Back to Journals » OncoTargets and Therapy » Volume 12
Cobalt Chloride Induced Apoptosis by Inhibiting GPC3 Expression via the HIF-1α/c-Myc Axis in HepG2 Cells
Authors Tong Y, Tong K, Zhu Q, Wu Y, Yang Y, Zhang J, Hu P, Yan S
Received 14 August 2019
Accepted for publication 15 November 2019
Published 5 December 2019 Volume 2019:12 Pages 10663—10670
DOI https://doi.org/10.2147/OTT.S227215
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Nicola Silvestris
Yaoyao Tong,1,2 Kun Tong,1,2 Qinghong Zhu,1 Yuqin Wu,3 Yi Yang,4 Jicai Zhang,1 Pei Hu,1,5 Shirong Yan2
1Department of Laboratory Medicine, Taihe Hospital, Hubei University of Medicine, Shiyan, Hubei, People’s Republic of China; 2Hubei Key Laboratory of Wudang Local Chinese Medicine Research, School of Pharmaceutical Sciences, Hubei University of Medicine, Shiyan, Hubei, People’s Republic of China; 3Department of Central Operating Room, Taihe Hospital, Hubei University of Medicine, Shiyan, Hubei, People’s Republic of China; 4Reproductive Medicine Centre, Taihe Hospital, Hubei University of Medicine, Shiyan, Hubei, People’s Republic of China; 5Hubei Key Laboratory of Embryonic Stem Cell Research, Hubei University of Medicine, Shiyan, Hubei, People’s Republic of China
Correspondence: Shirong Yan
Hubei Key Laboratory of Wudang Local Chinese Medicine Research, School of Pharmaceutical Sciences, Hubei University of Medicine, No. 30, South Renmin Road, Maojian District, Shiyan City, Hubei Province, People’s Republic of China
Email [email protected]
Pei Hu
Department of Laboratory Medicine, Taihe Hospital, Hubei University of Medicine, No. 32, South Renmin Road, Maojian District, Shiyan City, Hubei Province, People’s Republic of China
Email [email protected]
Purpose: To investigate the role of glypican-3 (GPC3) in cobalt chloride (CoCl2)-induced cell apoptosis in hepatocellular carcinoma.
Methods: HepG2 cells were treated with CoCl2 in the absence or presence of GPC3 plasmid transfection. Cell viability and apoptosis were assessed by MTT assay and flow cytometry, respectively. The expression of GPC3, hypoxia-inducible factor 1α (HIF-1α), c-myc, sp1, poly-ADP-ribose polymerase (PARP) and caspase-3 was determined by real-time PCR, Western blotting, and immunofluorescence after the cells were treated with different concentrations of CoCl2 or siRNA targeting HIF-1α.
Results: CoCl2 significantly inhibited the proliferation of HepG2 cells and induced apoptosis. Additionally, the expression of GPC3 mRNA and protein was decreased, and overexpression of GPC3 attenuated the tumour inhibiting effects. Further studies showed that CoCl2 increased the expression of HIF-1α while reducing the expression of sp1 and c-myc; knockdown of HIF-1α elevated the expression of GPC3, sp1, and c-myc.
Conclusion: CoCl2 inhibited the growth of HepG2 cells through downregulation of GPC3 expression via the HIF-1α/c-myc axis.
Keywords: cobalt chloride, c-myc, glypican-3, hepatocellular carcinoma, hypoxia-inducible factor 1α
Introduction
Hepatocellular carcinoma (HCC) is the most common malignancy, ranking third in morbidity and fifth in mortality among cancers worldwide. It is especially prevalent in Asia and sub-Saharan Africa.1 The complex mechanism underlying HCC carcinogenesis has yet to be elucidated for the development of effective targeted drugs. Therefore, the identification of the pathogenic mechanism of HCC is crucial for HCC therapy.
Tumour growth relies on the formation of new blood vessels to supply oxygen and nutrition. It has been demonstrated that oxygen deficiency modulates tumour growth, angiogenesis, vascular invasion, and metastasis by hypoxia-induced target genes,2 which is primarily mediated by hypoxia-inducible factor 1α (HIF-1α). When the tumour volume reaches 1–2 mm3, angiogenic factors, such as VEGF, are upregulated by HIF-1α and released to accelerate neovascularization.3 As a solid tumour, the hypoxic environment plays an important role in HCC tumour progression and metastasis.
Glypican-3 (GPC3) is a member of the heparan sulfate proteoglycan family, anchoring at the cell membrane by glycosylphosphatidylinositol. GPC3 is highly expressed in HCC tissues and has recently been identified as a novel potential HCC tumour biomarker.4–6 Several studies have shown that GPC3 plays a major role in HCC development and progression.7–10 However, the mechanism of hypoxia-mediated GPC3 regulation in HCC tissues is unknown. In the present study, CoCl2 was used to mimic cell hypoxia in HCC cells and investigate the effect of hypoxia on GPC3 expression, as well as explore the role of GPC3 in hypoxia-induced cell apoptosis in HCC.
Materials and Methods
Chemicals and Reagents
CoCl2 was purchased from Sigma-Aldrich (St. Louis, MO, USA). Primers for GAPDH, GPC3, and HIF-1α were synthesized by Sangon Biotech (Shanghai, China). The protease inhibitor was purchased from Roche (Mannheim, Germany). PowerUp™ SYBR™ Green Master Mix was purchased from Applied Biosystems (Foster City, CA, USA) Mouse anti-human monoclonal antibodies against β-actin and GPC3 were acquired from Santa Cruz Biotechnology (1:1000, Santa Cruz, CA, USA). Rabbit anti-human monoclonal antibodies against HIF-1α, c-myc, sp1, PARP and caspase-3 were obtained from Cell Signaling Technology (1:1000, Danvers, MA, USA). Anti-rabbit and anti-mouse IgG HRP-linked antibodies were procured from Cell Signaling Technology (1:2000, Danvers, MA, USA). RIPA lysis buffer was obtained from Beyotime Institute of Biotechnology (Shanghai, China).
Cell Culture
HepG2 cells were purchased from ATCC (Manassas, VA, USA) and maintained in DMEM medium (Gibco, Grand Island, NY, USA) with 10% foetal bovine serum (Gibco, Grand Island, NY, USA), 1% penicillin-streptomycin (10,000 U/mL penicillin and 10 mg/mL streptomycin) at 37 °C in a humidified atmosphere with 5% CO2. The cells were passaged using 0.25% trypsin (Gibco, Grand Island, NY, USA).
Cell Viability Assay
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-2H-tetrazolium bromide (MTT) (Beyotime Institute of Biotechnology, Shanghai, China) was used to assess cell viability according to the manufacturer’s instructions. Briefly, 2×104 HepG2 cells/well were seeded in 96-well plates and cultured for 24 h. The medium was replaced with 100 µL/well fresh medium containing various concentrations (0, 50, 100, and 200 μmol/L) of CoCl2 for 24 h. Then, 20 µL of 5 mg/mL MTT was added to each well and incubated at 37 °C for 4 h. Subsequently, the reaction was quenched by adding 150 µL DMSO, and the absorbance was measured at 490 nm with a microplate reader (Foster City, CA, USA).
Flow Cytometry
To confirm the effects on cell apoptosis, annexin V-fluorescein isothiocyanate (FITC)/propidium iodide (PI) double staining was performed with an annexin V-FITC apoptosis detection kit (BD Biosciences, Bedford, MA, USA) as according to the manufacturer’s instructions. Briefly, the cells were harvested and resuspended in 1× annexin V binding buffer at a concentration of 1×106 cells/mL. Then, 100 μL of this suspension was incubated with 5 μL FITC annexin V and 5 μL PI for 15 min at room temperature. The stained cells were analysed by flow cytometry (Beckman Coulter, CA, USA) within 1 h.
Real-Time PCR
Real-time PCR was performed as described previously.11 Total RNA was extracted using TRIzol reagent. Approximately 1 μg of RNA from each sample was used to synthesize cDNA using the PrimeScript™ RT reagent kit with gDNA Eraser (TakaraBio, Inc., Otsu, Japan). PCR was performed using PowerUp™ SYBR™ Green Master Mix on a StepOne Plus instrument (Applied Biosystems, Foster City, CA, USA) according to the following programme: 30 s at 95 °C and 60 s at 60 °C for 40 cycles. The PCR primers were as follows: GAPDH-F: 5ʹ-CTGGGCTACACTGAGCACC-3ʹ; GAPDH-R: 5ʹ-AAGTGGTCGTTGAGGGCAATG-3ʹ; GPC3-F: 5ʹ-ATTGGCAAGTTATGTGCCCAT-3ʹ; GPC3-R: 5ʹ-TTCGGCTGGATAAGGTTTCTTC-3ʹ; HIF-1α-F: 5ʹ-GAACG TCGAAAAGAAAAGTCTCG-3ʹ; and HIF-1α-R: 5ʹ-CCTTATCAAGATGCGAACTCACA-3ʹ. GAPDH was used to normalize mRNA expression. Quantification of the real-time PCR results was performed with the 2−ΔΔCT method.
Western Blot Analysis
HepG2 cells were treated with CoCl2 and harvested in RIPA lysis buffer (50 mM Tris-HCl pH 7.4, 0.5% sodium deoxycholate, 150 mM NaCl, 1% NP-40, and 0.1% sodium dodecyl sulfate) containing protease inhibitors. Then, the cells were lysed for 30 min on ice and subjected to centrifugation at 4 °C to collect the supernatant of the lysates. An equivalent amount of protein extracts was separated by SDS-PAGE and transferred to a nitrocellulose membrane. Subsequently, the membrane was blocked with 5% non-fat milk at room temperature and probed with primary antibodies overnight at 4 °C, followed by incubation with HRP-conjugated secondary antibodies at room temperature for 2 h. The immunoreactive proteins were visualized using an ECL kit (Millipore, Billerica, MA, USA).
Immunofluorescence and Laser Confocal Microscopy
HepG2 cells were cultured on glass chamber slides in the presence or absence of 200 μmol/L CoCl2 for 24 h, fixed with 4% paraformaldehyde, permeabilized with 0.3% Triton X-100, and blocked with 5% normal goat serum for 1 h at room temperature. Next, the cells were incubated with primary antibody (1:200) at 4 °C overnight, followed by probing with Alexa Fluor® 488-conjugated goat anti-rabbit or anti-mouse IgG (H+L) (1:100, ZSGB-BIO, Beijing, China). DAPI (Beyotime Institute of Biotechnology, Shanghai, China) was used for nuclei staining. The fluorescence intensity was analysed by confocal laser microscopy (Olympus Corporation, Japan).
Vector Construction and siRNA Transfection
The GPC3 plasmid was constructed in the pcDNA3.1(-) vector (Addgene, Watertown, MA, USA). The recombinant plasmid was verified by enzyme cleavage and sequencing analysis. The HIF-1α-targeting siRNA and control siRNA were synthesized by Guangzhou RiboBio (Guangzhou, China). HepG2 cells were seeded in 6-well plates and cultured overnight before plasmid or 20 nmol/L siRNA transfections were performed using Lipofectamine 3000TM (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s protocol.
Luciferase Reporter Assay
HepG2 cells were plated at a density of 2×104 cells/well in 24-well plates. Then, the cells that had been transfected with the c-myc luciferase reporter plasmid (Genomeditech, Shanghai, China) were cultured in the presence or absence of 200 μmol/L CoCl2 for 24 h. The cells were also co-transfected with pRL-TK (Renilla luciferase vector) for background normalization. The plasmid transfection was performed using LipofectamineTM 3000 transfection reagent. After 24 h, the cells were lysed, and luciferase activity was detected using the Genecopoeia Luc-Pair Duo-Luciferase Assay Kit (Genecopoeia, Inc., Shanghai, China) according to the instructions recommended by the manufacturer.
Statistical Analysis
All experiments were repeated at least two times. Data are presented as the mean ± standard error. Student’s t-test was used for data analysis using SPSS 17.0 software. P<0.05 was considered to be statistically significant.
Results
CoCl2 Induced Hypoxia Injury in HepG2 Cells by Inhibiting GPC3 Expression
It has been demonstrated that CoCl2 induces apoptosis in several types of tumour cells.12–14 To explore the effect of CoCl2 on HCC cells, HepG2 cells were treated with different concentrations of CoCl2 for 24 h, and then cell viability and apoptosis were assessed by MTT assay and flow cytometry, respectively. As shown in Figure 1, CoCl2 significantly repressed cell viability and induced cell apoptosis in a concentration-dependent manner, and cell apoptosis was further verified by the activation of caspase-3 and decreased expression of poly-ADP-ribose polymerase (PARP). The present study also confirmed that CoCl2 successfully induced hypoxia in HepG2 cells, indicated by increased expression of HIF-1α protein (Figure 2). Interestingly, the HIF-1α mRNA level was downregulated, which might be a negative feedback mechanism to maintain homeostasis of the HIF-1α protein level. Moreover, the expression of GPC3 was detected at both the mRNA and protein levels. Compared to the levels in the control group, 50–200 μmol/L CoCl2 treatment reduced the GPC3 mRNA level by more than 80%; accordingly, the protein level assessed by Western blotting and immunofluorescence was also significantly decreased in a concentration-dependent manner (Figure 2). Notably, immunofluorescence results suggested that CoCl2 also induced the translocation of GPC3 from the cytoplasm to the membrane, but the underlying mechanism remains to be investigated.
 |
Figure 1 CoCl2 inhibited HepG2 cell viability and induced cell apoptosis. (A) HepG2 cells were treated with different concentrations of CoCl2 for 24 h, and the cell viability was determined by MTT assay. (B) Cell apoptosis induced by CoCl2 for 24 h was assessed by flow cytometry. (C) Apoptosis rate of HepG2 cells induced by different concentrations of CoCl2. (D) Expression of PARP and caspase-3 induced by CoCl2 for 24 h was determined by Western blotting. *p<0.05 vs 0 μM. |
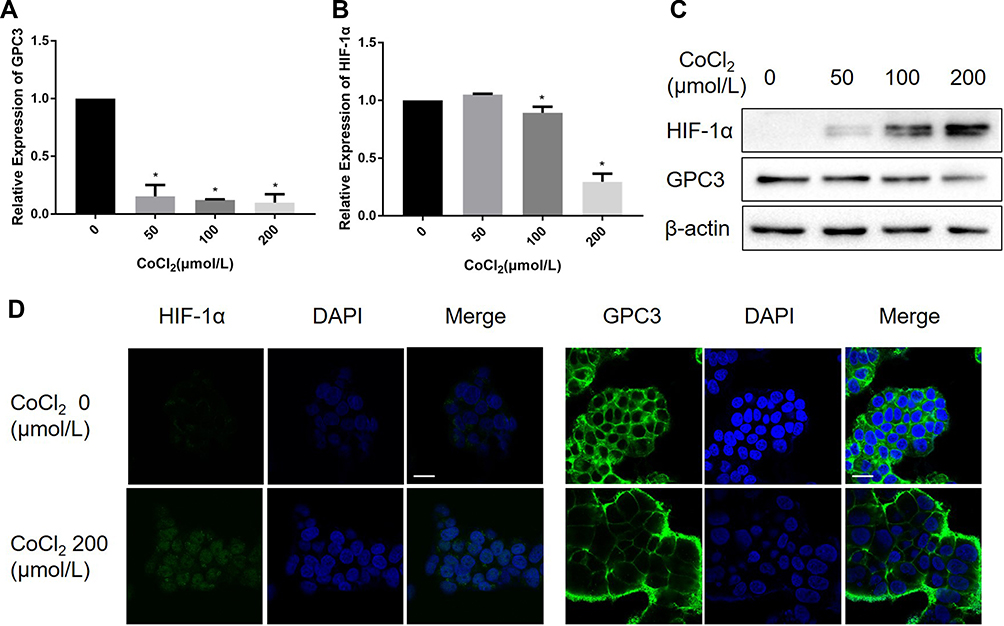 |
Figure 2 CoCl2 inhibited the expression of GPC3 in HepG2 cells. (A, B) HepG2 cells were treated with 50~200 μM CoCl2 for 24 h, and the mRNA levels of GPC3 and HIF-1α were evaluated by real-time PCR. (C) Protein expression of GPC3 and HIF-1α was determined by Western blotting. (D) Expression of GPC3 and HIF-1α in HepG2 cells was assessed by immunofluorescence, and the images were acquired by confocal laser microscopy. Scale bar=20μM. *p<0.05. |
CoCl2 Downregulated GPC3 Expression via the HIF-1α/c-Myc Axis
Accumulating evidence has shown that GPC3 is transcriptionally regulated by c-myc,15 and overexpression of c-myc induces GPC3 promoter-dependent luciferase activity and elevates GPC3 expression at both the mRNA and protein levels. Since CoCl2 decreased the expression of GPC3 mRNA, we hypothesized that CoCl2 might suppress the transcriptional activity of c-myc. Strikingly, the luciferase reporter data demonstrated that CoCl2 resulted in a 50% decline in the transcriptional activity of c-myc in HepG2 cells compared to that of the control group (Figure 3A). In addition, the expression of c-myc was significantly reduced by CoCl2 treatment in a concentration-dependent manner (Figure 3B). Additionally, the expression of sp1 was decreased by CoCl2. Furthermore, the interaction between HIF-1α and c-myc has been shown to play a pivotal role in malignant progression.16–18 Thus, to verify whether the HIF-1α/c-myc axis mediates the CoCl2-induced downregulation of GPC3, siRNA targeting HIF-1α was transfected into HepG2 cells, followed by 200 μM CoCl2 treatment. As shown in Figure 3C, knockdown of HIF-1α elevated the expression of GPC3, c-myc, and sp1, thereby indicating that the HIF-1α/c-myc axis mediated the inhibitory effect of CoCl2 on GPC3.
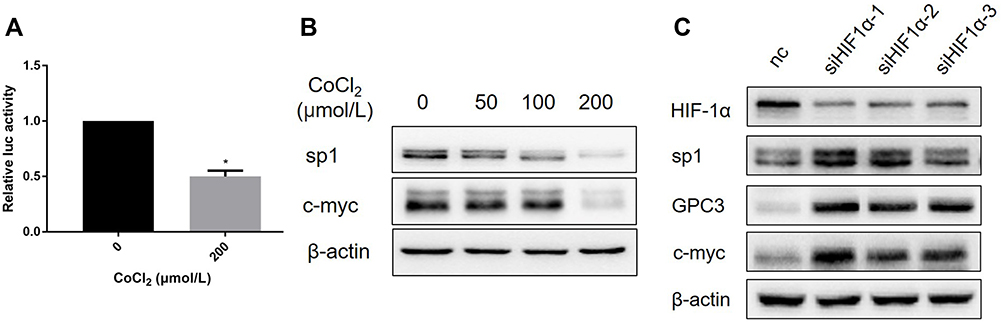 |
Figure 3 CoCl2 inhibited the expression of GPC3 via the HIF-1α/c-myc axis. (A) HepG2 cells transfected with the c-myc luciferase reporter plasmid were cultured in the presence or absence of CoCl2 for 24 h, and the Renilla luciferase reporter plasmid was used as a control. (B) HepG2 cells were treated with 50~200 μM CoCl2 for 24 h, and the expression of sp1 and c-myc was determined. (C) HepG2 cells were transfected with siRNA targeting HIF-1α, the control group was transfected with negative control (NC) siRNA, and all of the groups were stimulated with 200 μM CoCl2. The expression of GPC3, HIF-1α, c-myc, and sp1 was evaluated. *p<0.05. |
Overexpression of GPC3 Attenuated CoCl2-Induced Hypoxia Injury
Our previous studies have demonstrated that GPC3 promotes HepG2 cell proliferation and inhibits cell apoptosis through the Wnt/β-catenin signalling pathway,10 thereby suggesting a tumour-promoting effect of GPC3 on cell growth. To further explore the role of GPC3 in CoCl2-induced hypoxia injury, a GPC3 overexpression plasmid was constructed and transfected into HepG2 cells with or without CoCl2 treatment. As shown in Figure 4, GPC3 overexpression reversed cell proliferation and attenuated cell apoptosis induced by CoCl2 compared to those of the CoCl2 group.
 |
Figure 4 Overexpression of GPC3 attenuated cell apoptosis induced by CoCl2. Con, control. (A) HepG2 cells were transfected with the GPC3 plasmid or blank vector, and GPC3 expression was verified by Western blotting. (B) The transfected cells were treated with 200 μM CoCl2, and cell viability was assessed by MTT assay. (C) The transfected cells were treated with 200 μM CoCl2, and cell apoptosis was assessed. (D) Apoptosis rate of HepG2 cells in each group. (E) The transfected cells were treated with 200 μM CoCl2, and the expression of PARP and caspase-3 was determined by Western blotting. *p<0.05. |
Discussion
HCC is a common refractory tumour with high morbidity and mortality globally. Approximately 600,000 cases are diagnosed every year.19 Although surgical resection is the primary choice for HCC therapy, the majority of patients are diagnosed at a late stage with distant metastasis due to the concealed symptoms of the cancer. Thus, the opportunity for surgery or transplantation is missed, and the five-year survival rate of 3–11% is due to the lack of effective treatments.20
Although the pathogenesis of HCC is complicated, an array of critical genes or proteins have been identified as being involved in HCC tumour progression. Recent studies have shown that the level of GPC3 is drastically upregulated in HCC tissues,6,21–23 while a low level has been observed or remained undetected in normal hepatic cells and benign liver tumours. The upregulated expression of GPC3 displays a positive association with tumour size, histopathological differentiation, tumour invasion, and metastatic24 dysplasia in cirrhotic livers and could enrich the expression of HCC-related genes.25 Accumulating evidence has shown that GPC3 promotes HCC tumour growth via the Wnt/β-catenin signalling pathway,26,27 silences GPC3-induced cell apoptosis, and inhibits cell proliferation,28–31 indicating a critical role for GPC3 in HCC tumourigenesis and development.
Hypoxia is a distinct hallmark of HCC. HIF-1α is a hypoxia-induced transcription factor and a critical regulator of genes that are activated in responses to an oxygen-deficient environment. Under normoxic conditions, HIF-1α is hydroxylated by prolyl hydroxylase (PHD) and undergoes proteasomal degradation, while during hypoxia, HIF-1α is stabilized and cannot be hydroxylated by PHD, thereby preventing it from undergoing proteasomal degradation. Then, the accumulated HIF-1α is translocated to the nucleus, leading to the elevated expression of its target genes, which are involved in energy metabolism, cell proliferation, apoptosis, vascular remodelling, and erythropoiesis.32 Although cell survival is induced by the upregulation of HIF-1α target genes in the hypoxic microenvironment, rapid hypoxia could also induce irreversible cell damage.33
GPC3 is a critical oncoprotein in HCC; however, whether its expression is regulated by hypoxia remains unknown. Mimicking the oxygen-deficient environment in tumour cells by CoCl2 treatment is the canonical in vitro model. It was confirmed that CoCl2 blocks the oxygen signal and stabilizes the expression of HIF-1α by displacing Fe2+ in the proline hydroxylase cofactor,34 which led to a series of cell reactions induced by hypoxia, thereby making CoCl2 useful in hypoxia studies. In the current study, it was found that CoCl2 significantly reduced the expression of GPC3 at both the mRNA and protein levels in HepG2 cells and induced cell apoptosis in a dose-dependent manner. The overexpression of GPC3 attenuated CoCl2-induced cell apoptosis, indicating that CoCl2 induced cell apoptosis by inhibiting the expression of GPC3.
As expected, CoCl2 significantly elevated HIF-1α protein levels. To verify whether HIF-1α mediated the inhibitory effects induced by CoCl2, HIF-1α expression was silenced by siRNA. GPC3 expression was reversed by knockdown of HIF-1α in the presence of CoCl2, suggesting that GPC3 expression may be negatively regulated by HIF-1α. Our present study also demonstrated that the HIF-1α/c-myc axis mediates this biological process.
Both HIF-1α, c-myc and sp1 are crucial transcription factors regulating proliferation and metabolism in cancer cells. HIF-1α and c-myc partially modulate the complex pathways by acting alone in response to low oxygen and also function in concert to reprogram cell metabolism. Reportedly, crosstalk between HIF-1α and the proto-oncogene c-myc during hypoxia plays a major role in controlling genetic instability and cancer progression.35 Although HIF-1α and c-myc share certain common target genes, they exert inverse effects on cell proliferation, mitochondrial biogenesis, and DNA repair.17 Under normal oxygen conditions, c-myc interacts with sp1 and Max to form a transcriptional complex which drives the transcription of c-myc target genes.18 During oxygen deficiency, c-myc could be replaced by HIF-1α due to the high affinity of sp1 to HIF-1α36
Conclusions
Conclusively, the present study revealed that CoCl2 induced cell apoptosis via downregulated expression of GPC3. Further studies showed that the HIF-1α/c-myc axis mediates the inhibitory effects induced by CoCl2 and that knockdown of HIF-1α upregulates the expression of GPC3. This was the first evidence reporting that GPC3 is reduced by a hypoxia mimicking reagent and that GPC3 is negatively regulated by HIF-1α. Notably, although CoCl2 is commonly used to mimic hypoxia, cell metabolism is not consistent with oxygen deficit. Hypoxia is known to induce a more robust and extensive transcriptomic alteration than that of CoCl2,37 while the latter induces a more marked upregulation of hypoxia-induced genes, thereby necessitating further hypoxia studies.
Acknowledgements
The present study was supported by the Scientific and Technological Project of Shiyan City of Hubei Province (19Y43), the Cultivating Project for Young Scholars at Hubei University of Medicine (2018QDJZR07), and the Innovative Research Programme for Graduates of Hubei University of Medicine (YC2019034 and YC2019014).
Disclosure
The authors report no conflicts of interest in this work.
References
1. May M. Statistics: attacking an epidemic. Nature. 2014;509(7502):S50–51. doi:10.1038/509S50a
2. Xiong XX, Qiu XY, Hu DX, Chen XQ. Advances in hypoxia-mediated mechanisms in hepatocellular carcinoma. Mol Pharmacol. 2017;92(3):246–255. doi:10.1124/mol.116.107706
3. Huang F, Chen J, Lan R, et al. Hypoxia induced delta-Catenin to enhance mice hepatocellular carcinoma progression via Wnt signaling. Exp Cell Res. 2019;374(1):94–103. doi:10.1016/j.yexcr.2018.11.011
4. Lee HJ, Yeon JE, Suh SJ, et al. Clinical utility of plasma glypican-3 and osteopontin as biomarkers of hepatocellular carcinoma. Gut Liver. 2014;8(2):177–185. doi:10.5009/gnl.2014.8.2.177
5. Li J, Wang T, Jin B, et al. Diagnosis accuracy of serum glypican-3 level in patients with hepatocellular carcinoma: a systematic review with meta-analysis. Int J Biol Markers. 2018;1724600818784409.
6. Tahon AM, El-Ghanam MZ, Zaky S, et al. Significance of Glypican-3 in early detection of hepatocellular carcinoma in cirrhotic patients. J Gastrointest Cancer. 2018.
7. Dhungel B, Andrzejewski S, Jayachandran A, et al. Evaluation of the Glypican 3 promoter for transcriptional targeting of hepatocellular carcinoma. Gene Ther. 2018;25(2):115–128. doi:10.1038/s41434-018-0002-2
8. Dong Z, Yao M, Wang L, Yang J, Yao D. Down-regulating glypican-3 expression: molecular-targeted therapy for hepatocellular carcinoma. Mini Rev Med Chem. 2014;14(14):1183–1193. doi:10.2174/1389557515666150101105135
9. Hu P, Ke C, Guo X, et al. Both glypican-3/Wnt/beta-catenin signaling pathway and autophagy contributed to the inhibitory effect of curcumin on hepatocellular carcinoma. Digest Liver Dis. 2019;51(1):120–126. doi:10.1016/j.dld.2018.06.012
10. Li N, Wei L, Liu X, et al. A Frizzled-like cysteine-rich domain in Glypican-3 mediates Wnt binding and regulates hepatocellular carcinoma tumor growth in mice. Hepatology. 2019.
11. Hu P, Cheng B, He Y, Wei Z, Wu D, Meng Z. Autophagy suppresses proliferation of HepG2 cells via inhibiting glypican-3/wnt/beta-catenin signaling. Onco Targets Ther. 2018;11:193–200. doi:10.2147/OTT.S150520
12. Liu J, Zhu Y, Chen S, et al. Apocynin attenuates cobalt chloride-induced pheochromocytoma cell apoptosis by inhibiting P38-MAPK/Caspase-3 pathway. Cell Physiol Biochem. 2018;48(1):208–214. doi:10.1159/000491720
13. Cheng BC, Chen JT, Yang ST, Chio CC, Liu SH, Chen RM. Cobalt chloride treatment induces autophagic apoptosis in human glioma cells via a p53-dependent pathway. Int J Oncol. 2017;50(3):964–974. doi:10.3892/ijo.2017.3861
14. Lee M, Lapham A, Brimmell M, Wilkinson H, Packham G. Inhibition of proteasomal degradation of Mcl-1 by cobalt chloride suppresses cobalt chloride-induced apoptosis in HCT116 colorectal cancer cells. Apoptosis. 2008;13(8):972–982. doi:10.1007/s10495-008-0229-2
15. Li L, Jin R, Zhang X, et al. Oncogenic activation of glypican-3 by c-Myc in human hepatocellular carcinoma. Hepatology. 2012;56(4):1380–1390. doi:10.1002/hep.v56.4
16. Yoo YG, Hayashi M, Christensen J, Huang LE. An essential role of the HIF-1alpha-c-Myc axis in malignant progression. Ann N Y Acad Sci. 2009;1177:198–204. doi:10.1111/j.1749-6632.2009.05043.x
17. Podar K, Anderson KC. A therapeutic role for targeting c-Myc/Hif-1-dependent signaling pathways. Cell Cycle. 2010;9(9):1722–1728. doi:10.4161/cc.9.9.11358
18. Gordan JD, Thompson CB. and c-Myc: sibling rivals for control of cancer cell metabolism and proliferation. Cancer Cell. 2007;12(2):108–113. doi:10.1016/j.ccr.2007.07.006
19. Chen C, Lou T. Hypoxia inducible factors in hepatocellular carcinoma. Oncotarget. 2017;8(28):46691–46703. doi:10.18632/oncotarget.17358
20. Li YM, Liu ZY, Wang JC, et al. RIP3 deficiency recruits myeloid-derived suppressor cells to hepatocellular carcinoma through the CXCL1-CXCR2 axis. Hepatology. 2019.
21. Kawaida M, Yamazaki K, Tsujikawa H, et al. Diffuse and canalicular patterns of glypican-3 expression reflect malignancy of hepatocellular carcinoma. Pathol Int. 2019;69(3):125–134. doi:10.1111/pin.v69.3
22. Hamaoka M, Kobayashi T, Tanaka Y, Mashima H, Ohdan H. Clinical significance of glypican-3-positive circulating tumor cells of hepatocellular carcinoma patients: a prospective study. PLoS ONE. 2019;14(5):e0217586. doi:10.1371/journal.pone.0217586
23. Wang Z, Han YJ, Huang S, et al. Imaging the expression of glypican-3 in hepatocellular carcinoma by PET. Amino Acids. 2018;50(2):309–320. doi:10.1007/s00726-017-2517-z
24. Xue R, Feng J, Meng Q, et al. The significance of glypican-3 expression profiling in the tumor cellular origin theoretical system for hepatocellular carcinoma progression. J Gastroenterol Hepatol. 2017;32(8):1503–1511. doi:10.1111/jgh.2017.32.issue-8
25. Liu X, Wang SK, Zhang K, et al. Expression of glypican 3 enriches hepatocellular carcinoma development-related genes and associates with carcinogenesis in cirrhotic livers. Carcinogenesis. 2015;36(2):232–242. doi:10.1093/carcin/bgu245
26. Gao W, Ho M. The role of glypican-3 in regulating Wnt in hepatocellular carcinomas. Cancer Rep. 2011;1(1):14–19.
27. Zhou F, Shang W, Yu X, Tian J. Glypican-3: a promising biomarker for hepatocellular carcinoma diagnosis and treatment. Med Res Rev. 2018;38(2):741–767. doi:10.1002/med.21455
28. Qi XH, Wu D, Cui HX, et al. Silencing of the glypican-3 gene affects the biological behavior of human hepatocellular carcinoma cells. Mol Med Rep. 2014;10(6):3177–3184. doi:10.3892/mmr.2014.2600
29. Liu S, Li Y, Chen W, et al. Silencing glypican-3 expression induces apoptosis in human hepatocellular carcinoma cells. Biochem Biophys Res Commun. 2012;419(4):656–661. doi:10.1016/j.bbrc.2012.02.069
30. Miao HL, Lei CJ, Qiu ZD, et al. MicroRNA-520c-3p inhibits hepatocellular carcinoma cell proliferation and invasion through induction of cell apoptosis by targeting glypican-3. Hepatol Res. 2014;44(3):338–348. doi:10.1111/hepr.12121
31. Sun CK, Chua MS, He J, So SK. Suppression of glypican 3 inhibits growth of hepatocellular carcinoma cells through up-regulation of TGF-beta2. Neoplasia. 2011;13(8):735–747. doi:10.1593/neo.11664
32. Lin D, Wu J. Hypoxia inducible factor in hepatocellular carcinoma: a therapeutic target. World J Gastroenterol. 2015;21(42):12171–12178. doi:10.3748/wjg.v21.i42.12171
33. Wilson GK, Tennant DA, McKeating JA. Hypoxia inducible factors in liver disease and hepatocellular carcinoma: current understanding and future directions. J Hepatol. 2014;61(6):1397–1406. doi:10.1016/j.jhep.2014.08.025
34. Munoz-Sanchez J, Chanez-Cardenas ME. The use of cobalt chloride as a chemical hypoxia model. J Appl Toxicol. 2019;39(4):556–570. doi:10.1002/jat.3749
35. To KK, Huang LE. An efficient way of studying protein-protein interactions involving HIF-alpha, c-Myc, and Sp1. Methods Mol Biol. 2013;1012:77–84.
36. Fer N, Melillo G. The HIF-1alpha-c-Myc pathway and tumorigenesis: evading the apoptotic gate-keeper. Cell Cycle. 2011;10(19):3228. doi:10.4161/cc.10.19.17049
37. Zhigalova N, Artemov A, Mazur A, Prokhortchouk E. Transcriptome sequencing revealed differences in the response of renal cancer cells to hypoxia and CoCl 2 treatment. F1000Research. 2015;4:1518. doi:10.12688/f1000research
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.