Back to Journals » International Journal of Nanomedicine » Volume 10 » Issue 1
Codelivery of doxorubicin and curcumin with lipid nanoparticles results in improved efficacy of chemotherapy in liver cancer
Authors Zhao X, Chen Q, Liu W, Li Y ![]() , Tang H
, Tang H ![]() , Liu X, Yang X
, Liu X, Yang X
Received 27 August 2014
Accepted for publication 16 October 2014
Published 30 December 2014 Volume 2015:10(1) Pages 257—270
DOI https://doi.org/10.2147/IJN.S73322
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Thomas Webster
Xiaojing Zhao,1,2,* Qi Chen,3,* Wei Liu,1,2 Yusang Li,3 Hebin Tang,3 Xuhan Liu,1,2 Xiangliang Yang1,2
1College of Life Science and Technology, 2National Engineering Research Center for Nanomedicine, Huazhong University of Science and Technology, 3Department of Pharmacology, College of Pharmacy, South-Central University for Nationalities, Wuhan, People’s Republic of China
*These authors contributed equally to this work
Abstract: Liver cancer is a leading cause of cancer deaths worldwide. The combination therapy of cytotoxic and chemosensitizing agents loaded in nanoparticles has been highlighted as an effective treatment for different cancers. However, such studies in liver cancer remain very limited. In our study, we aim to develop a novel lipid nanoparticles loaded with doxorubicin (DOX) (an effective drug for liver cancer) and curcumin (Cur) (a chemosensitizer) simultaneously, and we examined the efficacy of chemotherapy in liver cancer. DOX and Cur codelivery lipid nanoparticles (DOX/Cur-NPs) were successfully prepared using a high-pressure microfluidics technique, showing a mean particle size of around 90 nm, a polydispersity index <0.3, and a zeta potential <−10 mV. The encapsulation efficacy was >90% for both DOX and Cur. The blank lipid nanoparticles were nontoxic, as determined by a cell cytotoxicity study in human normal liver cells L02 and liver cancer cells HepG2. In vitro DOX release studies revealed a sustained-release pattern until 48 hours in DOX/Cur-NPs. We found enhanced cytotoxicity and decreased inhibitory concentration (IC)50 in HepG2 cells and reduced cytotoxicity in L02 cells treated with DOX/Cur-NPs, suggesting the synergistic effects of DOX/Cur-NPs compared with free DOX and DOX nanoparticles (NPs). The optimal weight ratio of DOX and Cur was 1:1. Annexin-V-fluorescein isothiocyanate/propidium iodide double staining showed enhanced apoptosis in HepG2 cells treated with DOX/Cur-NPs compared with free DOX and DOX-NPs. An in vivo experiment showed the synergistic effect of DOX/Cur-NPs compared with DOX-NPs on liver tumor growth inhibition. Taken together, the simultaneous delivery of DOX and Cur by DOX/Cur-NPs might be a promising treatment for liver cancer.
Keywords: doxorubicin, curcumin, codelivery, liver cancer, cytotoxicity, tumor growth inhibition
Introduction
Liver cancer is a leading cause of cancer deaths worldwide.1 Since most patients are diagnosed at advanced stages, there is an urgent need for effective nonsurgical therapies, such as systemic chemotherapy.2 Currently, the most active single-agent drugs in liver cancer include doxorubicin (DOX), sorafenib, 5-fluorouracil, and cisplatin. However, the response rates are only 10% without significant impact on overall survival, which might be caused by the toxicity and poor response due to chemoresistance.3
Combination therapy is emerging as an important strategy for a better long-term prognosis with decreased side effects. For example, the combination of anticancer agents targeting multiple pathways, or a combination of chemotherapy with chemosensitizers has been shown to modulate different signaling pathways in cancer cells, which is beneficial to overcome multidrug resistance (MDR), maximize the therapeutic effect, and reduce side effects.4–6 Compared with a multiagent combination, the combination of cytotoxic and chemosensitizing agents exhibited better performance in the intracellular microenvironment, and are thus highlighted as more effective for tumor localization and for overcoming MDR.6,7
Despite the benefits of combination therapy, the nanoparticle (NP)-based targeted drug delivery (nanocarriers) has been designed to increase drug accumulation at tumor sites to improve permeability and retention,8 pharmacokinetic profiles, and reduce side effects.9,10 Therefore, the delivery of a chemotherapeutic agent and chemosensitizer using nanocarriers has been suggested as a novel and promising strategy in cancer treatment.11,12
DOX, an anthracycline antibiotic, is one of the most efficacious drugs in the treatment of liver cancer. However, the clinical application of DOX has been severely hindered because of its critical cardiotoxicity, narrow therapeutic window, and the development of MDR.13 In some cancer cell studies, improved MDR and increased apoptosis have been reported with treatment of the NP-based combination therapy of DOX and chemosensitizers (ie, TNF-related apoptosis-inducing ligand, verapamil, Bcl-2 small interfering [si]RNA, and P-gp siRNA).14–17
Curcumin (Cur), the polyphenol constituent of the perennial herb Curcuma longa, exhibits antioxidant, anti-inflammatory, antiangiogenic, antimicrobial, and anticancer activities.18 It also acts as a chemosensitizer to suppress the overexpression of P-glycoprotein to reverse MDR in ovarian adenocarcinoma cells.19 However, its extremely low water solubility and poor bioavailability have impeded its clinical use.20
To date, several studies have demonstrated the enhanced anticancer efficacy of the codelivery of DOX and Cur in nanocarriers, including polymeric NPs21–23 and liposomes24 in chronic myelogenous leukemia, lung, and breast cancers. However, no study has been performed in liver cancer. Moreover, the cytotoxicity of polymers, unsuitable drug release, and unavailability for large-scale production restrict the application of polymeric NPs.25 The main drawbacks of liposomes are their instability and ability to easily leak the loaded drug. Therefore, there is urgent need for more effective codelivery nanocarriers.
Lipid NPs have been demonstrated to successfully deliver DOX26,27 or Cur28,29 individually in previous studies due to good biocompatibility, high encapsulation efficacy (EE), sustained drug release, excellent stability, passive targeted delivery of anticancer drugs, and enhanced anticancer activity. We have successfully developed lipid NPs to deliver triptolide30 and podophyllotoxin31 in our previous studies. In the present work, we aim to develop lipid NPs to deliver DOX and Cur simultaneously, and to examine their efficacy on liver cancer in vitro and in vivo.
Materials and methods
Materials
DOX hydrochloride was purchased from Beijing Huafeng United Technology Co., Ltd. (Beijing, People’s Republic of China). Cur was purchased from Sinopharm Chemical Reagent Co., Ltd. (Shanghai, People’s Republic of China). Glyceryl distearate (Precirol ATO 5®) and medium-chain triglycerides (Labrafac™ Lipophile WL 1349) were kindly provided by Gattefossé (Genas, France). Polyoxyl 40 hydrogenated castor oil (Cremophor® RH 40) was obtained from BASF SE (Ludwigshafen, Germany). Soybean lecithin (Lipoid S75) was purchased from Lipoid GmbH (Ludwigshafen, Germany). Dulbecco’s Modified Eagle’s Medium (DMEM) and fetal bovine serum (FBS) were purchased from Gibco® (Thermo Fisher Scientific, Waltham, MA, USA). 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT), dimethyl sulphoxide (DMSO) of analytical reagent grade, and diethylnitrosamine (DEN) were obtained from Sigma-Aldrich Co. (St Louis, MO, USA). Acetonitrile (high-performance liquid chromatography [HPLC] grade) was purchased from Tedia Company (Fairfield, OH, USA). Ultrapure deionized water (Heal Force Bio-meditech Holdings Limited, Shanghai, People’s Republic of China) was used for all the experiments. All other reagents were of analytical grade.
Human normal liver cells (L02) and liver cancer cells (HepG2) were purchased from the Chinese Academy of Sciences (Shanghai, People’s Republic of China).
Male Kunming mice (18–22 g) were purchased from the Laboratory Animals Center of Tongji Medical College of Huazhong University of Science and Technology (Wuhan, People’s Republic of China). All studies on mice were approved by the Committee on the Ethics of Animal Experiments of the South-Central University for Nationalities, Wuhan, People’s Republic of China (permit number: 2012-SCUEC-AEC-005).
Preparation of DOX/Cur-NPs
DOX hydrochloride was converted to its free base, as described previously.32 NPs containing DOX and Cur were formulated using a high-pressure microfluidics technique. Briefly, the lipid phase, consisting of 300 mg of Precirol ATO 5, 100 mg of Labrafac Lipophile WL 1349, and 100 mg of Lipoid S75, was dissolved in ethanol and heated to 75°C. DOX (5 mg) and Cur (5 mg) were then added to the lipid sample. After removal of the solvent by rotary evaporation, 9.3 mL of preheated water (containing 250 mg of Cremophor RH 40) was added gradually to the hot and molten lipid sample and was gently magnetically stirred for 10 minutes. A coarse oil-in-water emulsion was formed by high-speed shearing via a Fluko® FA25 homogenizer (Fluko Equipment Shanghai Co., Ltd, Shanghai, People’s Republic of China) at 10,000 rpm for 30 seconds. The coarse emulsion was further homogenized for six cycles at 1,000 bar, using M-100PCE, a high-pressure microfluidics device (Microfluidics Corporation, Westwood, MA, USA). The hot dispersion was cooled down at 4°C and sterilized with a 0.45 μm cellulose acetate filter. Finally, the obtained material was stored in brown glass vessels at 4°C. NPs loaded with DOX (DOX-NPs), Cur (Cur-NPs), DOX and Cur at different weight ratios (DOX/Cur-NPs), and blank NPs were prepared using the same procedure.
Measurement of particle size and zeta potential
Particle size and zeta potential were measured by a Zetasizer Nano ZS90 (Malvern Instruments, Malvern, UK). Particle size (hydrodynamic diameter, nm) and polydispersity index (PDI) were determined by diluting 10 μL of NPs with 4 mL of ultrapure water. To determine the zeta potential, the NPs were diluted with ultrapure water until the conductivity of the dilute suspension was in the range of 40–50 μS/cm. All measurements were performed in triplicate.
Drug loading and encapsulation efficiency
The amount of DOX in DOX/Cur-NPs was determined with a Hitachi F-4500 fluorescence spectrophotometer (emission wavelength: 485 nm; Excitation wavelength: 590 nm; Hitachi Ltd., Tokyo, Japan). The amount of Cur was measured with a HPLC method using Agilent 1260 Infinity LC (Agilent Technologies, Santa Clara, CA, USA). HPLC analyses were performed on a Hypersil™ ODS-2 C18 column (250 mm ×4.6 mm; 5 μm). The mobile phases used were: acetonitrile and 4% (V/V) glacial acetic acid at the ratio of 45:55; temperature: 30°C; flow rate: 1.0 mL/minute; injection volume: 20 μL; ultraviolet detection: 420 nm. Briefly, 10 μL of NPs were diluted with 2 mL of methanol. The sample was mixed on a vortex for 30 seconds and centrifuged at 8,000 rpm for 5 minutes at 4°C. The amount of DOX or Cur in the supernatant (W1) was determined following the methods described earlier.
The EE of DOX or Cur was determined using the ultrafiltration method. A total of 0.5 mL of undiluted NPs were placed into the upper chamber of centrifugal filter tubes (molecular weight cut-off [MWCO]: 10 kDa; EMD Millipore, Billerica, MA, USA) and centrifuged for 30 minutes at 4,000 rpm. The amount of free DOX or Cur in the filtered aqueous phase (W2) was determined following the methods described earlier.
The drug loading (DL) and encapsulation efficiency (EE) were calculated by the following equations:
DL = (W1−W2)/W ×100%, | (1) |
EE = (W1−W2)/W1 ×100%, | (2) |
where W1 was the weight of the total drug in the NPs and W2 was the weight of the unencapsulated drug. W was the weight of the nanocarriers.
Transmission electron microscopy (TEM)
The morphology of DOX/Cur-NPs was observed by a TEM (3H-7000FA; Hitachi Ltd.). 100 μL of NPs were diluted with deionized water to 2 mL and sonicated for 1 minute. TEM samples were prepared by placing one drop of diluted NPs on a carbon-coated copper grid (400 mesh; Beijing Xinxing Braim Technology Co., Ltd, Beijing, People’s Republic of China) and allowing adsorption for 10 minutes. Then, the excess of liquid was blotted with a filter paper. After drying naturally for 1 hour, the grid was negatively stained with 2% phosphotungstic acid for 3 minutes and allowed to dry naturally. The dried specimen was observed with TEM at an acceleration voltage of 75 kV.
Differential scanning calorimetry (DSC)
A diamond differential scanning calorimeter (Diamond DSC; PerkinElmer Inc., Waltham, MA, USA) was employed to detect thermodynamic properties of DOX/Cur-NPs. The samples, including DOX, Cur, ATO 5, and a physical mixture of ATO 5, DOX, and Cur (60:1:1, w/w), lyophilized blank NPs, and lyophilized DOX/Cur-NPs (1:1) were weighted and placed in sealed aluminum crimp cell, and heated from 30°C to 230°C at a rate of 5°C/minute.
X-ray diffraction (XRD)
The crystallographic structure of DOX, Cur, ATO 5, the physical mixture, lyophilized blank NPs, and lyophilized DOX/Cur-NPs (1:1) were characterized by X-ray diffraction (XRD) (D8 Advance; Bruker Optik GmbH, Ettlingen, Germany). Samples were exposed to Cu-Ka radiation (40 kV; 40 mA) at a scan rate of 0.02°/second over the 2θ/minute range of 5°–50°.
In vitro release studies
In vitro release patterns of DOX and Cur were evaluated by the dialysis bag diffusion technique. A total of 1 mL of DOX-NPs, Cur-NPs, and DOX/Cur-NPs (2:1, 1:1, 1:2, respectively) were filled in dialysis bags (MWCO: 12–14 KDa) individually. The dialysis bags were immersed in 50 mL of PBS (0.01 M; pH 7.4) containing Tween® 80 (1%, w/v) and ethanol (20%, v/v) at 37°C in a QYC-200 shaker incubator (Shanghai CIMO Medical Equipment Manufacturing Co., Ltd, Shanghai, People’s Republic of China) at 100 rpm. Then, 1 mL sample of the released medium was withdrawn at 0.5 hours, 1 hour, 2 hours, 4 hours, 8 hours, 12 hours, 24 hours, and 48 hours, separately, and replaced with an equivalent volume of fresh release medium to maintain constant volume. The DOX and Cur content released from different samples were analyzed. All experiments were carried out in triplicate.
In vitro cytotoxicity
The cytotoxicity of blank NPs in L02 and HepG2 cells was evaluated by MTT assay. Briefly, cells (1×105 cells/mL) were seeded into 96-well plates in DMEM medium supplemented with 10% FBS, and then treated with 10–75,000 μg/mL of blank NPs or DMEM medium (control) for 48 hours. Then, 25 μL of MTT solution (5 mg/mL) was added to each well and the plate was incubated at 37°C for 3 hours. After incubation, DMSO was added and the absorbance was measured at 570 nm using a microplate reader (Infinite® 200 PRO; Tecan Group Ltd., Maennedorf, Switzerland). The inhibitory concentration (IC)50 values were calculated by GraphPad Prism 5 software (GraphPad Software, Inc., La Jolla, CA, USA).
Cell growth inhibition
The growth inhibition of free DOX, a physical mixture of DOX and Cur (DOX+Cur; 1:1), DOX-NPs, and DOX/Cur-NPs (2:1, 1:1, 1:2, respectively) were also evaluated by MTT assay, as described earlier. Untreated cells served as controls.
Apoptosis analysis
Apoptosis was measured by Annexin V-fluorescein isothiocyanate (FITC)/propidium iodide (PI) double staining. Briefly, HepG2 cells (1×105 cells/mL) were seeded in six-well plates and incubated with free DOX, DOX+Cur (1:1), DOX-NPs, and DOX/Cur-NPs (1:1) with an equivalent concentration of DOX of 1 μg/mL for 24 hours. Then, cells were treated with Annexin V-FITC/PI dye according to the manufacturer’s instructions (BestBio, Shanghai, People’s Republic of China). Cells incubated with PBS served as control. Finally, cells were observed under fluorescence microscopy (Nikon Ti series; Nikon Corporation, Tokyo, Japan).
In vivo tumor growth inhibition
A liver cancer model induced by DEN with modifications was employed according to a previous study.33 Male Kunming mice (18 to 22 g) were orally administrated the DEN solution (in sesame oil; 0.1 g/mL) at a dosage of 40 mg/kg once a week from 1–15 weeks. A total of 32 mice were randomly divided into four groups. Three groups of mice were treated with DEN for 15 weeks and then injected intravenously with saline solution, DOX-NPs (2 mg/kg), and DOX/Cur-NPs (1:1; equivalent to 2 mg/kg of DOX) from 16 weeks to 35 weeks, respectively. Mice without DEN treatment served as control. All mice were sacrificed at the 36th week. Livers were collected. A part of each liver, which was used for histopathological analysis, was fixed with 10% formalin, dehydrated, and paraffin embedded successively. The remaining part of the liver was stored at -80°C. The number of tumor nodules ≥1 mm in diameter on the surface of the livers, as well as the maximum nodule size (diameter, mm) were measured.
Histopathological analysis
Histopathological analysis was performed by sectioning the paraffin-embedded livers into slices of 5 μm thickness and stained with hematoxylin and eosin for microscopic examination (Ti50; Nikon Corporation).
Statistical analysis
One-way analysis of variance was used to determine statistical significance in terms of the cumulative release rate, cell viability, tumor number, and maximum tumor size. The results are expressed as means ± the standard error of the mean. P-values <0.05 were considered statistically significant. All statistical analyses were performed using SPSS version 19 (IBM Corporation, Armonk, NY, USA).
Results and discussion
Physical characterization of DOX/Cur-NPs
Particle size, PDI, and zeta potential of the prepared formulations detected by the Zetasizer are shown in Table 1 and Figure 1. The mean particle size for each formulation was approximately 90 nm. The PDI of each formulation was <0.3, indicating the homogeneous nature of the formulation. Figure 1A shows a narrow particle size distribution of DOX/Cur-NPs (1:1) with a span of 30–400 nm. No multiscattering phenomenon was observed.
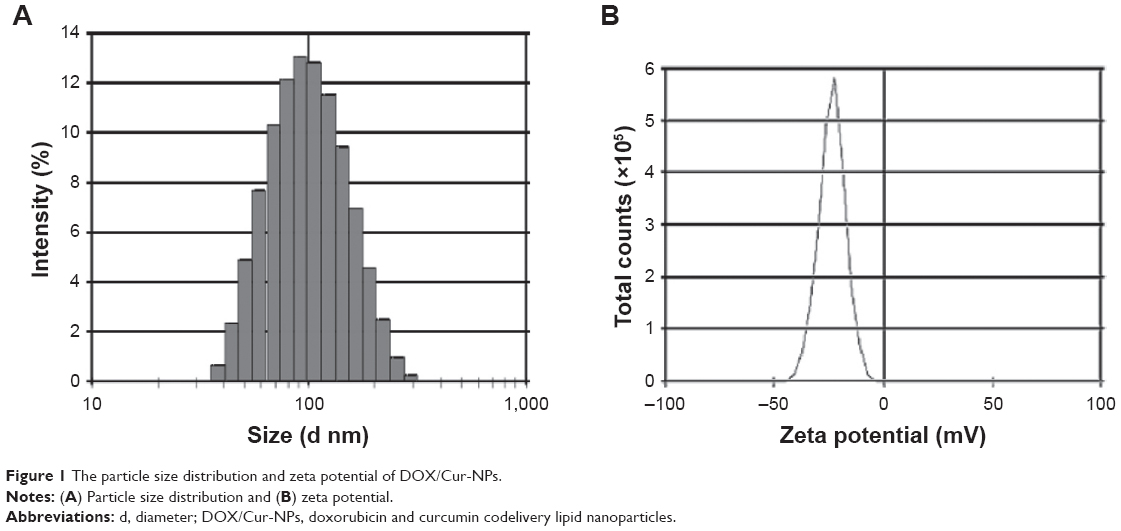 | Figure 1 The particle size distribution and zeta potential of DOX/Cur-NPs. |
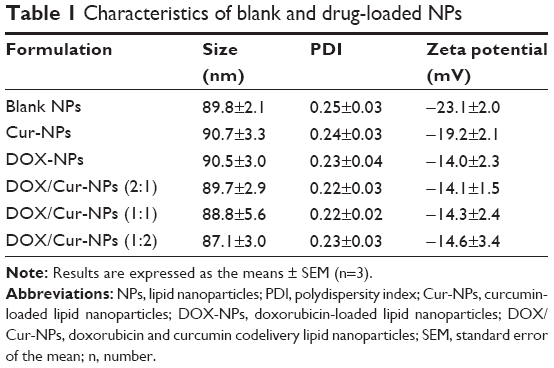 | Table 1 Characteristics of blank and drug-loaded NPs |
As shown in Figure 1B and Table 1, all formulations exhibit a negative charge of <−10 mV. The most likely origin of this negative charge is the presence of the anionic-type surfactant, soybean lecithin. Compared with blank NPs, we observed a slow increase in the zeta potential of Cur-NPs, which might be caused by a partial absorption of Cur on the NPs surface and the masking of negative charges in the surfactant. A significant increase in the zeta potential values for DOX-loaded NPs, as compared to blank NPs, could also be explained by the adsorption of DOX on the NPs surface. Although DOX contains an amino group with a positive charge, the zeta potential of DOX/Cur-NPs was slightly lower than that of DOX-NPs. Our hypothesis is that Cur facilitated the encapsulation of DOX in DOX/Cur-NPs, thereby mitigating the impact of DOX on surfactant negative charges.
As shown in Table 2, the loaded amount of DOX in DOX/Cur-NPs is comparable with that in DOX-NPs. The loaded amount of Cur in DOX/Cur-NPs is proportional to that of DOX. These results indicate the successful preparation of DOX/Cur-NPs.
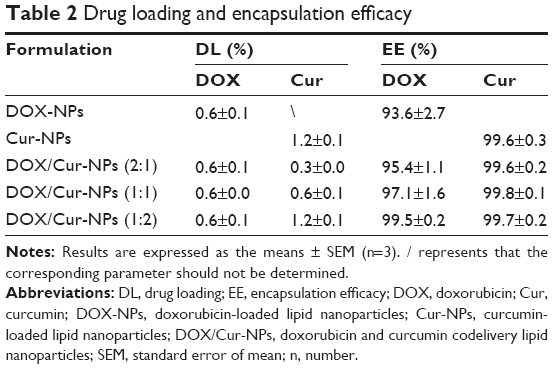 | Table 2 Drug loading and encapsulation efficacy |
The high EE (>99%) of Cur in each formulation (Table 2) may be related to Cur’s high affinity to lipids and low aqueous solubility.29 It is supported by the high EE when ATO 5 is used to incorporate the lipophilic drug Itraconazole into lipid NPs.34 The high EE may also be attributed to the massive crystal order disturbance caused by the incorporation of liquid lipid in solid lipid, resulting in a lipid matrix with crystal lattice defects, which provides enough space to accommodate drug molecules in the matrix.35
Similarly, we also observed high EE of DOX (>90%), which might be related to the conversion of DOX hydrochloride into a less soluble free base to facilitate DOX distribution into the oil phase.36 A slightly improved EE of DOX with the increase of Cur was detected, indicating a promoting effect of Cur on the encapsulation of DOX. This result verified the hypothesis in terms of the zeta potential measurement.
The morphological study of DOX/Cur-NPs using TEM (Figure 2) revealed that the DOX/Cur-NPs are spherical in shape with a smooth surface and uniform particle size (approximately 80 nm). The particle size and distribution by TEM correlated well with the results obtained from dynamic light scattering using a Zetasizer.
 | Figure 2 Transmission electron microscopy photographs of DOX/Cur-NPs. |
DSC was employed to investigate the thermal behavior of crystalline DOX/Cur-NPs. DSC thermograms were recorded for ATO 5; DOX; Cur; the physical mixture of ATO 5, DOX, and Cur (60:1:1); lyophilized blank NPs; and lyophilized DOX/Cur-NPs (Figure 3). The DSC curve of the physical mixture showed a melting peak of DOX at 217°C and a melting peak of Cur at 185°C. Furthermore, the presence of DOX and Cur in bulk lipid was confirmed by the decreased enthalpy of the physical mixture (151.17 J/g) compared with that in ATO 5 (157.05 J/g) (Table 3). These results indicated that DOX and Cur were not completely dissolved in ATO 5 and, therefore, remained in a crystalline state in the solid lipid. However, we did not observe a melting peak of DOX and Cur in the thermogram of the lyophilized DOX/Cur-NPs, indicating that DOX and Cur in NPs were in an amorphous state. A melting point depression (6.42°C) and a broadening peak (1.7-fold increase) in Precirol ATO 5 was observed when compared with those in blank NPs, which might be attributed to the colloidal size effect predicted by the Thomson equation.37 The DSC diagrams showed a lower enthalpy in DOX/Cur-NPs (51.32 J/g) than that in blank NPs (61.65 J/g), suggesting that miscible liquid lipid had an additional and greater effect with respect to colloidal size and surfactant to promote disordered arrangement and lattice defects.
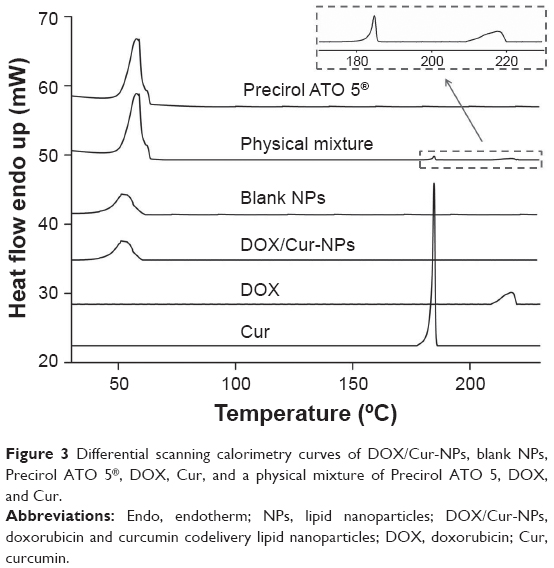 | Figure 3 Differential scanning calorimetry curves of DOX/Cur-NPs, blank NPs, Precirol ATO 5®, DOX, Cur, and a physical mixture of Precirol ATO 5, DOX, and Cur. |
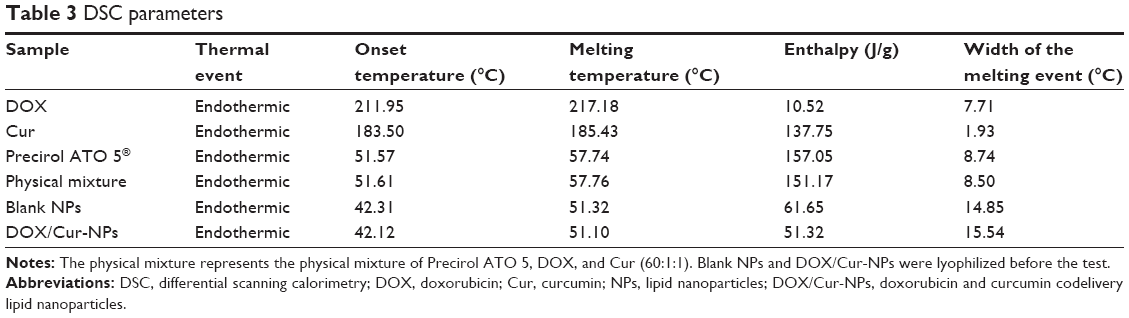 | Table 3 DSC parameters |
Overlaid XRD patterns of DOX, Cur, ATO 5, the physical mixture, lyophilized blank NPs, and lyophilized DOX/cur-NPs are shown in Figure 4. The XRD patterns of DOX and Cur exhibited sharp peaks at scattered angles, ranging from 15°–25° and from 8°–16°, respectively, indicating their crystalline nature. In the physical mixture, slight diffraction peaks of DOX and Cur around 16° were observed, revealing incomplete dissolution and the existence of a crystalline state of both drugs in bulk lipid. However, there were no characteristic peaks for DOX and Cur in DOX/Cur-NPs, suggesting the amorphous state of DOX and Cur in DOX/Cur-NPs. XRD patterns of the blank NPs and DOX/Cur-NPs were broader and much weaker than that of the bulk lipid. It indicated that ATO 5 in the NPs was partially recrystallized or less ordered due to the presence of liquid lipid. This DOX/Cur-NP XRD result was in agreement with that of DSC.
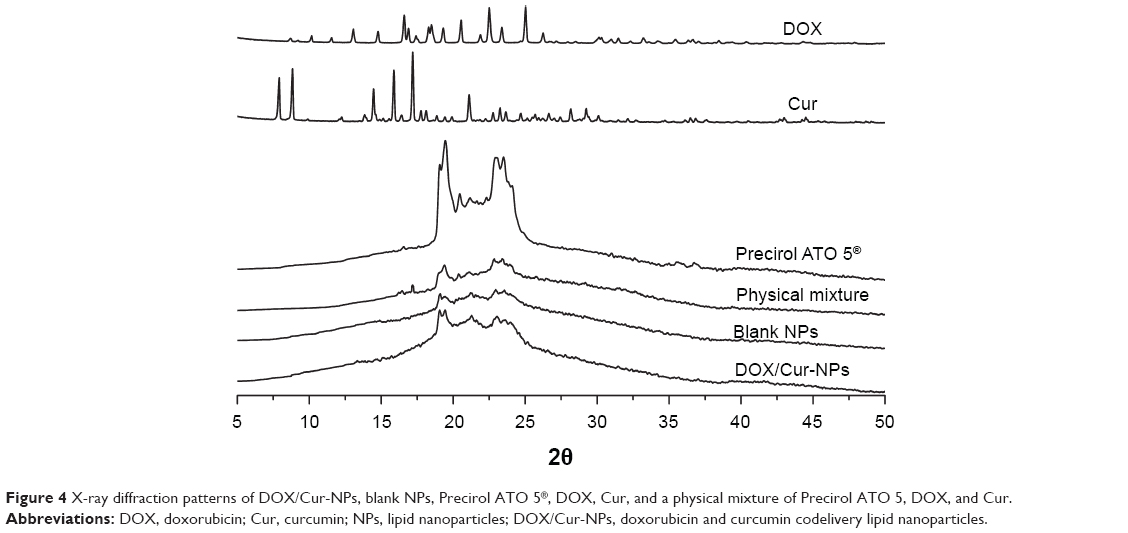 | Figure 4 X-ray diffraction patterns of DOX/Cur-NPs, blank NPs, Precirol ATO 5®, DOX, Cur, and a physical mixture of Precirol ATO 5, DOX, and Cur. |
In vitro drug release profile
The in vitro release profile in Figure 5A shows the biphasic release of DOX-NPs and DOX/Cur-NPs (2:1, 1:1, 1:2) with rapid release in the first 2 hours followed by sustained release until 48 hours. The initial rapid release could be ascribed to the DOX incorporated at the surface of the lipid NPs.38 The later sustained-release stage indicated that DOX might be stably retained in the lipid core before being slowly released by drug diffusion.39 However, this retardation was prominent in DOX/Cur-NPs when compared to DOX-NPs, with a concentration-dependent release observed for Cur. Compared with that in DOX-NPs (45.9%) at 2 hours, the drug release in DOX/Cur-NPs at 1:1 (33.3%; P<0.01) and 1:2 (28.1%, P<0.01) was significantly decreased. Similarly, the drug release in DOX/Cur-NPs (1:2) was significantly lower when compared with that in DOX/Cur-NPs 2:1 (39.0%; P<0.05). The accumulated drug release at 48 hours of DOX/Cur-NPs at 1:1 (55.7%; P<0.05) and 1:2 (53.5%; P<0.05) were significantly lower than that of DOX-NPs (64.9%). However, there was no significant difference in terms of drug release among DOX/Cur-NPs at 2:1, 1:1, and 1:2. One possible explanation for this is that the high EE of DOX in lipid NPs may contribute to decreased burst release and prolonged release. The prolonged release profile of Cur without burst release is presented in Figure 5B. No significant difference was observed in Cur release in Cur-NPs and DOX/Cur-NPs (2:1, 1:1, and 1:2) even at 48 hours (37.1%, 38.9%, 40.6%, and 43.0%, respective to the Cur release in Cur-NPs, DOX/Cur-NPs (2:1), DOX/Cur-NPs (1:1) and DOX/Cur-NPs (1:2).
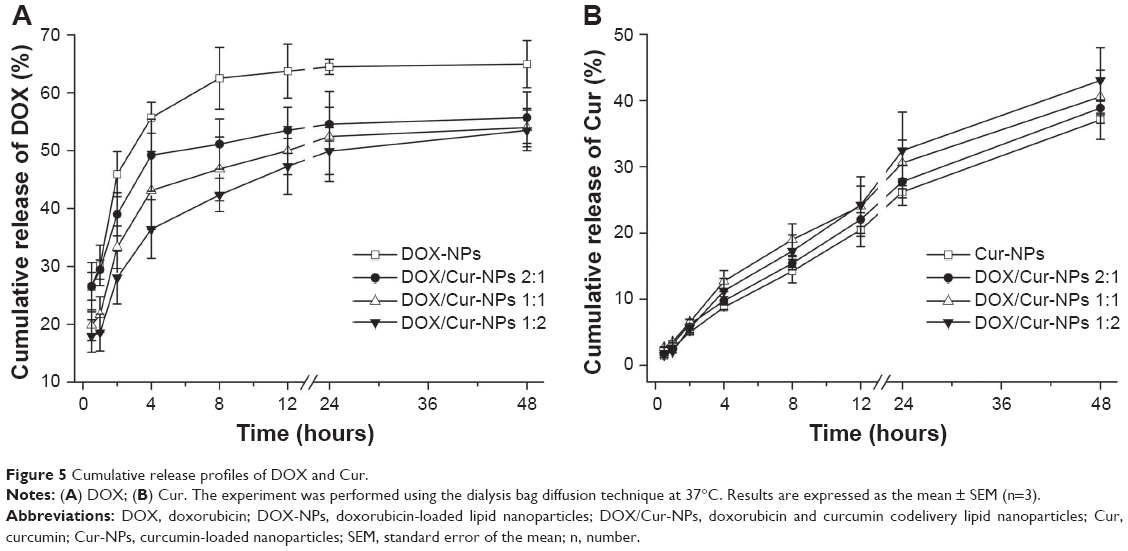 | Figure 5 Cumulative release profiles of DOX and Cur. |
The drug release data obtained were fitted into release kinetic models, including zero order, first order, Higuchi, and Ritger–Peppas equations40 (Table 4). The release of DOX from DOX-NPs calculated in the Ritger–Peppas equations was better than that calculated for others (r=0.9125 for Ritger–Peppas; r=0.7867 for Higuchi; r=0.6649 for first order; and r=0.6295 for zero order). Similarly, the release of DOX from DOX/Cur-NPs (2:1, 1:1, 1:2) calculated in Ritger–Peppas was also ascribed to higher r-values. For Cur, the Higuchi and Ritger–Peppas models fit well according to the higher r-values when compared with the others that are shown in Table 4. The lnt coefficient (“lnt” represents the natural logarithm of time) in the Ritger–Peppas equation of <0.45 indicated that DOX release from NPs was due to Fickian diffusion, while an lnt coefficient between 0.45 and 0.89 indicated that the release mechanism of Cur from NPs involved diffusion and matrix erosion.41
 | Table 4 The regression equation of the DOX and Cur in vitro release |
In vitro cytotoxicity of blank NPs
The cytotoxicity results of blank NPs in HepG2 and L02 cells are shown in Figure 6. We observed 99.5% cell viability in HepG2 with treatment of blank NPs at 0.6 mg/mL (Figure 6A), and 74.5% cell viability in L02 cells with treatment of blank NPs at 1.0 mg/mL (Figure 6B). The IC50 of blank NPs is 3.1 mg/mL in HepG2 and 4.0 mg/mL in L02 cells. These results suggested that the blank NPs were nontoxic.
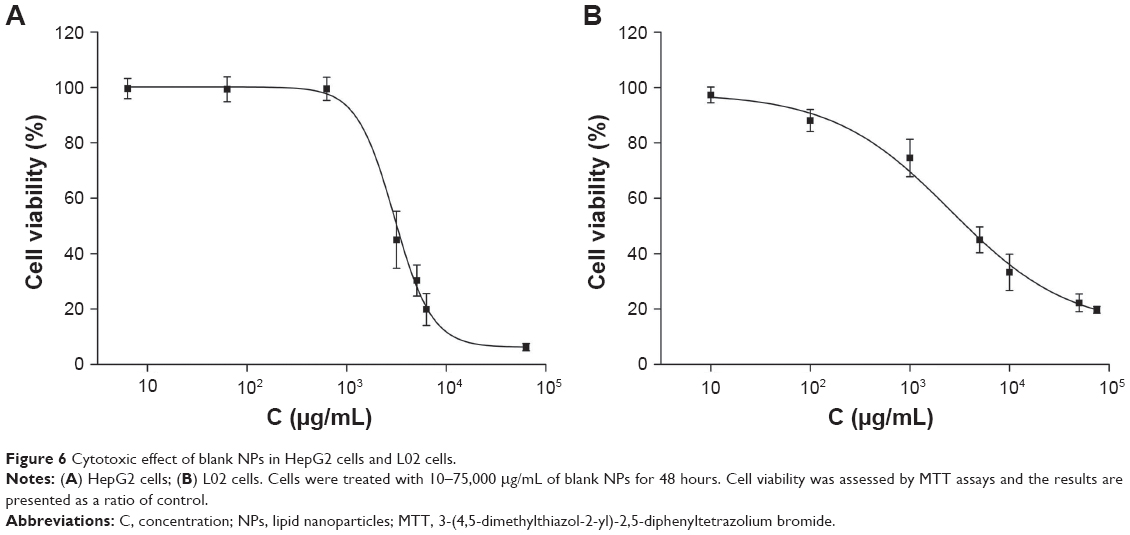 | Figure 6 Cytotoxic effect of blank NPs in HepG2 cells and L02 cells. |
Cell growth inhibition of drug-loaded NPs
A concentration-dependent decrease in cell viability was observed in HepG2 cells treated with free DOX, DOX+Cur (1:1), DOX-NPs, and DOX/Cur-NPs (2:1, 1:1, 1:2) (Figure 7A). Compared with free DOX, we observed higher cell cytotoxicity in DOX-NPs and DOX/Cur-NPs (2:1, 1:1, 1:2), which was in line with previous reports.42–45 In HepG2 cells, the IC50 of DOX was lower in DOX-NPs (1.16 μg/mL) when compared with that of free DOX (2.74 μg/mL), and that of DOX/Cur-NPs was lower (0.82 for 2:1; 0.42 for 1:1; 0.68 for 1:2) when compared with that of DOX+Cur (1.26 μg/mL) (Table 5), further confirming the improved cell cytotoxicity of DOX in NP-loaded formulations. The higher IC50 of Cur in HepG2 cells (8.84 μg/mL) than that of DOX suggests that Cur exhibits weaker cytotoxicity.46 However, Cur has been demonstrated to be a potent chemosensitizer that can induce synergistic effects with chemotherapeutic drugs against different cancer cell lines.19,21,24,47 In our study, DOX/Cur-NPs (2:1, 1:1, 1:2) showed a decrease (1.4-, 2.8-, and 1.7-fold, respectively) in IC50 when compared to that of DOX-NPs, further confirming the synergistic effects of DOX and Cur. Notably, DOX/Cur-NPs (1:1) displayed optimal effects on HepG2 cells according to the lower cell viability and IC50 compared with DOX/Cur-NPs (2:1 and 1:2).
 | Figure 7 Cell viability of HepG2 cells and L02 cells. |
 | Table 5 IC50 of DOX in HepG2 and L02 cells |
In Figure 7B, we observed the decreased cytotoxicity of DOX-NPs and DOX/Cur-NPs (2:1, 1:1, 1:2) when compared with that of free DOX in L02 cells, which might be caused by the incomplete release of DOX. The relative low release of DOX (65% after 48 hours) from lipid nanoparticles may reduce the adverse effect on L02 cells. Compared with the IC50 of DOX in DOX-NPs, DOX/Cur-NPs (2:1, 1:1, 1:2) showed increases (1.25-, 1.44-, and 1.38-fold, respectively), which may also be caused by the incomplete release of DOX. Notably, DOX/Cur-NPs (1:1) showed higher cell viability and IC50 than those of DOX/Cur-NPs (2:1) and (1:2). This result may be ascribed to the interaction between DOX and Cur in the NPs. Some studies indicated that the interaction between the chemotherapy drug and chemosensitizer are closely related to their ratio in nanocarriers. Ganta and Amiji19 studied the codelivery of paclitaxel and Cur with nanoemulsion; the results showed synergistic effects on the proliferation of SKOV3 cells at a weight ratio of 1:1. Barui et al24 reported the synergistically inhibitory effect of DOX and Cur on the cell growth of B16F10 tumor cells and human umbilical vein endothelial tumor cells by codelivery in liposomes at a molar ratio of 1:5. Similar results were found by the codelivery of DOX and Cur at the weight ratio of 1:1 and paclitaxel and tetrandrine at the molar ratio of 1:267 in methoxy polyethylene glycol-polycaprolactone nanoparticles (mPEG-PCL NPs).22,48 Thus, the higher cell viability and IC50 of DOX/Cur-NPs (1:1) suggested that 1:1 might be the optimal ratio of DOX and Cur in DOX/Cur-NPs.
Finally, the highest increase in cell viability and IC50 in L02 cells of DOX+Cur indicated that the mixture may have certain protective functions on the liver.
Apoptosis analysis
Annexin V-FITC and PI double staining was performed to determine the apoptosis in HepG2 cells. Untreated cells were primarily Annexin V-FITC- and PI-negative, indicating that they were viable and not undergoing apoptosis. After treatment with DOX/Cur-NPs (1:1), intense FITC green in the membrane and PI red in the nucleus were observed (Figure 8). In contrast, weak luminescence was observed from HepG2 cells treated with PBS, free DOX, DOX+Cur, and DOX-NPs, supporting the idea that DOX/Cur-NPs could induce enhanced apoptosis in HepG2 cells.
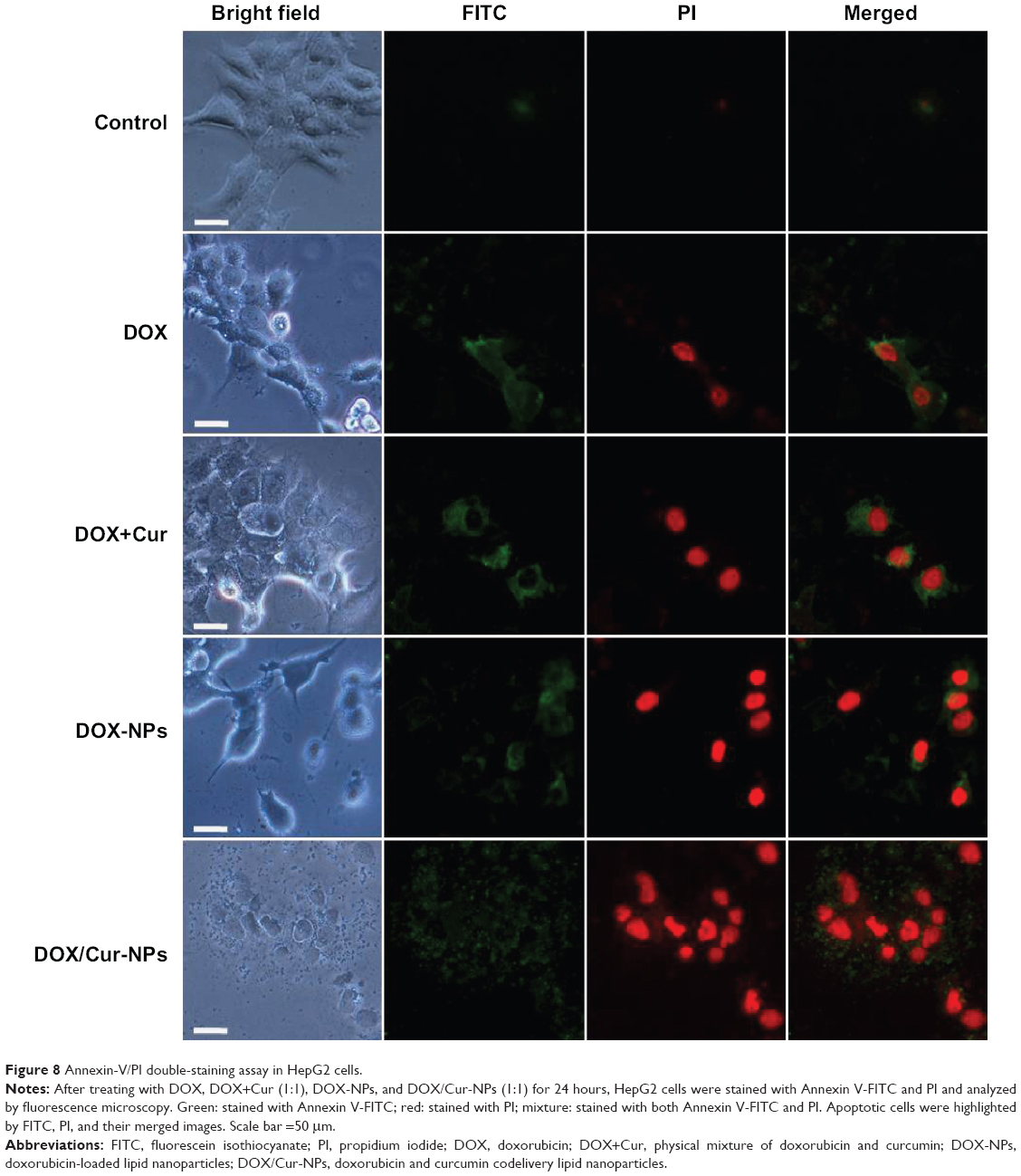 | Figure 8 Annexin-V/PI double-staining assay in HepG2 cells. |
In vivo tumor growth inhibition
To explore the efficacy of DOX/Cur-NPs on liver cancer in vivo, a well-established model induced by DEN was employed. Results of cell growth inhibition and apoptosis indicated that more effective efficacy was achieved by drug-loaded lipid NPs (DOX-NPs, DOX/Cur-NPs) than by the free base (free DOX, DOX+Cur). Thus, the efficacy of free DOX and DOX+Cur on liver cancer in vivo was not considered in this study. The optimal weight ratio of DOX and Cur was 1:1 according to the cell growth inhibition analysis; therefore, DOX/Cur-NPs (1:1) were employed for the in vivo tumor growth inhibition analysis. Several visible nodules appeared on the liver surface after treatment with DEN (Figure 9A). Treatment with DOX-NPs significantly reduced the number (Figure 9B; 4.2±0.4 to 2.4±0.5; P<0.05) and size (Figure 9C; diameter, 5.4±0.8 mm to 2.8±0.6 mm; P<0.05) (The pairs of values in the first set of parentheses correspond to the tumor number in saline group and DOX-NPs group, respectively. The pairs of values in the second set of parentheses correspond to the maximum tumor size in saline group and DOX-NPs group, respectively) of these nodules compared with the saline group, illustrating the anticancer efficacy of DOX. In the DOX/Cur-NPs group, the growth of tumor nodules was further significantly suppressed when compared with DOX-NPs (P<0.05), indicating the synergistic efficacy of DOX and Cur on liver cancer.
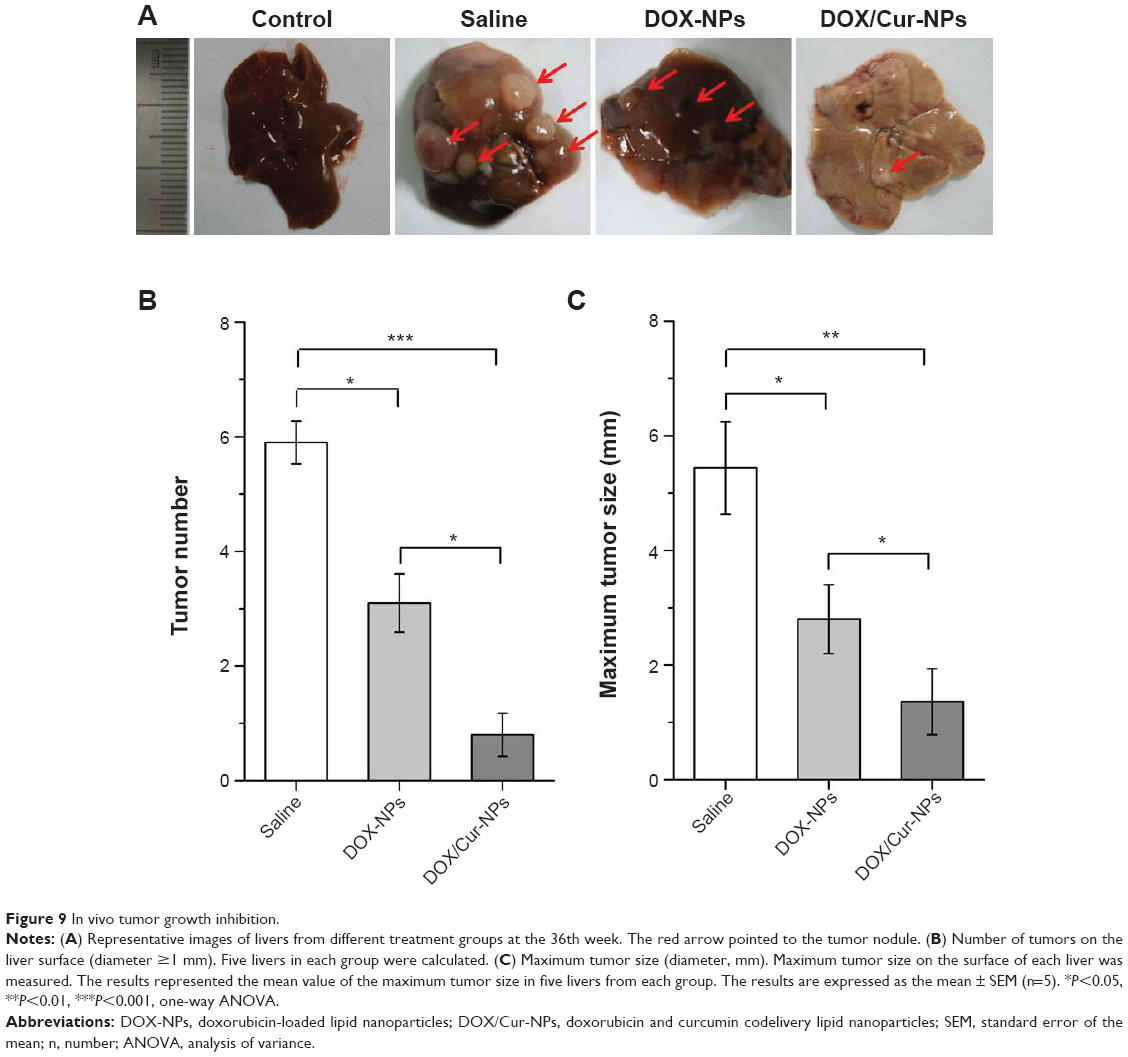 | Figure 9 In vivo tumor growth inhibition. |
Histopathological analysis
Histopathological examination showed well differentiated hepatocellular carcinoma of the trabecular type and severe hepatic steatosis in the saline group (Figure 10). Treatment with DOX-NPs reduced the differentiation of hyperplastic nodules, but their inflammatory cell infiltration and hepatocyte necrosis were noted in the adjacent tissues. However, treatment with DOX/Cur-NPs decreased the development of tumor nodules. Meanwhile, the adjacent tissues showed near normal architecture that was identical to the control group. These results indicated the enhanced antitumor effect of DOX by codelivery with Cur in lipid nanoparticles.
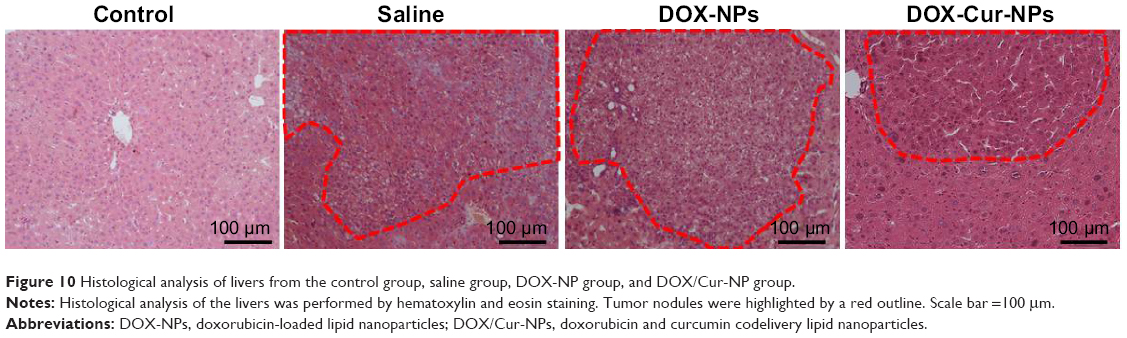 | Figure 10 Histological analysis of livers from the control group, saline group, DOX-NP group, and DOX/Cur-NP group. |
Conclusion
In the present study, DOX and Cur codelivery lipid NPs (DOX/Cur-NPs) were successfully prepared using a high-pressure microfluidics technique. The developed DOX/Cur-NPs were nontoxic, as determined by a cell cytotoxicity study. In vitro release studies revealed a sustained-release pattern of DOX/Cur-NPs when compared to DOX-NPs. Synergistic effects of DOX/Cur-NPs compared with DOX-NPs were found, as assessed by in vitro enhanced cytotoxicity and apoptosis in HepG2 cells, reduced cytotoxicity in L02 cells, and in vivo enhanced tumor growth inhibition. Our studies suggest that the simultaneous delivery of DOX and Cur by DOX/Cur-NPs might be a promising treatment for liver cancer.
Acknowledgments
This work was supported by the National Basic Research Program of China (973 Program, 2012CB932500), the National High Technology Research and Development Program of China (863 Program, SS2012AA022704), and the Natural Science Foundation of China (NSFC; 31170960 and 81101538). In addition, we would like to thank the Analytical and Testing Center of the Huazhong University of Science and Technology for the TEM, DSC, and XRD analysis.
Disclosure
The authors report no conflicts of interest in this work.
References
Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011;61(2):69–90. | ||
Villanueva A, Llovet JM. Targeted therapies for hepatocellular carcinoma. Gastroenterology. 2011;140(5):1410–1426. | ||
Asghar U, Meyer T. Are there opportunities for chemotherapy in the treatment of hepatocellular cancer? J Hepatol. 2012;56(3):686–695. | ||
Broxterman HJ, Georgopapadakou NH. Anticancer therapeutics: “Addictive” targets, multi-targeted drugs, new drug combinations. Drug Resist Updat. 2005;8(4):183–197. | ||
Greco F, Vicent MJ. Combination therapy: opportunities and challenges for polymer-drug conjugates as anticancer nanomedicines. Adv Drug Deliv Rev. 2009;61(13):1203–1213. | ||
Saraswathy M, Gong S. Different strategies to overcome multidrug resistance in cancer. Biotechnol Adv. 2013;31(8):1397–1407. | ||
Vinod BS, Maliekal TT, Anto RJ. Phytochemicals as chemosensitizers: from molecular mechanism to clinical significance. Antioxid Redox Signal. 2013;18(11):1307–1348. | ||
Matsumura Y, Maeda H. A new concept for macromolecular therapeutics in cancer chemotherapy: mechanism of tumoritropic accumulation of proteins and the antitumor agent smancs. Cancer Res. 1986;46(12 Pt 1):6387–6392. | ||
Peer D, Karp JM, Hong S, Farokhzad OC, Margalit R, Langer R. Nanocarriers as an emerging platform for cancer therapy. Nat Nanotechnol. 2007;2(12):751–760. | ||
Davis ME, Chen ZG, Shin DM. Nanoparticle therapeutics: an emerging treatment modality for cancer. Nat Rev Drug Discov. 2008;7(9):771–782. | ||
Hu CM, Aryal S, Zhang L. Nanoparticle-assisted combination therapies for effective cancer treatment. Ther Deliv. 2010;1(2):323–334. | ||
Livney YD, Assaraf YG. Rationally designed nanovehicles to overcome cancer chemoresistance. Adv Drug Deliv Rev. 2013;65(13–14):1716–1730. | ||
Deepa K, Singha S, Panda T. Doxorubicin nanoconjugates. J Nanosci Nanotechnol. 2014;14(1):892–904. | ||
Guo L, Fan L, Ren J, et al. A novel combination of TRAIL and doxorubicin enhances antitumor effect based on passive tumor-targeting of liposomes. Nanotechnology. 2011;22(26):265105. | ||
Liu Y, Li LL, Qi GB, Chen XG, Wang H. Dynamic disordering of liposomal cocktails and the spatio-temporal favorable release of cargoes to circumvent drug resistance. Biomaterials. 2014;35(10):3406–3415. | ||
Chen AM, Zhang M, Wei D, et al. Co-delivery of doxorubicin and Bcl-2 siRNA by mesoporous silica nanoparticles enhances the efficacy of chemotherapy in multidrug-resistant cancer cells. Small. 2009;5(23):2673–2677. | ||
Meng H, Liong M, Xia T, et al. Engineered design of mesoporous silica nanoparticles to deliver doxorubicin and P-glycoprotein siRNA to overcome drug resistance in a cancer cell line. ACS Nano. 2010;4(8):4539–4550. | ||
Wilken R, Veena MS, Wang MB, Srivatsan ES. Curcumin: A review of anti-cancer properties and therapeutic activity in head and neck squamous cell carcinoma. Mol Cancer. 2011;10:12. | ||
Ganta S, Amiji M. Coadministration of paclitaxel and curcumin in nanoemulsion formulations to overcome multidrug resistance in tumor cells. Mol Pharm. 2009;6(3):928–939. | ||
Sharma RA, Gescher AJ, Steward WP. Curcumin: the story so far. Eur J Cancer. 2005;41(13):1955–1968. | ||
Misra R, Sahoo SK. Coformulation of doxorubicin and curcumin in poly(D,L-lactide-co-glycolide) nanoparticles suppresses the development of multidrug resistance in K562 cells. Mol Pharm. 2011;8(3):852–866. | ||
Wang BL, Shen YM, Zhang QW, et al. Codelivery of curcumin and doxorubicin by MPEG-PCL results in improved efficacy of systemically administered chemotherapy in mice with lung cancer. Int J Nanomedicine. 2013;8:3521–3531. | ||
Duan J, Mansour HM, Zhang Y, et al. Reversion of multidrug resistance by co-encapsulation of doxorubicin and curcumin in chitosan/poly(butyl cyanoacrylate) nanoparticles. Int J Pharm. 2012;426(1–2):193–201. | ||
Barui S, Saha S, Mondal G, Haseena S, Chaudhuri A. Simultaneous delivery of doxorubicin and curcumin encapsulated in liposomes of pegylated RGDK-lipopeptide to tumor vasculature. Biomaterials. 2014;35(5):1643–1656. | ||
Calixto G, Bernegossi J, Fonseca-Santos B, Chorilli M. Nanotechnology-based drug delivery systems for treatment of oral cancer: a review. Int J Nanomedicine. 2014;9:3719–3735. | ||
Kang KW, Chun MK, Kim O, et al. Doxorubicin-loaded solid lipid nanoparticles to overcome multidrug resistance in cancer therapy. Nanomedicine. 2010;6(2):210–213. | ||
Wong HL, Rauth AM, Bendayan R, et al. A new polymer-lipid hybrid nanoparticle system increases cytotoxicity of doxorubicin against multidrug-resistant human breast cancer cells. Pharm Res. 2006;23(7):1574–1585. | ||
Vandita K, Shashi B, Santosh KG, Pal KI. Enhanced apoptotic effect of curcumin loaded solid lipid nanoparticles. Mol Pharm. 2012;9(12):3411–3421. | ||
Puglia C, Frasca G, Musumeci T, et al. Curcumin loaded NLC induces histone hypoacetylation in the CNS after intraperitoneal administration in mice. Eur J Pharm Biopharm. 2012;81(2):288–293. | ||
Zhang C, Peng F, Liu W, et al. Nanostructured lipid carriers as a novel oral delivery system for triptolide: induced changes in pharmacokinetics profile associated with reduced toxicity in male rats. Int J Nanomedicine. 2014;9:1049–1063. | ||
Chen H, Chang X, Du D, et al. Podophyllotoxin-loaded solid lipid nanoparticles for epidermal targeting. J Control Release. 2006;110(2):296–306. | ||
Lee ES, Na K, Bae YH. Doxorubicin loaded pH-sensitive polymeric micelles for reversal of resistant MCF-7 tumor. J Control Release. 2005;103(2):405–418. | ||
Iwai S, Murai T, Makino S, et al. High sensitivity of fatty liver Shionogi (FLS) mice to diethylnitrosamine hepatocarcinogenesis: comparison to C3H and C57 mice. Cancer Lett. 2007;246(1–2):115–121. | ||
Pardeike J, Weber S, Haber T, et al. Development of an itraconazole-loaded nanostructured lipid carrier (NLC) formulation for pulmonary application. Int J Pharm. 2011;419(1–2):329–338. | ||
Müller RH, Radtke M, Wissing SA. Solid lipid nanoparticles (SLN) and nanostructured lipid carriers (NLC) in cosmetic and dermatological preparations. Adv Drug Deliv Rev. 2002;54 Suppl 1:S131–S155. | ||
Wohlfart S, Khalansky AS, Gelperina S, et al. Efficient chemotherapy of rat glioblastoma using doxorubicin-loaded PLGA nanoparticles with different stabilizers. PLoS One. 2011;6(5):e19121. | ||
Hou D, Xie C, Huang K, Zhu C. The production and characteristics of solid lipid nanoparticles (SLNs). Biomaterials. 2003;24(10):1781–1785. | ||
Zhu RR, Qin LL, Wang M, et al. Preparation, characterization, and anti-tumor property of podophyllotoxin-loaded solid lipid nanoparticles. Nanotechnology. 2009;20(5):055702. | ||
Chen CC, Tsai TH, Huang ZR, Fang JY. Effects of lipophilic emulsifiers on the oral administration of lovastatin from nanostructured lipid carriers: physicochemical characterization and pharmacokinetics. Eur J Pharm Biopharm. 2010;74(3):474–482. | ||
Ye J, Wang A, Liu C, Chen Z, Zhang N. Anionic solid lipid nanoparticles supported on protamine/DNA complexes. Nanotechnology. 2008;19(28):285708. | ||
Ritger PL, Peppas NA. A simple equation for description of solute release II. Fickian and anomalous release from swellable devices. J Control Release. 1987;5(1):37–42. | ||
Barraud L, Merle P, Soma E, et al. Increase of doxorubicin sensitivity by doxorubicin-loading into nanoparticles for hepatocellular carcinoma cells in vitro and in vivo. J Hepatol. 2005;42(5):736–743. | ||
Thambi T, Deepagan VG, Yoon HY, et al. Hypoxia-responsive polymeric nanoparticles for tumor-targeted drug delivery. Biomaterials. 2014;35(5):1735–1743. | ||
Guo Y, Chu M, Tan S, et al. Chitosan-g-TPGS nanoparticles for anticancer drug delivery and overcoming multidrug resistance. Mol Pharm. 2014;11(1):59–70. | ||
Chiang WH, Huang WC, Chang CW, et al. Functionalized polymersomes with outlayered polyelectrolyte gels for potential tumor-targeted delivery of multimodal therapies and MR imaging. J Control Release. 2013;168(3):280–288. | ||
Fan H, Tian W, Ma X. Curcumin induces apoptosis of HepG2 cells via inhibiting fatty acid synthase. Target Oncol. 2014;9(3):279–286. | ||
Qian H, Yang Y, Wang X. Curcumin enhanced adriamycin-induced human liver-derived Hepatoma G2 cell death through activation of mitochondria-mediated apoptosis and autophagy. Eur J Pharm Sci. 2011;43(3):125–131. | ||
Li X, Lu X, Xu H, et al. Paclitaxel/tetrandrine coloaded nanoparticles effectively promote the apoptosis of gastric cancer cells based on “oxidation therapy”. Mol Pharm. 2012;9(2):222–229. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.