Back to Journals » Pharmacogenomics and Personalized Medicine » Volume 13
Comparison of Patient Susceptibility Genes Across Breast Cancer: Implications for Prognosis and Therapeutic Outcomes
Authors Peleg Hasson S ![]() , Menes T, Sonnenblick A
, Menes T, Sonnenblick A ![]()
Received 7 May 2020
Accepted for publication 20 July 2020
Published 27 July 2020 Volume 2020:13 Pages 227—238
DOI https://doi.org/10.2147/PGPM.S233485
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Martin H Bluth
Shira Peleg Hasson,1 Tehillah Menes,2 Amir Sonnenblick1
1Oncology Department, Tel Aviv Sourasky Medical Center and Sackler Faculty of Medicine, Tel Aviv University, Tel Aviv, Israel; 2Department of Surgery, Tel Aviv-Sourasky Medical Center and Sackler Faculty of Medicine, Tel Aviv University, Tel Aviv, Israel
Correspondence: Amir Sonnenblick Email [email protected]
Abstract: Hereditary breast cancer syndromes affect a small (10– 15% of cases) but significant group of patients. BRCA1 and BRCA2 are the most familiar and well-studied genes associated with inherited breast cancer. However, mutations in the high-penetrance genes, TP53, PTEN, CDH1, MSH1, MLH1, MSH6, PMS2, PALB2, and STK11, and in the moderate-penetrance genes, CHEK2, ATM, and BRIP1, also correlate with high lifetime risks of breast cancer and other malignancies as well. Advances in breast cancer genetics have led to an improved perception of diagnosis and screening strategies. The specific considerations and challenges involved in treating this unique population have become a fertile ground for research. Indeed, these genes and downstream molecular pathways have now become potential therapeutic targets in breast cancer patients, including those with BRCA1 or BRCA2 mutations. This review describes the variety of hereditary breast cancer genes, from their molecular origins to the prognosis and multidisciplinary clinical decision-making processes. Key publications and other reported recent clinical trials and guidelines are provided.
Keywords: breast cancer, BRCA, TP53, PTEN, CDH1, Lynch syndrome
Introduction
Breast cancer is the major cause of cancer death among women worldwide.1 Most breast cancer cases are sporadic rather than inherited. Approximately 10–15% of breast cancer cases are associated with hereditary syndromes, and the majority of them will carry a deleterious mutation in BRCA1/22,3 (Figure 1). Patients with pathogenic mutations other than BRCA can be detected by commercial multigene panel testing. These genes may be related to highly penetrant syndromes, such as Cowden (PTEN) and Li-Fraumeni (p53). Mutation carriers in one of these genes have a lifetime breast cancer risk above 50%.4–6 The relatively low cost of gene sequencing has allowed widespread use of panel testing that examines a variety of cancer-causing genes. When analyzing test results of more than 60,000 patients with breast cancer, and after excluding BRCA-positive patients and those with syndromic genes (PTEN, TP53), more than 6% of them were found to have a pathogenic mutation in other genes, including CHEK2, PALB2 and ATM. High or moderately increased risks were associated with pathogenic variants in PALB2 (odd ratio [OR], 7.46), RAD51D (OR, 3.07), ATM (OR, 2.78), BARD1 (OR, 2.16), and CHEK2 (OR, 1.48).7 The increased use of panel testing is directly associated with escalation in the diagnosis of patients with mutations in moderate- and low-penetrance genes. While extensive data are available for BRCA1/2 mutation carriers, information on less common genes and their management is limited.
 |
Figure 1 Breast cancer patients with genetic mutations. Approximately 10–15% of breast cancer cases are associated with hereditary syndromes, and the majority of them will carry a deleterious mutation in BRCA1 and BRCA2. Other rare highly penetrant syndromes are Cowden (PTEN) and Li-Fraumeni (p53). After excluding BRCA-positive and syndromic genes, there are pathogenic mutation in other genes. |
In this review, we aim to describe clinical implications, management and prognosis of patients with known germline gene mutations, including those in moderate-penetrance genes.
High-Penetrance Genes in Breast Cancer
BRCA
BRCA1/2 pathogenic mutations are associated with elevated risk for breast, ovarian8 and peritoneal cancer in women, with breast cancer and prostate cancer in men, and, to a lesser degree, with pancreatic cancer. BRCA1/2 mutations exhibit an autosomal dominant pattern of transmission, and they are rare in the general population (1 to 500), but they account for more than 5% of all breast cancer cases.9 The frequency of BRCA1/2 approximates 1 in 40 in the Ashkenazi Jewish population.10 Penetrance is variable, even within families with the same variant.11,12 In general, the cumulative breast cancer risk up to age 80 years is above 50% for BRCA1/2 carriers.3,13,14 Moreover, they also bear a significant increase in the risk of contralateral breast cancers,3,15 which is greater with earlier age at diagnosis, and BRCA1 (rather than BRCA2) pathogenic mutation.16 A protective effect was reported in women undergoing bilateral salpingo-oophorectomy,3 however, a more recent publication suggested that this association is a result of bias and does not actually exist.17 BRCA mutation carriers commonly develop breast cancer at a young age18 and bilateral disease is more frequent than in non-carriers.19 BRCA1 breast cancer is often “triple negative” (TNBC).20 The prognosis of BRCA-associated breast cancers, however, is relatively similar to that of sporadic breast cancers,21 with inconsistent data from meta-analyses showing worse overall survival (OS) on the one hand22,23 and some suggesting better OS among triple negative breast cancer patients on the other hand.24 In addition, BRCA1/2 mutation carriers have an estimated 8–62% lifetime risk for ovarian cancer.8
Therapeutic Outcomes
Surgery
A bilateral mastectomy reduces this risk of developing breast cancer in healthy carriers, but an overall survival effect in comparison to surveillance has not been clearly demonstrated.25 A recent report suggested a survival benefit in BRCA1 carriers.26 Contralateral prophylactic mastectomy is a risk-reducing procedure performed in patients diagnosed with a unilateral breast cancer. While there is no clear survival benefit for most BRCA1/2 breast cancer patients,25,27-29 some evidence suggests a diseases-free survival (DFS) and OS benefit in younger patients with early-stage disease.30,31 A meta-analysis including 4 studies (N=2555) reported an association between contralateral prophylactic mastectomy and reduced mortality,30 however those studies bear some bias since healthier women are selected for risk-reducing surgery. Interestingly, the rates of performing a contralateral prophylactic mastectomy have increased over the past few years.31,32 The NCCN guidelines panel recommends considering risk-reducing mastectomy on a case-by-case basis. When counseling patients on contralateral risk-reducing mastectomy, one should consider the various risks relevant to the specific patient, such as the prognosis of the present breast cancer and the likelihood of developing a contralateral breast cancer, as well as the risks associated with the surgery itself. A risk-reducing salpingo-oophorectomy (BSO) is recommended after completion of childbearing.33
Radiation
The risk of recurrent cancer in ipsilateral breast is roughly 15% in 10 years in BRCA1/2 mutation carriers who had undergone breast lumpectomy. This risk is relatively lower than that of the contralateral breast34 as a result of irradiation of the affected breast.34,35 Therefore, ipsilateral whole breast radiation is strongly recommended to all BRCA1/2 mutation carriers undergoing breast-conserving surgery. This reduction in risk also raised the hypothesis that radiation to the contralateral breast may have a risk-reducing role in BRCA1/2 carriers wishing to refrain from undergoing a prophylactic mastectomy. A non-randomized Phase II trial that evaluated prophylactic breast irradiation for the contralateral breast in BRCA mutation carriers showed reduction in the incidence of breast cancer (P= 0.011).36
Treatment in the Neoadjuvant Setting
The contribution of platinum-based therapy to pathologic complete (pCR) response was explored in women with BRCA1/2 mutations and TNBC. All patients with TNBC were more likely to achieve a pCR with the addition of carboplatin to neoadjuvant chemotherapy. However, patients without a germline BRCA mutation exhibited a much larger increase in pCR rate than BRCA-mutated patients.37,38 For example, in the BrighTNess trial, the addition of carboplatin increased the pCR rate among BRCA-wild type patients from 29% to 59% compared to an increase from 41% to 50% in BRCA carriers.39 Moreover, in the outcomes analysis from the GeparSixto trial, the absolute improvement in DFS with the addition of carboplatin was greater among BRCA-wildtype patients compared to BRCA-mutated patients.40 A systematic review of TNBC patients undergoing neoadjuvant treatment reported that platinum is associated with significantly increased pCR in BRCA1/2 wild type patients, but not n BRCA1/2-mutation carriers.41 This phenomenon may be attributed to the higher sensitivity of tumors with BRCA1/2 mutation to other alkylating chemotherapies, such as cyclophosphamide. Therefore, decisions regarding the addition of platinum agents in the neo-adjuvant setting should be individualized regardless of BRCA status and with consideration of clinical response to chemotherapy and initial stage.
In cells with a BRCA1/2 mutation, inhibition of poly (ADP-ribose) polymerase (PARP) causes irreversible DNA damage.42 In the neoadjuvant setting, the use of single-agent oral talazoparib for 6 months resulted in 53% of the participants achieving pCR.43 This strategy may be more widely adopted in the future, pending the results of larger trials. Since there are different resistance mechanisms to PARP inhibitors and chemotherapy (including platinum),44 future strategies may incorporate sequential use of PARP inhibitors in the neoadjuvant setting, with the addition of chemotherapy to patients not achieving pCR.
Treatment in Metastatic Disease
The use of the PARP inhibitor Olaparib in BRCA1/2-metastatic patients with triple-negative breast cancer was assessed in the OlympiAD trial. Patients randomly assigned to Olaparib experienced an improved PFS compared to those assigned to chemotherapy.45 A recent update of that trial suggested an OS benefit for olaparib in patients who had not received chemotherapy for metastatic disease, with longer OS compared with chemotherapy-treated patients.46 Similarly, talazoparib improved PFS compared to single-agent chemotherapy in the TNBC subgroup of the EMBRACA trial.47 Interestingly, the EMBRACA trial showed improvement in PFS in HR positive patients as well. In the BROCADE trial, patients with metastatic HER2-negative and a germline BRCA mutation demonstrated improved PFS with the addition of the PARP inhibitor Veliparib to chemotherapy (comprised of carboplatin/paclitaxel).48 The TNT randomized trial compared docetaxel with carboplatin as a first-line treatment in women with metastatic TNBC. The overall response rates were similar, but carboplatin resulted in a higher response rate (68% versus 33%) and improved PFS (6.8 versus 4.4 months) among BRCA1/2-mutation carriers.49
Overall, these studies clearly demonstrate the role of PARP inhibitors and platinum agents in the treatment of metastatic BRCA1/2 carriers.
TP53 – Li Fraumeni Syndrome
Li Fraumeni syndrome (LFS) is a rare, highly penetrant, autosomal dominant syndrome associated with a germline mutation in the tumor protein p53 gene.50 It constitutes approximately 1% of all hereditary breast cancer cases.51 p53 mutation carriers are at increased risk of developing various cancers, including soft tissue sarcomas, premenopausal breast cancers, tumors of the brain, lung, skin, pancreas, adrenal cortex, and leukemia in childhood or early adulthood,52 with a lifetime cancer incidence of nearly 100%.53 The lifetime risk of breast cancer in female mutation carriers approaches 50% by age 60 years. The mean age at onset is approximately 35 years, and a first diagnosis of breast cancer is rare after 50.54
There are no data on the benefit of risk-reduction mastectomies, although they seem like a reasonable option when extrapolating data from BRCA1/2 carriers. The NCCN guidelines panel recommends a case-by-case discussion that considers family history, life expectancy and reconstructive options.
Therapeutic Outcomes
Radiation
Carriers are at increased risk of developing secondary malignancies in radiation fields.55,56 Women with LFS who develop breast cancer are generally recommended to undergo mastectomy rather than lumpectomy and radiation, given the risks of radiation-induced malignancies in this syndrome.
Systemic Treatment
HER2 is positive in 64% to 83% of breast cancers among TP53 carriers.57 Patients with TP53 mutations have limited response to chemotherapy in both the neoadjuvant and adjuvant settings.58,59 Clinical decisions regarding chemotherapy in this patient population should be made on a case-by-case basis while considering the limited response and the secondary malignancy risk.
PTEN (Cowden Syndrome)
The germline PTEN mutations detected in Cowden syndrome clinically present as multiple hamartomas and contain an increased risk for breast, thyroid, endometrium, kidney and colorectal malignancies.60 The PTEN gene is a negative regulator of the phosphoinositide-3-kinase (PI3K), and a mammalian target of rapamycin (mTOR) signaling pathways, which are known to be involved in cell proliferation, cell cycle progression, and apoptosis.61
A decrease in PTEN expression may be associated with poor outcomes in BC.62,63 There are no data on the benefit of risk-reducing mastectomies although they seem to be a reasonable option when extrapolating data from BRCA1/2 carriers. The NCCN guidelines panel recommends a case-by-case discussion that considers family history, life expectancy and reconstructive options.
Therapeutic Outcomes
Systemic Therapy
Trials that evaluated the addition of Everolimus to chemotherapy and trastuzumab reported a potential benefit in women with PTEN mutations.64 Studies that evaluated the addition of AKT inhibitor treatment to chemotherapy in TNBC reported an association between PTEN alterations and response to AKT inhibitor treatment, both in the early and advanced settings.65,66 It was recently suggested that the PTEN gene has a role in maintaining genomic stability67,68 and PARP inhibitor sensitization.69–71 A phase II trial that evaluated Talazoparib (PARPi) for BRCA1/2 wild type HER2- breast cancer patients with PTEN gene mutations exhibited response or stability.72
CDH1 (Hereditary Diffuse Gastric Cancer Syndrome)
Hereditary diffuse gastric cancer syndrome is associated with germline pathogenic variants in the cadherin 1 gene (CDH1).73,74 The lifetime cumulative risk for advanced diffuse-type gastric cancer is 70% for males and 56% for females by the age of 80 years.75 Germline CDH1 mutations are associated with the development of lobular breast cancer in women, with a cumulative lifetime risk estimated to be as high as 50% to 60%.73,76,77 CDH1 somatic mutations do not impact prognosis of lobular breast cancer patients, however, the presence of CDH1 plus ERBB2 mutations leads to worse prognosis.78
Therapeutic Outcomes
Systemic Therapy
Invasive lobular carcinoma (ILC) is relatively resistant to chemotherapy compared to invasive ductal carcinoma. The cause might be the luminal A phenotype. Indeed, adjuvant endocrine therapy is preferred for this patient population.79 Since the PI3K pathway is activated upon E-cadherin loss, lobular cancer cells were hypothesized to be sensitive to AKT inhibitors.80 Therefore, it is reasonable to consider the use of AKT inhibitors in the metastatic setting. More clinical trials are warranted, perhaps with combinations of effective hormonal treatments.
PALB2
Partner and localizer of BRCA2 (PALB2) is a breast cancer susceptibility gene that encodes the BRCA2-interacting protein.81,83 PALB2 bi-allelic mutations cause Fanconi anemia and predispose to pediatric malignancies, including medulloblastoma, Wilm’s tumor and acute myeloid leukemia.82 Mono-allelic mutations of PALB2 cause familial breast and pancreatic cancer,83,84 prostate cancer in men, and ovarian cancer in women.85 Mutation carriers have a cumulative risk of breast cancer ranging between 33% to 58% by age 70 years.86,88 PALB2 is considered to be a moderate- to high-risk gene associated with hereditary breast cancer.85–87 Breast cancer risk associated with a PALB2 pathogenic variant appears to be influenced by a family history of breast cancer and other environmental factors. Women with no family history of breast cancer have a cumulative risk of 33%, compared to 58% in women with two or more family members with breast cancer. Breast cancers in patients with PALB2 mutations have phenotypic characteristics relatively similar to BRCA1/2 mutant tumors: 50% are grade III, 40% are triple-negative phenotype, 58% are estrogen receptor-negative and 93% are HER2-negative.88
Systemic Therapy
Increased sensitivity to PARP inhibitors in PALB2-deficient cells demonstrates the synthetic lethal interaction between PALB2 loss and DNA-damaging agents.89 Furthermore, one preclinical study demonstrated response in PLAB2-mutated cells to other DNA-damaging agents, such as platinum.90 These preliminary findings warrant validation in clinical studies.
STK11 (LKB1, Peutz-Jeghers Syndrome)
Peutz-Jeghers syndrome (PJS) is a rare disorder associated with pathogenic variants in the serine/threonine kinase 11 gene (STK11, also called LKB1).91 Mucocutaneous pigmented lesions occur in about 95% of affected patients. Additionally, hamartomatous polyps in the gastrointestinal tract are hallmark features.92 This syndrome is associated with an elevated risk for breast cancer.92 The cumulative risk of breast cancer is approximately 55%, and the diagnosis tends to occur at a younger age (mean 37 years).5
MSH1, MLH1, MSH6, PMS2, and EPCAM (Lynch Syndrome)
Lynch syndrome, also known as hereditary nonpolyposis colon cancer (HNPCC), is associated with pathogenic variants in mismatch repair genes. The primary cancers associated with Lynch syndrome involve the colon, endometrium, ovaries and stomach.93–96 No statistical increase in breast cancer risk was found in 13 out of 21 studies, an increased risk was reported in 8 and 1 prospective study identified a fourfold increased risk for breast cancer.96–98 Subsequent studies have found that the breast cancer risk may vary based on genotype. For example, in a study on MLH1 and MSH2 families in the United Kingdom, the cumulative breast cancer risk until age 70 years in MLH1 carriers was 18.6%, whereas the comparable risk in MSH2 carriers was 11.2%.99 In another study on pathogenic variants among women with Lynch syndrome, breast cancer risks were elevated in MSH6 and PMS2 carriers but not in MLH1 or MSH2 carriers.100,102
Systemic Therapy
Mismatch repair errors characteristic of the MSI phenotype are rare in breast cancer,101 being found in fewer than 2% of cases.102 High numbers of MSI tumors have been recently found to be susceptible to immunotherapy,103 leading to the use of pembrolizumab (an anti-PD-1 agent) for high MSI metastatic solid tumors.104
Moderate Penetrance Genes for Breast Cancer
CHEK2
The checkpoint kinase 2 (CHEK2) gene is associated with a DNA damage repair response.105 Several CHEK2 variants have been identified, including single polymorphism (1100delC) which has emerged as being associated with low-to-moderate penetrance breast cancer sensitivity.106 The 1100delC variant is also linked to increased risk for colorectal cancer, especially with a family history of colon cancer.107 Other common malignancies associated with CHEK2 include male breast cancer,108 stomach, prostate, kidney and thyroid cancer, and also sarcoma.109,110 The 1100delC variant is associated with a two- to threefold increased risk of breast cancer, predominantly among caucasian women of northern or eastern european descent.46,111-115 The cumulative risk of breast cancer to age 80 years in women with this variant is about 32%, whereas the cumulative risk to age 49 years is about 6%.85
Therapeutic Outcomes
Systemic Treatment
Mutations in the CHEK2 gene have been associated with a lack of benefit from anthracycline in breast cancer.116,117 The H371Y variant was associated with better response.118 In clinical studies of women with the CHEK2 1100delC mutation, no differences in response to chemotherapy were observed when compared to non-carriers.118,119
Radiation
Patients with a CHEK2 mutation are not characterized by any distinct radiosensitivity.120 Therefore, planning modifying radiotherapy for women with breast cancer who have this mutation is unwarranted.121
ATM
The ataxia-telangiectasia mutated (ATM) gene encodes a protein kinase involved in DNA repair. Heterozygotic carriers are at a twofold increased risk of developing breast cancer than non-carriers, with a cumulative lifetime breast cancer risk of about 30%.85,122-124 ATM pathogenic variants have also been associated with increased risks for pancreatic and ovarian cancer.93,125
Rare pathogenic variants in the ATM gene may be associated with a substantially higher risk of breast cancer.123,126 The risk of second primary breast cancer is not clear.127
Therapeutic Outcomes
Radiation
Radiation toxicity in ATM mutation carriers is a subject of controversy. Upon activation by ionizing radiation, ATM phosphorylates the proteins that control the pathways involved in DNA repair, including BRCA.128 Studies have suggested that radiation exposure may cause contralateral breast cancer in women who carry deleterious ATM missense variants.129 However, the rarity of these variants implies that ATM mutations could explain only a small fraction of second primary breast cancers.129 Contrarily, no evidence of breast radiation toxicity in ATM mutation carriers has been reported, suggesting that breast-conserving therapy can be safely considered in this patient population.130 Moreover, a meta-analysis including 5 studies showed that radiation therapy is safe in ATM mutation carriers diagnosed with cancer.131 Currently, NCCN and ESMO guidelines do not recommend against radiation in this patient population.132
Systemic Treatment
ATM mutations may potentially have increased sensitivity for platinum chemotherapy. However, in vitro killing of tumor cells by platinum drugs has not been demonstrated.133 Checkpoint kinase 1 (Chk1) is downstream of ATM in the DNA damage–induced cell-cycle arrest. Therefore, inhibitors of Chk1 may act as chemosensitizers in ATM-mutated cancers.134
BRIP1
BRIP1 (BRCA1 interacting protein C-terminal helicase 1) is a DNA repair gene that interacts with BRCA1.137 BRIP1 is a protein that supports BRCA1 to repair damaged DNA. BRIP1 inactivating mutations are hypothesized to be associated with a marginal increased risk of breast cancer, and have more frequently been linked with a moderately increased risk of ovarian cancer.136–139 It has been postulated that BRIP1 is a potential target for PARP inhibitors and platinum agents.135
Conclusion
In this review, we summarized common breast cancer hereditary syndromes, including BRCA1/2, as well as the less common culpable genes, TP53, PTEN, CDH1, MSH1, MLH1, MSH6, PMS2, PALB2, STK11, CHEK2, ATM, and BRIP1, that are frequently diagnosed in accessible multigene panels. We presented an overview of gene carrier prognosis, and indicated our recommendations for clinical decision-making with regard to surgery, radiation and systemic treatment (Table 1 and Figure 2). In this unique population of breast cancer patients, clinicians must strongly consider the patient’s family history, life expectancy and any risk for other secondary malignancies. Participation in clinical trials should be encouraged. Moreover, patients are strongly advised to attend multidisciplinary clinics for surveillance, risk reduction and primary prevention.
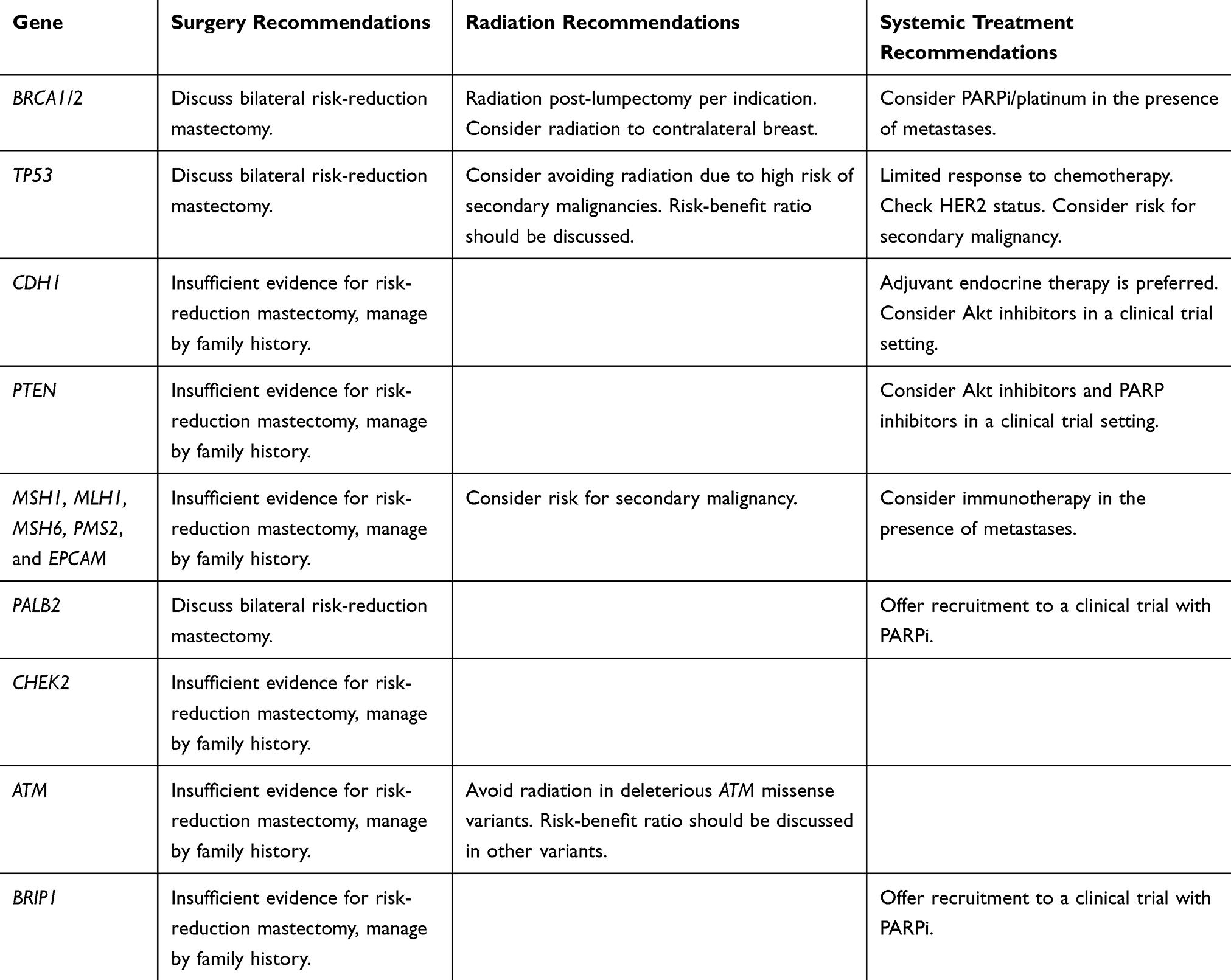 |
Table 1 Common Genes and Clinical Recommendations |
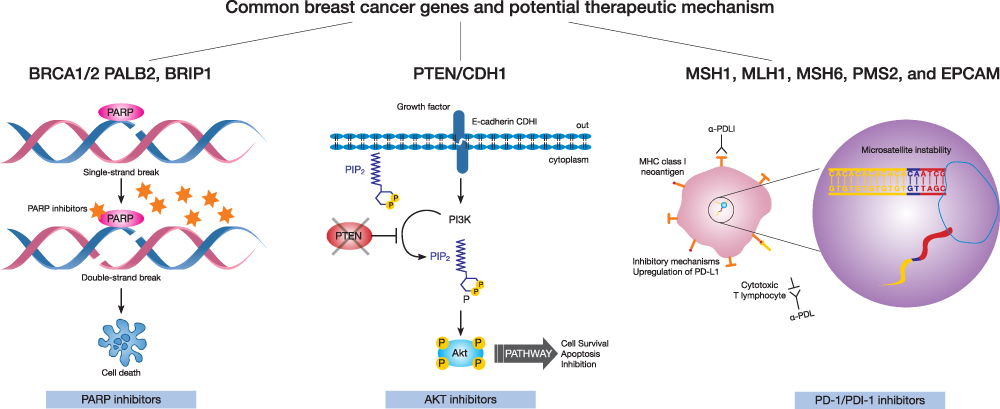 |
Figure 2 Breast cancer susceptibility genes and potential therapeutic mechanisms. In Patients with BRCA1/2, PALB2 and BRIP1 mutations, PARPi and platinum should be considered. In patients with PTEN and CDH1 mutations AKT inhibitors in a clinical trial setting may be reasonable. In patients with Lynch syndrome immunotherapy has an emerging role. |
Acknowledgments
We would like to thank Esther Eshkol for English editing.
Disclosure
Dr Amir Sonnenblick reports personal fees from Eli lilly, Pfizer, and Roche; grants from Novartis, outside the submitted work. The authors report no other conflict of interest in this work.
References
1. Ferlay J, Colombet M, Soerjomataram I, et al. Estimating the global cancer incidence and mortality in 2018: GLOBOCAN sources and methods. Int J Cancer. 2019;144(8):1941–1953. doi:10.1002/ijc.31937
2. Buys SS, Sandbach JF, Gammon A, et al. A study of over 35,000 women with breast cancer tested with a 25-gene panel of hereditary cancer genes. Cancer. 2017;123(10):1721–1730. doi:10.1002/cncr.30498
3. Kuchenbaecker KB, Hopper JL, Barnes DR, et al. Risks of breast, ovarian, and contralateral breast cancer for BRCA1 and BRCA2 mutation carriers. JAMA. 2017;317(23):2402–2416. doi:10.1001/jama.2017.7112
4. Hisada M, Garber JE, Li FP, Fung CY, Fraumeni JF. Multiple primary cancers in families with Li-Fraumeni syndrome. J Natl Cancer Inst. 1998;90(8):606–611. doi:10.1093/jnci/90.8.606
5. Giardiello FM, Brensinger JD, Tersmette AC, et al. Very high risk of cancer in familial Peutz-Jeghers syndrome. Gastroenterology. 2000;119(6):1447–1453. doi:10.1053/gast.2000.20228
6. Tan M-H, Mester JL, Ngeow J, Rybicki LA, Orloff MS, Eng C. Lifetime cancer risks in individuals with germline PTEN mutations. Clin Cancer Res. 2012;18(2):400–407. doi:10.1158/1078-0432.CCR-11-2283
7. Couch FJ, Shimelis H, Hu C, et al. Associations between cancer predisposition testing panel genes and breast cancer. JAMA Oncol. 2017;3(9):1190–1196. doi:10.1001/jamaoncol.2017.0424
8. Finch A, Beiner M, Lubinski J, et al. Salpingo-oophorectomy and the risk of ovarian, fallopian tube, and peritoneal cancers in women with a BRCA1 or BRCA2 mutation. JAMA. 2006;296(2):185–192. doi:10.1001/jama.296.2.185
9. Anglian Breast Cancer Study Group. Prevalence and penetrance of BRCA1 and BRCA2 mutations in a population-based series of breast cancer cases. Br J Cancer. 2000;83(10):1301–1308. doi:10.1054/bjoc.2000.1407
10. Metcalfe KA, Poll A, Royer R, et al. Screening for founder mutations in BRCA1 and BRCA2 in unselected Jewish women. J Clin Oncol. 2010;28(3):387–391. doi:10.1200/JCO.2009.25.0712
11. Abeliovich D, Kaduri L, Lerer I, et al. The founder mutations 185delAG and 5382insC in BRCA1 and 6174delT in BRCA2 appear in 60% of ovarian cancer and 30% of early-onset breast cancer patients among Ashkenazi women. Am J Hum Genet. 1997;60(3):505–514.
12. Nelson HD, Pappas M, Cantor A, Haney E, Holmes R. Risk assessment, genetic counseling, and genetic testing for BRCA-related cancer in women: updated evidence report and systematic review for the US preventive services task force. JAMA. 2019;322(7):666–685. doi:10.1001/jama.2019.8430
13. Antoniou A, Pharoah PDP, Narod S, et al. Average risks of breast and ovarian cancer associated with BRCA1 or BRCA2 mutations detected in case series unselected for family history: a combined analysis of 22 studies. Am J Hum Genet. 2003;72(5):1117–1130. doi:10.1086/375033
14. Chen S, Parmigiani G. Meta-analysis of BRCA1 and BRCA2 penetrance. J Clin Oncol. 2007;25(11):1329–1333. doi:10.1200/JCO.2006.09.1066
15. van den Broek AJ, Van ’T Veer LJ, Hooning MJ, et al. Impact of age at primary breast cancer on contralateral breast cancer risk in BRCA1/2 mutation carriers. J Clin Oncol. 2016;34(5):409–418. doi:10.1200/JCO.2015.62.3942
16. Metcalfe K, Gershman S, Lynch HT, et al. Predictors of contralateral breast cancer in BRCA1 and BRCA2 mutation carriers. Br J Cancer. 2011;104(9):1384–1392. doi:10.1038/bjc.2011.120
17. Heemskerk-Gerritsen BAM, Seynaeve C, van Asperen CJ, et al. Breast cancer risk after salpingo-oophorectomy in healthy BRCA1/2 mutation carriers: revisiting the evidence for risk reduction. J Natl Cancer Inst. 2015;107:5. doi:10.1093/jnci/djv033
18. Gonzalez-Angulo AM, Timms KM, Liu S, et al. Incidence and outcome of BRCA mutations in unselected patients with triple receptor-negative breast cancer. Clin Cancer Res. 2011;17(5):1082–1089. doi:10.1158/1078-0432.CCR-10-2560
19. King M-C, Marks JH, Mandell JB, New York Breast Cancer Study Group. Breast and ovarian cancer risks due to inherited mutations in BRCA1 and BRCA2. Science. 2003;302(5645):643–646. doi:10.1126/science.1088759
20. Atchley DP, Albarracin CT, Lopez A, et al. Clinical and pathologic characteristics of patients with BRCA-positive and BRCA-negative breast cancer. J Clin Oncol. 2008;26(26):4282–4288. doi:10.1200/JCO.2008.16.6231
21. Bordeleau L, Panchal S, Goodwin P. Prognosis of BRCA-associated breast cancer: a summary of evidence. Breast Cancer Res Treat. 2010;119(1):13–24. doi:10.1007/s10549-009-0566-z
22. Zhong Q, Peng H-L, Zhao X, Zhang L, Hwang W-T. Effects of BRCA1- and BRCA2-related mutations on ovarian and breast cancer survival: a meta-analysis. Clin Cancer Res. 2015;21(1):211–220. doi:10.1158/1078-0432.CCR-14-1816
23. van den Broek AJ, Schmidt MK, van ’T Veer LJ, Tollenaar RAEM, van Leeuwen FE. Worse breast cancer prognosis of BRCA1/BRCA2 mutation carriers: what’s the evidence? A systematic review with meta-analysis. PLoS One. 2015;10(3):e0120189. doi:10.1371/journal.pone.0120189
24. Baretta Z, Mocellin S, Goldin E, Olopade OI, Huo D. Effect of BRCA germline mutations on breast cancer prognosis: a systematic review and meta-analysis. Medicine. 2016;95(40):e4975. doi:10.1097/MD.0000000000004975
25. Lostumbo L, Carbine NE, Wallace J. Prophylactic mastectomy for the prevention of breast cancer. Cochrane Database Syst Rev. 2010;11:CD002748. doi:10.1002/14651858.CD002748.pub3
26. Heemskerk-Gerritsen BAM, Jager A, Koppert LB, et al. Survival after bilateral risk-reducing mastectomy in healthy BRCA1 and BRCA2 mutation carriers. Breast Cancer Res Treat. 2019;177(3):723–733. doi:10.1007/s10549-019-05345-2
27. Bedrosian I, Hu C-Y, Chang GJ. Population-based study of contralateral prophylactic mastectomy and survival outcomes of breast cancer patients. J Natl Cancer Inst. 2010;102(6):401–409. doi:10.1093/jnci/djq018
28. Metcalfe K, Gershman S, Ghadirian P, et al. Contralateral mastectomy and survival after breast cancer in carriers of BRCA1 and BRCA2 mutations: retrospective analysis. BMJ. 2014;348:g226. doi:10.1136/bmj.g226
29. Herrinton LJ, Barlow WE, Yu O, et al. Efficacy of prophylactic mastectomy in women with unilateral breast cancer: a cancer research network project. J Clin Oncol. 2005;23(19):4275–4286. doi:10.1200/JCO.2005.10.080
30. Li X, You R, Wang X, et al. Effectiveness of prophylactic surgeries in BRCA1 or BRCA2 mutation carriers: a meta-analysis and systematic review. Clin Cancer Res. 2016;22(15):3971–3981. doi:10.1158/1078-0432.CCR-15-1465
31. King TA, Sakr R, Patil S, et al. Clinical management factors contribute to the decision for contralateral prophylactic mastectomy. J Clin Oncol. 2011;29(16):2158–2164. doi:10.1200/JCO.2010.29.4041
32. Zeichner SB, Zeichner SB, Ruiz AL, Markward NJ, Rodriguez E. Improved long-term survival with contralateral prophylactic mastectomy among young women. Asian Pac J Cancer Prev. 2014;15(3):1155–1162. doi:10.7314/apjcp.2014.15.3.1155
33. Rebbeck TR, Kauff ND, Domchek SM. Meta-analysis of risk reduction estimates associated with risk-reducing salpingo-oophorectomy in BRCA1 or BRCA2 mutation carriers. J Natl Cancer Inst. 2009;101(2):80–87. doi:10.1093/jnci/djn442
34. Pierce LJ, Levin AM, Rebbeck TR, et al. Ten-year multi-institutional results of breast-conserving surgery and radiotherapy in BRCA1/2-associated stage I/II breast cancer. J Clin Oncol. 2006;24(16):2437–2443. doi:10.1200/JCO.2005.02.7888
35. Metcalfe K, Lynch HT, Ghadirian P, et al. Contralateral breast cancer in BRCA1 and BRCA2 mutation carriers. J Clin Oncol. 2004;22(12):2328–2335. doi:10.1200/JCO.2004.04.033
36. Evron E, Ben-David AM, Goldberg H, et al. Prophylactic irradiation to the contralateral breast for BRCA mutation carriers with early-stage breast cancer. Ann Oncol. 2019;30(3):412–417. doi:10.1093/annonc/mdy515
37. Sikov WM, Berry DA, Perou CM, et al. Impact of the addition of carboplatin and/or bevacizumab to neoadjuvant once-per-week paclitaxel followed by dose-dense doxorubicin and cyclophosphamide on pathologic complete response rates in stage II to III triple-negative breast cancer: CALGB 40603 (Alliance). J Clin Oncol. 2015;33(1):13–21. doi:10.1200/JCO.2014.57.0572
38. Hahnen E, Lederer B, Hauke J, et al. Germline mutation status, pathological complete response, and disease-free survival in triple-negative breast cancer: secondary analysis of the GeparSixto randomized clinical trial. JAMA Oncol. 2017;3(10):1378–1385. doi:10.1001/jamaoncol.2017.1007
39. Loibl S, O’Shaughnessy J, Untch M, et al. Addition of the PARP inhibitor veliparib plus carboplatin or carboplatin alone to standard neoadjuvant chemotherapy in triple-negative breast cancer (BrighTNess): a randomised, Phase 3 trial. Lancet Oncol. 2018;19(4):497–509. doi:10.1016/S1470-2045(18)30111-6
40. Loibl S, Weber KE, Timms KM, et al. Survival analysis of carboplatin added to an anthracycline/taxane-based neoadjuvant chemotherapy and HRD score as predictor of response-final results from GeparSixto. Ann Oncol. 2018;29(12):2341–2347. doi:10.1093/annonc/mdy460
41. Poggio F, Bruzzone M, Ceppi M, et al. Platinum-based neoadjuvant chemotherapy in triple-negative breast cancer: a systematic review and meta-analysis. Ann Oncol. 2018;29(7):1497–1508. doi:10.1093/annonc/mdy127
42. Lord CJ, Ashworth A. PARP inhibitors: synthetic lethality in the clinic. Science. 2017;355(6330):1152–1158. doi:10.1126/science.aam7344
43. Litton JK, Scoggins ME, Hess KR, et al. Neoadjuvant talazoparib for patients with operable breast cancer with a germline BRCA pathogenic variant. J Clin Oncol. 2020;38(5):388–394. doi:10.1200/JCO.19.01304
44. Sonnenblick A, de Azambuja E, Azim HA, Piccart M. An update on PARP inhibitors–moving to the adjuvant setting. Nat Rev Clin Oncol. 2015;12(1):27–41. doi:10.1038/nrclinonc.2014.163
45. Robson M, Im S-A, Senkus E, et al. Olaparib for metastatic breast cancer in patients with a germline BRCA mutation. N Engl J Med. 2017;377(6):523–533. doi:10.1056/NEJMoa1706450
46. Robson ME, Tung N, Conte P, et al. OlympiAD final overall survival and tolerability results: olaparib versus chemotherapy treatment of physician’s choice in patients with a germline BRCA mutation and HER2-negative metastatic breast cancer. Ann Oncol. 2019;30(4):558–566. doi:10.1093/annonc/mdz012
47. Litton JK, Rugo HS, Ettl J, et al. Talazoparib in patients with advanced breast cancer and a germline BRCA mutation. N Engl J Med. 2018;379(8):753–763. doi:10.1056/NEJMoa1802905
48. A phase 3 randomized, placebo-controlled trial of carboplatin and paclitaxel with or without veliparib (ABT-888) in HER2-negative metastatic or locally advanced unresectable BRCA-associated breast cancer - full text view - ClinicalTrials.gov. Available from: https://clinicaltrials.gov/ct2/show/NCT02163694.
49. Tutt A, Tovey H, Cheang MCU, et al. Carboplatin in BRCA1/2-mutated and triple-negative breast cancer BRCAness subgroups: the TNT trial. Nat Med. 2018;24(5):628–637. doi:10.1038/s41591-018-0009-7
50. Damineni S, Rao VR, Kumar S, et al. Germline mutations of TP53 gene in breast cancer. Tumour Biol. 2014;35(9):9219–9227. doi:10.1007/s13277-014-2176-6
51. Sidransky D, Tokino T, Helzlsouer K, et al. Inherited p53 gene mutations in breast cancer. Cancer Res. 1992;52(10):2984–2986.
52. Malkin D. Li-fraumeni syndrome. Genes Cancer. 2011;2(4):475–484. doi:10.1177/1947601911413466
53. Mai PL, Best AF, Peters JA, et al. Risks of first and subsequent cancers among TP53 mutation carriers in the National Cancer Institute Li-Fraumeni syndrome cohort. Cancer. 2016;122(23):3673–3681. doi:10.1002/cncr.30248
54. McBride KA, Ballinger ML, Killick E, et al. Li-Fraumeni syndrome: cancer risk assessment and clinical management. Nat Rev Clin Oncol. 2014;11(5):260–271. doi:10.1038/nrclinonc.2014.41
55. Limacher JM, Frebourg T, Natarajan-Ame S, Bergerat JP. Two metachronous tumors in the radiotherapy fields of a patient with Li-Fraumeni syndrome. Int J Cancer. 2001;96(4):238–242. doi:10.1002/ijc.1021
56. Heymann S, Delaloge S, Rahal A, et al. Radio-induced malignancies after breast cancer postoperative radiotherapy in patients with Li-Fraumeni syndrome. Radiat Oncol. 2010;5:104. doi:10.1186/1748-717X-5-104
57. Rath MG, Masciari S, Gelman R, et al. Prevalence of germline TP53 mutations in HER2+ breast cancer patients. Breast Cancer Res Treat. 2013;139(1):193–198. doi:10.1007/s10549-012-2375-z
58. Kappel S, Janschek E, Wolf B, et al. TP53 germline mutation may affect response to anticancer treatments: analysis of an intensively treated Li-Fraumeni family. Breast Cancer Res Treat. 2015;151(3):671–678. doi:10.1007/s10549-015-3424-1
59. Lowe SW, Ruley HE, Jacks T, Housman DE. p53-dependent apoptosis modulates the cytotoxicity of anticancer agents. Cell. 1993;74(6):957–967. doi:10.1016/0092-8674(93)90719-7
60. Yehia L, Ngeow J, Eng C. PTEN-opathies: from biological insights to evidence-based precision medicine. J Clin Invest. 2019;129(2):452–464. doi:10.1172/JCI121277
61. Stambolic V, Suzuki A, de la Pompa JL, et al. Negative regulation of PKB/Akt-dependent cell survival by the tumor suppressor PTEN. Cell. 1998;95(1):29–39. doi:10.1016/s0092-8674(00)81780-8
62. Luen SJ, Asher R, Lee CK, et al. Association of somatic driver alterations with prognosis in postmenopausal, hormone receptor-positive, HER2-negative early breast cancer: a secondary analysis of the BIG 1-98 randomized clinical trial. JAMA Oncol. 2018;4(10):1335–1343. doi:10.1001/jamaoncol.2018.1778
63. Stern HM, Gardner H, Burzykowski T, et al. PTEN loss is associated with worse outcome in HER2-amplified breast cancer patients but is not associated with trastuzumab resistance. Clin Cancer Res. 2015;21(9):2065–2074. doi:10.1158/1078-0432.CCR-14-2993
64. André F, Hurvitz S, Fasolo A, et al. Molecular alterations and everolimus efficacy in human epidermal growth factor receptor 2-overexpressing metastatic breast cancers: combined exploratory biomarker analysis from BOLERO-1 and BOLERO-3. J Clin Oncol. 2016;34(18):2115–2124. doi:10.1200/JCO.2015.63.9161
65. Kim S-B, Dent R, Im S-A, et al. Ipatasertib plus paclitaxel versus placebo plus paclitaxel as first-line therapy for metastatic triple-negative breast cancer (LOTUS): a multicentre, randomised, double-blind, placebo-controlled, Phase 2 trial. Lancet Oncol. 2017;18(10):1360–1372. doi:10.1016/S1470-2045(17)30450-3
66. Oliveira M, Saura C, Nuciforo P, et al. FAIRLANE, a double-blind placebo-controlled randomized phase II trial of neoadjuvant ipatasertib plus paclitaxel for early triple-negative breast cancer. Ann Oncol. 2019;30(8):1289–1297. doi:10.1093/annonc/mdz177
67. Shen WH, Balajee AS, Wang J, et al. Essential role for nuclear PTEN in maintaining chromosomal integrity. Cell. 2007;128(1):157–170. doi:10.1016/j.cell.2006.11.042
68. Bassi C, Ho J, Srikumar T, et al. Nuclear PTEN controls DNA repair and sensitivity to genotoxic stress. Science. 2013;341(6144):395–399. doi:10.1126/science.1236188
69. Minami D, Takigawa N, Takeda H, et al. Synergistic effect of olaparib with combination of cisplatin on PTEN-deficient lung cancer cells. Mol Cancer Res. 2013;11(2):140–148. doi:10.1158/1541-7786.MCR-12-0401
70. Lin F, de Gooijer MC, Roig EM, et al. ABCB1, ABCG2, and PTEN determine the response of glioblastoma to temozolomide and ABT-888 therapy. Clin Cancer Res. 2014;20(10):2703–2713. doi:10.1158/1078-0432.CCR-14-0084
71. Mendes-Pereira AM, Martin SA, Brough R, et al. Synthetic lethal targeting of PTEN mutant cells with PARP inhibitors. EMBO Mol Med. 2009;1(6–7):315–322. doi:10.1002/emmm.200900041
72. Gruber JJ, Afghahi A, Hatton A, Scott D, McMillan A, Ford JM, Telli ML. Talazoparib beyond BRCA: a phase II trial of talazoparib monotherapy in BRCA1 and BRCA2 wild-type patients with advanced HER2-negative breast cancer or other solid tumors with a mutation in homologous recombination (HR) pathway genes. J Clin Oncol. 2019. Available from: https://ascopubs.org/doi/abs/10.1200/JCO.2019.37.15_suppl.3006. Accessed April 7, 2020.
73. Fitzgerald RC, Hardwick R, Huntsman D, et al. Hereditary diffuse gastric cancer: updated consensus guidelines for clinical management and directions for future research. J Med Genet. 2010;47(7):436–444. doi:10.1136/jmg.2009.074237
74. Guilford P, Humar B, Blair V. Hereditary diffuse gastric cancer: translation of CDH1 germline mutations into clinical practice. Gastric Cancer. 2010;13(1):1–10. doi:10.1007/s10120-009-0531-x
75. van der Post RS, Vogelaar IP, Carneiro F, et al. Hereditary diffuse gastric cancer: updated clinical guidelines with an emphasis on germline CDH1 mutation carriers. J Med Genet. 2015;52(6):361–374. doi:10.1136/jmedgenet-2015-103094
76. Hansford S, Kaurah P, Li-Chang H, et al. Hereditary diffuse gastric cancer syndrome: CDH1 mutations and beyond. JAMA Oncol. 2015;1(1):23–32. doi:10.1001/jamaoncol.2014.168
77. Kaurah P, MacMillan A, Boyd N, et al. Founder and recurrent CDH1 mutations in families with hereditary diffuse gastric cancer. JAMA. 2007;297(21):2360–2372. doi:10.1001/jama.297.21.2360
78. Ping Z, Siegal GP, Harada S, et al. ERBB2 mutation is associated with a worse prognosis in patients with CDH1 altered invasive lobular cancer of the breast. Oncotarget. 2016;7(49):80655–80663. doi:10.18632/oncotarget.13019
79. Colleoni M, Russo L, Dellapasqua S. Adjuvant therapies for special types of breast cancer. Breast. 2011;20(Suppl 3):S153–S157. doi:10.1016/S0960-9776(11)70315-0
80. Teo K, Gómez-Cuadrado L, Tenhagen M, et al. E-cadherin loss induces targetable autocrine activation of growth factor signalling in lobular breast cancer. Sci Rep. 2018;8(1):15454. doi:10.1038/s41598-018-33525-5
81. Rahman N, Seal S, Thompson D, et al. PALB2, which encodes a BRCA2-interacting protein, is a breast cancer susceptibility gene. Nat Genet. 2007;39(2):165–167. doi:10.1038/ng1959
82. Reid S, Schindler D, Hanenberg H, et al. Biallelic mutations in PALB2 cause fanconi anemia subtype FA-N and predispose to childhood cancer. Nat Genet. 2007;39(2):162–164. doi:10.1038/ng1947
83. Casadei S, Norquist BM, Walsh T, et al. Contribution of inherited mutations in the BRCA2-interacting protein PALB2 to familial breast cancer. Cancer Res. 2011;71(6):2222–2229. doi:10.1158/0008-5472.CAN-10-3958
84. Yang X, Leslie G, Doroszuk A, et al. Cancer Risks associated with germline PALB2 pathogenic variants: an International Study of 524 families. J Clin Oncol. 2020;38(7):674–685. doi:10.1200/JCO.19.01907
85. Tung N, Domchek SM, Stadler Z, et al. Counselling framework for moderate-penetrance cancer-susceptibility mutations. Nat Rev Clin Oncol. 2016;13(9):581–588. doi:10.1038/nrclinonc.2016.90
86. Antoniou AC, Casadei S, Heikkinen T, et al. Breast-cancer risk in families with mutations in PALB2. N Engl J Med. 2014;371(6):497–506. doi:10.1056/NEJMoa1400382
87. Catucci I, Milgrom R, Kushnir A, et al. Germline mutations in BRIP1 and PALB2 in Jewish high cancer risk families. Fam Cancer. 2012;11(3):483–491. doi:10.1007/s10689-012-9540-8
88. Tischkowitz M, Xia B. PALB2/FANCN: recombining cancer and fanconi anemia. Cancer Res. 2010;70(19):7353–7359. doi:10.1158/0008-5472.CAN-10-1012
89. Buisson R, Dion-Côté A-M, Coulombe Y, et al. Cooperation of breast cancer proteins PALB2 and piccolo BRCA2 in stimulating homologous recombination. Nat Struct Mol Biol. 2010;17(10):1247–1254. doi:10.1038/nsmb.1915
90. Villarroel MC, Rajeshkumar NV, Garrido-Laguna I, et al. Personalizing cancer treatment in the age of global genomic analyses: PALB2 gene mutations and the response to DNA damaging agents in pancreatic cancer. Mol Cancer Ther. 2011;10(1):3–8. doi:10.1158/1535-7163.MCT-10-0893
91. Hemminki A, Markie D, Tomlinson I, et al. A serine/threonine kinase gene defective in Peutz-Jeghers syndrome. Nature. 1998;391(6663):184–187. doi:10.1038/34432
92. Beggs AD, Latchford AR, Vasen HFA, et al. Peutz-Jeghers syndrome: a systematic review and recommendations for management. Gut. 2010;59(7):975–986. doi:10.1136/gut.2009.198499
93. Lu H-M, Li S, Black MH, et al. Association of breast and ovarian cancers with predisposition genes identified by large-scale sequencing. JAMA Oncol. 2019;5(1):51–57. doi:10.1001/jamaoncol.2018.2956
94. Walsh MD, Buchanan DD, Cummings MC, et al. Lynch syndrome-associated breast cancers: clinicopathologic characteristics of a case series from the colon cancer family registry. Clin Cancer Res. 2010;16(7):2214–2224. doi:10.1158/1078-0432.CCR-09-3058
95. Shulman LP. Hereditary breast and ovarian cancer (HBOC): clinical features and counseling for BRCA1 and BRCA2, lynch syndrome, cowden syndrome, and Li-Fraumeni syndrome. Obstet Gynecol Clin North Am. 2010;37(1):109–133, Table of Contents. doi:10.1016/j.ogc.2010.03.003
96. Win AK, Young JP, Lindor NM, et al. Colorectal and other cancer risks for carriers and noncarriers from families with a DNA mismatch repair gene mutation: a prospective cohort study. J Clin Oncol. 2012;30(9):958–964. doi:10.1200/JCO.2011.39.5590
97. Win AK, Lindor NM, Jenkins MA. Risk of breast cancer in lynch syndrome: a systematic review. Breast Cancer Res. 2013;15(2):R27. doi:10.1186/bcr3405
98. Win AK, Macinnis RJ, Dowty JG, Jenkins MA. Criteria and prediction models for mismatch repair gene mutations: a review. J Med Genet. 2013;50(12):785–793. doi:10.1136/jmedgenet-2013-101803
99. Harkness EF, Barrow E, Newton K, et al. Lynch syndrome caused by MLH1 mutations is associated with an increased risk of breast cancer: a cohort study. J Med Genet. 2015;52(8):553–556. doi:10.1136/jmedgenet-2015-103216
100. Roberts ME, Jackson SA, Susswein LR, et al. MSH6 and PMS2 germ-line pathogenic variants implicated in lynch syndrome are associated with breast cancer. Genet Med. 2018;20(10):1167–1174. doi:10.1038/gim.2017.254f
101. Anbazhagan R, Fujii H, Gabrielson E. Microsatellite instability is uncommon in breast cancer. Clin Cancer Res. 1999;5(4):839–844.
102. Cortes-Ciriano I, Lee S, Park W-Y, Kim T-M, Park PJ. A molecular portrait of microsatellite instability across multiple cancers. Nat Commun. 2017;8:15180. doi:10.1038/ncomms15180
103. Le DT, Durham JN, Smith KN, et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science. 2017;357(6349):409–413. doi:10.1126/science.aan6733
104. Cancer Drugs Approved Based on Biomarkers and Not Tumor Type-FDA Approval of Pembrolizumab for Mismatch Repair-Deficient Solid Cancers. Abstract - Europe PMC. Available from: https://europepmc.org/article/med/29285544.
105. Moldovan G-L, D’Andrea AD. How the fanconi anemia pathway guards the genome. Annu Rev Genet. 2009;43:223–249. doi:10.1146/annurev-genet-102108-134222
106. Kuschel B, Auranen A, Gregory CS, et al. Common polymorphisms in checkpoint kinase 2 are not associated with breast cancer risk. Cancer Epidemiol Biomarkers Prev. 2003;12(8):809–812.
107. Xiang H, Geng X, Ge W, Li H. Meta-analysis of CHEK2 1100delC variant and colorectal cancer susceptibility. Eur J Cancer. 2011;47(17):2546–2551. doi:10.1016/j.ejca.2011.03.025
108. Meijers-Heijboer H, van den Ouweland A, Klijn J, et al. Low-penetrance susceptibility to breast cancer due to CHEK2(*)1100delC in noncarriers of BRCA1 or BRCA2 mutations. Nat Genet. 2002;31(1):55–59. doi:10.1038/ng879
109. Näslund-Koch C, Nordestgaard BG, Bojesen SE. Increased risk for other cancers in addition to breast cancer for CHEK2*1100delC heterozygotes estimated from the Copenhagen General Population Study. J Clin Oncol. 2016;34(11):1208–1216. doi:10.1200/JCO.2015.63.3594
110. Cybulski C, Górski B, Huzarski T, et al. CHEK2 is a multiorgan cancer susceptibility gene. Am J Hum Genet. 2004;75(6):1131–1135. doi:10.1086/426403
111. Schmidt MK, Tollenaar RAEM, de Kemp SR, et al. Breast cancer survival and tumor characteristics in premenopausal women carrying the CHEK2*1100delC germline mutation. J Clin Oncol. 2007;25(1):64–69. doi:10.1200/JCO.2006.06.3024
112. Guénard F, Pedneault CS-L, Ouellette G, et al. Evaluation of the contribution of the three breast cancer susceptibility genes CHEK2, STK11, and PALB2 in non-BRCA1/2 French Canadian families with high risk of breast cancer. Genet Test Mol Biomarkers. 2010;14(4):515–526. doi:10.1089/gtmb.2010.0027
113. Weischer M, Nordestgaard BG, Pharoah P, et al. CHEK2*1100delC heterozygosity in women with breast cancer associated with early death, breast cancer-specific death, and increased risk of a second breast cancer. J Clin Oncol. 2012;30(35):4308–4316. doi:10.1200/JCO.2012.42.7336
114. Schmidt MK, Hogervorst F, van Hien R, et al. Age- and tumor subtype-specific breast cancer risk estimates for CHEK2*1100delC Carriers. J Clin Oncol. 2016;34(23):2750–2760. doi:10.1200/JCO.2016.66.5844
115. Cybulski C, Wokołorczyk D, Jakubowska A, et al. Risk of breast cancer in women with a CHEK2 mutation with and without a family history of breast cancer. J Clin Oncol. 2011;29(28):3747–3752. doi:10.1200/JCO.2010.34.0778
116. Knappskog S, Chrisanthar R, Løkkevik E, et al. Low expression levels of ATM may substitute for CHEK2/TP53 mutations predicting resistance towards anthracycline and mitomycin chemotherapy in breast cancer. Breast Cancer Res. 2012;14(2):R47. doi:10.1186/bcr3147
117. Chrisanthar R, Knappskog S, Løkkevik E, et al. CHEK2 mutations affecting kinase activity together with mutations in TP53 indicate a functional pathway associated with resistance to epirubicin in primary breast cancer. PLoS One. 2008;3(8):e3062. doi:10.1371/journal.pone.0003062
118. Kriege M, Hollestelle A, Jager A, et al. Survival and contralateral breast cancer in CHEK2 1100delC breast cancer patients: impact of adjuvant chemotherapy. Br J Cancer. 2014;111(5):1004–1013. doi:10.1038/bjc.2014.306
119. Kriege M, Jager A, Hollestelle A, et al. Sensitivity to systemic therapy for metastatic breast cancer in CHEK2 1100delC mutation carriers. J Cancer Res Clin Oncol. 2015;141(10):1879–1887. doi:10.1007/s00432-015-1981-7
120. Baeyens A, Claes K, Willems P, De Ruyck K, Thierens H, Vral A. Chromosomal radiosensitivity of breast cancer with a CHEK2 mutation. Cancer Genet Cytogenet. 2005;163(2):106–112. doi:10.1016/j.cancergencyto.2005.04.017
121. Reiner AS, Robson ME, Mellemkjær L, et al. Radiation treatment, ATM, BRCA1/2, and CHEK2*1100delC pathogenic variants and risk of contralateral breast cancer. J Natl Cancer Inst. 2020. doi:10.1093/jnci/djaa031
122. Renwick A, Thompson D, Seal S, et al. ATM mutations that cause ataxia-telangiectasia are breast cancer susceptibility alleles. Nat Genet. 2006;38(8):873–875. doi:10.1038/ng1837
123. Chenevix-Trench G, Spurdle AB, Gatei M, et al. Dominant negative ATM mutations in breast cancer families. J Natl Cancer Inst. 2002;94(3):205–215. doi:10.1093/jnci/94.3.205
124. Paglia LL, Laugé A, Weber J, et al. ATM germline mutations in women with familial breast cancer and a relative with haematological malignancy. Breast Cancer Res Treat. 2010;119(2):443–452. doi:10.1007/s10549-009-0396-z
125. Roberts NJ, Jiao Y, Yu J, et al. ATM mutations in patients with hereditary pancreatic cancer. Cancer Discov. 2012;2(1):41–46. doi:10.1158/2159-8290.CD-11-0194
126. Mansfield SA, Pilarski R, Agnese DM. ATM mutations for surgeons. Fam Cancer. 2017;16(3):407–410. doi:10.1007/s10689-016-9959-4
127. Bernstein JL, Concannon P. ATM, radiation, and the risk of second primary breast cancer. Int J Radiat Biol. 2017;93(10):1121–1127. doi:10.1080/09553002.2017.1344363
128. Cortez D, Wang Y, Qin J, Elledge SJ. Requirement of ATM-dependent phosphorylation of brca1 in the DNA damage response to double-strand breaks. Science. 1999;286(5442):1162–1166. doi:10.1126/science.286.5442.1162
129. Bernstein JL, Haile RW, Stovall M, et al. Radiation exposure, the ATM gene, and contralateral breast cancer in the women’s environmental cancer and Radiation Epidemiology Study. J Natl Cancer Inst. 2010;102(7):475–483. doi:10.1093/jnci/djq055
130. Modlin LA, Flynn J, Zhang Z, et al. Breast radiotherapy among ATM-mutation carriers. JCO. 2019;37(15_suppl):1504. doi:10.1200/JCO.2019.37.15_suppl.1504
131. van Os NJH, Roeleveld N, Weemaes CMR, et al. Health risks for ataxia-telangiectasia mutated heterozygotes: a systematic review, meta-analysis and evidence-based guideline. Clin Genet. 2016;90(2):105–117. doi:10.1111/cge.12710
132. Paluch-Shimon S, Pagani O, Partridge AH, et al. ESO-ESMO 3rd international consensus guidelines for breast cancer in young women (BCY3). Breast. 2017;35:203–217. doi:10.1016/j.breast.2017.07.017
133. Teng P, Bateman NW, Darcy KM, et al. Pharmacologic inhibition of ATR and ATM offers clinically important distinctions to enhancing platinum or radiation response in ovarian, endometrial, and cervical cancer cells. Gynecol Oncol. 2015;136(3):554–561. doi:10.1016/j.ygyno.2014.12.035
134. Ma Z, Yao G, Zhou B, Fan Y, Gao S, Feng X. The Chk1 inhibitor AZD7762 sensitises p53 mutant breast cancer cells to radiation in vitro and in vivo. Mol Med Rep. 2012;6(4):897–903. doi:10.3892/mmr.2012.999
135. Bridge WL, Vandenberg CJ, Franklin RJ, Hiom K. The BRIP1 helicase functions independently of BRCA1 in the Fanconi anemia pathway for DNA crosslink repair. Nat Genet. 2005;37(9):953–957. doi:10.1038/ng1627
136. Seal S, Thompson D, Renwick A, et al. Truncating mutations in the Fanconi anemia J gene BRIP1 are low-penetrance breast cancer susceptibility alleles. Nat Genet. 2006;38(11):1239–1241. doi:10.1038/ng1902
137. Rafnar T, Gudbjartsson DF, Sulem P, et al. Mutations in BRIP1 confer high risk of ovarian cancer. Nat Genet. 2011;43(11):1104–1107. doi:10.1038/ng.955
138. Song H, Ramus SJ, Kjaer SK, et al. Tagging single nucleotide polymorphisms in the BRIP1 gene and susceptibility to breast and ovarian cancer. PLoS One. 2007;2(3):e268. doi:10.1371/journal.pone.0000268
139. Easton DF, Lesueur F, Decker B, et al. No evidence that protein truncating variants in BRIP1 are associated with breast cancer risk: implications for gene panel testing. J Med Genet. 2016;53(5):298–309. doi:10.1136/jmedgenet-2015-103529
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.