Back to Journals » International Journal of Nanomedicine » Volume 9 » Issue 1
Comparison of in vitro dialysis release methods of loperamide-encapsulated liposomal gel for topical drug delivery
Authors Hua S ![]()
Received 11 October 2013
Accepted for publication 6 December 2013
Published 30 January 2014 Volume 2014:9(1) Pages 735—744
DOI https://doi.org/10.2147/IJN.S55805
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 5
Susan Hua
The School of Biomedical Sciences and Pharmacy, The University of Newcastle, Callaghan, NSW, Australia
Background: The purpose of this study was to determine the most appropriate dialysis equilibrium method to assess liposomal gel formulations containing hydrophobic drugs, to give the most accurate indication of drug release.
Methods: Loperamide hydrochloride-encapsulated liposomes, composed of L-α-phosphatidylcholine and cholesterol (molar ratio of 2:1), were prepared according to the method of dried lipid film hydration. The liposomes were incorporated into a carbopol gel (0.5%, weight/weight). The release of the drug from the nanoparticles was assessed using a number of variations of the dialysis technique, taking into account solubility parameters and formulation. Method 1 (below saturation point) and Method 2 (above saturation point) used a dilution method to evaluate how drug concentration and solubility affects the in vitro drug-release profile of loperamide hydrochloride, while Methods 3 (below saturation point) and 4 (above saturation point) evaluated how drug concentration and the gel base affect the release profile.
Results: In Method 1, the liposomes showed a rapid release of just over 60% in the first 3 hours and then a slower, sustained release to just over 70% at 24 hours. Method 2 showed a gradual, sustained release profile with the liposomes with 55% release at 24 hours. In Method 3, the liposomes showed a rapid burst release of 98% at 2 hours. In Method 4, the liposomal gel had a rapid release of 60% within 3 hours and then a more gradual, sustained release with 86% release at 24 hours. The free drug suspension in Methods 2 and 4 showed a limited release across the dialysis membrane, in comparison to Methods 1 and 3, which showed a complete release in a timely manner.
Conclusion: This study has demonstrated that the actual method used for equilibrium dialysis plays a significant role in determining the true characteristics of a topical nanoformulation, with Method 3 providing the most accurate indication of the release of a hydrophobic drug from a topical liposomal formulation.
Keywords: liposomes, gel, in vitro drug release, transdermal, topical, loperamide
Introduction
Liposomes are small vesicles consisting of one or more concentric lipid bilayers enclosing discrete aqueous spaces. The unique ability of liposomes to entrap drugs both in an aqueous and a lipid phase make such delivery systems attractive for hydrophilic and hydrophobic drugs. Hydrophobic molecules are intercalated within the bilayer membrane, and hydrophilic molecules can be entrapped in the internal aqueous region.1 In recent years, liposomes have gained increasing attention for topical preparations, as the skin offers a lot of advantages for the administration of such systems. The aim of topical administration of liposomes is either for dermal drug delivery with an optimal localized effect or transdermal drug delivery with the goal of systemic absorption. Liposomes offer a number of advantages in dermal and transdermal drug delivery as they have a high solubilization capacity and penetration-enhancing effect, even for highly lipophilic drugs.2 There are several positive results regarding the potential of liposomal carrier systems for targeted skin delivery as well as for transdermal drug delivery.2
The kinetics of drug release from a liposomal formulation is a critical part of the rational design of drug delivery systems, as it is a major determinant on the efficacy of delivery of the carrier in vivo and the subsequent release of the free drug. An in vitro release profile reveals important information on the structure and behavior of the formulation, possible interactions between the drug and carrier composition, and their influence on the rate and mechanism of drug release.3–5 In comparison to parenteral drug delivery, not much attention has been devoted to the development of a reliable in vitro release technique for topical liposomal formulations, especially those encapsulating hydrophobic compounds. The dialysis release method is a well-established and useful technique to study in vitro release from micro- and nano-particulate delivery systems. In this method, drug-loaded carriers are physically separated from the bulk media by a dialysis membrane, and the release is generally assessed from the outer bulk over time.3,6 This technique has been used to study a variety of formulations, including liposomes and nanoparticles,7–15 and it is almost exclusively used in the literature for the measurement of release kinetics.6 It is a well-established technique, and although still widely used in the literature, it has been shown to suffer from significant limitations; therefore, it provides an inaccurate indication of the release kinetics of nanocarrier formulations.6,16 The hypothesis of this study is that the dialysis method can still be used to provide a reliable indication of the true release of hydrophobic drugs from topical liposomal formulations; however, it requires specific parameters in the design of the release assay. This study will evaluate a number of variations of the dialysis technique, taking into account solubility parameters and formulation to compare in vitro release profiles of the loperamide-encapsulated liposomal gel, which is a highly hydrophobic drug. This study will aim to determine the most appropriate dialysis equilibrium method to assess liposomal gel formulations containing hydrophobic drugs to give the most accurate indication of a release of the drug from the delivery system.
Materials and methods
Materials
The L-α-phosphatidylcholine (EPC) was purchased from Avanti Polar Lipids (Alabaster, AL, USA). Cholesterol, loperamide hydrochloride (HCl), and triethanolamine were purchased from Sigma-Aldrich (St Louis, MO, USA). The carbomer 940 NF resin was purchased from PCCA (Houston, Texas, USA). All other chemicals and solvents were of at least analytical grade.
Preparation of conventional liposomes
Conventional liposomes were prepared according to the method of dried lipid film hydration. Briefly, 16 mg EPC (Avanti Polar Lipids), 4 mg cholesterol (Sigma-Aldrich) (molar ratio of 2:1) and 4 mg loperamide HCl (Sigma-Aldrich) were solubilized in 6 mL chloroform:methanol (2:1, volume/volume) in a 50 mL round-bottomed flask and dried by rotary evaporation under reduced pressure (100 mbar, 15 minutes, 40°C). The resultant thin lipid film was hydrated with the addition of 1 mL of phosphate- buffered saline (PBS) (pH 6.5) and resuspended in a 40°C water bath. The resultant multilamellar dispersions were reduced in size and lamellarity by probe sonication (60 amps, 5 minutes) at 40°C. The size distribution of the liposomal dispersion was determined by dynamic laser light scattering (Zetasizer Nano S, Malvern Instruments, Malvern, UK). The unencapsulated drug was removed from the liposome suspension using Slide-A-Lyzer Dialysis Cassettes, 10K MWCO (Thermo Fisher Scientific, Waltham, MA, USA). The entrapment efficiency percentage was determined by disrupting the vesicles, using ethanol, and evaluating the loperamide HCl concentration using high-performance liquid chromatography (HPLC). Liposomes were stored at 4°C and were used the following day. All chemicals and solvents were of at least analytical grade.
Preparation of carbopol gel
The gel (0.5%, weight/weight [w/w]) was prepared by dispersing carbomer 940 NF resin (PCCA) (0.5 g) in distilled water (88 g), in which glycerol (10 g) was previously added. The mixture was stirred until thickening occurred and then neutralized by the dropwise addition of 50% (w/w) triethanolamine to achieve a transparent gel of pH 5.5. Liposomes were mixed into the carbopol gel by mechanical stirring for 5 minutes.
Evaluation of loperamide HCl concentration by HPLC
The concentration of loperamide HCl was determined by HPLC using the 1200 series HPLC system (Agilent Technologies, Santa Clara, CA, USA). The HPLC consisted of a binary pump, autoinjector, column oven, and the ultraviolet–visible detector. The data were integrated using the Agilent ChemStation for LC Systems (version 02/06); Agilent Technologies (Waldbronn, Germany). The separation was performed using a Thermo Scientific Hypersil™ BDS C18 column (150 × 4.6 mm, 5 μm), and the wavelength of detection was 210 nm. The mobile phase consisted of 50% acetonitrile, 5% isopropanol, and a 45% buffer (0.05 M NaH2PO4). This was pumped through the column at a flow rate of 1.5 mL/minute. The column was maintained at 25°C. Calibration curves were established by plotting the standard concentrations of loperamide HCl dissolved in PBS pH 6.5 versus the area under the curve. The loperamide HCl release percentage was obtained according to:
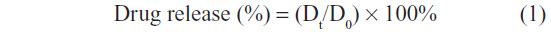
where Dt and D0 indicate the amount of drug released from the liposome suspension at certain intervals and the total amount of drug in the liposome suspension, respectively. At the end of the study, the liposome samples were recovered from the dialysis system and lysed with ethanol for analysis for loperamide HCl content by HPLC.
In vitro dialysis release study
Method 1: modified liposome drug release assay accounting for solubility parameters
To circumvent potential solubility issues of loperamide HCl across the dialysis membrane, a modified assay was developed to assess the true release of loperamide HCl from the liposomes in a gel formulation without surpassing the saturation point. In brief, 50 μL of the 4 mg/mL loperamide HCl-encapsulated liposome suspension (equivalent to 200 μg loperamide HCl) was added in a dialysis bag (molecular weight cut off [MWCO] 10 kDa, Thermo Fisher Scientific) with 1 mL of carbopol gel (0.5%, w/w) and 9.95 mL of PBS, pH 6.5. The dialysis system was suspended in a release volume of 40 mL PBS, pH 6.5, at 37°C and rotated at 200 rpm (1:4 dilution between the donor and acceptor compartments). For control groups, 4 mg of loperamide HCl was dissolved in 200 mL PBS, pH 7.4, and 10 mL of this solution (equivalent to 200 μg loperamide HCl) was placed in a dialysis bag with 1 mL of carbopol gel (0.5%, w/w), and stability was assessed using the dialysis method described. At scheduled intervals, 200 μL of the release medium was collected for the HPLC assay. The same volume of fresh PBS buffer at the same temperature was added immediately to maintain a constant release volume. The length of the dialysis tubing was kept consistent for all methods to ensure that the surface area available for dialysis remained constant. To ensure that a 1:4 dilution between the donor and acceptor compartments provided sink conditions, a 1:10 dilution study was also conducted where the release volume was set at 100 mL PBS, pH 6.5.
Method 2: conventional drug release assay (above loperamide HCl saturation point)
This method does not take into account the solubility issues associated with hydrophobic drugs in an aqueous solution. In brief, 200 μL of the 4 mg/mL loperamide HCl-encapsulated liposome suspension (equivalent to 800 μg loperamide HCl) was added in a dialysis bag (MWCO 10 kDa, Thermo Fisher Scientific) with 1 mL of carbopol gel (0.5%, w/w) and 9.8 mL of PBS, pH 6.5. The dialysis system was suspended in a release volume of 40 mL PBS at 37°C and rotated at 200 rpm (1:4 dilution between donor and acceptor compartments). For control groups, 800 μg of loperamide HCl was added to the 10 mL PBS, pH 6.5, in a dialysis bag with 1 mL of carbopol gel (0.5%, w/w). The stability was assessed using the dialysis method described. At scheduled intervals, 200 μL of the release medium was collected for the HPLC assay. The same volume of fresh PBS buffer at the same temperature was added immediately to maintain constant release volume. The length of the dialysis tubing was kept consistent for all methods to ensure that the surface area available for dialysis remained constant. To ensure that a 1:4 dilution between the donor and acceptor compartments provided sink conditions, a 1:10 dilution study was also conducted where the release volume was set at 100 mL PBS pH 6.5.
Method 3: drug release assay from gel containing free drug solution
In brief, 5 μL of 4 mg/mL loperamide HCl-encapsulated liposome suspension (equivalent to 20 μg loperamide HCl) was mixed with 1 mL of carbopol gel (0.5%, w/w) and added in a dialysis bag. The dialysis system was suspended in a release volume of 10 mL PBS, pH 6.5, at 37°C and rotated at 200 rpm. For control groups, 1 mL of carbopol gel (0.5%, w/w) containing 20 μg of free drug in solution was placed in a dialysis bag, and stability was assessed using the dialysis method described. This is the concentration in which loperamide HCl is soluble in the base of the gel during formulation. The drug-loaded gel was spread thinly onto the membrane surface within the dialysis tubing to mimic topical administration. At scheduled intervals, 50 μL of the release medium was collected for HPLC assay. The same volume of fresh PBS buffer at the same temperature was added immediately to maintain constant release volume. The length of the dialysis tubing was kept consistent for all methods to ensure that the surface area available for dialysis remained constant.
Method 4: drug release assay from gel containing drug suspension
In brief, 200 μL of 4 mg/mL loperamide HCl-encapsulated liposome suspension (equivalent to 0.8 mg loperamide HCl) was mixed with 1 mL of carbopol gel (0.5%, w/w) and added in a dialysis bag (MWCO 10 kDa; Thermo Fisher Scientific). The dialysis system was suspended in a release volume of 40 mL PBS, pH 6.5, at 37°C and rotated at 200 rpm. For control groups, 0.8 mg of loperamide HCl was mixed with 1 mL of carbopol gel (0.5%, w/w) and placed in a dialysis bag. Stability was assessed using the dialysis method described. The drug-loaded gel was spread thinly onto the membrane surface within the dialysis tubing to mimic topical administration. At scheduled intervals, 200 μL of the release medium was collected for the HPLC assay. The same volume of fresh PBS buffer at the same temperature was added immediately to maintain constant release volume. The length of the dialysis tubing was kept consistent for all methods to ensure that the surface area available for dialysis remained constant.
Results
Dispersion properties
The loperamide HCl encapsulated liposomes had a mean particle size of 103±3 nm and a polydispersity index of 0.228±0.075. The low polydispersity indices indicate that the mean particle size is a reasonable indicator of the size of the majority of the particles in the dispersions. This procedure resulted in high loperamide HCl encapsulation efficiency of >99%, which equated to 4.011±0.089 mg/mL of loperamide HCl encapsulated in the liposome suspension.
Method 1: modified liposome drug release assay accounting for solubility parameters
Loperamide HCl has a maximum solubility of 4 mg in 200 mL of PBS (pH 6.5, 20 μg/mL). This pH was used as the skin’s pH is closely regulated around 5.5 to 6.5. Figure 1 shows the percentage of cumulative drug release after 24 hours of the liposome formulation and the control loperamide HCl solution. The diffusion of free drug through the dialysis membrane from the control was more than 80% in the first 6 hours and complete by 12 hours, demonstrating that the release of loperamide HCl was not limited by the dialysis membrane. The in vitro release profile of the liposomes showed a rapid release of just more than 60% in the first 3 hours and then a slower, sustained release of loperamide HCl from the nanocarriers to just more than 70% at 24 hours.
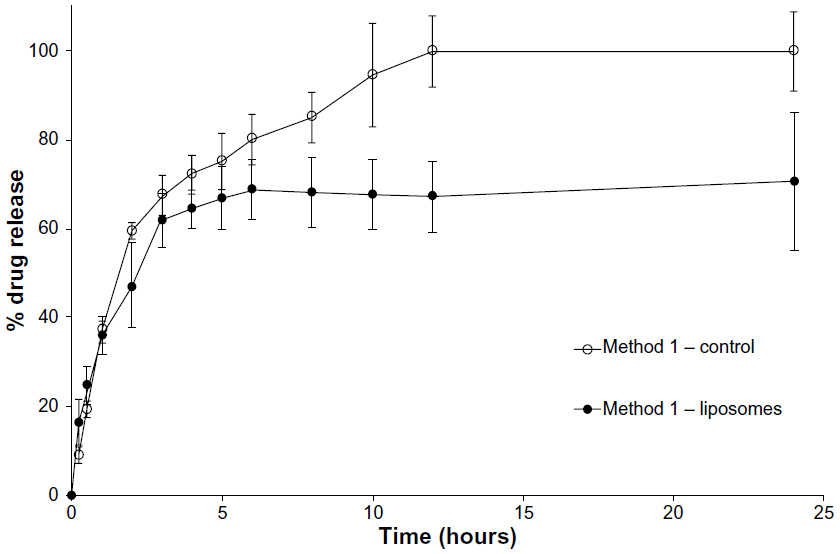 | Figure 1 Method 1 (1:4 dilution). |
Figure 2 shows the drug release profile of loperamide HCl at a 1:10 dilution between the donor and acceptor compartment. A rapid release of 67% occurred in the first 5 hours and then a sustained release of drug from the liposomes of up to 73% at 24 hours. The control release profile shows complete diffusion of the free drug through the dialysis membrane within 10 hours.
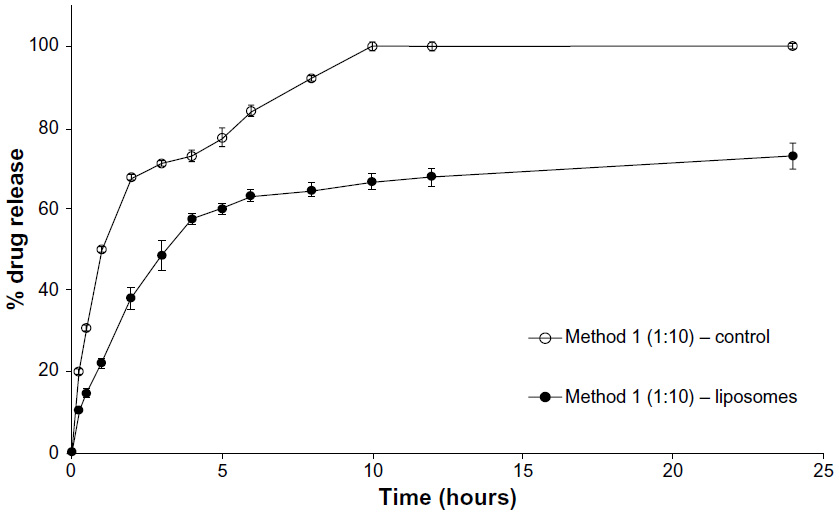 | Figure 2 Method 1 (1:10 dilution). |
Method 2: conventional drug release assay (above loperamide HCl saturation point)
Figure 3 shows the drug release profile of loperamide HCl as the free drug suspension and in liposomes over 24 hours at a concentration above the solubility of the drug in PBS (pH 6.5). In this set of experiments, 800 μg of loperamide HCl free drug suspension (80 μg/mL) or loperamide HCl-encapsulated liposome suspension was dialyzed into the release volume. The equilibrium concentration following release into the dialysis medium equated to 20 μg/mL. The liposome release profile demonstrates a gradual, sustained release of loperamide HCl from the nanocarriers of up to 55% at 24 hours. The control release profile shows a limitation in the release of the free drug suspension across the dialysis membrane, with only 61% release at 24 hours.
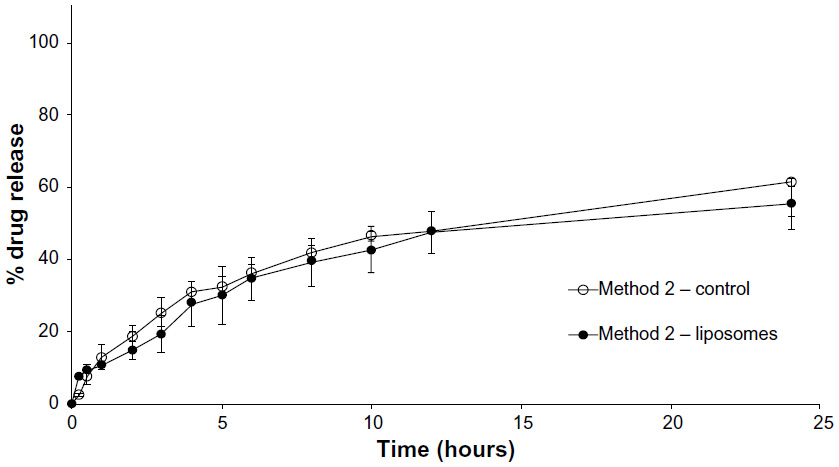 | Figure 3 Method 2 (1:4 dilution). |
Figure 4 shows the drug release profile of loperamide HCl at a 1:10 dilution between the donor and acceptor compartment. The graph shows a slow and gradual release of drug from liposomes over the time course of the study, with a maximum release of 57% at 24 hours. The control release profile shows a limitation in the release of the loperamide HCl suspension across the dialysis membrane, with 51% release at 24 hours.
 | Figure 4 Method 2 (1:10 dilution). |
Method 3: drug release assay from gel formulation containing free drug solution
Figure 5 shows the drug release profile of loperamide HCl as a solution in carbopol gel and in liposomal gel over 24 hours at a concentration below the solubility of the drug in PBS (pH 6.5, 20 μg/mL). The in vitro release profile of the liposomes showed a rapid burst release of 98% at 2 hours and a complete release by 4 hours. The control free drug solution in gel showed a rapid and complete release by 1 hour.
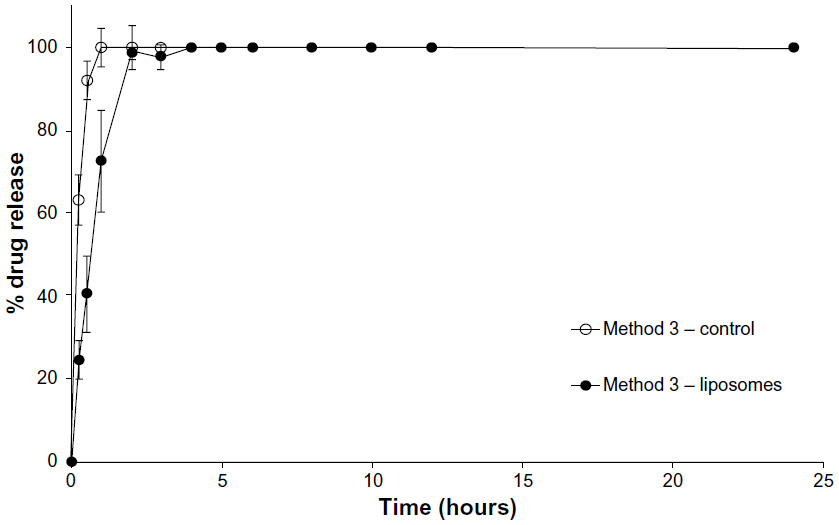 | Figure 5 Method 3. |
Method 4: drug release assay from gel formulation containing drug suspension
This study followed a similar procedure to Method 3; however, the concentration of loperamide HCl was above the solubility of the drug in PBS (pH 6.5, 800 μg/mL). Figure 6 shows the drug release profile of loperamide HCl as a suspension in carbopol gel and in liposomal gel over 24 hours. The liposomal gel release profile demonstrates a rapid release of 60% within 3 hours and then a more gradual, sustained release with 86% release at 24 hours. The control release profile shows a limitation in the release of the free drug suspension across the dialysis membrane, with 51% release at 24 hours.
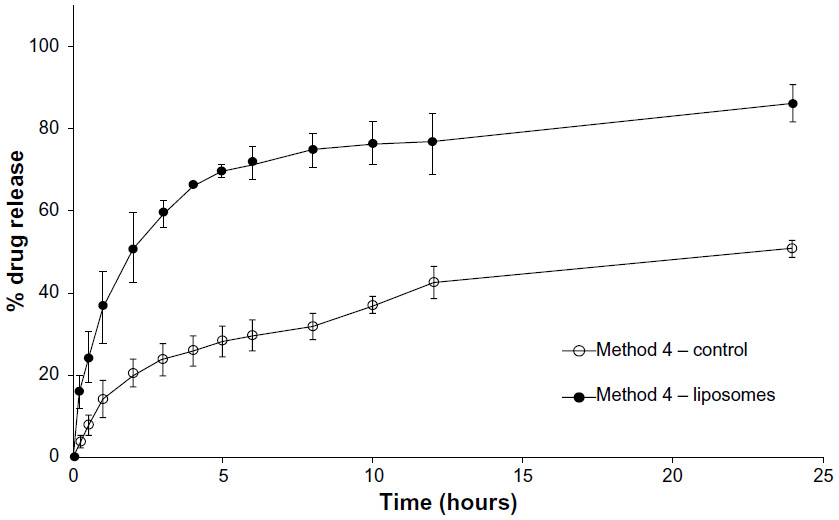 | Figure 6 Method 4. |
Discussion
Topical nanoformulations are of interest to increase the efficacy of therapeutic compounds by increasing their penetration and half-life when administered to the skin.2 Drug release and stability studies are important in determining the release potential of the drug from the carrier and the stability of the nanoformulation. The dialysis method is used widely because sampling and media replacement are convenient due to the physical separation of the liposomes from the outer media by a dialyzing membrane.4,6 This technique in particular mimics in vivo conditions where the nanoparticles are immobilized upon administration, such as following dermal, transdermal, subcutaneous, or intramuscular administration.3 Various modifications of the basic technique have been employed to assess drug release, especially for the use of hydrophobic drugs, including adjusting the dialysis media based on drug solubility and stability.8 However, the addition of surfactants or solvents to increase the lipophilicity of the dialysis medium may potentially interfere with the structure and stability of the nanoparticles themselves, therefore affecting the release profile.16 This study has evaluated various modifications of the basic dialysis method to determine if the drug concentration, solubility, or the gel formulation influences the release profile of the nanoformulation. This will establish the most accurate method for assessing in vitro drug release from topical formulations.
Methods 1 and 2 evaluated how drug concentration and solubility affect the in vitro drug release profile of the hydrophobic drug, loperamide HCl. In this set of experiments, the liposomal gel dispersion inside the dialysis tubing was diluted with media to measure the subsequent release of the drug from the nanoparticles into the surrounding free solution. This dilution has been reported to be necessary to measure drug release from colloidal delivery systems, which is often overlooked in studies where methods, such as equilibrium dialysis, are employed.16 Consequently, release is often dictated by membrane transport effects, making it difficult to reconcile the results obtained in terms of release of the drug from the delivery system.16
Using this dilution method, Figure 1 (Method 1) shows a relatively rapid release of loperamide HCl over the first few hours and then a slower release phase over the remainder of the study. This is consistent with the biphasic release profiles of liposomal dispersions.8 The burst effect varies with the liposome type and lipid composition. The liposomes in this study were composed of the low lipid-phase transition temperature lipid, EPC, and cholesterol. Therefore, at a dialyzing temperature of 37°C, it is expected for the drug to be released from the nanoparticles. Figure 3 (Method 2), however, appears to indicate that the release of loperamide HCl from the liposomal gel is more of a gradual, sustained release that takes place over the entire 24 hours.
By looking at the release profile of the control group, it is clear how drug solubility affects the release profile in this two-compartment dialysis system. Method 1 was conducted below the saturation point of the hydrophobic drug; therefore, the control release profile shows a complete release of the free drug solution across the dialysis membrane, which confirms that loperamide HCl is able to run through the cellulose membrane tubing freely (Figure 1). This method is a more reliable indicator of drug release from the nanoparticles using the dilution method.
Method 2 was conducted above the saturation point, with the dialysis of a free drug suspension. The control release profile shows a limitation in the release of the free drug across the dialysis membrane (Figure 3). This is due to the fact that when the concentration of the free drug is above the saturation point and, therefore, remains mostly as solid drug particles, the rate of drug release from within the dialysis tube into the acceptor compartment is dependent on the solubility of the drug particles within the volume of buffer in the donor compartment. Hence, Method 2 is not an accurate indicator of drug release, as lipophilic drugs (especially above the saturation point) will be under partition control.
To confirm that sink conditions were maintained across all experiments, the release studies were conducted at 1:4 and 1:10 ratio between the volume of buffer inside the dialysis membrane (containing the nanoparticles) to that of the acceptor compartment. This factor is important to provide a driving force for drug transport to the outside and to maintain sink conditions. The results indicate similar drug release profiles at 1:4 and 1:10 ratio for both Methods 1 (Figures 1 and 2) and 2 (Figures 3 and 4), indicating that the sink conditions were maintained.
The next step was to determine whether dilution within the donor compartment is actually necessary to measure drug release from colloidal delivery systems for topical formulations. The dialysis method is known to suffer from membrane-limited diffusion of the free drug from the donor compartment to the acceptor compartment.3,16 The concentration of drug in the acceptor compartment lags significantly behind that of the donor compartment, and it has been suggested not to be a useful indicator of the drug release from colloidal particles over times shorter than days.16 In comparison to the intravenous parenteral formulations where the colloidal nanoparticles are significantly diluted following systemic administration, topical formulations are not predisposed to the same conditions.
Methods 3 and 4 evaluated how the drug concentration and the gel base affect the in vitro drug release profile of loperamide HCl. The drug-loaded gel was spread thinly onto the membrane surface within the dialysis tubing to mimic topical administration. Method 3 was conducted below the saturation point of the hydrophobic drug. The results demonstrated a rapid release of loperamide HCl from the liposomes, with the majority of encapsulated drug released within 2 hours of dialysis at 37°C (Figure 5). Similarly, the control group containing free drug in solution incorporated within the gel base showed a rapid release across the dialysis membrane (Figure 5). This result is consistent with the pressure ultrafiltration method used by Boyd,16 published in 2003, to support the finding of a rapid burst release profile of the lipophilic drug, diazepam, when encapsulated with cubosomes. The equilibrium dialysis method has been previously reported to incorrectly indicate sustained drug release from cubosomes, liposomes, and other nanoparticles.6,16
Conversely, when the concentration of the loperamide HCl was above the saturation point, the drug release profile of the liposomal formulation shows a similar biphasic release as compared to Method 1 (Figures 1 and 2), with a rapid release phase within the first few hours and then a sustained release phase for the remainder of the study (Figure 6). The release profile for the control group, containing solid loperamide HCl mixed into the gel base, closely resembles the release profile of the control group in Method 2 (Figures 3 and 4). The limitation in the release of the free drug across the dialysis membrane is clearly evident. Therefore, this method does not give an accurate indication of drug release of a hydrophobic drug from nanoparticles.
This nondilution method is commonly used to assess drug release from topical liposomal gel formulations. A number of studies using this method have reported their formulation to have controlled release kinetics, even when using low-phase transition temperature lipids and hydrophobic drugs. For example, in 2010 Gupta et al7 reported extremely slow, sustained release of the hydrophobic drug, fluconazole, from a liposomal gel composed of EPC and cholesterol (molar ratio of 2.33:1) over a period of greater than 24 hours. The fluconazole release from plain gel was more than 80% in the first 12 hours, at similar concentrations as the liposomal gel. This sustained release was attributed to the difference between bilayer compositions, and the effective diffusion double barrier consisted of both gel and vesicular lamellae.7 A similar observation was reported by Nounou et al,8 which studied the in vitro release of the hydrophobic drug, dibucaine base, from liposomal dispersions and gels, using the dialysis method. The in vitro release study showed no burst effect, but it did show, rather, a sustained release activity over the 12-hour time frame.8 These results are not in line with those reported in this current study. Potential differences may include surface area of the dialysis membrane, thickness of the gel sample within the donor compartment, or use of surfactants or solvents to assist the dissolution of the free drug within the donor compartment.
Conclusion
The equilibrium dialysis method is one of the most widely used and simplest experiments to conduct, to ascertain important information about the stability and release profile of drug-loaded nanoparticles. Although it has been suggested that this method may provide misleading results for the in vitro release of hydrophobic drugs from colloidal drug delivery systems,6,16 this study has shown that it is still a practical assay to assess topical colloidal formulations when using the optimal method. This study has shown that drug concentration, drug solubility, and the gel base of the formulation are important considerations when conducting a drug release study using the dialysis method. Methods 1 and 3 are more accurate indicators of the true release of hydrophobic drugs from nanoparticles, as they are conducted below the saturation point of the drug. The contested issue is whether a dilution method is a better indicator of true release from a topical formulation than a nondilution method. This does question the importance of the gel base of the formulation in influencing the release profile of the drug from the nanoparticles when administered to the skin. Usually, the gel acts merely as a vehicle for the nanoparticles when rubbed into the skin, and the nanoparticles themselves then interact with the sink conditions provided by the skin. As the composition of the liposomes in this study contains EPC (with a low lipid phase transition temperature) and cholesterol at a standard molar ratio, a rapid burst release of drug from the nanoparticles is expected at an incubation temperature of 37°C, which is more in line with the nondilution method (Method 3). Therefore, this study shows that Method 3 provides the most accurate indication of release kinetics from topical liposomal formulations containing hydrophobic drugs.
In summary, this study has demonstrated that the actual method used for equilibrium dialysis plays a significant role in determining the true characteristics of a topical nanoformulation.
Acknowledgment
The author wishes to thank The Pharmacy Research Trust of New South Wales for providing financial support for the research.
Disclosure
The author reports no conflicts of interest in this work.
References
Allen TM, Cullis PR. Liposomal drug delivery systems: from concept to clinical applications. Adv Drug Del Rev. 2013;65(1):36–48. | |
Pierre MB, Dos Santos Miranda Costa I. Liposomal systems as drug delivery vehicles for dermal and transdermal applications. Arch Dermatol Res. 2011;303(9):607–621. | |
D’Souza SS, DeLuca PP. Methods to assess in vitro drug release from injectable polymeric particulate systems. Pharm Res. 2006;23(3):460–474. | |
Hitzman CJ, Wiedmann TS, Dai H, Elmquist WF. Measurement of drug release from microcarriers by microdialysis. J Pharm Sci. 2005;94(7):1456–1466. | |
Washington C. Drug release from microdisperse systems: a critical review. Int J Pharm. 1990;58(1):1–12. | |
Wallace SJ, Li J, Nation RL, Boyd BJ. Drug release from nanomedicines: Selection of appropriate encapsulation and release methodology. Drug Deliv Transl Res. 2012;2(4):284–292. | |
Gupta M, Goyal AK, Paliwal SR, et al. Development and characterization of effective topical liposomal system for localized treatment of cutaneous candidiasis. J Liposome Res. 2010;20(4):341–350. | |
Nounou MM, El-Khordagui LK, Khalafallah NA, Khalil SA. In vitro release of hydrophilic and hydrophobic drugs from liposomal dispersions and gels. Acta Pharm. 2006;56(3):311–324. | |
Saarinen-Savolainen P, Järvinen T, Taipale H, Urtti A. Method for evaluating drug release from liposomes in sink conditions. Int J Pharm. 1997;159(1):27–33. | |
Parsaee S, Sarbolouki MN, Parnianpour M. In vitro release of diclofenac diethylammonium from lipid-based formulations. Int J Pharm. 2002;241(1):185–190. | |
Leo E, Cameroni R, Forni F. Dynamic dialysis for the drug release evaluation from doxorubicin-gelatin nanoparticle conjugates. Int J Pharm. 1999;180(1):23–30. | |
Panwar P, Pandey B, Lakhera PC, Singh KP. Preparation, characterization, and in vitro release study of albendazole-encapsulated nanosize liposomes. Int J Nanomedicine. 2010;5:101–108. | |
Lansakara-P DS, Rodriguez BL, Cui Z. Synthesis and in vitro evaluation of novel lipophilic monophosphorylated gemcitabine derivatives and their nanoparticles. Int J Pharm. 2012;429(1–2):123–134. | |
Hua S, Cabot PJ. Targeted nanoparticles that mimic immune cells in pain control inducing analgesic and anti-inflammatory actions: a potential novel treatment of acute and chronic pain condition. Pain Physician. 2013;16(3):E199–E216. | |
Wang Y, Tu S, Pinchuk AN, Xiong MP. Active drug encapsulation and release kinetics from hydrogel-in-liposome nanoparticles. J Colloid Interface Sci. 2013;406:247–255. | |
Boyd BJ. Characterisation of drug release from cubosomes using the pressure ultrafiltration method. Int J Pharm. 2003;260(2):239–247. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.