Back to Journals » International Journal of Women's Health » Volume 17
Comprehensive Analysis of Vaginal and Gut Microbiome Alterations in Endometriosis Patients
Authors Zhao Y, Hu X, Li C, Huang J, Guo K, Pan Q, Yu Z
Received 1 September 2025
Accepted for publication 4 December 2025
Published 30 December 2025 Volume 2025:17 Pages 5775—5786
DOI https://doi.org/10.2147/IJWH.S561386
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Everett Magann
Yiming Zhao,1,2,* Xinyu Hu,1,* Chunyan Li,2 Jing Huang,3 Ke Guo,4 Qiong Pan,1 Zheng Yu2
1Department of Obstetrics and Gynecology, the Third Xiangya Hospital of Central South University, Changsha, People’s Republic of China; 2Human Microbiome and Health Group, Department of Microbiology, Xiangya School of Basic Medical Sciences, Central South University, Changsha, Hunan, People’s Republic of China; 3Department of Parasitology, Xiangya School of Basic Medical Sciences, Central South University, Changsha, Hunan, People’s Republic of China; 4Department of Neurology, the Third Xiangya Hospital of Central South University, Changsha, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Qiong Pan, Email [email protected] Zheng Yu, Email [email protected]
Purpose: Endometriosis (EMS) is a chronic gynecological disorder with unclear pathogenesis. While the vaginal and gut microbiomes are known to influence EMS, few studies have analyzed both microbiomes integrally. This study aims to characterize the vaginal and gut microbiome profiles in EMS patients and evaluate their diagnostic potential.
Patients and Methods: We conducted metagenomic sequencing on 22 paired vaginal and fecal samples from EMS patients and controls. Microbial composition, diversity, and metabolic pathways were analyzed. Machine learning models were employed to assess the predictive performance of microbiome features in EMS diagnosis.
Results: EMS patients exhibited pronounced shifts in the vaginal microbiome, characterized by reduced Lactobacillus and increased Bifidobacterium and Gardnerella, which correlated with elevated luteinizing hormone (LH) and follicle-stimulating hormone (FSH) levels. The gut microbiome displayed decreased diversity, with a depletion of beneficial taxa such as Ruminococcus and Prevotella, alongside an enrichment of Dialister. Metabolic pathways in both microbial communities were significantly altered. Machine learning analyses demonstrated that gut microbiome features outperformed both vaginal microbiome and hormonal indices in predicting EMS, highlighting their strong diagnostic potential.
Conclusion: This study underscores the pivotal role of the gut microbiota in EMS and elucidates the complex interplay between microbial dysbiosis and disease pathogenesis. Our findings indicate that gut microbiome signatures may serve as superior diagnostic biomarkers for EMS, thereby paving the way for microbiome-based diagnostic and therapeutic strategies.
Keywords: vaginal microbiome, gut microbiome, endometriosis, metagenomic sequencing
Introduction
Endometriosis (EMS) is a chronic gynecological disorder which can lead to pelvic pain, dysmenorrhea, and infertility.1 Despite its significant impact on women’s health, the pathogenesis of endometriosis remains poorly understood. The retrograde menstruation theory, which posits that viable endometrial cells are transported to the pelvic cavity, is widely regarded as the most plausible explanation for the development of EMS.2 However, the fact that only a small part of women develop EMS despite the near-universal occurrence of retrograde menstruation to some degree, implies that additional factors are at play.3 Recent studies have reported that genetic,4 immunological,5 hormonal,6 and environmental7 factors can influence the development of EMS.
More and more studies have highlighted the potential role of the human microbiome,8 particularly the vaginal and gut microbiota.9 Alterations in the composition and function of these microbial communities, known as dysbiosis, have been associated with the development and progression of numerous health conditions. Emerging evidence suggests that dysbiosis of the vaginal and gut microbiomes may contribute to the pathophysiology of EMS.10 The vaginal microbiome, predominantly composed of Lactobacillus, plays a vital role in maintaining vaginal health through the production of lactic acid and other antimicrobial substances.11 Some studies suggest a potential link between bacterial vaginosis (BV) and endometriosis. BV, characterized by a depletion of Lactobacillus and overgrowth of anaerobes, may alter the vaginal and uterine immune microenvironment, facilitating ascending infections and chronic inflammation. Such dysbiosis has been associated with an increased risk of endometriosis or its complications, including tubo-ovarian abscesses in affected women.12 A meta-analysis also indicated that vaginal microecological imbalance might contribute to the pathogenesis of endometriosis.13 These findings highlight the importance of considering BV and vaginal dysbiosis in studies of endometriosis development. Disruption of this balance has been linked to increased susceptibility to infections and inflammatory conditions, potentially influencing the inflammatory environment associated with EMS. Similarly, the gut microbiome is integral to immune function,14 hormone regulation,15 and maintaining the integrity of the intestinal barrier.16 Alterations in gut microbiota composition and function have been implicated in systemic inflammation, altered estrogen metabolism, and increased intestinal permeability, all of which are factors believed to be involved in the pathogenesis of EMS. Moreover, some studies have shown that gut dysbiosis can lead to elevated circulating estrogen levels.17 Increased estrogen exposure can stimulate the growth of ectopic endometriotic foci and enhance their inflammatory activity.18 Therefore, the gut microbiome, through its role in the regulation of the estrogen cycle may be associated with EMS.
Most studies have focused either on the vaginal or gut microbiome independently; however, integrated analysis of both microbiomes could provide a more comprehensive understanding of their roles in EMS. Here, we collected 22 paired vaginal and fecal samples from 11 participants, and integrated vaginal and gut microbiome analysis using several machine-learning methods. By employing metagenomic sequencing technologies and multiple-bioinformatics analyses, we characterized the specific microbial composition and dysbiotic patterns associated with EMS. Furthermore, we demonstrated that the gut microbiome plays a more prominent role in the diagnosis of EMS compared with the vaginal microbiome and sex hormones. This work provides new insights into the role of vaginal and gut microbiota in the mechanism of EMS and contributes to the growing body of knowledge on the intricate interplay between the microbiome and human health.
Materials and Methods
Participant and Sample Collection
Five women with histologically confirmed endometriosis (EMS) were enrolled as the study group, and six asymptomatic women attending routine well-woman visits served as controls (Non-EMS). Exclusion criteria included: (1) use of antibiotics, immunosuppressive agents, or probiotics within the previous three months; (2) history of HPV infection or other clinical signs of infection; (3) body mass index >30 kg/m2; (4) presence of gastrointestinal or systemic diseases; (5) history of malignancy; and (6) menstruation, sexual activity within 48 hours before sampling, or vaginal interventions such as lavage, medication, or lubricant use. All participants underwent sex hormone testing to assess endocrine status, and EMS cases were staged according to the American Society for Reproductive Medicine (ASRM) classification19 for patients with EMS were evaluated by the professional clinician blindly. The studies involving participants were reviewed and approved by The Third Xiangya Hospital of Central South University and performed under the relevant guidelines and regulations (IRB number 22224). The studies were conducted in accordance with local legislation and institutional requirements. All study-related procedures followed relevant guidelines and regulations. All participants provided written informed consent and completed a questionnaire to collect baseline demographic information (Table S1). The detailed description of the ASRM scores and stages of endometriosis in each patient can be found in Table S2.
Vaginal discharge samples were collected using sterile swabs. Each swab was carefully inserted into the vagina and rotated to obtain sufficient discharge from the vaginal walls. The swab was then placed into a sterile freezing tube and transported to the laboratory using a biological sample transport box. Participants were also provided with sterile stool collection kits and instructions to collect a fecal sample from their first morning bowel movement. Fecal samples were collected from the middle and distal portions, placed into sterile tubes containing preservation solution, and all samples were stored at −80 °C until further processing. All participants were sampled during the follicular phase of their menstrual cycle to minimize hormonal variation that could influence the microbiota.
DNA Extraction and Metagenomic Sequencing
All genomic DNA was extracted from vaginal and fecal samples using the E.Z.N.A. Soil DNA Kit (Omega Bio-Tek, USA). The extracted DNA was eluted in elution buffer and stored at −20 °C. DNA yield was quantified using a full-spectrum ultraviolet spectrophotometer (Amersham Biosciences, USA), and DNA purity was assessed by agarose gel electrophoresis for both metagenomic DNA and PCR products. High-quality genomic DNA was subsequently fragmented into 200–500 bp fragments using an ultrasonicator, and the fragment size distribution was confirmed by agarose gel electrophoresis (Bio-Rad, USA). The resulting fragments were then recovered using the QIAquick Gel Extraction Kit. Following successful library construction, the products were purified and sequenced on the Illumina NovaSeq 6000 platform (Shanghai Biozeron Biotech. Co., Ltd., China). Illumina PE libraries were constructed, and the obtained sequencing data underwent quality control.
Sequencing Data Processing and Taxonomic Profiling
After obtaining the sequencing reads, all raw sequences underwent quality control. Low-quality reads (average quality <35 or containing more than 16 ambiguous bases) were removed using Fastp20 (version 0.21.0). Duplicate reads were eliminated using FastUniq21 (version 1.1.0). Sequences of human origin were filtered out by mapping to the human reference genome (hg38) using Bowtie222 (version 2.3.5) with the sensitive mode. Subsequently, the clean reads were subjected to a k-mer based algorithm for microbial profiling using Kraken223 (version 2.1.3). To improve annotation accuracy and reduce the error rate, the results were further calibrated using Bracken (version 2.9). To reduce false positives in obtaining data, only the taxonomic units with reads of more than 10 were retained and rarefied to the minimum read counts using the R package “picante”.24 The Shannon-Wiener index was calculated to assess the alpha diversity of microbial communities. Simultaneously, Bray-Curtis distance metrics were computed based on species relative abundances and visualized using Principal Coordinates Analysis (PCoA) with the R package “Vegan”.
Functional Profiling and Gene Source Tracking
To study the microbial functions, all clean reads were also assembled to contigs using Megahit25 (version 1.2.9) with default settings. Only the contigs longer than 500 bp were retained to perform the gene prediction with MetaGeneMark26 (version 3.38). After that, Hmmer27 (version 3.3.1) with the AntiFam database28 was applied to remove spurious genes. Next, a gene catalog was constructed using MMseqs229 (version 13.45111) with a minimum sequence identity of >95% and a clustering threshold of >90%. Functional annotation of the gene catalog was performed using eggNOG-mapper30 (version 2.0.1). For each gene, reads from the respective sample were mapped using BWA31 (version 0.7.17), and the gene abundance was calculated according to the mapped read number divided by the gene length. The different genes were identified by the R package “DESeq2” (Version 1.26.0) between the two groups. In addition, Kraken223 (version 2.1.3) was performed to determine the taxonomic classification of the gene.
Detection of Microbial Markers Using Machine Learning Methods
To investigate the effect of the microbial and functional composition on the models, each top four variables were obtained from all microbial and functional multiple variables using the PCoA algorithm. The data of differential taxonomy and genes were collected following the methods mentioned before. The Random Forest analysis was performed with default settings in python using the “scikit-learn” package. The Ordinary Least Square (OLS) model by the package “statsmodels” in python was used to study the correlation between features and ASRM staging. The area under the curve (AUC) analysis was performed using the R package “pROC”.32
Analysis of Public Dataset
Additional data from NCBI (https://www.ncbi.nlm.nih.gov/) were incorporated into the analysis. Totally, 88 public sequencing data obtained from vagina or feces of EMS and Non-EMS participants were collected. The details are shown in Table S3. The downloaded 16S rRNA gene sequence is screened with low quality (Discards bases with Phred score <4, and removes sequences with any ambiguous bases), merged, truncated to 200 bp, and then the chimera was removed, as referred to our previous article.33 Afterwards, the remaining sequences were clustered into operational taxonomic unit (OTU). To reduce false positives of obtaining data, only the OTUs mapped reads more than 20 were retained. The OTUs with adjusted p-values <0.05 were classified as differential OTUs using the R package “DESeq2” (Version 1.26.0). And top four variables were obtained from all microbial variables using the PCoA algorithm. Differential OTUs and four variables (PCoA1, PCoA2, PCoA3, PCoA4) were used to fit the machine learning models (Random Forest using “scikit-learn” and AUC analysis using “pROC”).
Other Statistical Analysis
For parametric feature-wise multivariable testing, we used MaAsLin2 with default parameters,34 which finds associations between microbial relative abundance and metadata. MaAsLin2 employs a transformed generalized linear model to iteratively associate each feature with relevant covariates. The baseline data were obtained using the R package “compareGroups”.35 Linear Discriminant Analysis Effect Size (LefSe) analysis was performed to identify significant gut microbial features between groups. The data were processed using “lefse” (Version 1.1.01) python package,36 and an effect size threshold of three was applied. All statistical tests used the “scipy” or “statsmodels” package in Python. Most data visualization was applied using the R package “ggplot2” R or the “matplotlib” package in Python. To evaluate whether the sample size was sufficient to detect meaningful biological effects, we performed a power analysis using the “statsmodels” and “numpy” python packages. And the detailed results are in Table S4.
Results
Study Design and Clinical Characteristics
To investigate alterations in the vaginal and gut microbiomes of patients with endometriosis (EMS), a total of 22 paired vaginal and fecal samples from 11 women (EMS and Non-EMS groups) were included in this study. The overall workflow of this study was shown in Figure 1A. There were no significant differences in age, BMI, smoking status and other baseline characteristics between the EMS and Non-EMS groups (all p-value >0.05) (details of all individuals are presented in Table S1). Meanwhile, some sex hormone indexes were also collected. Among patients with EMS, the lower estradiol (E2) but higher luteinizing hormone (LH) and follicle-stimulating hormone (FSH) were observed, which demonstrated the disorder of sex hormone secretion in the patients with EMS (Figure 1B). Next, both vaginal and gut specimens were subjected to shotgun metagenomic sequencing to characterize the respective microbiomes. By correlating the obtained sex hormone indices with overall variations in the vaginal and gut microbial compositions, we observed that different hormone indices were associated with the microbiome to varying degrees (Figure 1C). Anti-Müllerian hormone (AMH) and progesterone (P) had the largest explanatory power for the interindividual variation in vaginal and gut microbiota, respectively, which further suggested the microbiome at different body axes with unsimilar relationships with dynamic sex hormone level (Figure 1C).
 |
Figure 1 Overview of the study cohort. (A) The workflow schematic diagram for this study. (B) The proportion of five sex hormone indexes in two groups. (C) The effect sizes of five sex hormone indexes on patients’ microbiota are evaluated by permutational multivariate analysis of variance (Adonis, permutations=999). Abbreviations: AMH, Anti-Müllerian hormone; P, progesterone; E2, estradiol; LH, luteinizing hormone; FSH, follicle-stimulating hormone. |
Dysbiosis of Vaginal Microbiome in Patients with Endometriosis
By analyzing the metagenomic data, Firmicutes was found to be the predominant phylum in both groups; however, we found Actinobacteria was enriched in Non-EMS group, whereas Bacteroidota showed a higher relative abundance in EMS group (Figure S1). The microbial composition of the reproductive tract is typically classified into five community state types (CST I–V), of which four types (CST I, II, III, and V) are dominated by Lactobacillus.37 Lactobacillus plays an important role in the reproductive tract by maintaining a low pH in healthy woman vagina.38 The distribution of Lactobacillus in two groups was next estimated, and it showed that the proportion of Lactobacillus in reproductive tract decreased compared with Non-EMS group (Figure 2A). According to Amsel’s criteria,39 the alteration of microbiome in vaginal environment and the decrease of overall pH may be related to BV. However, the relative abundance of other anaerobic bacteria, such as Bifidobacterium and Gardnerella, was increased (Figure 2B), indicating a disruption of the vaginal microbiota. Next, we associated the vaginal microbiota with groups and sex hormone indexes using MaAslin2. As expected, the Lactobacillus was related with Non-EMS; however, other bacteria, some of known opportunistic pathogens, such as Klebsiella, Gardnerella, Streptococcus, and Staphylococcus were associated with the EMS group (Figure 2C). In addition, different vaginal bacteria exhibited consistent synergistic associations with several hormones. For example, LH and FSH, which were elevated in EMS (Figure 1B), were correlated with nearly all vaginal bacteria except Lactobacillus (Figure 2C).
 |
Figure 2 Vaginal microbiome disorder in patients with EMS. (A) The relative abundance of Lactobacillus distribution in two groups, and its distribution in another article classified in CST-IV is also shown with grey color. (B) The proportion of genus in two groups, only the top three genus are shown. (C) The specific association between vaginal microbiota and different sex hormone indexes. The associations were identified using MaAsLin2. (D) The significantly different genes count in different COG classes at level 1. (E) The taxonomic classification of all significant different genes between two groups. Different color represents different COG levels (Metabolism, Information store, Cellular processes and signaling). The size of circles represents the different gene count, and the top three microbes with most different genes are marked with bold font. |
We also investigated alterations in the functional composition of the vaginal microbiome and identified a total of 1,165 significantly differential genes (False discovery rate, FDR < 0.05) between the two groups (Figure 2D). Notably, nearly half of these genes (48.84%) were annotated to metabolic functions (Figure 2D), suggesting a potential link between the vaginal microbiome and an unhealthy vaginal environment. Furthermore, gene source tracking analysis revealed that the main differential genes originated from Bifidobacterium, Lactobacillus, and Gardnerella (Figure 2E). Collectively, these results indicate a potential relationship between vaginal microbiome dysbiosis and EMS.
Altered Gut Microbiome in the Patients with Endometriosis
Next, a comparative analysis of the gut microbiome was conducted. The Shannon-Wiener and Gini-Simpson indices were significantly lower in the EMS group compared to the Non-EMS group, indicating reduced gut microbial diversity in patients with EMS (Figure 3A). In addition, the microbial composition in the EMS group was significantly different with that in the Non-EMS group (Figure 3B, p-value=0.03). To further understand the differences of gut microbiota between the two groups, the Linear Discriminant Analysis Effect Size (LEfSe) analysis was performed (Figure 3C). The result showed that the relative abundance of some core gut microbes, which play an important role in the intestine, such as Ruminococcus, Oscillibacter, and Prevotella decreased. However, Dialister, a kind of microbe which was found to be a biomarker of cervical cancer,40 was enriched in the EMS group, which suggested the change of gut microbiota was associated with vaginal health (Figure 3D).
 |
Figure 3 Gut microbial composition and function in patients with EMS. (A) The Shannon-Wiener and Gini-Simpson indexed are calculated to estimate the alpha-diversity. (*: 0.01 < p-value < 0.05). (B) The PCoA based on intestinal microbial composition and the analysis of similarity (ANOSIM) is applied to test the difference between two groups. (C) The LEfSe analysis is used to detect different microbial biomarkers between two groups, and only the features with LDA score more than three were shown. (D) The different microbial biomarkers detected using LEfSe at genus level are shown. (E) All genes are shown in the scatter plot. The genes with adjusted p-value (FDR) less than 0.05 are colored. (F) The enrichment analysis of differential genes, and only the top 20 KEGG pathways with least adjusted p-value are shown. Different color means various significantly different gene counts. (G) The taxonomic classification of all significant different genes, and the only top 10 genus with most gene counts are shown to visualize. (H) The function of differential genes which are tracked from Dialister are shown. |
Although the obvious taxonomic remodeling was detected in EMS group (Figure 3B), the overall functional genes seemed to be with relatively little change (Figure S2). However, there were still 13,466 under-represented and 14,317 over-represented genes found in the EMS group (Figure 3E). These differential genes were enriched in many vital metabolism pathways, including starch, sucrose, amino and nucleotide sugar, purine, pyrimidine, cysteine, methionine, galactose metabolisms (Figure 3F), which were implicated in host metabolic health. Gene source tracking analysis revealed that the differential genes were primarily derived from a variety of gut microbes, including Bacteroides, Prevotella, Oscillibacter, and other common gut bacteria. Not surprisingly, most of these microbes coincided with the differential microbiota detected using LEfSe (Figure 3G). In addition, some differential genes from Dialister were annotated as formate-based reductases involved in purine and pyrimidine metabolism pathways (Figure 3H). This highlights that the altered functional spectrum of intestinal microbes has the potential to influence host metabolism.
Gut Microbiome as a Predictor of Endometriosis
To explore the predictive potential of the vaginal and gut microbiome for detecting of EMS patients, we constructed prediction models using five types of data sets (differential taxonomy, differential genes, microbial composition, functional composition and sex hormone index) with the total 25,814 features derived from the both vaginal and fecal sequencing data (Figure 4A). To evaluate the contributions of different features to model performance, we employed the Random Forest (RF) algorithm and assessed the area under the curve (AUC) of the receiver operating characteristic (ROC). In the modeling results, traditional sex hormone indices showed poor performance; none had an RF importance score above zero, indicating they had less ability to classify EMS (Figure 4B). However, we found the features from gut microbiome had better prediction performance (Figure 4C and Figure S3). And additional public data was also confirmed the better potential of prediction using gut microbiome compared to vaginal microbiome (Figure S4).
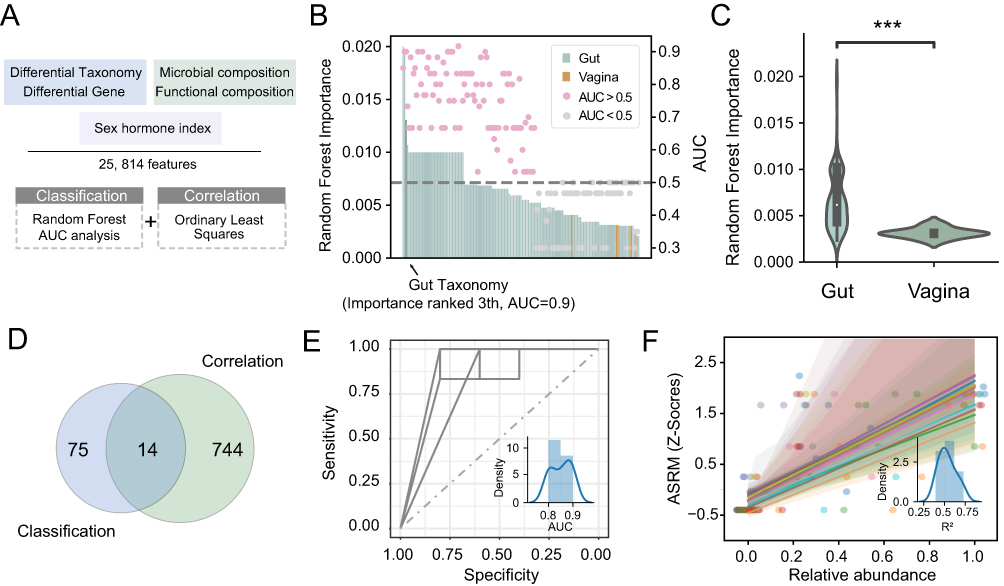 |
Figure 4 Machine learning methods revealed microbial markers for diagnosis of EMS. (A) Pipeline of the integrated methods for diagnostic EMS markers recognition. The workflow schematic diagram for this study. (B) The Random Forest algorithm and AUC analysis are used to detect potential markers for classification EMS patients. (C) The proportion of Importance index from Random Forest algorithm for different features are shown (***: p-value < 0.001). (D) The Venn plot shows the overlap of the markers detected for classification between two groups and the markers detected for reflecting the correlation. The features with the Random Forest Importance index more than zero and AUC more than 0.5 are detected as markers for classification. And the features with p-value less than 0.05 are remained as markers of correlation. There are 14 features detected as both markers for classification and correlation. The receiver operating characteristic curve (ROC) of them are shown in (E), and the relationship of them and ASRM staging are shown in (F). The subplot shows their AUC and R2 distribution, respectively. |
Next, the predictive ability of microbial features for estimating EMS severity was evaluated using ordinary least-squares regression analysis. A total of 758 features showed p-values <0.05, and 14 of these were also identified as valid markers (RF importance >0 and AUC >0.5) for EMS classification (Figure 4D). Notably, these 14 features demonstrated strong predictive performance, with AUCs ranging from 0.8 to 0.9 (mean AUC = 0.85; ROC plotted by combining all 14 features, Figure 4E). Additionally, these features closely corresponded with ASRM staging (Figure 4F). Collectively, these results indicate that gut microbiome features provide superior predictive power for EMS compared with the vaginal microbiome and sex hormone indices.
Discussion
In this study, we reported a study on the alteration of vaginal and gut microbes in patients with endometriosis (EMS). By using metagenomic sequencing on vaginal and fecal samples from two groups (EMS and Non-EMS) of participants, we deeply investigated the microbiome alteration related with EMS. The microbiome from reproductive tract and gut were combined for comprehensive analysis, and the microbial disorder was found both in vagina and gut from the patients with EMS. In addition, by introducing the machine-learning methods, we found gut microbiota had the highest predictive ability among all features to detect EMS patients. These results demonstrated the paramount role of the gut microbiome in EMS diagnosis and provided the insights to understand the mechanism under the development of EMS.
Firstly, we observed the luteinizing hormone (LH) and follicle-stimulating hormone (FSH) increased in the patients with EMS, which was consistent with previous study.41 In addition, the estradiol (E2) was reported as a critical mediator related with the balance of immunity in EMS,41 and we also found it also fluctuated in our study. For the vaginal microbiome, results showed that the dominant bacteria in the EMS group shifted from Lactobacillus to Bifidobacterium and Gardnerella compared with the Non-EMS group. Lactobacillus is an important commensal species that helps maintain a healthy vaginal pH and metabolic environment.42,43 As for the function of Bifidobacterium in vagina, there were contradictions in some studies.44,45 Gardnerella was a vaginal bacterium that associated with bacterial vaginosis46,47 and host inflammation.48 Comparison with previous studies confirmed the disruption of vaginal microbiota in patients with EMS. In addition, decreased Lactobacillus and increased Gardnerella, Klebsiella, Streptococcus, and Staphylococcus were associated with elevated LH and FSH levels, which may be explained by vaginal microbiota dysbiosis. Furthermore, differential genes in the vaginal microbiome were enriched in metabolic pathways, suggesting that the altered vaginal microbiota may exert metabolic effects in patients with EMS.
Next, we examined alterations in the gut microbiome between EMS and Non-EMS groups. Alpha diversity of the gut microbiota was significantly reduced in EMS patients, a finding consistent with previous reports by Agnes et al.49 In addition, we observed significant alterations in gut microbial composition between the two groups. The relative abundances of Ruminococcus, Oscillibacter, Prevotella, Paraprevotella, and Monoglobus were decreased in EMS patients compared with Non-EMS individuals. In contrast, Dialister was the only taxon enriched in the EMS group and has been previously reported as an EMS-associated biomarker.40 Moreover, most of the decreased microbes are reported as core or probiotic gut microbiota.50–52 We also found that the altered genes were enriched in key pathways potentially affecting gut microbial stability. Notably, differential genes from Dialister, which was enriched in EMS, were involved in formate metabolism, a pathway related to gut health and host immunity.53,54 Moreover, it has been reported that women with EMS have a 50% increased risk of inflammatory bowel disease, highlighting the potential link between gut health and EMS.55 Similarly, another study reported that oral gavage of feces from mice with EMS could restore endometriotic lesion growth and inflammation.56 It can therefore be assumed that the altered gut microbiome was relative with EMS.
Finally, we introduced some machine learning methods to explore the important effects for predictive models from different features. The Random Forest model can select a random subset of variable to fit the small sample size data and provided accurate results.57 At the same time, regression analysis can better measure the relationship between different microbial variables and disease severity.58 Using both of them, gut microbial features detected to classify and predict the ASRM staging of EMS had a good predictive performance. Although current results provide valuable preliminary evidence, a limitation of this study lies in its modest sample size. Although we introduced additional public samples (Table S3 and Figure S4), to strengthen the feasibility of overall analysis, the associations should not be interpreted as conclusive evidence of causality. We recommend it as exploratory and hypothesis-generating, laying the ground work for future validations. In addition to bacterial taxa, the microbiome also includes eukaryotic microbes such as yeasts. Recent studies have reported a prevalence of Candida in patients with EMS, suggesting that fungal dysbiosis may coexist with bacterial alterations and contribute to local inflammation.59 While our current analysis focused primarily on bacterial features, future work incorporating fungal components of the vaginal microbiota may provide further insights into their role in EMS pathophysiology.
Conclusion
This study provides a comprehensive analysis of the alterations in the vaginal and gut microbiome in patients with endometriosis (EMS). Through metagenomic sequencing of vaginal and fecal samples, we identified significant dysbiosis in both microbiomes of EMS patients. The functional analysis revealed that these microbial changes were associated with disruptions in vital metabolic pathways, indicating that gut dysbiosis may contribute to the systemic inflammation and altered estrogen metabolism implicated in EMS pathogenesis. Importantly, our machine learning models demonstrated that gut microbiome features provided superior predictive power for EMS diagnosis compared to vaginal microbiome and sex hormone indices. This underscores the potential of gut microbiota as a biomarker for EMS and highlights the critical role of gut health in the disease. By shedding light on the intricate relationship between the microbiome and EMS, this study contributes to the growing body of knowledge on the multifactorial nature of endometriosis and opens new avenues for research and clinical intervention.
Abbreviation
EMS, Endometriosis; LH, luteinizing hormone; FSH, Follicle-stimulating hormone; ASRM, American Society for Reproductive Medicine; AMH, Anti-Müllerian hormone; E2, Estradiol; P, Progesterone; PCoA, Principal co-ordinates analysis; OLS, The Ordinary Least Square; AUC, Area under the curve; RF, Random Forest model.
Data Sharing Statement
The raw sequence data reported in this paper have been deposited in the Genome Sequence Archive60 in the National Genomics Data Center, China National Center for Bioinformation/Beijing Institute of Genomics, Chinese Academy of Sciences, under accession number CRA017871 that are publicly accessible at https://bigd.big.ac.cn/gsa.
Ethical Approval
The studies involving participants were reviewed and approved by The Third Xiangya Hospital of Central South University and performed under the relevant guidelines and regulations (IRB number 22224). This study was conducted in accordance with the principles of the Declaration of Helsinki. Informed consent was obtained from all subjects involved in the study. Written informed consent has been obtained from the patients to publish this paper.
Acknowledgments
We express our gratitude to Syeda Sundas Batool for the valuable assistance in the preparation of this manuscript.
Author Contributions
Yiming Zhao and Xinyu Hu contributed equally to this work and share first authorship. All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This work was funded by the National Natural Science Foundation of China (32170071), the Central South University Innovation-Driven Research Programme (No. 2023CXQD059), and the Natural Science Foundation of Hunan Province (2022JJ30898).
Disclosure
The author(s) report no conflicts of interest in this work.
References
1. Taylor HS, Kotlyar AM, Flores VA. Endometriosis is a chronic systemic disease: clinical challenges and novel innovations. Lancet. 2021;397(10276):839–852. doi:10.1016/S0140-6736(21)00389-5
2. Ahn SH, Monsanto SP, Miller C, Singh SS, Thomas R, Tayade C. Pathophysiology and immune dysfunction in endometriosis. Biomed Res Int. 2015;2015(1):795976. doi:10.1155/2015/795976
3. Halme J, Hammond MG, Hulka JF, Raj SG, Talbert LM. Retrograde menstruation in healthy women and in patients with endometriosis. Obstetrics Gynecol. 1984;64(2):151–154.
4. Simpson JL, Elias S, Malinak LR, Buttram VC Jr. Heritable aspects of endometriosis: i. Genetic Stud Ame J Obstet Gynecol. 1980;137(3):327–331. doi:10.1016/0002-9378(80)90917-5
5. Vinatier D, Dufour P, Oosterlynck D. Immunological aspects of endometriosis. Human Reproduction Update. 1996;2(5):371. doi:10.1093/humupd/2.5.371
6. Gurates B, Bulun SE. Endometriosis: the ultimate hormonal disease. Seminars Reprod Med. 2003;21(02):125–134.
7. Bellelis P, Podgaec S, Abrão MS. Environmental factors and endometriosis. Rev Assoc Med Bras. 2011;57(4):456–461. doi:10.1590/S0104-42302011000400022
8. Turnbaugh PJ, Ley RE, Hamady M, Fraser-Liggett CM, Knight R, Gordon JI. The human microbiome project. Nature. 2007;449(7164):804–810. doi:10.1038/nature06244
9. Ma B, Forney LJ, Ravel J. Vaginal microbiome: rethinking health and disease. Ann Rev Microbiol. 2012;66(1):371–389. doi:10.1146/annurev-micro-092611-150157
10. Leonardi M, Hicks C, El‐Assaad F, El‐Omar E, Condous G. Endometriosis and the microbiome: a systematic review. BJOG. 2020;127(2):239–249. doi:10.1111/1471-0528.15916
11. Delaney ML, Onderdonk AB. Microbiology, group PS. Nugent score related to vaginal culture in pregnant women. Obstetrics Gynecol. 2001;98(1):79–84.
12. Kavoussi SK, Pearlman MD, Burke WM. Gynecology, endometrioma complicated by tubo‐ovarian abscess in a woman with bacterial vaginosis. Infect Dis Obstetr Gynecol. 2006;2006(1):084140.
13. Qing X, Xie M, Liu P, et al. Correlation between dysbiosis of vaginal microecology and endometriosis: a systematic review and meta-analysis. PLoS One. 2024;19(7):e0306780. doi:10.1371/journal.pone.0306780
14. Cerf-Bensussan N, Gaboriau-Routhiau V. The immune system and the gut microbiota: friends or foes? Nat Rev Immunol. 2010;10(10):735–744. doi:10.1038/nri2850
15. Org E, Mehrabian M, Parks BW, et al. Sex differences and hormonal effects on gut microbiota composition in mice. Gut Microbes. 2016;7(4):313–322. doi:10.1080/19490976.2016.1203502
16. Chelakkot C, Ghim J, Ryu SH. Mechanisms regulating intestinal barrier integrity and its pathological implications. Exp Mol Med. 2018;50(8):1–9. doi:10.1038/s12276-018-0126-x
17. Flores R, Shi J, Fuhrman B, et al. Fecal microbial determinants of fecal and systemic estrogens and estrogen metabolites: a cross-sectional study. J Transl Med. 2012;10(1):1–11. doi:10.1186/1479-5876-10-253
18. Plottel CS, Blaser MJ. Microbiome and malignancy. Cell Host Microbe. 2011;10(4):324–335. doi:10.1016/j.chom.2011.10.003
19. Canis M, Donnez JG, Guzick DS, et al. Revised American society for reproductive medicine classification of endometriosis: 1996. Fertil Sterility. 1997;67(5):817–821. doi:10.1016/S0015-0282(97)81391-X
20. Chen S, Zhou Y, Chen Y, Gu J. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics. 2018;34(17):i884–i890. doi:10.1093/bioinformatics/bty560
21. Xu H, Luo X, Qian J, et al. FastUniq: a fast de novo duplicates removal tool for paired short reads. PLoS One. 2012;7(12):e52249. doi:10.1371/journal.pone.0052249
22. Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9(4):357–359. doi:10.1038/nmeth.1923
23. Lu J, Rincon N, Wood DE, et al. Metagenome analysis using the Kraken software suite. Nature Protocols. 2022;17(12):2815–2839. doi:10.1038/s41596-022-00738-y
24. Kembel SW, Cowan PD, Helmus MR, et al. Picante: r tools for integrating phylogenies and ecology. Bioinformatics. 2010;26(11):1463–1464. doi:10.1093/bioinformatics/btq166
25. Li D, Liu CM, Luo R, Sadakane K, Lam TW. MEGAHIT: an ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics. 2015;31(10):1674–1676. doi:10.1093/bioinformatics/btv033
26. Zhu W, Lomsadze A, Borodovsky M. Ab initio gene identification in metagenomic sequences. Nucleic Acids Res. 2010;38(12):e132–e132. doi:10.1093/nar/gkq275
27. Wheeler TJ, Eddy SR. nhmmer: DNA homology search with profile HMMs. Bioinformatics. 2013;29(19):2487–2489. doi:10.1093/bioinformatics/btt403
28. Eberhardt RY, Haft DH, Punta M, Martin M, O’Donovan C, Bateman A. AntiFam: a tool to help identify spurious ORFs in protein annotation. Database. 2012;2012:bas003. doi:10.1093/database/bas003
29. Steinegger M, Soding J. MMseqs2 enables sensitive protein sequence searching for the analysis of massive data sets. Nat Biotechnol. 2017;35(11):1026–1028. doi:10.1038/nbt.3988
30. Huerta-Cepas J, Szklarczyk D, Heller D, et al. eggNOG 5.0: a hierarchical, functionally and phylogenetically annotated orthology resource based on 5090 organisms and 2502 viruses. Nucleic Acids Res. 2019;47(D1):D309–D314. doi:10.1093/nar/gky1085
31. Li H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv preprint arXiv:13033997. 2013.
32. Robin X, Turck N, Hainard A, et al. pROC: an open-source package for R and S+ to analyze and compare ROC curves. BMC Bioinf. 2011;12(1):1–8. doi:10.1186/1471-2105-12-77
33. Zhao Y, Chen L, Yao S, et al. Genome-centric investigation of the potential succession pattern in gut microbiota and altered functions under high-protein diet. Curr Res Food Sci. 2023;7:100600. doi:10.1016/j.crfs.2023.100600
34. Mallick H, Rahnavard A, McIver LJ, et al. Multivariable association discovery in population-scale meta-omics studies. PLOS Computat Biolog. 2021;17(11):e1009442. doi:10.1371/journal.pcbi.1009442
35. Subirana I, Sanz H, Vila J. Building bivariate tables: the comparegroups package for R. J Statist Software. 2014;57(12):1–16. doi:10.18637/jss.v057.i12
36. Segata N, Izard J, Waldron L, et al. Metagenomic biomarker discovery and explanation. Genome Biology. 2011;12(6):R60. doi:10.1186/gb-2011-12-6-r60
37. Ravel J, Gajer P, Abdo Z, et al. Vaginal microbiome of reproductive-age women. Proc Natl Acad Sci. 2011;108(supplement_1):4680–4687. doi:10.1073/pnas.1002611107
38. Aldunate M, Srbinovski D, Hearps AC, et al. Antimicrobial and immune modulatory effects of lactic acid and short chain fatty acids produced by vaginal microbiota associated with eubiosis and bacterial vaginosis. Front Physiol. 2015;6:164. doi:10.3389/fphys.2015.00164
39. Mohammadzadeh F, Dolatian M, Jorjani M, Alavi Majd H. Majd HAJGjohs. Diagnostic value of Amsel’s clinical criteria for diagnosis of bacterial vaginosis. Global Journal of Health Science. 2014;7(3):8. doi:10.5539/gjhs.v7n3p8
40. Sims TT, Colbert LE, Zheng J, et al. Gut microbial diversity and genus-level differences identified in cervical cancer patients versus healthy controls. Gynecologic Oncol. 2019;155(2):237–244. doi:10.1016/j.ygyno.2019.09.002
41. Romanski PA, Brady PC, Farland LV, Thomas AM, Hornstein MD. The effect of endometriosis on the antimüllerian hormone level in the infertile population. J Assist Reprod Genetics. 2019;36(6):1179–1184. doi:10.1007/s10815-019-01450-9
42. Petrova MI, Lievens E, Malik S, Imholz N, Lebeer S. Lactobacillus species as biomarkers and agents that can promote various aspects of vaginal health. Front Physiol. 2015;6:81. doi:10.3389/fphys.2015.00081
43. Chee WJY, Chew SY, Than LTL. Vaginal microbiota and the potential of Lactobacillus derivatives in maintaining vaginal health. Microb Cell Fact. 2020;19(1):203. doi:10.1186/s12934-020-01464-4
44. Freitas AC, Hill JE. Quantification, isolation and characterization of bifidobacterium from the vaginal microbiomes of reproductive aged women. Anaerobe. 2017;47:145–156. doi:10.1016/j.anaerobe.2017.05.012
45. Giordani B, Melgoza LM, Parolin C, et al. Vaginal bifidobacterium breve for preventing urogenital infections: development of delayed release mucoadhesive oral tablets. Int J Pharm. 2018;550(1–2):455–462. doi:10.1016/j.ijpharm.2018.09.003
46. Schwebke JR, Muzny CA, Josey WE. Role of gardnerella vaginalis in the pathogenesis of bacterial vaginosis: a conceptual model. J Infect Dis. 2014;210(3):338–343. doi:10.1093/infdis/jiu089
47. Morris M, Nicoll A, Simms I, Wilson J, Catchpole M. Catchpole MJBJoO, bacterial vaginosis: a public health review. BJOG. 2001;108(5):439–450. doi:10.1111/j.1471-0528.2001.00124.x
48. Hardy L, Jespers V, Abdellati S, et al. A fruitful alliance: the synergy between atopobium vaginae and gardnerella vaginalis in bacterial vaginosis-associated biofilm. Sexually Transmit Infect. 2016;92(7):487–491. doi:10.1136/sextrans-2015-052475
49. Svensson A, Brunkwall L, Roth B, Orho-Melander M, Ohlsson B. Associations between endometriosis and gut microbiota. Reprod Sci. 2021;28(8):2367–2377. doi:10.1007/s43032-021-00506-5
50. Maeda Y, Takeda K. Role of gut microbiota in rheumatoid arthritis. J Clin Med. 2017;6(6):60. doi:10.3390/jcm6060060
51. Yeoh YK, Sun Y, LYT I, et al. Prevotella species in the human gut is primarily comprised of prevotella copri, prevotella stercorea and related lineages. Sci Rep. 2022;12(1):9055. doi:10.1038/s41598-022-12721-4
52. Zhao Y, Li C, Wu K, et al. Exploring the impact of short term travel on gut microbiota and probiotic bacteria mediated stability. Biomedicines. 2024;12(7):1378. doi:10.3390/biomedicines12071378
53. Ternes D, Tsenkova M, Pozdeev VI, et al. The gut microbial metabolite formate exacerbates colorectal cancer progression. Nat Metab. 2022;4(4):458–475. doi:10.1038/s42255-022-00558-0
54. Pietzke M, Meiser J, Vazquez A. Formate metabolism in health and disease. Mol Metabol. 2020;33:23–37. doi:10.1016/j.molmet.2019.05.012
55. Jess T, Frisch M, Jørgensen KT, Pedersen BV, Nielsen NM. Increased risk of inflammatory bowel disease in women with endometriosis: a nationwide Danish cohort study. Gut. 2012;61(9):1279–1283. doi:10.1136/gutjnl-2011-301095
56. Chadchan SB, Cheng M, Parnell LA, et al. Antibiotic therapy with metronidazole reduces endometriosis disease progression in mice: a potential role for gut microbiota. Hum Reprod. 2019;34(6):1106–1116. doi:10.1093/humrep/dez041
57. Belk A, Xu ZZ, Carter DO, et al. Microbiome data accurately predicts the postmortem interval using random forest regression models. Genes. 2018;9(2):104. doi:10.3390/genes9020104
58. Zhao Y, Yi J, Xiang J, et al. Exploration of lung mycobiome in the patients with non-small-cell lung cancer. BMC Microbiol. 2023;23(1):81. doi:10.1186/s12866-023-02790-4
59. Bauşic AIG, Creţoiu SM, Bauşic V, et al. The role of gut dysbiosis in endometriosis’ diagnosis and treatment approaches–case report. Romanian J Morphol Embryol. 2023;64(2):263. doi:10.47162/RJME.64.2.17
60. Chen T, Chen X, Zhang S, et al. The genome sequence archive family: toward explosive data growth and diverse data types. Genomics Proteom Bioinformat. 2021;19(4):578–583. doi:10.1016/j.gpb.2021.08.001
 © 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.