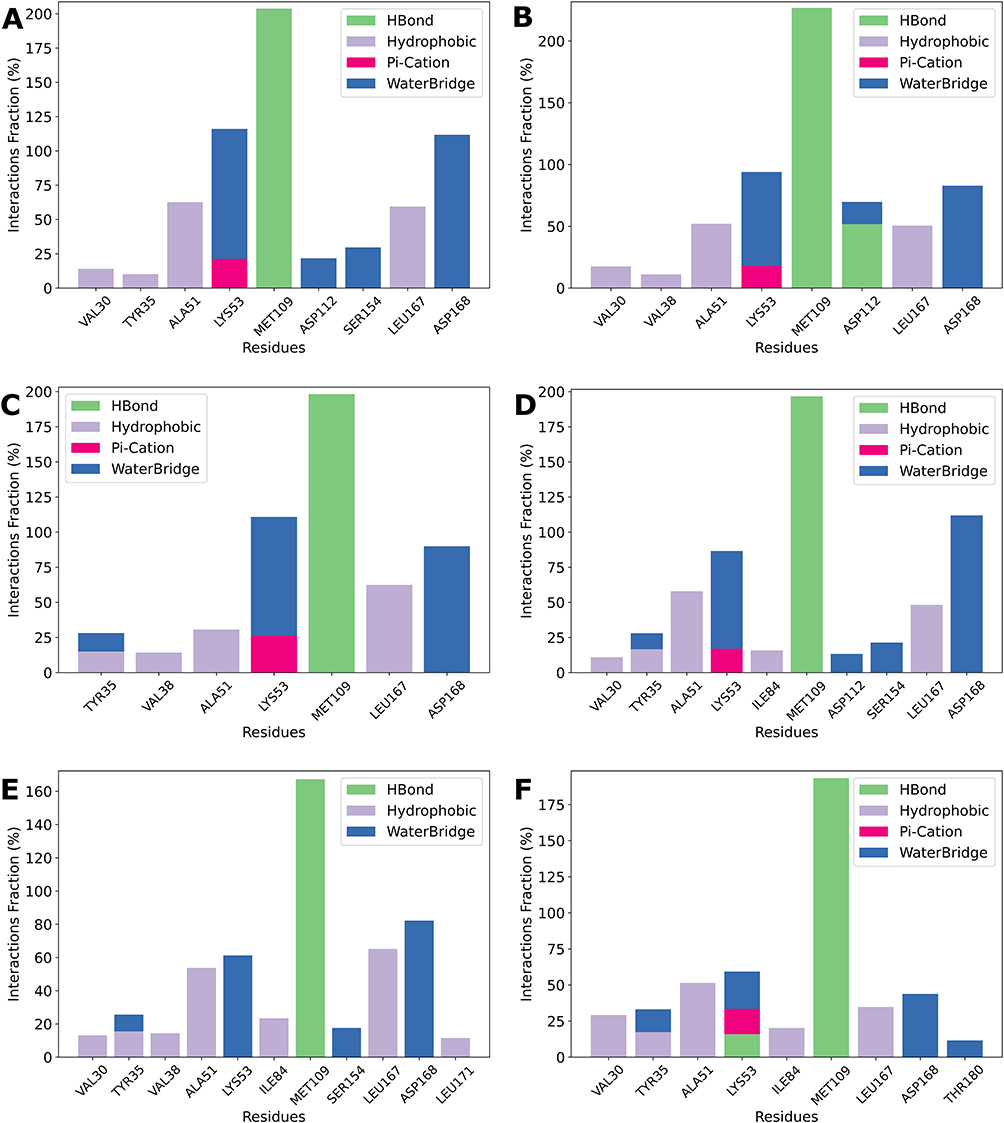 |
Figure 6 Binding Interaction Profiles of Top Ligands with Key Residues. Protein-ligand interactions observed for six compounds, with only interactions occurring more than 10% of the simulation time shown. Subplots (A–F) correspond to Compounds 26, 28, 47, 48, 50, and 53, respectively. Interaction types include hydrogen bonds (green), hydrophobic contacts (purple), π-cation interactions (pink), and water bridges (blue). The x-axis lists interacting residues, while the y-axis represents the interaction frequency as a percentage. Note that the scale of the y-axis varies between plots. |
Across all six compounds, MET109 consistently emerges as a key residue, exhibiting strong H-bonding interactions. This is particularly evident in Compounds 26, 28, 47, and 48 (Figure 6A–D), where interaction fractions approach or exceed 200%, suggesting the formation of two stable hydrogen bonds. This highlights MET109’s potentially central role in ligand stabilization. In addition, several other residues contribute through distinct interaction types, further highlighting their critical role in receptor-ligand binding. ALA51 frequently engages in hydrophobic interactions, stabilizing the ligand for approximately 50–60% of the simulation time across all compounds, except for Compound 47 (Figure 6C), where it only interacts with the ligand for approximately 30% of the simulation duration.
LYS53 consistently forms water bridges, and is particularly substantial in Compounds 26, 28, 47, and 48 (~60–80%, Figure 6A–D). In addition, π-cation interactions between the positively charged side chain of Lys53 and the aromatic π system of the ligand are observed in most cases (~20–30%). This interaction is much less frequent for Compound 50 (Figure 6E), where it occurs in less than 10% of the simulation time and is therefore excluded from the plot. LEU167 primarily contributes through hydrophobic interactions, maintaining interaction frequencies of ~50% for Compounds 26, 28, and 48 (Figure 6A, and D), ~60% for Compounds 47 and 50 (Figure 6C and E), but dropping to ~35% for Compound 53 (Figure 6F). Finally, ASP168 consistently forms water bridges across all compounds, though the interaction duration varies. For Compounds 26 and 48 (Figure 6A and D), interaction fractions exceed 100%, suggesting stable and recurring interactions. In Compounds 28, 47, and 50 (Figure 6B, and E), ASP168 interacts ~80% of the time, whereas for Compound 53 (Figure 6F), the interaction is less prominent, occurring during less than 50% of the simulation time.
ADME Characterization
For the most potent compounds, 26, 28, 47, 48, 50 and 53 key physicochemical and ADME properties were experimentally measured to further assess their developability. These included solubility, lipophilicity (logD), plasma protein binding, plasma recovery, Caco-2 permeability, plasma half-life, intrinsic clearance, and CYP3A inhibition. Compound 54 was considered for testing, but not available for further analysis. Table 4 summarizes the primary findings, while Table S2.A–K in the supplementary materials contains the complete dataset.
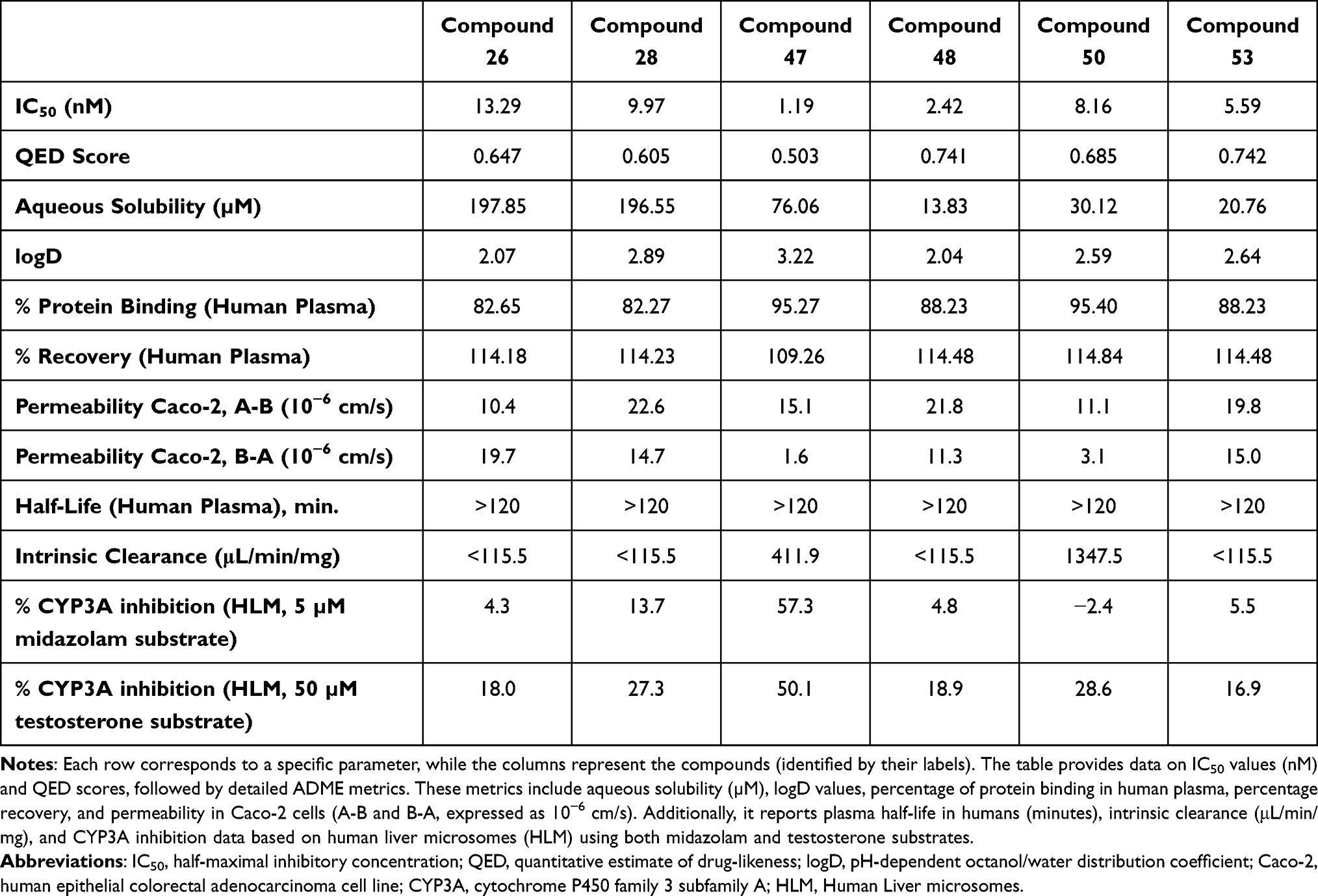 |
Table 4 Measured Physicochemical and ADME Properties for the Top Compounds |
The ADME properties of the six potent compounds were assessed in vitro, focusing on solubility, permeability, and metabolic stability. Aqueous solubility varied widely, with Compounds 26 and 28 exhibiting high solubility (>190 µM), while Compound 47 showed moderate solubility (76 µM). In contrast, Compounds 48, 50 and 53 showed rather poor solubility (~14–30 µM), which may limit their oral bioavailability. Lipophilicity, measured as logD (2.04–3.22) at pH 7.4, indicated moderate membrane permeability. Additionally, all compounds displayed high plasma protein binding (>80%), which could reduce free drug availability.
Caco-2 permeability assays revealed Compound 28 and Compound 53 had high A-B permeability (~20 × 10−6 cm/s), while Compound 26 and 50 exhibited lower permeability (~11 × 10−6 cm/s), indicating moderate absorption potential. Plasma stability was high for all, with half-lives exceeding 120 minutes. Intrinsic clearance varied substantially, with Compound 50 showed rapid metabolism (Cl_int = 1347.5 µL/min/mg), while Compound 26 and Compound 48 demonstrated lower clearance, indicating extended systemic exposure. CYP3A inhibition was moderate for Compound 47 (~57%), while others exhibited low interaction risks.
Intracellular Target Engagement
To evaluate the intracellular target engagement, Compounds 26 and 48 were assessed using the MAPK14 (p38α) NanoBRET assay. The resulting dose-response curves are shown in Figure 7.
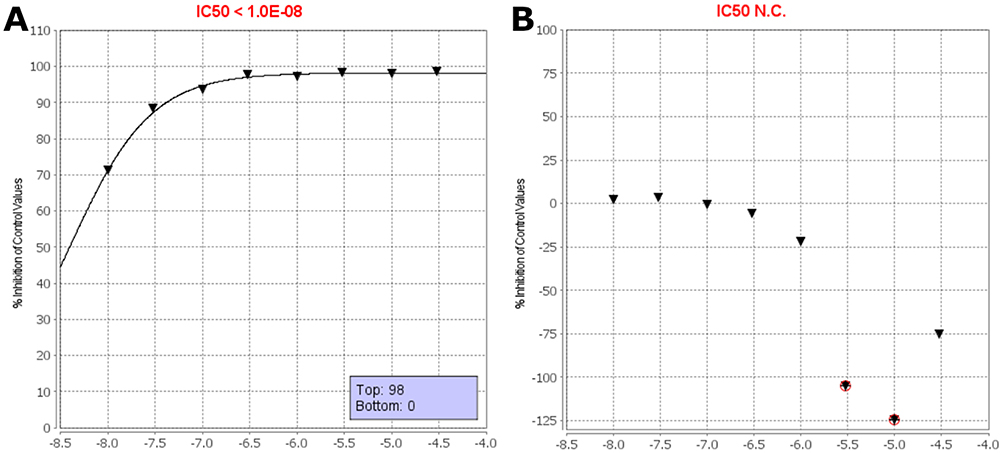 |
Figure 7 NanoBRET IC50 assays. (A) Dose-response curve for Compound 26, showing potent target engagement with an IC50 < 10 nM. (B) Compound 48, which showed no measurable activity across the tested concentration range. The y-axis indicates percent inhibition of p38α binding, and the x-axis shows the log-transformed concentration (M). Black triangles indicate the mean percent inhibition of control from two independent measurements at each concentration. Red circles denote individual data points that were flagged and excluded from the IC50 calculation. |
Compound 26 demonstrated potent cellular binding, with a measured IC50 of <10 nM, confirming strong engagement of p38α in a live-cell context. Although the exact IC50 could not be determined due to the assay’s lower detection limit, significant inhibition was observed even at the lowest tested concentration (10 nM), with 71.4% inhibition of control signal. The inhibition further increased to >98% at higher concentrations, confirming effective and saturable binding to intracellular p38α (Figure 7A).
In contrast, Compound 48 showed no measurable target engagement across the tested concentration range. The dose–response curve (Figure 7B) displayed minimal inhibition (<5%), and in some cases, high concentrations resulted in apparent negative values—likely due to non-specific effects or assay variability. As such, an IC50 value could not be calculated for this compound.
These results highlight Compound 26’s strong potential for further development, demonstrating not only high biochemical potency but also effective intracellular engagement of p38α. In contrast, the absence of measurable activity for Compound 48 may reflect poor cell permeability, metabolic instability, or insufficient binding affinity under cellular conditions. These findings underscore the value of early cellular assays for identifying compounds with true intracellular activity and prioritizing candidates for further optimization.
Kinome Profiling
The kinase selectivity of Compound 26 was evaluated using a large-scale kinase binding assay that quantitatively measures kinase engagement via displacement of a reporter probe across a panel of 468 kinases. The results are visualized using phylogenetic tree maps (Figure 8), providing a visual representation of kinase engagement at different concentrations. The complete dataset of binding values across the entire kinase panel is provided in the Supporting Information (Table S3). To assess selectivity, Compound 26 was tested at 100 nM and 1000 nM, and its selectivity scores (S-values) were calculated. These scores, S(35), S(10), and S(1), represent the fraction of kinases for which binding was reduced to ≤35%, 10%, or 1% of control levels, respectively. Lower selectivity scores indicate a more selective compound, as they reflect reduced interaction with off-target kinases. A summary of these selectivity scores is presented in Table 5. By evaluating these scores at multiple concentrations, this analysis provides insights into the dose-dependent effects of Compound 26.
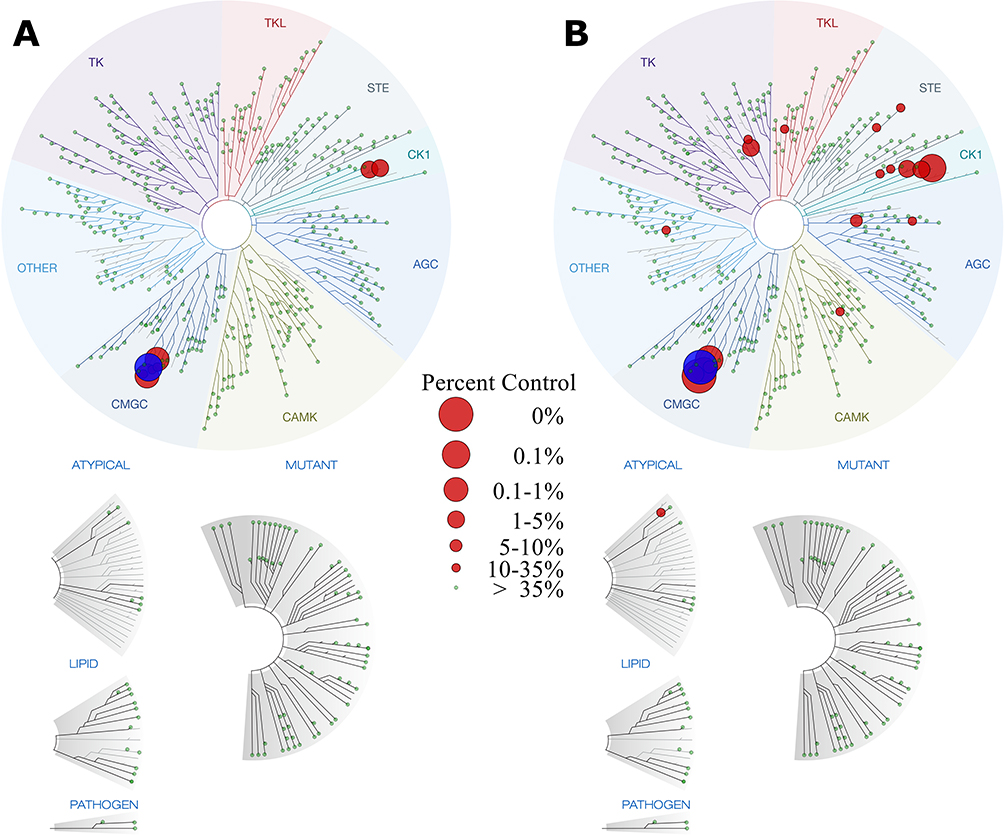 |
Figure 8 Kinase binding interaction map for Compound 26. Subplot (A) represents kinase binding interactions at 100 nM, while subplot (B) shows interactions at 1000 nM. Each circle corresponds to an individual kinase, with larger circles indicating stronger binding by Compound 26. The main target, p38α (MAPK14) is highlighted in blue for clarity. |
 |
Table 5 Kinase Selectivity Scores for Compound 26 |
At 100 nM, Compound 26 demonstrated high selectivity, with a limited number of kinases being bound. The selectivity scores at this concentration were S(35) = 0.020, S(10) = 0.012, and S(1) = 0.007, indicating that only a small fraction of the kinome was affected. The corresponding kinase interaction map (Figure 8A) supports this observation, showing minimal off-target binding and reinforcing the compound’s preference for the p38 family of kinases.
When the concentration was increased to 1000 nM, a slight increase in kinase engagement was observed, as expected due to the higher compound exposure. The selectivity scores at this concentration were S(35) = 0.052, S(10) = 0.027, and S(1) = 0.012, suggesting that while more kinases were affected, the compound remained relatively selective. The phylogenetic kinome profile map (Figure 8B) illustrates this shift, showing additional interactions, though still concentrated within a defined subset of kinases. Importantly, even at this higher concentration, Compound 26 did not display broad promiscuity, further supporting its targeted kinase inhibition profile.
A comparison of selectivity scores across both concentrations is summarized in Table 5. The data indicate that Compound 26 maintains a strong preference for a limited number of kinases, with only a modest increase in off-target interactions at a tenfold higher concentration. The relatively low S(35) and S(10) scores across both conditions suggest that Compound 26 is unlikely to cause widespread kinase inhibition, which is a desirable property for minimizing off-target effects in therapeutic applications.
Conclusion
This study demonstrates an integrated and effective approach to early-stage drug discovery, combining advanced computational screening, SAR analysis, and experimental validation to identify and optimize potent p38α kinase inhibitors. By leveraging consensus docking through the ESSENCE-Dock framework, we efficiently prioritized candidate molecules with reduced false-positive rates and enriched hit quality. This led to the identification of several promising compounds, including Compound 24, which featured a novel scaffold and high potency.
The subsequent optimization efforts, guided by a consensus fingerprint similarity approach and Butina clustering, led to the discovery of a series of highly potent compounds, including Compounds 28, 47, 48, 50, and 53, all of which demonstrated IC50 values below 10 nM. The identified scaffold also exhibited promising Absorption, Distribution, Metabolism, and Excretion (ADME) characteristics, with high plasma stability and favorable solubility/metabolic profiles, indicating strong potential for good bioavailability, metabolic stability, and overall drug-likeness. Additionally, the scaffold exhibited favorable chemical properties, including efficient synthetic pathways that yielded high outputs, likely making it well-suited for large-scale production and subsequent optimization efforts. Molecular dynamics confirmed stable binding interactions with key residues (eg, MET109, ASP168). Compound 26 emerged as a particularly promising candidate, exhibiting strong kinase selectivity in a 468-kinase kinome panel and potent intracellular target engagement as confirmed by NanoBRET assays. SAR analysis using MolSHAP further suggested potential R-group contributions to potency. Although the SAR analysis presented here is based on a relatively limited dataset and early-stage exploration of the chemical space around the scaffold, the observed trends nevertheless provide useful guidance for future optimization of activity, selectivity, and drug-like properties. Future work will focus on expanding the explored chemical space around this scaffold and evaluating additional analogues to further refine the SAR and optimize pharmacological and pharmacokinetic properties.
Given the well-established role of p38α signaling in inflammatory pathways and neurodegenerative disease progression, the identification of a potent and selective scaffold may provide a useful starting point for the development of next-generation inhibitors targeting these indications. Together, these findings support the therapeutic potential of this new scaffold class for targeting p38α in inflammatory and neurodegenerative disorders and highlight the value of integrating computational and experimental strategies in modern drug discovery.
Acknowledgments
Jochem Nelen was funded by Cátedra Villapharma-UCAM. José Manuel Villalgordo-Soto is affiliated with Eurofins-Villapharma, a company focused on synthesis of novel compounds for drug discovery. As part of a collaboration, Laure Breuils, an employee of Eurofins-Cerep S.A., assisted with the NanoBRET assay, which was provided free of charge. Supercomputing resources in this work have been supported by the Plataforma Andaluza de Bioinformática of the University of Málaga. Powered@NLHPC: This research was partially supported by the supercomputing infrastructure of the NLHPC (CCSS210001). The authors gratefully acknowledge the Eurofins-Villapharma Analysis and ADME teams for their substantial experimental contributions, expert analytical support, and valuable scientific guidance. This paper has been uploaded to Chemrxiv.org as a preprint: https://chemrxiv.org/engage/chemrxiv/article-details/6857d9ab3ba0887c33f1e41c.
Disclosure
José Manuel Villalgordo-Soto is part of Eurofins-Villapharma Research SLU. Laure Breuils is employed by Eurofins-CEREP S.A. The authors report no other financial or non-financial competing interests relevant to the work described in this manuscript.
References
1. Cuenda A, Rousseau S. p38 MAP-Kinases pathway regulation, function and role in human diseases. Biochim Biophys Acta Mol Cell Res. 2007;1773(8):1358–27. doi:10.1016/j.bbamcr.2007.03.010
2. Canovas B, Nebreda AR. Diversity and versatility of p38 kinase signalling in health and disease. Nat Rev Mol Cell Biol. 2021;22(5):346–366. doi:10.1038/s41580-020-00322-w
3. Awasthi A, Raju MB, Rahman MA. Current insights of inhibitors of p38 mitogen-activated protein kinase in inflammation. Med Chem Shariqah United Arab Emir. 2021;17(6):555–575. doi:10.2174/1573406416666200227122849
4. Gupta J, Nebreda AR. Roles of p38α mitogen-activated protein kinase in mouse models of inflammatory diseases and cancer. FEBS J. 2015;282(10):1841–1857. doi:10.1111/febs.13250
5. Valipour M, Mohammadi M, Valipour H. CNS-active p38α MAPK inhibitors for the management of neuroinflammatory diseases: medicinal chemical properties and therapeutic capabilities. Mol Neurobiol. 2024;61(7):3911–3933. doi:10.1007/s12035-023-03829-3
6. Wydra VR, Ditzinger RB, Seidler NJ, Hacker FW, Laufer SA. A patent review of MAPK inhibitors (2018 – present). Expert Opin Ther Pat. 2023;33(6):421–444. doi:10.1080/13543776.2023.2242584
7. Lee JC, Laydon JT, McDonnell PC, et al. A protein kinase involved in the regulation of inflammatory cytokine biosynthesis. Nature. 1994;372(6508):739–746. doi:10.1038/372739a0
8. Pargellis C, Tong L, Churchill L, et al. Inhibition of p38 MAP kinase by utilizing a novel allosteric binding site. Nat Struct Biol. 2002;9(4):268–272. doi:10.1038/nsb770
9. Aston NM, Bamborough P, Buckton JB, et al. p38α mitogen-activated protein kinase inhibitors: optimization of a series of biphenylamides to give a molecule suitable for clinical progression. J Med Chem. 2009;52(20):6257–6269. doi:10.1021/jm9004779
10. Pfizer. A randomized, double-blind, placebo-controlled 2-way crossover study to evaluate the efficacy, safety and tolerability of PF-03715455 administered twice daily by inhalation for 4 weeks in subjects with moderate to severe Chronic Obstructive Pulmonary Disease (COPD). clinicaltrials.gov; 2016. Available from: https://clinicaltrials.gov/study/NCT02366637.
11. Koeberle SC, Romir J, Fischer S, et al. Skepinone-L is a selective p38 mitogen-activated protein kinase inhibitor. Nat Chem Biol. 2012;8(2):141–143. doi:10.1038/nchembio.761
12. Rudalska R, Harbig J, Forster M, et al. First-in-class ultralong-target-residence-time p38α inhibitors as a mitosis-targeted therapy for colorectal cancer. Nat Cancer. 2025;6(2):259–277. doi:10.1038/s43018-024-00899-7
13. Roy SM, Grum-Tokars VL, Schavocky JP, et al. Targeting human central nervous system protein kinases: an isoform selective p38αMAPK inhibitor that attenuates disease progression in Alzheimer’s disease mouse models. ACS Chem Neurosci. 2015;6(4):666–680. doi:10.1021/acschemneuro.5b00002
14. Frazier HN, Braun DJ, Bailey CS, et al. A small molecule p38α MAPK inhibitor, MW150, attenuates behavioral deficits and neuronal dysfunction in a mouse model of mixed amyloid and vascular pathologies. Brain Behav Immun Health. 2024;40:100826. doi:10.1016/j.bbih.2024.100826
15. Yang F, Zhao LJ, Xu Q, Zhao J. The journey of p38 MAP kinase inhibitors: from bench to bedside in treating inflammatory diseases. Eur J Med Chem. 2024;280:116950. doi:10.1016/j.ejmech.2024.116950
16. Zheng Q, Li S, Wang A, et al. p38 mitogen-activated protein kinase: functions and targeted therapy in diseases. MedComm Oncol. 2023;2(3):e53. doi:10.1002/mog2.53
17. Igea A, Nebreda AR. The stress kinase p38α as a target for cancer therapy. Cancer Res. 2015;75(19):3997–4002. doi:10.1158/0008-5472.CAN-15-0173
18. Madkour MM, Anbar HS, El-Gamal MI. Current status and future prospects of p38α/MAPK14 kinase and its inhibitors. Eur J Med Chem. 2021;213:113216. doi:10.1016/j.ejmech.2021.113216
19. Nelen J, Carmena-Bargueño M, Martínez-Cortés C, Rodríguez-Martínez A, Villalgordo-Soto JM, Pérez-Sánchez H. ESSENCE-Dock: a consensus-based approach to enhance virtual screening enrichment in drug discovery. J Chem Inf Model. 2024;64(5):1605–1614. doi:10.1021/acs.jcim.3c01982
20. Stroganov OV, Novikov FN, Stroylov VS, Kulkov V, Chilov GG. Lead finder: an approach to improve accuracy of protein-ligand docking, binding energy estimation, and virtual screening. J Chem Inf Model. 2008;48(12):2371–2385. doi:10.1021/ci800166p
21. McNutt AT, Francoeur P, Aggarwal R, et al. GNINA 1.0: molecular docking with deep learning. J Cheminformatics. 2021;13(1):43. doi:10.1186/s13321-021-00522-2
22. Corso G, Stärk H, Jing B, Barzilay R, Jaakkola T. DiffDock: diffusion steps, twists, and turns for molecular docking. arXiv. 2023:
23. Riniker S, Landrum GA. Better informed distance geometry: using what we know to improve conformation generation. J Chem Inf Model. 2015;55(12):2562–2574. doi:10.1021/acs.jcim.5b00654
24. Molconvert | chemaxon Docs. Available from: https://docs.chemaxon.com/display/docs/Molconvert.md.
25. Morris GM, Huey R, Lindstrom W, et al. AutoDock4 and AutoDockTools4: automated docking with selective receptor flexibility. J Comput Chem. 2009;30(16):2785–2791. doi:10.1002/jcc.21256
26. Schrödinger Release. 2024-3: maestro, Schrödinger, LLC, New York, NY, 2024.
27. Dyckman AJ, Li T, Pitt S, et al. Discovery of pyrrolo[2,1-f][1,2,4]triazine C6-ketones as potent, orally active p38α MAP kinase inhibitors. Bioorg Med Chem Lett. 2011;21(15):4633–4637. doi:10.1016/j.bmcl.2011.05.091
28. Lu C, Wu C, Ghoreishi D, et al. OPLS4: improving force field accuracy on challenging regimes of chemical space. J Chem Theory Comput. 2021;17(7):4291–4300. doi:10.1021/acs.jctc.1c00302
29. Gasteiger J, Marsili M. A new model for calculating atomic charges in molecules. Tetrahedron Lett. 1978;19(34):3181–3184. doi:10.1016/S0040-4039(01)94977-9
30. bio-hpc. bio-hpc/metascreener. 2025. Available from: https://github.com/bio-hpc/metascreener.
31. Nelen J. Jnelen/DiffDockHPC. 2024. Available from: https://github.com/Jnelen/DiffDockHPC.
32. Nelen J. Jnelen/ConFiLiS. 2025. Available from: https://github.com/Jnelen/ConFiLiS.
33. Rogers D, Hahn M. Extended-Connectivity Fingerprints. J Chem Inf Model. 2010;50(5):742–754. doi:10.1021/ci100050t
34. PubChem. PubChem Substructure Fingerprint. 2009. Available from: https://ftp.ncbi.nlm.nih.gov/pubchem/specifications/pubchem_fingerprints.pdf.
35. Gedeck P, Rohde B, Bartels C. QSAR − how good is it in practice? Comparison of descriptor sets on an unbiased cross section of corporate data sets. J Chem Inf Model. 2006;46(5):1924–1936. doi:10.1021/ci050413p
36. Ji H, Deng H, Lu H, Zhang Z. Predicting a molecular fingerprint from an electron ionization mass spectrum with deep neural networks. Anal Chem. 2020;92(13):8649–8653. doi:10.1021/acs.analchem.0c01450
37. Butina D. Unsupervised data base clustering based on Daylight’s fingerprint and tanimoto similarity: a fast and automated way to cluster small and large data sets. J Chem Inf Comput Sci. 1999;39(4):747–750. doi:10.1021/ci9803381
38. Sydow D, Morger A, Driller M, Volkamer A. TeachOpenCADD: a teaching platform for computer-aided drug design using open source packages and data. J Cheminformatics. 2019;11(1):29. doi:10.1186/s13321-019-0351-x
39. Tian T, Li S, Fang M, Zhao D, Zeng J. MolSHAP: interpreting quantitative structure–activity relationships using shapley values of R-Groups. J Chem Inf Model. 2024;64(7):2236–2249. doi:10.1021/acs.jcim.3c00465
40. Lu W, Zhang J, Huang W, et al. DynamicBind: predicting ligand-specific protein-ligand complex structure with a deep equivariant generative model. Nat Commun. 2024;15(1):1071. doi:10.1038/s41467-024-45461-2
41. Varadi M, Anyango S, Deshpande M, et al. AlphaFold Protein Structure Database: massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res. 2022;50(D1):D439–D444. doi:10.1093/nar/gkab1061
42. Schrödinger Release. 2024-3: protein Preparation Wizard; Epik, Schrödinger, LLC, New York, NY, 2024; Impact, Schrödinger, LLC, New York, NY; Prime, Schrödinger, LLC, New York, NY, 2024.
43. Nelen J. Jnelen/DynamicBindHPC. 2024. Available from: https://github.com/Jnelen/DynamicBindHPC.
44. Schrödinger Release. 2024-3: desmond Molecular Dynamics System, D. E. Shaw Research, New York, NY, 2024; Maestro-Desmond Interoperability Tools, Schrödinger, New York, NY, 2024.
45. Wentsch HK, Walter NM, Bührmann M, et al. Optimized target residence time: type I inhibitors for p38α MAP kinase with improved binding kinetics through direct interaction with the R-Spine. Angew Chem Int Ed. 2017;56(19):5363–5367. doi:10.1002/anie.201701185
46. Bickerton GR, Paolini GV, Besnard J, Muresan S, Hopkins AL. Quantifying the chemical beauty of drugs. Nat Chem. 2012;4(2):90–98. doi:10.1038/nchem.1243
47. Lundberg S, Lee SI. A unified approach to interpreting model predictions. arXiv. 2017:
48. Adasme MF, Linnemann KL, Bolz SN, et al. PLIP 2021: expanding the scope of the protein–ligand interaction profiler to DNA and RNA. Nucleic Acids Res. 2021;49(W1):W530–W534. doi:10.1093/nar/gkab294
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.