Back to Journals » Drug Design, Development and Therapy » Volume 9
Complete genome-wide screening and subtractive genomic approach revealed new virulence factors, potential drug targets against bio-war pathogen Brucella melitensis 16M
Authors Pradeepkiran JA, Sainath DB, Kumar KK, Bhaskar M, P RB
Received 3 November 2014
Accepted for publication 4 December 2014
Published 19 March 2015 Volume 2015:9 Pages 1691—1706
DOI https://doi.org/10.2147/DDDT.S76948
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Shu-Feng Zhou
Jangampalli Adi Pradeepkiran,1* Sri Bhashyam Sainath,2,3* Konidala Kranthi Kumar,1 Matcha Bhaskar1
1Division of Animal Biotechnology, Department of Zoology, Sri Venkateswara University, Tirupati, India; 2CIMAR/CIIMAR, Centro Interdisciplinar de Investigação Marinha e Ambiental, Universidade do Porto, Rua dos Bragas, Porto, Portugal, 3Department of Biotechnology, Vikrama Simhapuri University, Nellore, Andhra Pradesh, India
*These authors contributed equally to this work
Abstract: Brucella melitensis 16M is a Gram-negative coccobacillus that infects both animals and humans. It causes a disease known as brucellosis, which is characterized by acute febrile illness in humans and causes abortions in livestock. To prevent and control brucellosis, identification of putative drug targets is crucial. The present study aimed to identify drug targets in B. melitensis 16M by using a subtractive genomic approach. We used available database repositories (Database of Essential Genes, Kyoto Encyclopedia of Genes and Genomes Automatic Annotation Server, and Kyoto Encyclopedia of Genes and Genomes) to identify putative genes that are nonhomologous to humans and essential for pathogen B. melitensis 16M. The results revealed that among 3 Mb genome size of pathogen, 53 putative characterized and 13 uncharacterized hypothetical genes were identified; further, from Basic Local Alignment Search Tool protein analysis, one hypothetical protein showed a close resemblance (50%) to Silicibacter pomeroyi DUF1285 family protein (2RE3). A further homology model of the target was constructed using MODELLER 9.12 and optimized through variable target function method by molecular dynamics optimization with simulating annealing. The stereochemical quality of the restrained model was evaluated by PROCHECK, VERIFY-3D, ERRAT, and WHATIF servers. Furthermore, structure-based virtual screening was carried out against the predicted active site of the respective protein using the glycerol structural analogs from the PubChem database. We identified five best inhibitors with strong affinities, stable interactions, and also with reliable drug-like properties. Hence, these leads might be used as the most effective inhibitors of modeled protein. The outcome of the present work of virtual screening of putative gene targets might facilitate design of potential drugs for better treatment against brucellosis.
Keywords: Brucella melitensis 16M, homology modeling, putative genes, structure based virtual screening, subtractive genomic approach, targets
Introduction
Identification of potential drug targets is one of the critical factors for effective therapy against pathogen-mediated diseases. Nowadays, the role of computational strategies in the identification of drug targets for harmful pathogens is one of the growing areas in the field of therapeutic medicine.1 The other plausible reason for the intervention of computational approaches is to avoid unnecessary risks from research experiments related to bio-war pathogens like Anthrax, Clostridium, and Brucella, and at the same time provide prospective research feasibilities to unveil drug targets in harmful pathogens.2 Moreover, through a subtractive genomic approach, the possible virulence factors responsible for pathogenesis in humans can be effectively screened.3
Brucella melitensis 16M is categorized under the bio-war pathogen list. It causes a disease known as brucellosis, which severely affects the livestock production and management people who are in close contact with domestic animals.4 The genus Brucella consists of six species, out of which four species (ie, B. melitensis, B. suis, B. abortus, and B. canis) have the ability to infect humans. Among the four species, B. melitensis 16M is highly pathogenic to humans.5 B. melitensis 16M is a Gram-negative, coccobacillus, nonmotile, facultative, intracellular pathogen. It causes abortion in cattle, goats, and sheep and a febrile illness (undulant fever) in humans. Brucellosis is associated with many symptoms in humans, such as weight loss, intermittent fever, liver and spleen disorders, neurological problems, reproductive abnormalities, and heart-related problems.6 Thus, it seems apparent that brucellosis targets vital organs such as liver, spleen, heart, testis, and brain, thereby negatively affecting their functions.7 Brucella genomes exhibit some peculiar characteristic features, such as less divergence between the species8,9 and also great stability with high GC content (57%) at the genomic level.10 They also exhibit high similarity with the plant pathogenic bacteria Pseudomonas.11 Although brucellosis is a commonly occurring disease, the structural and functional aspects of virulence factors and the exact mechanism(s) of virulence factors mediating brucellosis are not well defined. Therefore, identification of such virulence factors is crucial for drug therapy strategies to control brucellosis.
The route for transmission of brucellosis is believed to be due to the consumption of contaminated dairy products. It has been indicated that the primary source of Brucella infection is the consumption of unpasteurized dairy products from infected animals.12 It has also been reported that contact with contaminated products of aborted animals significantly influences the transmission of brucellosis to humans,13 while airborne transmission of bacteria to humans has also been documented in clinical laboratories and abattoirs.14 Therefore, it seems apparent that approaches to control brucellosis are of prime importance. Recently, molecular techniques coupled with genomic and proteomic in silico strategies provided valuable information related to pathogens. The promising means of identification of novel drug targets is to detect bacterial genes that are nonhomologues of human genes and are essential for the survival of the pathogens in the host. Such an approach is classically known as the subtractive genomic strategy. In the present study, we identified genes that are very specific to pathogen and nonhomologous to humans in the genome of B. melitensis 16M by using subtractive genomic analysis. This strategy provides 1) mechanistic possibilities of proteins involved in the brucellosis and 2) rapid potential drug target identification, thereby greatly facilitating the search for new antibiotics. In conclusion, the results of the present study pinpoint the utility of the subtractive genomic approach using large genomic databases for in silico systematic drug target identification in the postgenomic era.
Materials and methods
The whole procedure carried out in order to construct a schematic diagram is shown in Figure 1.
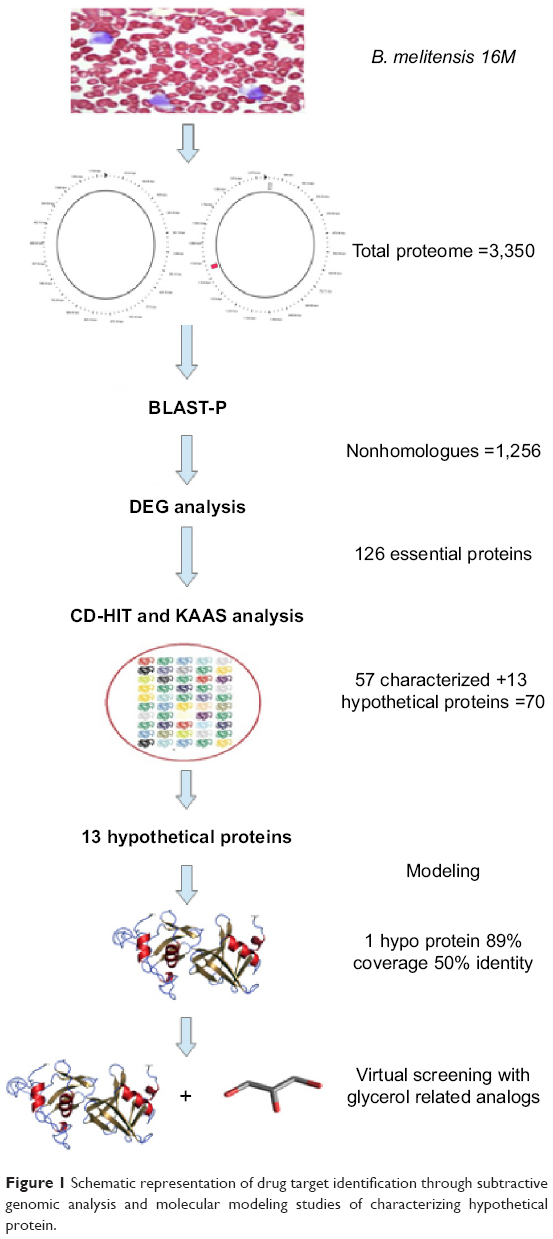 | Figure 1 Schematic representation of drug target identification through subtractive genomic analysis and molecular modeling studies of characterizing hypothetical protein. |
Screening of nonhomologues
The complete genome sequence of B. melitensis 16M was retrieved from the National Center for Biotechnology Information (NCBI) through a sequence retrieval system with accession numbers NC_003317.1 and NC_003318.1.15 The genome sequence was distributed in two circular chromosomes with 32 kb. We screened a total of 3,350 protein sequences of B. melitensis 16M for the identification of nonhomologue sequences by computing against Homo sapiens. The NCBI Basic Local Alignment Search Tool protein (BLAST-P) analysis was carried out by excluding the human homologues with a 50% bit score, E-value 10−3, cutoff value ≤25% identity, and ≤50% query coverage.16,17
Identification of essential genes and their paralogues
The selected sequences of B. melitensis 16M were subjected to Database of Essential Genes (DEG) analysis for the identification of essential sequences.18 The parameter was set with the minimum cutoff E-value ≤1 for screening the reliable essential genes.19 The recognized essential genes were subjected to CD-HIT web server to discard the genes consisting of either paralogues or duplicate sequences. The screening process was done with default parameters available with the server and the limit was set to 0.6 (60%) sequence identity.20
Identification of orthologues and unique pathways
The predicted essential and nonhomologue genes were subjected to Kyoto Encyclopedia of Genes and Genomes (KEGG) Automatic Annotation Server (KAAS) analysis for the elimination of human orthologues, and KEGG analysis was used for the identification of unique pathways in B. melitensis 16M.21 The identified nonhomologue essential genes were classified as enzymes and proteins with KAAS analysis with KEGG orthology (KO) numbers of respective metabolic pathways.22
Prediction of subcellular localization
The localization of proteins also plays a vital role in pathogenesis. Hence, we used the PSORTb v3 server to identify the subcellular localization of essential genes.23
Screening of hypothetical virulence
The virulent nature of uncharacterized genes was predicted by the VirulentPred online analysis tool (http://bioinfo.icgeb.res.in/virulent/). The server depends on position-specific iterated BLAST (psi-blast) to generate the possible matches with Position Specific Scoring Matrix algorithm by five-fold cross-validation technique.24
Motif-based screening of hypothetical proteins
The 13 hypothetical proteins were subjected to Motif Scan (http://myhits.isb-sib.ch/cgi-bin/motif_scan) for the identification of conserved motifs within functional domains in the present study. The proteins containing the highest conserved motifs were selected for further studies.25
Molecular features of hypothetical target protein
The hypothetical protein molecular features, such as physicochemical properties like molecular weight, isoelectric point, negatively and positively charged amino acid residues, extinction coefficients,26 aliphatic index,27 instability index,28 grand average of hydropathicity,29 transmembrane helical regions, and signal peptides of the target primary sequence, were calculated by using ExPASy ProtParam Proteomics server (http://web.expasy.org/protparam/),30 Tied Mixture Hidden Markov Model v2.0 server,31 and SignalP v4.1 server,32 respectively.
Secondary structure prediction
The secondary structure prediction of target hypothetical protein was carried out by using the Self-Optimized Prediction Method with Alignment server.33 The predicted results were also confined with WHATIF secondary structure analysis, and the outcome end results were analyzed.
Homology modeling and optimization
The preferred essential protein sequence was subjected to blastp against Protein Data Bank to find a suitable template for homology modeling.34 Homology modeling of target mature proteins is done in MODELLER 9.12 through comparative modeling by using python scripts. The alignment of the target and the template was carried out using align 2D and then 100 three-dimensional (3D) structures were generated.35 The best model was selected based on low discrete optimized protein energy values. The selected model was optimized using variable target function method (VTFM) with conjugate gradients (CGs) and then refined by molecular dynamics (MD) with simulated annealing (SA) parameter. The VTFM optimization with 500 maximum iterations and MD optimization with slow level mode was carried out, and the whole cycle was repeated two times to generate an optimized conformation of the model.36 The optimized model was evaluated by Ramachandran plot, verify_3D, ERROT, and WHATIF servers.
Prediction of active site
Proteins are composed of amino acids, which interact with small molecules like substrates, ligands, peptides, and deoxyribonucleic acid (DNA) within their domains to carry out their specific function. Therefore, it seems apparent that prediction of protein domains is crucial to understand protein function. Thus, 3D structural domains and active site residues were identified by a Computed Atlas of Surface Topography of Proteins (CASTp) server.37
Identification of small molecule inhibitors of target protein
In the present study, Silicibacter pomeroyi DUF1285 family protein (PDB: 2RE3) was selected as a template to build the model for hypothetical protein 5 (gene accession number NP_539378.1). Moreover, a ligand glycerol is present in the active site of 2RE3 structure. Hence, the structural analogs of glycerol were selected for the structure-based virtual screening studies. The coordinates of the lead molecules were retrieved from the PubChem BioAssay database with the glycerol Chemical Identifier (CID)-751.38 The errors in the identified leads were solved by lead optimization in PyRx, including OpenBable, and ligand energy minimization interface with united force field with a limit of 500 iterations for each ligand. The energy-minimized ligands were converted into AutoDock ligand format (.pdbqt) and prepared as a data set.
Prediction of drug likeness
Based on the Lipinski rule of five, the drug likeness of the ligands was analyzed by molecular property explorer: ie, MolSoft server (http://www.molsoft.com/mprop/) and PubChem ligand property information database.
Virtual screening
Virtual screening was carried out by using 54 minimized leads against the predicted binding site of the optimized conformation of the target protein through AutoDock Vina39 in PyRx software. Active site dimensions were considered as grid size (XYZ axis) to dock with ligands, based on the XYZ grid sizes of modeled protein we calculate the 10 maximum posses for each ligand to fit the grid. The ligand forms hydrogen bond interactions, which affect the stabilization of protein. Hence, the atomic involvement of hydrogen bond formation with active site amino acid residues with bond angles and bond distance was calculated.
Results and discussion
Drug targets, screening
B. melitensis consists of two chromosomes where chromosome-I and -II comprise 2,211 and 1,139 genes, respectively (two cutoff values: ie, ≤25% identity and ≤50% query). The outcome of blastp results showed 1,256 nonhomologous genes where 1,139 genes belong to chromosome-I and 117 genes belong to chromosome-II. Further, we subjected 1,256 genes to the DEG database to screen unique nonhomologous genes from chromosomes of B. melitensis. The results of the DEG database indicated that 77 out of 1,139 genes from chromosome-I and 49 out of 117 genes from chromosome-II were nonhomologous unique genes of pathogen. Thus, a total of 126 nonhomologous essential unique genes were identified in the chromosomes of B. melitensis 16M genome.
It is well known that something is important or something is considered an important element for the genes that mediate common metabolic activities both in the host and bacteria.40 In order to know the level of gene redundancy, we screened 126 genes using the CD-HIT server. Fifty-three genes out of 126 exhibited redundancy. The cutoff values were used with sequence identity (60%) and default algorithm parameters, respectively. Hence, the remaining nonredundant 73 genes were subjected to KAAS analysis for the identification of the orthologues that shared common properties between host and pathogen. The results showed that three genes among 73 (KO 03030 [DNA replication], KO 03420 [nucleotide excision repair], and KO 03430 [mismatch repair]) exhibit common properties in both host and pathogen. So, for further analysis, we excluded the orthologue genes. Therefore, these three genes were excluded for further analysis. After eliminating these common genes, a total number of 70 putative genes (57 characterized +13 hypothetical proteins =70) were subjected to KEGG analysis to classifying essential genes. The results indicated that all the final potential drug targets were clustered into 14 different metabolic pathways (Table 1 and Figure 2).
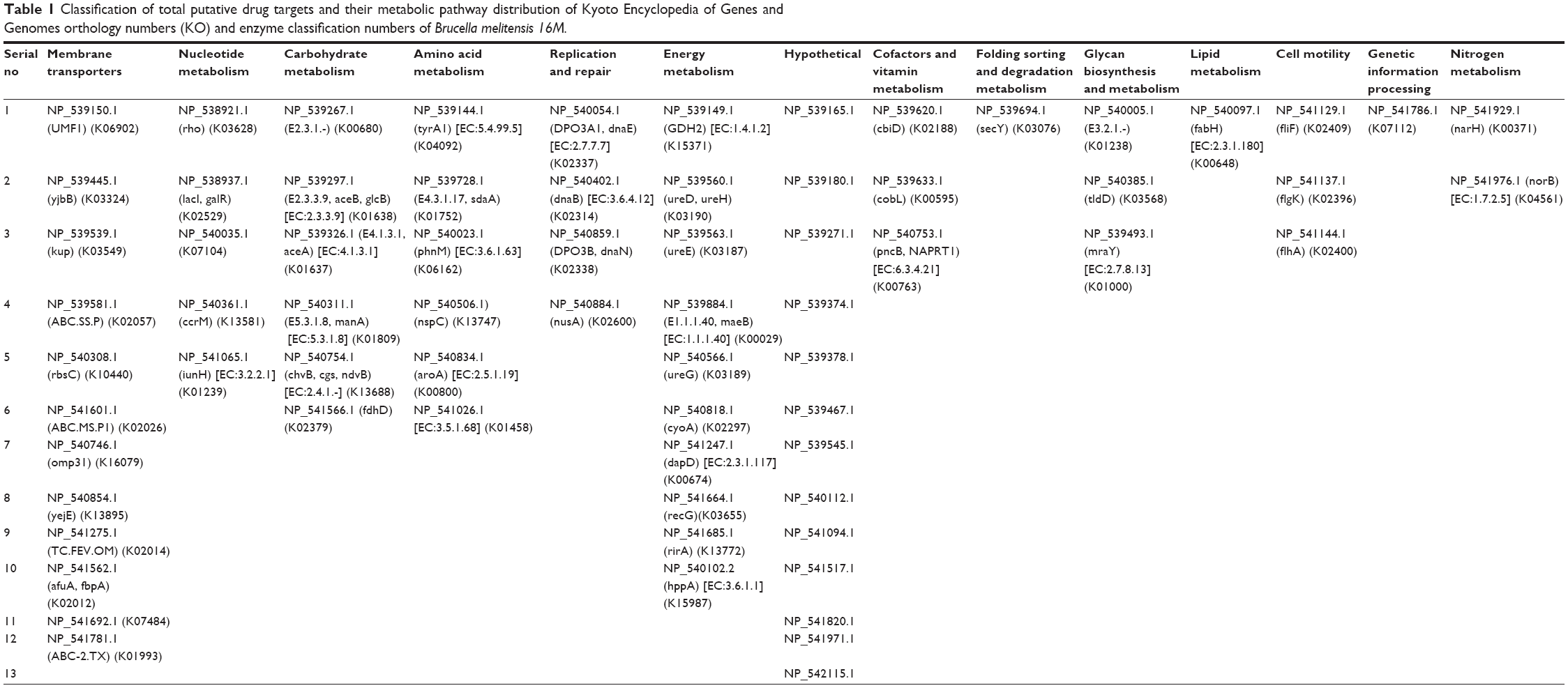 | Table 1 Classification of total putative drug targets and their metabolic pathway distribution of Kyoto Encyclopedia of Genes and Genomes orthology numbers (KO) and enzyme classification numbers of Brucella melitensis 16M. |
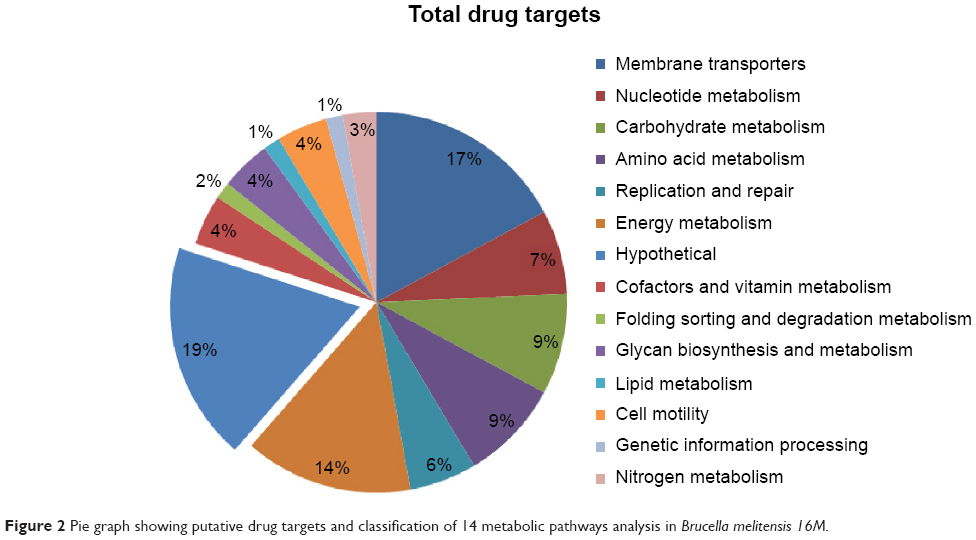 | Figure 2 Pie graph showing putative drug targets and classification of 14 metabolic pathways analysis in Brucella melitensis 16M. |
Characterized drug targets
Proteins involved in membrane transport systems and metabolic cascades are crucial for the survival of bacteria in a host environment. Thus, understanding genes that code for proteins might be crucial for effective identification of drug targets.
Membrane drug targets
It has been reported that 12 membrane transporters play vital roles in pathogenesis.41 The membrane proteins, such as unknown major facilitator protein-1 (UMF1), Na+/potassium transporting symporter (yjbB), and KUP system potassium uptake protein (kup), mediate transport of electrolytes and for the energy-consuming processes. Three sugar transporters (ABC.SS.P, rbsC, and ABC.MS.P1) are also considered important for pathogenesis and also to maintain the isotonic conditions and to overcome stress conditions. Earlier, it has been indicated that ABC transport system of B. melitensis 16M shows the highest number of nutrient importers as compared with other Brucella species.42 This suggests that the mode of nutrient transport in B. melitensis is different from that of other Brucella species. Thus, the presence of an amino acid ABC transporter system might be used as a characteristic feature to differentiate Brucella species. Moreover, blocking these transport systems may prevent bacterial survival and thereby pathogenesis. Therefore, an ABC system might be used as one of the drug targets to prevent pathogenesis of B. melitensis.43
In recent studies it has also been indicated that the drug targets against membrane transporters were crucial to prevent pathogenesis of B. melitensis 16M.44 Previously, it has been indicated that “microcin C” specific to B. melitensis 16M specifically blocks multiple sugar transport system permease protein (ABC.MS.P1) transport systems.45,46 Moreover, studies related to sugar transport and ATP binding cassette involved in the uptake of sugar moieties indicated that the drug targets against permease protein turns might prevent pathogenesis of B. melitensis 16M.47 Past reports have shown that HlyD family secretion protein (ABC-2.TX), iron (III) transport system substrate-binding proteins (afuA, fbpA), and iron complex outer membrane receptor protein (TC.FEV.OM) are crucial for the survival of B. melitensis.48,49 Therefore, targeting these proteins might also offer efficacious potential drug targets against this pathogen.50
Lipid metabolism proteins as drug targets
Lipopolysaccharides (LPS) are the chief constituents of cell wall synthesis, which imparts the pathogenic characters to the bacteria and helps in differentiation of bacteria. In B. melitensis 16M, glycan biosynthesis and metabolism contains tldD, mraY, TldD, and phospho-N-acetylmuramoyl-pentapeptide-transferase proteins in which LPS metabolic pathways are distinctly expressed in Gram-negative bacteria. In general, a bacterial secretion system has the ability to transport bacterial toxins to outer membrane in the host environment.51 However, such a secretary system in B. melitensis 16M is yet to be characterized.
Nucleotide metabolism proteins as drug targets
Nucleotide metabolism plays an important role in the cell survival and reproduction of prokaryotes. In the B. melitensis 16M strain, five proteins have been identified that play important roles in nucleotide metabolism. These proteins include GTP binding protein Rho factor (rho), LacI family transcriptional regulators (lacI, galR), methyltranferase enzyme (ccrM), ionosine–uridine preferring nucleoside hydrolase (iunH), and K0714. Among the five proteins, Rho, lacI, and galR are involved in transcription regulation, whereas ccrM protects the bacteria from the external nuclease drug analogs.52 On the other hand, iunH catalyzes the hydrolysis of the entire glycosidic bond in case of mismatches. Therefore, proteins such as Rho, lacI, and galR proteins and ccrM may be used as potential drug targets, as blocking these targets may inhibit transcription or defense mechanisms, respectively, and thereby affect the survival of the pathogen.
Carbohydrate metabolism proteins as drug targets
The carbohydrate metabolism is an important and essential metabolism for bacterial surveillance and bacterial multiplication. Carbohydrates are the rich carbon sources and substances essential for the organism and are to be supplied as nutrients. In B. melitensis 16M, six targets involved in carbohydrate metabolism, among those five considered as enzymes i.e. acetyltransferase (E2.3.1), malate synthase (E2.3.3.9, aceB, glcB), isocitratelyase (E4.1.3.1, aceA), mannose-6-phosphate isomerase (E5.3.1.8, manA), and one cyclic beta-1,2-glycan synthase (chvB, cgs, ndvB) are potent B. melitensis 16M virulence factors that facilitate the persistence in the host environment by masking the innate immune responses in the host53 and another one target identified as protein i.e. formate dehydrogenase accessory protein (fdhD), which regulates the formate dehydrogenase activity for the bacteria.
Amino acid metabolism proteins as drug targets
Six drug targets involved in amino acid metabolism are already mentioned in Table 1, ie, chorismatemutase (tyrA1), L-serine dehydratase (E4.3.1.17, sdaA), alpha-D-ribose 1-methylphosphonate 5-triphosphate diphosphatase (phnM), carboxy norspermidine decarboxylase (nspC), 3-phosphoshikimate 1-carboxyvinyltransferase (aroA), and an enzyme, N-formylglutamate deformylase (EC:3.5.1.68), are involved where they support the pathogenesis54 by producing the building blocks of macromolecules.
Genetic information and processing drug targets
Replication is an important mechanism for pathogens to transfer the genetic information to progeny and existence. There are four essential drug targets identified in repair and replication metabolism of B. melitensis 16M: ie, DNA polymerase III subunit alpha and beta (DPO3A1, dnaE and DPO3B, dnaN), replicative DNA helicase (dnaB), and utilization substance protein A (nusA).
Cofactor proteins as drug targets
There are three cofactors involved in vitamin and cobalt metabolisms, cobalt-precorrin-5B (C1)-methyltransferase (cbiD), precorrin-6Y C5,15-methyltransferase (decarboxylating) (cobL), and nicotinate phosphoribosyl transferase (pncB, NAPRT1), whereas three flagellar proteins, ie, flagellar M-ring protein (fliF), flagellar hook-associated protein 1 (flgK), and flagellar biosynthesis protein (flhA), were identified in cell motility metabolism as potential drug targets in this pathogen. The two nitrogen-metabolizing enzymes nitrate reductase beta subunit (narH) and nitric oxide reductase subunit B (norB) are considered as potential drug targets, and also, as they closely resemble plant pathogens, B. melitensis 16M is classified as α-proteobacteria.55
Energy metabolism proteins as drug targets
Energy metabolism is crucial for bacterial survival in the host environment. In B. melitensis 16M, the genes responsible for the synthesis of ATPs, NADPH, cytochrome o, and ATP binding cassette proteins are primarily involved in the biochemical reactions for energy generation and thereby the survival of pathogens. To overcome acidic conditions, these bacteria possess urease accessory proteins such as ureD, ureH, ureE, and ureG.56 Proteins such as cytochrome o ubiquinol oxidase subunit II (cyoA), ATP-dependent DNA helicase RecG (recG), Rrf2 family transcriptional regulators, and iron-responsive regulator (rirA) are involved in catabolic and anabolic aspects, thereby sustaining energy generation for the organism’s survival. Therefore, therapeutic strategies against these proteins might affect the survival of pathogens. By virtue of consideration of all these facts, it seems apparent that membrane channels and receptor molecules play vital roles in the pathogenesis of B. melitensis 16M. Earlier, a few studies addressed the therapeutic efficacy of few drugs that particularly blocked membrane and receptor proteins and thereby the survival of the pathogen. Although the results are promising, none of the drugs is target specific, indicating the putative role of other proteins in the survival of pathogens. Surprisingly, we identified (uncharacterized) hypothetical proteins in the genome of B. melitensis 16M through a subtractive genomic approach.
Localization of drug targets
Once the metabolic involvement has been determined in the pathogenesis, the localization of these proteins was determined using PSORTb v3 server, which provides valuable information regarding a protein’s function, to annotate genomes, to design proteomics experiments, and – particularly in the case of bacterial pathogen proteins – to identify potential diagnostic, drug, and vaccine targets. The results of PSORTb indicated that among the 70 genes unique to pathogen, 57 sequences were observed to be characterized and the remaining 13 genes are considered as hypothetical elements. This study revealed that the total target genes were located in cytoplasmic, membrane, and periplasmic regions of Brucella.
VirulentPred analysis (prediction of prokaryotic virulent)
The finalized potential drug targets of B. melitensis 16M are scanned against VirulentPred, a bacterial virulent protein prediction tool, to unveil the functional characterization of bacterial proteins based on virulence factors. These factors are the disease-causing elements found in bacteria that play a key role in pathogenesis. Most of the bacterial-induced infections are mediated by common classical virulence factors like pili, fimbriae, or virulence plasmids, secreted proteases, exotoxins, endotoxins, and secretary systems like class I, class II, class III, and class IV. These virulence factors also transport the bacterial toxins in outer membrane to the host environment. VirulentPred database predicted 13 hypothetical proteins with their virulence nature.
Selection of hypothetical protein as a potential drug target
Further analysis using the Motif Scan database revealed that among 13 hypothetical proteins, one hypothetical protein, NP_539378.1, possesses the highest conserved motif regions based on the normalized score formula: ie, N_Score = log10 DB_size-log10 E-value. The selected protein contains eight motif patterns: ie, two protein kinase C (PKC) phosphorylation motifs present in between 15–17 and 134–136 regions, two casein kinase 2 phosphorilation sites at 15–18 and 103–106 regions, tyrosine phosphorilation site at 92–100 position, two MYRISTYL motifs at 75–80 and 78–81, and AMIDATION motif at 78–81 positions, respectively (Table 2).
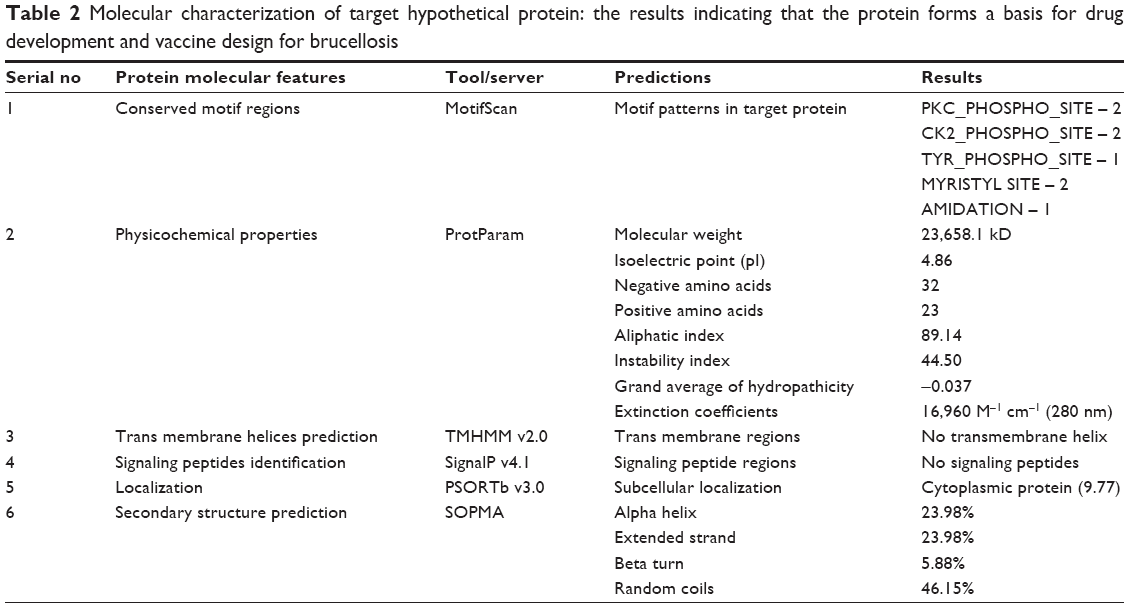 | Table 2 Molecular characterization of target hypothetical protein: the results indicating that the protein forms a basis for drug development and vaccine design for brucellosis |
The protein repeat profile was observed to be matching well with protein prenyltransferases alpha subunit repeat profile (PS51147) with 1–29 amino acid residues that are present in N-terminal domain region, and may also actively participate in ligand binding interactions.
In fact, five phosphorylation sites were observed, among which two of them are casein kinase II (CKII), another two are protein kinase C (PKC), and the remaining one is tyrosine kinase phosphorylation sites. In addition to these phosphorylation sites, one amidation and one myristoylation site were also identified. All these sites were observed to be surrounding different regions of the target protein with 37.5% identity match of repeat profiles with protein prenyltransferases alpha subunit and major vault protein, which helps prenylation, posttranslational functional property, intracellular transport mechanism, and introduction of some peptide extensions. The major vault protein was also useful to target vaults to the cell surface receptor. The present hypothetical protein contains unknown functional domain “DUF1285” in bacteria and contains an overall number of domains of unknown function, which were widely distributed throughout the genome; these domains of unknown function are conserved and essential for bacterial pathogens.57
The aforementioned functional prospects of repeat profiles and also blastp results revealed the reasonable coordinate structure with 80% query coverage and 50% identity and were reliable score matches for constructing the 3D structure target protein. Hence, the nonhuman homologue uncharacterized (NP_539378.1) protein is considered as a potential drug target in B. melitensis 16M.
Characterization of hypothetical target protein
The selected uncharacterized modeled protein (accession number: NP_539378.1) is characterized and summarized in Table 2. The characterization was mainly based on physicochemical properties using a ProtParam tool. The mature protein contains 221 amino acids length with molecular weight 23,658.1 kD. It contains 32 negatively charged residues (Asp + Glu) and 23 positively charged residues (Arg + Lys). Moreover, isoelectric point of the selected protein was about 4.86 and the extinction coefficient at 280 nm was found to be 16,960 M−1 cm−1. The other parameters, such as instability index, aliphatic index, and grand average of hydropathicity, were found to be 44.50, 89.14, and −0.037, respectively. Thus, based on these characteristics, the protein might be unstable and hydropathic in nature. Further, the protein secondary structure was predicted from SOPMA and WHATIF servers. The secondary structure consists of alpha helix, extended strand, beta turn, and random coil in 23.98%, 23.98%, 5.88%, and 46.15% proportions, respectively. These results indicate that the secondary structure of the selected protein form mostly coils and may specifically involve mutations through interaction with chemical factors and cause drug resistance.58 Here, the absence of transmembrane helix from TMHMM server and SignalP 4.0 prediction showed that the target protein contains no signal peptides (Table 2), and hence the protein would definitely an unsecreted cytoplasmic protein. With this interpretation and kinase phosphorylation activity, the protein could conclusively play a pivotal role in the cellular processes of Brucella.
Homology modeling and optimization
The blastp results revealed a perfectly reliable template: ie, crystal structure of a DUF1285 family protein (SPO_0140) from Silicibacter pomeroyi DSS-3 at 2.50 Å resolutions (PDB: 2RE3),59 shared 89% query coverage, and 50% identity with 1e-53 E-value. Align 2D results showed a high-level sequence similarity between target and template. Based on alignment, 100 basic models were generated for target protein by using MODELLER 9.12 comparative modeling, and the missing side chains were added and aligned from WHATIF server. However, the basic modeling was unable to optimize the conformation due to disorganizations in the spatial restraints of amino acid residues. Hence, the modeled protein was optimized in the MODELLER VTFM method by applying the 500 steps steepest descent algorithm of conjugated gradients and then refined by using MD with SA by applying 1,000 steps minimization. The resultant models were sorted by discrete optimized protein energy scoring function. The stereochemical quality of protein model was assessed by Structural Analysis and Verification Server (SAVES) criteria and PROCHECK (for analyzing the residue-by-residue geometry and overall structure geometry), ERRAT (analyzes the statistics of nonbonded interactions between different atom types and plots), and VERIFY_3D (determines the compatibility of an atomic model [3D] with its own amino acid sequence [1D]). The overall information provided that the model number 50 (M50) consists of low discrete optimized protein energy score (ie, −20314.10547 with 1243.77490 molpdf energy). The z-score of modeled protein evaluated by ProSA-web server and the model consisted of −5.98. Hence, we selected M50 for further studies. M50 contains four helices, 13 sheets, and two coils. Furthermore, the optimized structure of the selected hypothetical protein was evaluated for its stereochemical quality by using Ramachandran plot, Verify_3D, ERROT, and WHATIF. Ramachandran plot calculation showed only one amino acid, GLU95, located in disallowed regions, but maximum amino acids were present in most favored regions (Figure 3).
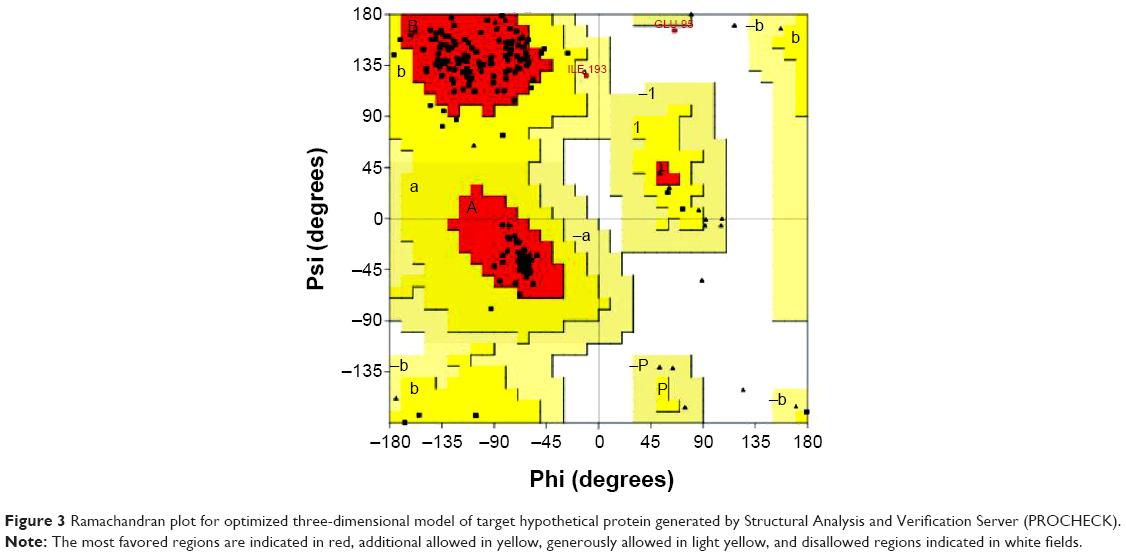 | Figure 3 Ramachandran plot for optimized three-dimensional model of target hypothetical protein generated by Structural Analysis and Verification Server (PROCHECK). |
The most favored regions are indicated in red, additional allowed in yellow, generously allowed in light yellow, and disallowed regions indicated in white fields. The other parameters, such as protein error value from ERROT, showed an increased quality factor 68 to 72 after optimization, and Verify_3D showed compatibility score above zero (83.7% 3D–1D values), indicating that the protein contains favored side chain environments and good fold regions. The evaluated scores of target protein before and after optimization are illustrated in Table 3. The average energy grids for all amino acid residues were shown at allowed regions and protein secondary conformation and superimposed structure with the template are shown in Figure 4A and B. All these results strongly encourage the protein model as good and more reliable for the docking studies.60
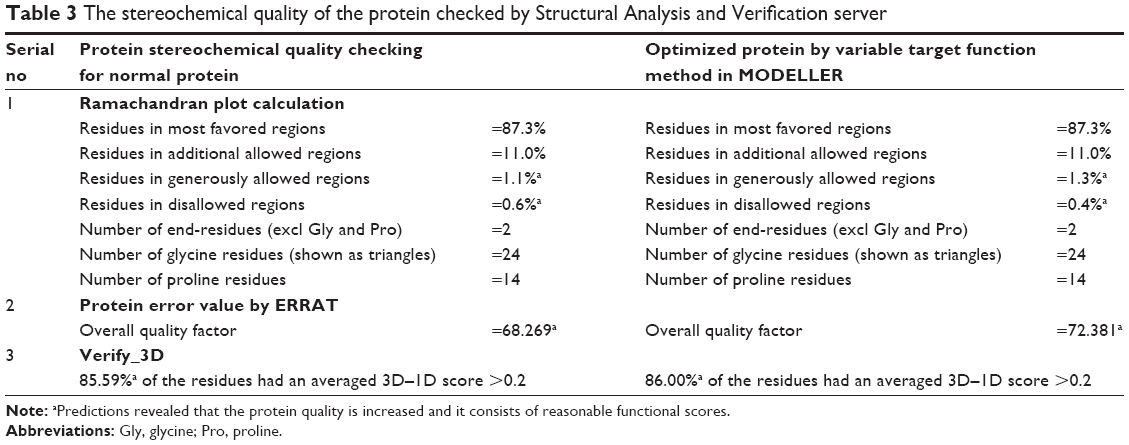 | Table 3 The stereochemical quality of the protein checked by Structural Analysis and Verification server |
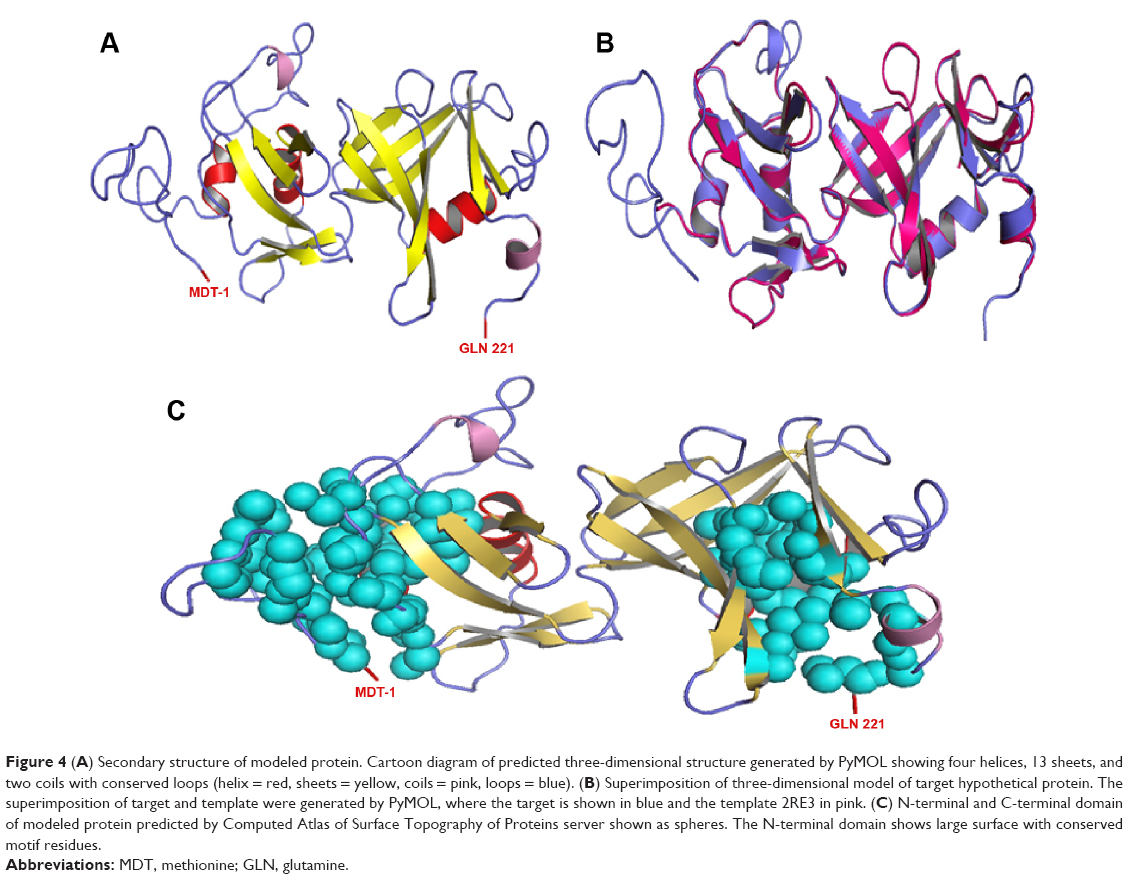 | Figure 4 (A) Secondary structure of modeled protein. Cartoon diagram of predicted three-dimensional structure generated by PyMOL showing four helices, 13 sheets, and two coils with conserved loops (helix = red, sheets = yellow, coils = pink, loops = blue). (B) Superimposition of three-dimensional model of target hypothetical protein. The superimposition of target and template were generated by PyMOL, where the target is shown in blue and the template 2RE3 in pink. (C) N-terminal and C-terminal domain of modeled protein predicted by Computed Atlas of Surface Topography of Proteins server shown as spheres. The N-terminal domain shows large surface with conserved motif residues. |
Prediction of active site
The CASTp results showed two reasonable binding pockets with N and C-terminals of the protein model. N-terminal domain showed good prospective active residues for ligand binding and functional modifications. The cassette binding cavity will start with methionine and ends at valine from amino acid positions 1 to 108. Thus, a pocket from methionine to valine was actively involved in the binding of lead molecules (Figure 4C). Hence, we selected the N terminal binding pocket of the target protein for further docking simulations.
Identification of small molecule inhibitors of target protein
PubChem helps to filter small molecules, which act like drugs with their bioactivity and structural identity with glycerol about ≤90%. Fifty-four compounds were identified from PubChem and were screened for their drug likeness by applying the Lipinski rule. Hydrogen atoms were added to each ligand and optimized through energy minimization (united force field), and the atom restraints were rectified by PyRx OpenBable software.
Prediction of drug likeness
All the leads showed the Lipinski rule of five and good ADMET properties and compared with the positive control for confining through PubChem bioactive database property explorer. The rule of five or Lipinski rule of five defines four simple physicochemical parameter ranges of orally active compounds, like molecular weight, logP, H-bond donors, H-bond acceptors (MWT_500, logP_5, H-bond donors_5, and H-bond acceptors_10). So prediction of drug-like nontoxic compounds is important for modern drug discovery. They are mainly obtained from the repositories of the Modern Drug Data Report (MDDR), Comprehensive Medicinal Chemistry (CMC), and Derwent Word Drug Index (WDI).61,62 Herein, the glycerol-related nontoxic bioactive compounds were identified based on the rule of five predictions from MolSoft. The drug-like inhibitors for target were further optimized, along with hydrogens, before being subjected to the virtual screening process.
Virtual screening and docking
The template used for the construction of the homology model was found to contain glycerol and methionine ligands. Where the methionine is common, amino acid is present in all the proteins normally located at the C-terminal end of the proteins; hence, we shifted over to glycerol-related ligand screening from the PubChem database.63 The structure-based virtual screening was done through AutoDock Vina in PyRx software, to identify docking energies for each ligand with 10 maximum fitting process. Root-mean-square deviation calculations were carried out through lamarkin geometric algorithm. Active site grid dimensions were set as X=24.1349, Y=9.1808, and Z=42.7728 for center and total size dimensions were set as X=48.598933968, Y=40.7985145569, and Z=64.6560720539. The virtual screening of glycerol structural analogs revealed that five compounds showed best affinity with positive control. The ligands trimethylolethane (6502), trimethylolphosphine (76001), bis (hydroxymethyl) phosphinic acid (237875), CHEMBL85846 (44319866), and 2-amino-2-methyl-1, 3-propanediol (1531) showed the binding affinities of −5.2, −5.0, −5.0, −5.0, and −4.9 (kcal/Mol), respectively. Docking results and hydrogen bond interactions with these ligands and their bond angles, bond lengths, and atoms involved in these interactions were analyzed and are illustrated in Table 4.
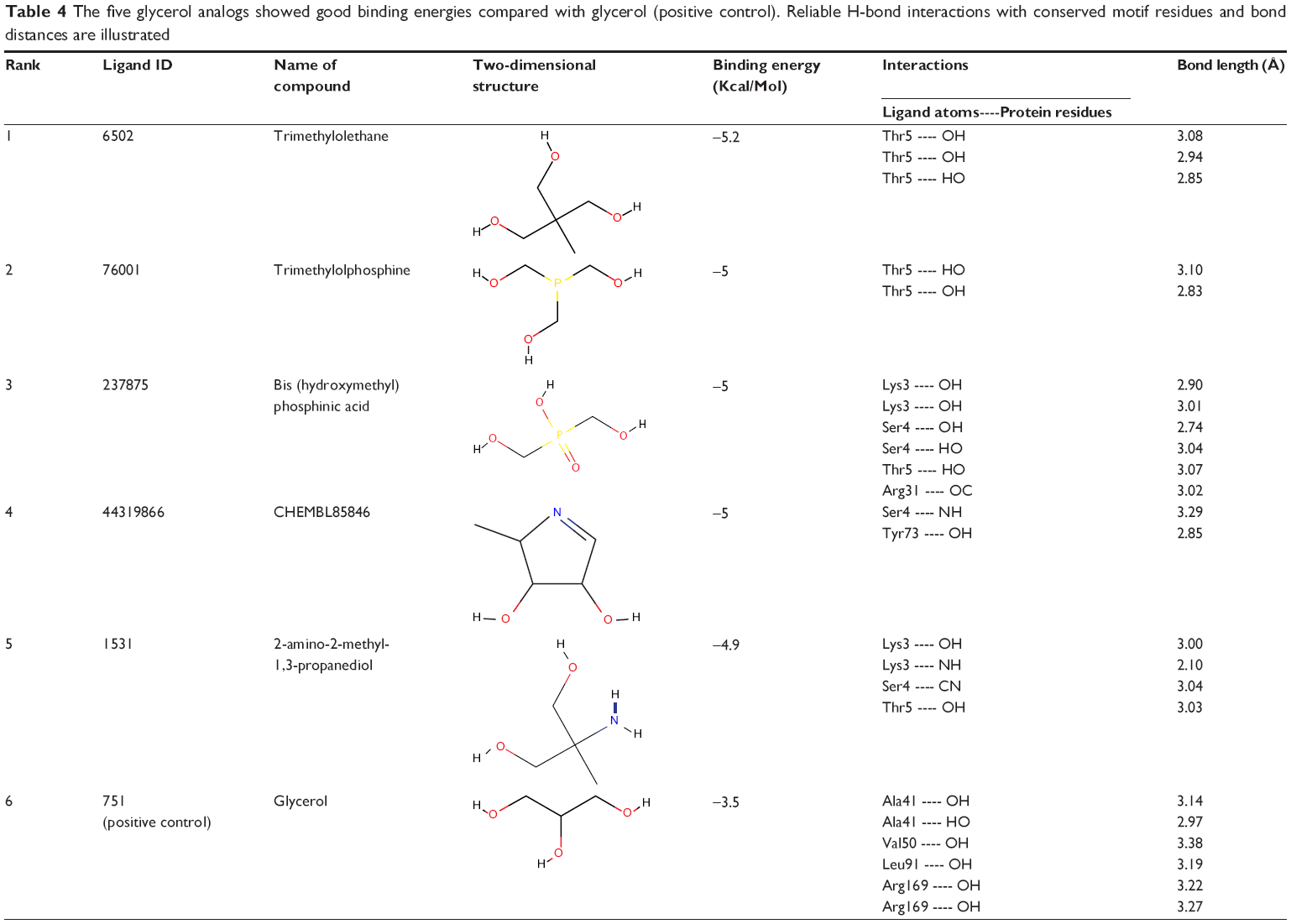 | Table 4 The five glycerol analogs showed good binding energies compared with glycerol (positive control). Reliable H-bond interactions with conserved motif residues and bond distances are illustrated |
Simulation analysis also revealed that all the leads have the ability to interact with N-terminal ligand binding domain loop. The amino acid residues that actively participated in the interaction were as follows: Lys3, Ser4, Thr5, Arg31, and Tyr73 by means of C----O, O----H, N----O, N----H, and CA----N atoms. The hydrogen bond formation was majorly observed among all the glycerol analogs with almost equal bond distances. Trimethylolethane, 2-amino-2-methyl-1, 3-propanediol, Bis (hydroxymethyl) phosphinic acid, and CHEMBL85846 showed three, four, six, and two hydrogen bond interactions, respectively. Residues such as Thr5 with trimethylolethane, Lys3, Ser4, and Thr5 with 2-amino-2-methyl-1, 3-propanediol, Lys3, Ser4, Thr5, and Arg31 with Bis (hydroxymethyl) phosphinic acid and Ser4 and Tyr73 with CHEMBL85846 were associated with the hydrogen bond formation. Moreover, all the aforementioned glycerol analogs showed reliable affinities with selected hypothetical protein (Figure 5A–E).
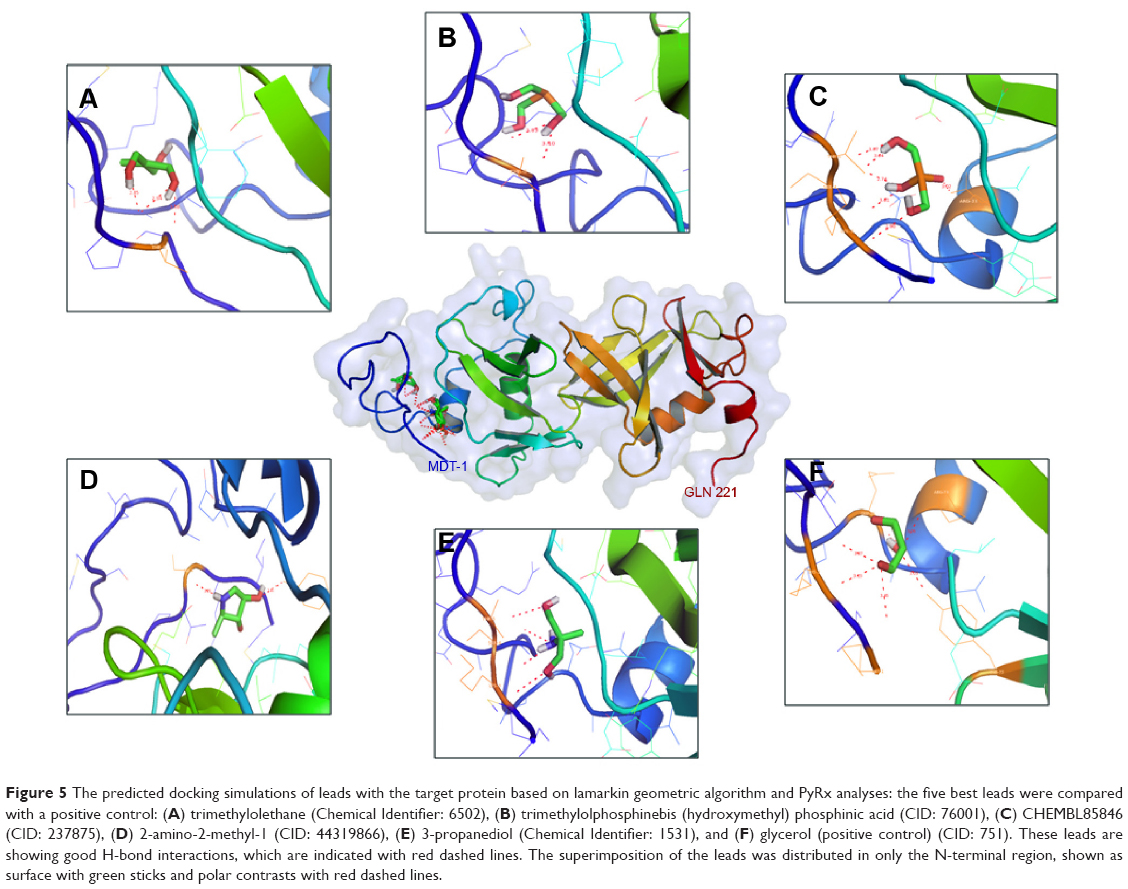 | Figure 5 The predicted docking simulations of leads with the target protein based on lamarkin geometric algorithm and PyRx analyses: the five best leads were compared with a positive control: (A) trimethylolethane (Chemical Identifier: 6502), (B) trimethylolphosphinebis (hydroxymethyl) phosphinic acid (CID: 76001), (C) CHEMBL85846 (CID: 237875), (D) 2-amino-2-methyl-1 (CID: 44319866), (E) 3-propanediol (Chemical Identifier: 1531), and (F) glycerol (positive control) (CID: 751). These leads are showing good H-bond interactions, which are indicated with red dashed lines. The superimposition of the leads was distributed in only the N-terminal region, shown as surface with green sticks and polar contrasts with red dashed lines. |
The other active site amino acid residues also participated in hydrophobic interactions with ligands. All these interactions reflected better affinity levels among the target and ligands than with the positive control glycerol (Figure 5F). Thus, the in silico method adopted (genomic and proteomic databases) in the present study helped us to identify the potential drug target and the target-specific leads using the computational software and online tools for brucellosis. This method reduces cost and time in designing safer and more effective drugs, as well as analyzing the drug likeness prior to clinical trials.
Conclusion
In conclusion, we identified 53 putative characterized and 13 uncharacterized hypothetical proteins through subtractive genomic analysis. Among the hypothetical proteins, one hypothetical protein 5 was selected based on the conserved motifs within the domain. Hence, the 3D structure was modeled and evaluated. Furthermore, for the structure-based virtual screening against the target protein using PyRx AutoDock Vina, we identified five best inhibitors: ie, trimethylolethane, trimethylolphosphine, bis (hydroxymethyl) phosphinic acid, CHEMBL85846, and 2-amino-2-methyl-1, 3-propanediol, which showed good binding orientations and strong affinities within the active site. Further, in vitro and in vivo studies are required to evaluate the prospective drug activity and efficacy of the proposed leads against B. melitensis 16M.
Acknowledgment
We would like to thank Dr Y Nanda Kumar, Scientist-C, Ponducheri Central University, India for his valuable suggestions.
Disclosure
The authors have declared that no competing interests exist.
References
Lehmann JS, Fouts DE, Haft DH, et al. Pathogenomic inference of virulence-associated genes in Leptospira interrogans. PLoS Negl Trop Dis. 2013;7(10):e2468. | ||
Thavaselvam D, Vijayaraghavan R. Biological warfare agents. J Pharm Bioallied Sci. 2010;2(3):179–188. | ||
Fraser CM, Eisen JA, Nelson KE, Paulsen IT, Salzberg SL. The value of complete microbial genome sequencing (you get what you pay for). J Bacteriol. 2002;184(23):6403–6405. | ||
Sam IC, Karunakaran R, Kamarulzaman A, et al. large exposure to Brucella melitensis in a diagnostic laboratory. J Hosp Infect. 2012;80:321–325. | ||
Rajashekara G, Glasner JD, Glover DA, Splitter GA. Comparative whole-genome hybridization reveals genomic islands in Brucella species. J Bacteriol. 2004;186:5040–5051. | ||
Gyuranecz M, Szeredi L, Ronai Z, et al. Detection of Brucella canis-induced reproductive diseases in a kennel. J Vet Diagn Invest. 2011;23(1):143–147. | ||
Pappas G, Akritidis N, Bosilkovski M, Tsianos E. Brucellosis. N Engl J Med. 2005;352(22):2325–2336. | ||
Foster JT, Beckstrom-Sternberg SM, Pearson T, et al. Whole-genome-based phylogeny and divergence of the genus Brucella. J Bacteriol. 2009;191:2864–2870. | ||
Kiran JA, Chakravarthi VP, Kumar YN, Rekha SS, Kruti SS, Bhaskar M. Comparison and correlation of Simple Sequence Repeats distribution in genomes of Brucella species. Bioinformation. 2011;6(5):179–182. | ||
Ratushna VG, Weller JW, Gibas CJ. Secondary structure in the target as a confounding factor in synthetic oligomer microarray design. BMC Genomics. 2005;6:31. | ||
Aujoulat F, Romano-Bertrand S, Masnou A, Marchandin H, Jumas-Bilak E. Niches population structure and genome reduction in Ochrobactrum intermedium: clues to technology-driven emergence of pathogens. PLoS One. 2014;9(1):e83376. | ||
Franco MP, Mulder M, Gilman RH, Smits HL. Human brucellosis. Lancet Infect Dis. 2007;7:775–786. | ||
Fernandez-Prada CM, Zelazowska EB, Nikolich M, et al. Interactions between Brucella melitensis and human phagocytes: bacterial surface O-polysaccharide inhibits phagocytosis, bacterial killing, and subsequent host cell apoptosis. Infect Immun. 2003;71:2110–2119. | ||
Billard E, Cazevieille C, Dornand J, Gross A. High susceptibility of human dendritic cells to invasion by the intracellular pathogens Brucella suis, B. abortus, and B. melitensis. Infect Immun. 2005;73(12):8418–8424. | ||
Paulsen IT, Seshadri R, Nelson KE, et al. The Brucella suis genome reveals fundamental similarities between animal and plant pathogens and symbionts. Proc Natl Acad Sci. 2002;99:13148–13153. | ||
Aloy P, Querol E, Aviles FX, Sternberg MJ. Automated structure-based prediction of functional sites in proteins: applications to assessing the validity of inheriting protein function from homology in genome annotation and to protein docking. J Mol Biol. 2001;311:395–408. | ||
Su F, Ou HY, Tao F, Tang H, Xu P. PSP: rapid identification of orthologous coding genes under positive selection across multiple closely related prokaryotic genomes. BMC Genomics. 2013;14:924. | ||
Luo H, Lin Y, Gao F, Zhang CT, Zhang R. DEG 10, an update of the Database of Essential Genes that includes both protein-coding genes and noncoding genomic elements. Nucleic Acids Res. 2014;42:D574–D580. | ||
Umamaheswari A, Pradhan D, Priyadarshini IV, Subramanyam G. In silico putative drug targets in Leptospira interrogans and homology modeling of UDP-N-acetylglucosamine 1-carboxyvinyltransferase MurA. Gen Med. 2008;2:295. | ||
Huang Y, Niu B, Gao Y, Fu L, Li W. CD-HIT suite: a web server for clustering and comparing biological sequences. Bioinformatics. 2010;26:680–682. | ||
Kanehisa M, Goto S. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 2000;28:27–30. | ||
Kanehisa M, Goto S, Sato Y, Kawashima M, Furumichi M, Tanabe M. Data, information, knowledge and principle: back to metabolism in KEGG. Nucleic Acids Res. 2014;42:D199–D205. | ||
Lu Z, Szafron D, Greiner R, et al. Predicting subcellular localization of proteins using machine-learned classifiers. Bioinformatics. 2004;20:547–556. | ||
Garg A, Gupta D. VirulentPred: a SVM based prediction method for virulent proteins in bacterial pathogens. BMC Bioinformatics. 2008;9:62. | ||
Lodish H, Berk A, Zipursky SL, Matsudaira P, Baltimore D, Darnell J. Hierarchical structure of proteins. In: Molecular Cell Biology. 4th ed. New York: W. H. Freeman; 2000. | ||
Gill SC, von Hippel PH. Calculation of protein extinction coefficients from amino acid sequence data. Anal Biochem. 1989;182:319–326. | ||
Guruprasad K, Reddy BV, Pandit MW. Correlation between stability of a protein and its dipeptide composition: a novel approach for predicting in vivo stability of a protein from its primary sequence. Protein Eng. 1990;4:155–161. | ||
Ikai A. Thermostability and aliphatic index of globular proteins. J Biochem. 1980;88:1895–1898. | ||
Kyte J, Doolittle RF. A simple method for displaying the hydropathic character of a protein. J Mol Biol. 1982;157:105–132. | ||
Wilkins MR, Gasteiger E, Bairoch A, et al. Protein identification and analysis tools in the ExPASy server. Methods Mol Biol. 1999;112:531–552. | ||
Krogh A, Larsson B, von Heijne G, Sonnhammer EL. Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J Mol Biol. 2001;305(3):567–580. | ||
Petersen TN, Brunak S, von Heijne G, Nielsen H. SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat Methods. 2011;8(10):785–786. | ||
Geourjon C, Deléage G. SOPMA: significant improvement in protein secondary structure prediction by consensus prediction from multiple alignments. Comp Applications Biosci. 1995;11(6):681–684. | ||
Butt AM, Batool M, Tong Y. Homology modeling, comparative genomics and functional annotation of Mycoplasma genitalium hypothetical protein MG_237. Bioinformation. 2011;7(6):299–303. | ||
Sehar U, Mehmood MA, Nawaz S, Gill SS, Saqib A. Three dimensional (3D) structure prediction and substrate-protein interaction study of the chitin binding protein CBP24 from B. thuringiensis. Bioinformation. 2013;9:725–729. | ||
Sali A, Blundell TL. Comparative protein modelling by satisfaction of spatial restraints. J Mol Biol. 1993;234:779–815. | ||
Binkowski TA, Naghibzadeh S, Liang J. CASTp: Computed Atlas of Surface Topography of proteins. Nucleic Acids Res. 2003;31:3352–3355. | ||
Wang Y, Suzek T, Zhang J, et al. PubChem BioAssay: 2014 update. Nucleic Acids Res. 2014;42:D1075–D1082. | ||
Trott O, Olson, AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem. 2010;31(2):455–461. | ||
Bhaskar A, Chawla M, Mehta M, et al. Reengineering redox sensitive GFP to measure mycothiol redox potential of Mycobacterium tuberculosis during infection. PLoS Pathog. 2014;10:e1003902. | ||
Shanmugham B, Pan A. Identification and characterization of potential therapeutic candidates in emerging human pathogen Mycobacterium abscessus: a novel hierarchical in silico approach. PLoS One. 2013;8:e59126. | ||
Jenner DC, Dassa E, Whatmore AM, Atkins HS. ATP-binding cassette systems of Brucella. Comp Funct Genomics. 2009;354649. | ||
Ratushna VG, Sturgill DM, Ramamoorthy S, Reichow SA, He Y. Molecular targets for rapid identification of Brucella spp. BMC Microbiol. 2006;6:13. | ||
He Y. Analyses of Brucella pathogenesis, host immunity, and vaccine targets using systems biology and bioinformatics. Front Cell Infect Microbiol. 2012;2:2. | ||
Macklaim JM, Gloor GB, Anukam KC, Cribby S, Reid G. At the crossroads of vaginal health and disease, the genome sequence of Lactobacillus iners AB-1. Proc Natl Acad Sci U S A. 2011;108:4688–4695. | ||
Eschenbrenner M, Wagner MA, Horn TA, Kraycer JA, Mujer CV. Comparative proteome analysis of Brucella melitensis vaccine strain Rev 1 and a virulent strain, 16M. J Bacteriol. 2002;184:4962–4970. | ||
Bhattacharyya A, Stilwagen S, Reznik G, et al. Draft sequencing and comparative genomics of Xylella fastidiosa strains reveal novel biological insights. Genome Res. 2002;12:1556–1563. | ||
Henderson IR, Navarro-Garcia F, Desvaux M, Fernandez RC, Ala Aldeen D. Type V protein secretion pathway: the autotransporter story. Microbiol Mol Biol Rev. 2004;68:692–744. | ||
Fronzes R, Christie PJ, Waksman G. The structural biology of type IV secretion systems. Nat Rev Microbiol. 2009;7:703–714. | ||
Terwagne M, Mirabella A, Lemaire J, Deschamps C, De Bolle X, Letesson JJ. Quorum sensing and self-quorum quenching in the intracellular pathogen Brucella melitensis. PLoS One. 2013;8:e82514. | ||
Beeckman DS, Vanrompay DC. Bacterial secretion systems with an emphasis on the chlamydial type III secretion system. Curr Issues Mol Biol. 2010;12:17–41. | ||
Gonzalez D, Collier J. DNA methylation by CcrM activates the transcription of two genes required for the division of Caulobacter crescentus. Mol Microbiol. 2013;88:203–218. | ||
Arellano-Reynoso B, Lapaque N, Salcedo S, et al. Cyclic beta-1,2-glucan is a Brucella virulence factor required for intracellular survival. Nat Immunol. 2005;6:618–625. | ||
Guay DR. Drug forecast: the peptide deformylase inhibitors as antibacterial agents. Ther Clin Risk Manag. 2007;3:513–525. | ||
Atack JM, Srikhanta YN, Djoko KY, et al. Characterization of an ntrX mutant of Neisseria gonorrhoeae reveals a response regulator that controls expression of respiratory enzymes in oxidase-positive proteobacteria. J Bacteriol. 2013;195:2632–2641. | ||
Koper TE, El-Sheikh AF, Norton JM, Klotz MG. Urease-encoding genes in ammonia-oxidizing bacteria. Appl Environ Microbiol. 2004;70:2342–2348. | ||
Goodacre NF, Gerloff DL, Uetz P. Protein domains of unknown function are essential in bacteria. MBio. 2001;5:e 00744–13. | ||
Hanks SK, Quinn AM, Hunter T. The protein kinase family: conserved features and deduced phylogeny of the catalytic domains. Science. 1988;241(4861):42–52. | ||
Paradis-Bleau C, Kritikos G, Orlova K, Typas A, Bernhardt TG. A genome-wide screen for bacterial envelope biogenesis mutants identifies a novel factor involved in cell wall precursor metabolism. PLoS Genet. 2014;10:e1004056. | ||
Han GW, Bakolitsa C, Miller MD, et al. Structures of the first representatives of Pfam family PF06938 (DUF1285) reveal a new fold with repeated structural motifs and possible involvement in signal transduction. Acta Crystallogr Sect F Struct Biol Cryst Commun. 2010;66:1218–1225. | ||
Frishman D, Argos P. Knowledge-based protein secondary structure assignment. Proteins. 1995;23:566–579. | ||
Oprea TI. Property distribution of drug-related chemical databases. J Comput Aided Mol Des. 2000;14:251–264. | ||
Butkiewicz M, Lowe EW Jr, Mueller R, et al. Benchmarking ligand-based virtual High-Throughput Screening with the PubChem database. Molecules. 2013;18(1):735–756. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.