Back to Journals » Therapeutics and Clinical Risk Management » Volume 11
Clinical use of pasireotide for Cushing's disease in adults
Authors Ceccato F, Scaroni C, Boscaro M
Received 30 August 2014
Accepted for publication 6 October 2014
Published 17 March 2015 Volume 2015:11 Pages 425—434
DOI https://doi.org/10.2147/TCRM.S37314
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Garry Walsh
Filippo Ceccato, Carla Scaroni, Marco Boscaro
Endocrinology Unit, Department of Medicine, Padova University Hospital, Padova, Italy
Cushing’s disease: Excessive corticotroph hormone levels sustained by an adrenocorticotropic hormone-secreting pituitary adenoma lead to a severe clinical condition caused by excess cortisol secretion, called Cushing’s disease (CD). Neurosurgery and radiotherapy are used to treat the pituitary adenoma directly, but new medical treatments targeting the corticotroph cells have recently become available.
Pasireotide: This is a novel multireceptor ligand somatostatin (SST) analog with a high binding affinity for SST receptor 5, the predominant receptor in human corticotroph adenomas that is not downregulated by high cortisol levels (as SST receptor 2 is). Pasireotide has been recently approved by the European Medical Agency and the US Food and Drug Administration for treating adults with CD with recurrent hypercortisolism after surgery, or for whom surgery is not an option. A dose of 600–1,200 µg twice a day can normalize urinary free cortisol levels after 3 months of treatment in up to 28% of patients, reducing their blood pressure and improving their weight, lipid profile, and quality of life. Combining pasireotide with cabergoline to achieve a greater hormone response can normalize cortisol secretion in 50% of patients, and adding ketoconazole induces biochemical control in most patients with CD.
Safety and hyperglycemia: The adverse effects of pasireotide are similar to those of other SST analogs, including diarrhea, nausea, and biliary sludge or gallstones. Hyperglycemia is common during pasireotide treatment, which affects the secretion of pancreatic insulin and intestinal glucagon-like peptide 1. Self-monitoring is essential to achieve good metabolic control, and endocrinologists should first administer metformin if insulin resistance is evident and then add dipeptidyl peptidase 4 inhibitors/glucagon-like peptide 1 receptor agonists or insulin.
Conclusion: In recent years, medical treatment with pasireotide has been proposed as monotherapy for adults with CD characterized by mild to moderate hypercortisolemia, as well as in combination with other available therapies. It is generally well-tolerated, but endocrinologists need to monitor glucose levels to ensure prompt treatment.
Keywords: Cushing’s disease, pasireotide, medical therapy, hyperglycemia, safety
Introduction
Cushing’s syndrome (CS) is a severe clinical condition caused by excess cortisol secretion. It is classified as adrenocorticotropic hormone (ACTH)-independent in 15% of cases (adrenal adenoma or carcinoma) and ACTH-dependent in the remaining 85% of patients, whose cortisol excess is stimulated by uncontrolled ACTH secretion from either an ectopic or a pituitary source of ACTH. The latter case applies to the condition known as Cushing’s disease (CD), which accounts for about 70% of cases of endogenous chronic hypercortisolism.1,2 CS is rare, and patients may present with several characteristic signs or symptoms or with a by-no-means-clear clinical picture, and sometimes only isolated signs. Screening strategies adopted for specific populations (eg, in cases of diabetes mellitus or osteoporosis) lead to the diagnosis of a higher number of cases of CS, making endogenous hypercortisolism not so uncommon (ie, 1% among patients with type 2 diabetes and 5% among subjects with osteoporosis).3,4 The Endocrine Society Clinical Practice Guidelines1 recommend screening for CS with 24 hour urinary free cortisol (UFC) excretion (which has high diagnostic accuracy when performed using liquid chromatography-tandem mass spectrometry),5 late-night salivary cortisol assays,6 or the 1 mg dexamethasone suppression test (with a recommended cutoff for serum cortisol of 50 nmol/L to avoid false-negative results).1
The mortality rate for patients with CD is higher than for age- and sex-matched controls because chronic cortisol excess leads to metabolic and cardiovascular diseases (diabetes mellitus, obesity, hypertension, ischemic attacks), hypercoagulability, and susceptibility to opportunistic infections.7,8
The goals of treatment for CD are to normalize cortisol levels, reverse the clinical features of the disease, and remove the ACTH-producing pituitary adenoma while preserving pituitary function.2 These results may be achieved in several ways, including neurosurgery, radiotherapy, and/or medical therapy. Although neurosurgery is generally considered the first-line option, the reported overall remission rate in literature is 65%–90%, and recurrences are not uncommon (reported in up to 30%–40% of patients, even several years after successful surgery).9–12 Repeated surgery is associated with even lower remission rates and, obviously, a higher risk for hypopituitarism.10,13 Some patients have clearly unresectable invasive tumors (eg, locally aggressive macroadenomas or cavernous sinus involvement) or may be at high surgical risk (eg, elderly patients with comorbidities or cases whose severe hypercortisolism carries a high anesthetic risk). Radiotherapy has traditionally been considered an option for persistent or recurrent hypercortisolism after surgery, but it can take from 3 to 10 years for its therapeutic effects to be felt, and brain irradiation may have adverse effects, including pituitary deficiency.14,15 The last chance is bilateral adrenalectomy, which achieves an immediate reduction in cortisol levels but necessitates lifelong gluco- and mineral-corticoid replacement therapy and has a strong effect on quality of life (QoL) and mortality,16 as well as carrying a risk for the ACTH-secreting pituitary adenoma growing excessively (so-called Nelson’s syndrome).17
In this complex scenario of treatment options, it may be difficult for clinical endocrinologists to make the most appropriate choice. Medical treatments are increasingly used, especially when surgery fails or is not indicated or while waiting for radiotherapy to take effect.
Medical treatment for CD
Two different types of medical approach to CD can be considered: adrenal- or pituitary-directed drugs (as summarized in Table 1). There has recently been much interest in agents that target the pituitary corticotroph cells, which contain receptors and transcription factors that interact with dopamine, somatostatin (SST), or retinoic acid.2,12,13
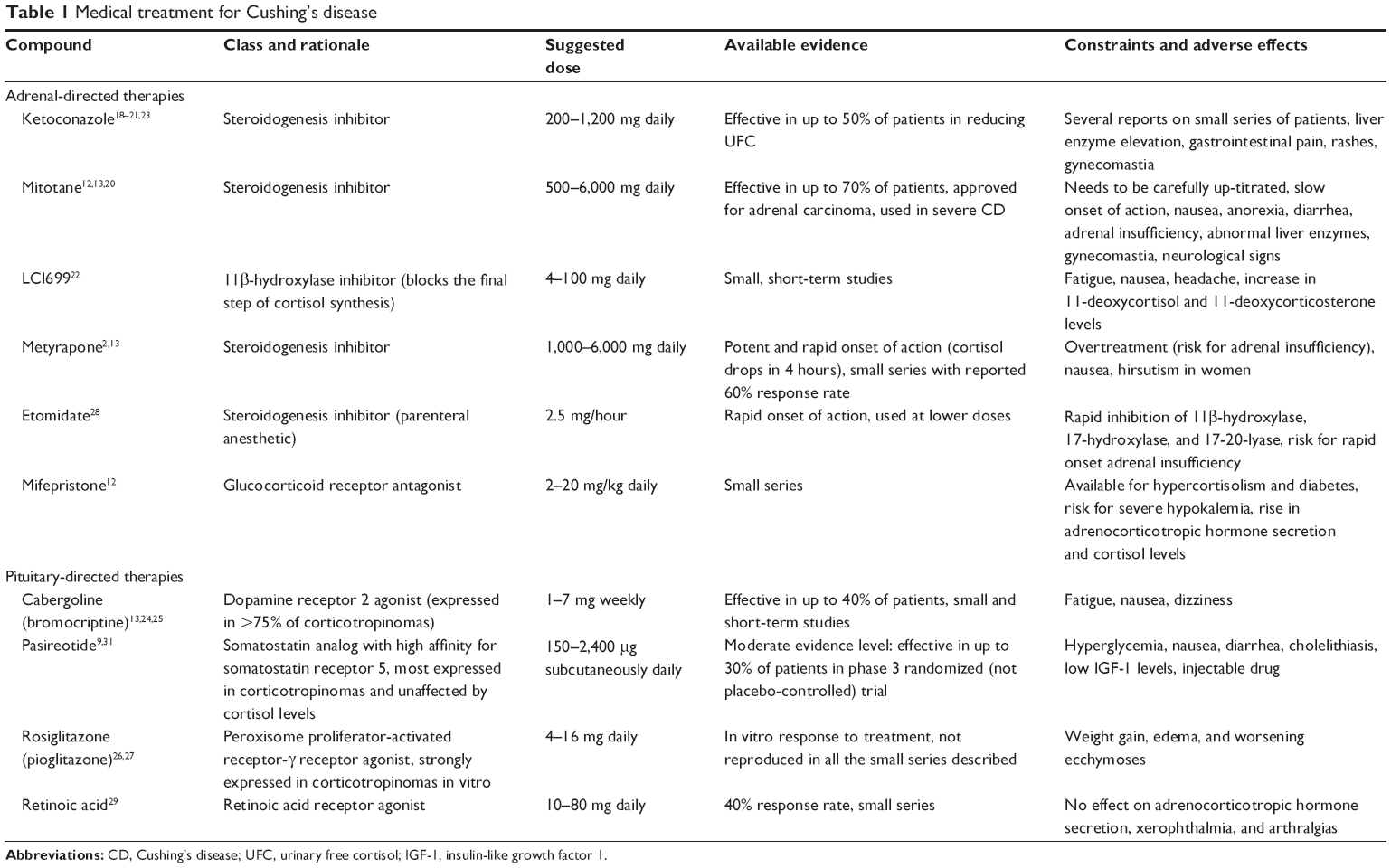 | Table 1 Medical treatment for Cushing’s disease |
Adrenal-directed or steroid-targeting medical therapies have been developed to limit cortisol secretion by the adrenal glands or its action through inhibition of the glucocorticoid receptor.18–23 The current available compounds are ketoconazole, metyrapone, mitotane, etomidate, mifepristone, and LCI699.18–23 They can be used in all forms of CS, even before surgery or when no pituitary adenoma is apparent on conventional imaging.18 Steroidogenesis inhibitors are most effective medical agents and achieve normocortisolemia in a majority of patients.18–23 These compounds can be administered alone or in combination to reduce cortisol secretion quickly in severe forms of CS,20 but they are unable to control ACTH secretion or influence pituitary adenoma volume.
Several pituitary-targeting therapies have been proposed in recent years, including dopamine or peroxisome proliferator-activated receptor gamma (PPARγ) agonists24–28 and retinoic acid.29 Dopamine agonists (especially cabergoline, an oral agent) are increasingly used because the dopamine D2 receptor is expressed in more than 75% of corticotroph adenoma cells and is only slightly downregulated in the presence of high cortisol levels, but few studies have claimed that cabergoline may be effective in treating CD,21,24,25 enabling up to 40% of patients to achieve normal UFC levels.12,24 Cabergoline causes relatively mild adverse effects (primarily asthenia), and small trials have suggested that combination therapy with ketoconazole increases the two drugs’ effectiveness.12,19,21 As for the use of retinoic acid to treat CD, suggested in the light of in vitro studies,30 a recently performed prospective study involving retinoic acid up to 80 mg daily indicated it is able to normalize UFC, at least in some patients.29
Pasireotide is a novel SST analog that has recently been proposed as a medical treatment option for CD, improving outcomes and increasing the choice of treatments.9,31 It is the first agent to have been approved by both the European Medical Agency and the US Food and Drug Administration for treating adult patients with CD with recurrent hypercortisolism after surgery or for whom surgery is not an option.2 Pasireotide is also the only medical treatment available that directly targets ACTH secretion and pituitary adenoma, and it has proved effective in the biochemical control of cortisol levels, achieving a clinical improvement in patients with CD.2
A systematic review was conducted on CD patients’ rate of response to medical therapy with a view to pooling a number of patients sufficient to compare the efficacy of several treatments (as some reports described small series of cases).13 Using well-known criteria to assess the level of evidence, the review showed that only pasireotide could boast a moderate level of evidence of its efficacy (with a response rate up to 28% demonstrated in a randomized trial), whereas the level of evidence was low for the effectiveness of the other medical treatments considered, even though some reportedly achieved higher response rates in controlling cortisol secretion (about 75% for metyrapone and mitotane, 50% for ketoconazole, and 25%–50% for cabergoline).13
Pasireotide pharmacokinetics and SST receptors
Naïve SST is a cyclopeptide with 14 or 28 amino acids and a very short half-life (about 3 minutes), expressed in many tissues from the central nervous system to the gastrointestinal tract.32 Naïve SST binds with a high affinity to all SST receptors (SSTRs) expressed on the target tissues: five SSTRs have been cloned in human tissue, with a 39%–57% sequence homology.32 The majority of corticotroph adenomas express SSTR-2 and SSTR-5 mRNAs, but the membrane density of SSTR-2 is lowered by hypercortisolism, whereas that of SSTR-5 is unaffected by high cortisol levels,33 and that is why SSTR-5s are predominantly expressed in the pituitary corticotroph cells of patients with active CD.
Octreotide and lanreotide (the first-generation SST analogs) have a high affinity for SSTR-2 and a low affinity for SSTR-5, with a broad clinical efficacy in acromegaly and neuroendocrine tumors.34,35
In 2002, a drug design approach was developed to identify a new stable SST analog with a view to discovering a drug showing a universal SSTR binding profile.32 In this pharmacologic study, incorporating a stable cyclohexapeptide template in modified unnatural amino acids resulted in the identification of a novel compound, which was named pasireotide (SOM230).32 It is a multireceptor ligand SST analog with a high binding affinity for four of the five known SSTRs (SSTR-1, SSTR-2, SSTR-3, and SSTR-5), and its 40-fold higher affinity for SSTR-5 explains its potential role in CD.31,36 Pasireotide is characterized by a rapid absorption after subcutaneous injection, a long half-life, and an extensive distribution in healthy volunteers.37 The pharmacokinetics of pasireotide have been assessed using multiple doses (from 2.5 to 1,200 μg) in healthy individuals, and all parameters tend to be approximately dose-proportional: the drug’s half-life is 1–2 hours in the α phase and 7–11 hours in the β phase, and its peak concentrations early (0.25–0.5 hours) after its administration suggest a rapid absorption.37
UFC levels after pasireotide treatment in CD
Two clinical trials have been published in the literature to date on the effectiveness of pasireotide in CD.
In 2008, the first phase 2, proof-of-concept, open-label, single-group study was conducted to explore the efficacy of short-term pasireotide therapy (for 15 days) in 39 patients with CD with active disease who had received no radiotherapy (that might have a confounding effect on any remission).31 In this study, patients self-administered pasireotide 600 μg twice a day subcutaneously at 9 am and 9 pm. Steady-state plasma concentrations of pasireotide were achieved within 5 days of treatment, and serum pasireotide levels were higher in responders than in nonresponders. Considering the 29/39 patients included in the primary efficacy study, pasireotide treatment reduced UFC levels in 76% of cases (22 patients) and normalized UFC in 17% (5 patients), with a mean 45% reduction from baseline levels.31
In 2012, the first results of a double-blind phase 3 study were published with a view to gaining the final approval of the European Medical Agency.9 This study involved 162 CD patients with baseline UFC 1.5 times the upper limit of normality (ULN) or higher who were randomized to receive pasireotide 600 μg (82 patients) or 900 μg (80 patients) twice a day. At month 3, there was a first-dose titration: Patients with UFC less than 2 times the ULN and below their baseline levels continued to receive their randomly assigned dose of pasireotide; all the others received an additional 300 μg twice a day (up to a maximum of 1,200 μg twice a day). At month 6, patients entered an open-label phase that extended to month 12: If their UFC levels were higher than ULN, their dose of pasireotide could be increased by 300 μg twice a day at any time (up to the maximum dose of 1,200 μg twice a day). Patients’ UFC levels dropped quickly, with a median reduction of approximately 50% after 2 months of therapy, and remained stable at 6 and 12 months. After 6 months of therapy (data available for 103 patients, as depicted in Figure 1), 29% of the patients treated with pasireotide 900 μg twice a day and 16% of those given pasireotide 600 μg twice a day had normalized UFC levels, and nearly 50% of patients had at least experienced a substantial reduction vis-a-vis their baseline UFC levels, even considering the high variability of individual responses.9 After 12 months of pasireotide (completed by 77 patients, 48% of the initial cohort), the treatment’s efficacy was much the same as at the sixth month: 13% of patients in the 600 μg group and 25% of those in the 900 μg group had normalized UFC levels.9 Although the strength of this study was partially limited by the lack of any comparison with a control group and a group treated with another medical treatment approved for use in CD, it nonetheless prompted the final approval of pasireotide for the treatment of CD in European countries and the United States.
 | Figure 1 Change in urinary free cortisol levels from baseline to month 6 in those patients enrolled in a phase 3 trial. |
The first study identified a trend suggesting that lower baseline UFC levels predicted a response to pasireotide.31 In the second clinical trial, half of the patients with “mild” hypercortisolism (defined as baseline UFC levels less than twice the ULN) treated with pasireotide 900 μg twice a day returned to normal UFC levels, and patients with baseline UFC levels less than five times the ULN more frequently achieved a complete biochemical response.9 A randomization bias emerged in this second trial, in that there was a higher proportion of patients whose mean UFC level at the baseline was more than five times the ULN in the group given 600 μg twice a day of pasireotide (48%; median UFC, 730 nmol/24 hours) than in the group given 900 μg (28%; median UFC, 487 nmol/L).9
In another small series, tested without any strict protocol design or inclusion/exclusion criteria, pasireotide proved effective in restoring normal UFC levels in patients with CD.38,39 Late-night salivary cortisol is a simple, noninvasive biomarker of endogenous cortisol rhythm that has recently been proposed as a useful tool for following up patients receiving medical treatment with pasireotide, which is able to reduce the levels of this parameter within 2 weeks.40 Moreover, there are some case reports about long-term efficacy (up to 5 years) of pasireotide in CD in a few selected patients, even in those with worsening of glycemic control.38,39
Pasireotide in CD: effects on clinical signs and symptoms
CD becomes manifest with several well-known clinical signs and symptoms that are particularly pronounced in cases of active disease. A phase 3 trial investigated not only the effect of pasireotide on UFC levels but also the clinical implications of their reduction. The most important effects seen up to the twelfth month of treatment concerned blood pressure (systolic, −6 mmHg, and diastolic, −4 mmHg), body weight (−7 kg), and lipid profile (low-density lipoprotein cholesterol, −15 mg/dL, and triglyceride levels, −2 mg/dL).9,41
CD negatively affects patients’ health-related QoL because the high cortisol levels associated with this condition can induce major depression and anxiety disorders.42 The CushingQoL has recently been proposed as a tool for measuring QoL in patients with CD. It is a self-administered questionnaire with twelve items that explore areas of personal life, including difficulty sleeping; wound healing and bruising; irritability, mood swings, and anger; self-confidence; physical changes; ability to participate in activities; interaction with friends and family; memory issues; and concern about future health.43 The score is the sum of all the item responses and, after standardization, can range from 0 (worst QoL) to 100 (best QoL).43 In the phase 3 study on pasireotide, patients’ CushingQoL scores improved by 11 points after 12 months of treatment, confirming the drug’s positive effects on the clinical signs and symptoms of CD, especially among patients achieving a complete or partial biochemical control.44
Combination therapies
Because a single medical treatment proved unable to normalize UFC levels in the majority of patients, some authors have reported treating small series of CD patients with combinations of adrenal- and pituitary-directed drugs, such as cabergoline associated with ketoconazole.19,21 There has also been an interesting report of ACTH secretion being inhibited in vitro in a cellular model of CD after activation of the dopamine receptor type 2 promoter by retinoic acid.30 The adverse effects of single drugs may also be contained by reducing their compound dosage in combination treatment schedules.
In a synergistic prospective study, Feelders et al described 17 patients treated for 80 days in a subsequent trial that involved changes of treatment only for patients whose UFC levels did not return to normal.45 All patients began with 100 μg pasireotide subcutaneously three times a day, and this dose was increased to 250 μg subcutaneously three times a day 2 weeks later. After 1 month, cabergoline 0.5 mg every other day was added, increasing to 1 mg every other day after 5 days and to 1.5 mg every other day 10 days later. After 2 months, ketoconazole was added at a dose of 600 mg daily. In this interesting step-by-step trial, pasireotide monotherapy normalized UFC levels in five of the 17 patients; adding cabergoline led to UFC level normalization in nine of 17 patients, and adding ketoconazole induced biochemical control in 15 of 17 cases.45 Although this was only a small series, it clearly points to the scarcely explored potential advantages of combination therapies (including pasireotide). Endocrinologists must bear in mind, however, that both pasireotide and ketoconazole can prolong the QT interval, so the association should be used with caution in patients who are at risk for arrhythmia.
Pasireotide safety and tolerability
The adverse effect profile of pasireotide is similar to that of other SST analogs and includes mainly mild-to-moderate events and transient injection-site reactions, apart from an increase in the frequency of hyperglycemia or overt diabetes mellitus.9,31
The most commonly reported drug-related adverse effects, most of them graded as 1 or 2, were gastrointestinal disorders (54%), mainly involving diarrhea (44%–58%), nausea (23%–52%), and abdominal pain (18%–24%).9,31 In the series described by Boscaro et al, 13% of patients experienced hypotension and asthenia (symptoms consistent with adrenocortical insufficiency),31 and 8% of the patients in the second study likewise developed hypocortisolism-related adverse events,9 which regressed with a temporary dose reduction or suspension of the treatment; both trials deduced hypoadrenalism from patients’ clinical pictures, however, and provided no data on their cortisol levels. SST analogs are known to have the potential to affect the QT interval, although only 2% of patients receiving pasireotide had a new-onset prolongation of Fridericia’s corrected QT interval more than 480 msec, which never warranted the suspension of the treatment.9 Among the patients with a normal gallbladder on baseline ultrasound, sludge became detectable in 7%, and 20% developed gallstones (requiring cholecystectomy in 4%) while receiving pasireotide treatment.9
Hyperglycemia occurring in patients with active CD treated with pasireotide has the odd feature of being secondary both to cortisol excess and to pasireotide treatment. Numerous pathophysiological mechanisms underlie glucocorticoid-induced hyperglycemia and the onset of diabetes, the most important being a lower insulin activity in peripheral tissues, leading to a greater insulin resistance. Cortisol excess also partially inhibits insulin release by the pancreatic beta cells, causing an increase in appetite.46 Pasireotide-induced hyperglycemia was reported in up to 36% of patients in the first series31 and in up to 73% of the patients involved in the phase 3 clinical trial,9 figures higher than those reported in patients treated with first-generation SST analogs (octreotide and lanreotide). In addition, 6% of patients discontinued pasireotide treatment because of hyperglycemia-related adverse events or uncontrolled diabetes mellitus.9 Their mean glycated hemoglobin (HbA1c) levels rose from 5.8% at the baseline to 7.3% after 6 and 12 months of pasireotide treatment, irrespective of its dosage.9 The pathophysiology of pasireotide-induced hyperglycemia has been investigated in mechanistic studies in healthy volunteers:47 The drug affects the secretion not only of pancreatic insulin but also of intestinal glucagon-like peptide (GLP)-1 and glucose-dependent insulin tropic peptide, possibly through SSTR inactivation at the pancreatic and enteroendocrine cell level, whereas hepatic and peripheral insulin sensitivity is unaffected. New antidiabetic agents such as dipeptidyl-peptidase (DPP)-4 inhibitors (eg, sitagliptin, vildagliptin, saxagliptin, linagliptin) and GLP-1 receptor agonists (eg, liraglutide, exenatide) may consequently be the most effective means for reducing pasireotide-associated hyperglycemia, which is related to a diminished insulinotropic incretin effect.8
Although declining cortisol levels reduced the cortisol-derived tendency toward hyperglycemia, blood glucose and HbA1c levels increased soon after pasireotide treatment was begun, and 46% of patients were subsequently begun on glucose-lowering medication.9
It is important to know that pasireotide can raise glucose levels even in healthy young men with a normal glucose metabolism. In a randomized placebo-controlled trial involving more than 70 healthy males, the glucose levels measured 2 hours later in subjects given less than 100 μg pasireotide were much the same as in those given a placebo, but the mean glucose levels were higher in participants administered more than 200 μg pasireotide than in those who received a placebo.37 Six hours after the injection of pasireotide, only patients given more than 600 μg revealed higher glucose levels, and their glucose metabolism returned to normal 24 hours later.37 In another study that tested incretin in healthy patients given pasireotide, an oral glucose tolerance test was performed at the baseline and 1 week after participants had been randomly assigned to receive pasireotide 600 μg twice a day subcutaneously alone or together with oral metformin 500 mg twice a day, oral nateglinide 60 mg three times a day, oral vildagliptin 50 mg twice a day, or liraglutide 0.6 mg subcutaneously four times a day.47 After 1 week, the post-oral glucose tolerance test plasma glucose area under the curve increased by 69% with pasireotide; this increase was reduced by 13%, 29%, 45%, and 72%, respectively, when metformin, nateglinide, vildagliptin, and liraglutide were coadministered,47 suggesting the use of associated incretin drugs as a glucose-lowering treatment in this particular form of diabetes mellitus.
Recommendations for managing pasireotide-induced hyperglycemia
Given the observation that a significant number of patients with CD treated with pasireotide will develop hyperglycemia, a group of European experts on CD and diabetes mellitus recently met to propose some recommendations for managing pasireotide-induced hyperglycemia.8
First of all, they suggested that endocrinologists and patients must all be aware of the risk for hyperglycemia in CD and of the altered glucose control induced by pasireotide treatment. All patients should be accurately informed and closely monitored.
Patients with a normal glucose metabolism (before pasireotide treatment) should self-monitor their fasting and postprandial glucose levels by taking several blood glucose measurements in a day (eg, before and 2 hours after each meal), twice during the first week of treatment and once a week thereafter. Patients with impaired fasting glucose and/or impaired glucose tolerance or who are known to be receiving treatment for overt diabetes mellitus must be closely monitored on a daily basis when they start treatment with pasireotide, and if necessary, their antidiabetic treatment should be adjusted or changed to control their new glucose levels.8
The management of pasireotide-induced hyperglycemia should be based on the currently recommended treatment algorithms for type 2 diabetes mellitus.48 If a patient with active CD is a young adult at high cardiovascular risk (with hypertension, hyperglycemia, atherosclerosis, dyslipidemia, central obesity, and so on),7 clinicians must carefully monitor their glucose control and define individual glycemic targets: a HbA1c level lower than 7.5% (58 mmol/mol) is appropriate, unless the treating clinician perceives a risk for hypoglycemia. It is mandatory to educate patients to make the necessary lifestyle changes, modify their diet, and get more exercise. As pasireotide-related hyperglycemia is associated with a lower insulin secretion and incretin response, treatment should preferentially address these two pathophysiological mechanisms. In patients with evidence of insulin resistance, medical treatment should begin with metformin as the first-line therapy, unless it is contraindicated or poorly tolerated.8,48 If metformin alone fails to achieve and maintain glycemic control, combination therapy with an incretin-based drug is recommended. Consistent with the pathophysiological mechanisms behind pasireotide-induced hyperglycemia, therapy with a DPP4-inhibitor may be administered as a second-step combination. If glycemic targets are still not reached with this combination, then the DPP-4 inhibitor can be replaced with a GLP-1 receptor agonist. In this regard, GLP-1 analogs have demonstrated the best HbA1c-lowering effect without increasing the risk for hypoglycemia and may facilitate a reduction in body weight (which is desirable in CD). In contrast, one of the main drawbacks affecting patient compliance is that, similar to pasireotide, GLP-1 analogs have to be injected and the number of daily injections required may induce patients to abandon this treatment. If blood glucose levels are still not under control, then insulin therapy becomes necessary. Initial combination therapies involving metformin with a once-daily administration of a long-acting basal insulin analog (glargine or detemir) are recommended, and if individual HbA1c targets are not met, or postprandial glucose levels are not controlled, then prandial insulin therapy ultimately has to be established (as summarized in Figure 2).8,48 Endocrinologists should add a DDP-4 inhibitor or a GLP-1 receptor agonist in case of hyperglycemia (or increase in HbA1c levels) in those patients with CD with known insulin-treated diabetes mellitus before pasireotide treatment.
 | Figure 2 Suggested management of pasireotide-induced hyperglycemia. In patients with insulin-treated diabetes mellitus (DM) consider adding a DDP-4 inhibitor or a GLP receptor agonist if hyperglycemia or HbA1c levels are more than 58 mmol/mol. |
Sulfonylureas or PPARγ agonist treatments may also theoretically be possible but are not recommended: sulfonylureas induce weight gain and carry a risk for hypoglycemia (because of glucose-independent insulin secretion), and PPARγ agonists are associated with an increase in body weight, fluid retention, and bone fractures, even though rosiglitazone is reportedly capable of controlling both hyperglycemia and hypercortisolism.26
Further progress: pasireotide long-acting release
Similar to octreotide, pasireotide first became available for subcutaneous injection, but in cases of chronic disease, multiple daily subcutaneous injections may be inconvenient for patients. To avoid this issue, octreotide long-acting release (LAR) and lanreotide autogel are now available, both of which can be administered monthly in cases of acromegaly or patients with neuroendocrine tumors.35,49 This is available also for pasireotide, and the LAR formulation is expected to produce fewer peak-to-trough fluctuations than the subcutaneous formulation, which could reduce the drug’s adverse effects, such as nausea and hyperglycemia.
Pasireotide is developed in di-aspartate form for subcutaneous administration and in pamoate form for intramuscular injection in the LAR formulation.50 In healthy subjects, administering 40–60 mg pasireotide LAR induced initially increasing serum concentrations that peaked after 10–24 hours, followed by a decrease that lasted for a week, followed by a second serum peak 20 days later coinciding with the slow-release phase of the LAR formulation.50 Pasireotide LAR thus showed a dose-proportional, extended-release profile suitable for monthly administration, as studied in patients with acromegaly and neuroendocrine tumors,35,49 and a phase 3 trial is still underway on its use in CD.
Conclusion and practical guidelines
Medical treatment for CD should be recommended to patients who have refused surgery or when it has proved unsuccessful, or for those awaiting the effects of radiotherapy, or in the event of recurrences. Pasireotide is a novel pituitary-directed drug, a SST analog with a broad range of effects on almost all SSTRs, that has recently been approved for use in adult patients with CD. As a monotherapy at 600–900 μg twice a day, it has been proved effective in up to 30% of cases, but in combination with other pituitary- or adrenal-targeting compounds, it may induce cortisol reduction or normalization in almost all patients. The most common adverse effect of pasireotide is hyperglycemia, which can be treated effectively with incretin-based drugs.
To optimize CD management, pasireotide treatment could be the optimal choice in CD patients without severe hypercortisolism and with normal or slightly impaired glucose metabolism.
Disclosure
The authors report no conflicts of interest in this work.
References
Nieman LK, Biller BM, Findling JW, et al. The diagnosis of Cushing’s syndrome: an Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab. 2008;93(5):1526–1540. | ||
Colao A, Boscaro M, Ferone D, Casanueva FF. Managing Cushing’s disease: the state of the art. Endocrine. 2014;47(1):9–20. | ||
Chiodini I, Mascia ML, Muscarella S, et al. Subclinical hypercortisolism among outpatients referred for osteoporosis. Ann Intern Med. 2007;147(8):541–548. | ||
Terzolo M, Reimondo G, Chiodini I, et al. Screening of Cushing’s syndrome in outpatients with type 2 diabetes: results of a prospective multicentric study in Italy. J Clin Endocrinol Metab. 2012;97(10):3467–3475. | ||
Ceccato F, Antonelli G, Barbot M, et al. The diagnostic performance of urinary free cortisol is better than the cortisol:cortisone ratio in detecting de novo Cushing’s syndrome: the use of a LC–MS/MS method in routine clinical practice. Eur J Endocrinol. 2014;171(1):1–7. | ||
Ceccato F, Barbot M, Zilio M, et al. Performance of salivary cortisol in the diagnosis of Cushing’s syndrome, adrenal incidentaloma, and adrenal insufficiency. Eur J Endocrinol. 2013;169(1):31–36. | ||
Boscaro M, Arnaldi G. Approach to the patient with possible Cushing’s syndrome. J Clin Endocrinol Metab. 2009;94(9):3121–3131. | ||
Colao A, De Block C, Gaztambide MS, Kumar S, Seufert J, Casanueva FF. Managing hyperglycemia in patients with Cushing’s disease treated with pasireotide: medical expert recommendations. Pituitary. 2014;17(2):180–186. | ||
Colao A, Petersenn S, Newell-Price J, et al; Pasireotide B2305 Study Group. A 12-month phase 3 study of pasireotide in Cushing’s disease. N Engl J Med. 2012;366(10):914–924. | ||
Dimopoulou C, Schopohl J, Rachinger W, et al. Long-term remission and recurrence rates after first and second transsphenoidal surgery for Cushing’s disease: care reality in the Munich Metropolitan Region. Eur J Endocrinol. 2014;170(2):283–292. | ||
Barbot M, Albiger N, Koutroumpi S, et al. Predicting late recurrence in surgically treated patients with Cushing’s disease. Clin Endocrinol (Oxf). 2013;79(3):394–401. | ||
Nieman LK. Update in the medical therapy of Cushing’s disease. Curr Opin Endocrinol Diabetes Obes. 2013;20(4):330–334. | ||
Gadelha MR, Vieira Neto L. Efficacy of medical treatment in Cushing’s disease: a systematic review. Clin Endocrinol (Oxf). 2014;80(1):1–12. | ||
Devin JK, Allen GS, Cmelak AJ, Duggan DM, Blevins LS. The efficacy of linear accelerator radiosurgery in the management of patients with Cushing’s disease. Stereotact Funct Neurosurg. 2004;82(5–6):254–262. | ||
Losa M, Picozzi P, Redaelli MG, Laurenzi A, Mortini P. Pituitary radiotherapy for Cushing’s disease. Neuroendocrinology. 2010;92(suppl 1):107–110. | ||
Husebye ES, Allolio B, Arlt W, et al. Consensus statement on the diagnosis, treatment and follow-up of patients with primary adrenal insufficiency. J Intern Med. 2014;275(2):104–115. | ||
Biller BM, Grossman AB, Stewart PM, et al. Treatment of adrenocorticotropin-dependent Cushing’s syndrome: a consensus statement. J Clin Endocrinol Metab. 2008;93(7):2454–2462. | ||
Castinetti F, Morange I, Jaquet P, Conte-Devolx B, Brue T. Ketoconazole revisited: a preoperative or postoperative treatment in Cushing’s disease. Eur J Endocrinol. 2008;158(1):91–99. | ||
Vilar L, Naves LA, Azevedo MF, et al. Effectiveness of cabergoline in monotherapy and combined with ketoconazole in the management of Cushing’s disease. Pituitary. 2010;13(2):123–129. | ||
Kamenický P, Droumaguet C, Salenave S, et al. Mitotane, metyrapone, and ketoconazole combination therapy as an alternative to rescue adrenalectomy for severe ACTH-dependent Cushing’s syndrome. J Clin Endocrinol Metab. 2011;96(9):2796–2804. | ||
Barbot M, Albiger N, Ceccato F, et al. Combination therapy for Cushing’s disease: effectiveness of two schedules of treatment. Should we start with cabergoline or ketoconazole? Pituitary. 2014;17(2):109–117. | ||
Bertagna X, Pivonello R, Fleseriu M, et al. LCI699, a potent 11β-hydroxylase inhibitor, normalizes urinary cortisol in patients with Cushing’s disease: results from a multicenter, proof-of-concept study. J Clin Endocrinol Metab. 2014;99(4):1375–1383. | ||
Castinetti F, Guignat L, Giraud P, et al. Ketoconazole in Cushing’s disease: is it worth a try? J Clin Endocrinol Metab. 2014;99(5):1623–1630. | ||
Pivonello R, De Martino MC, Cappabianca P, et al. The medical treatment of Cushing’s disease: effectiveness of chronic treatment with the dopamine agonist cabergoline in patients unsuccessfully treated by surgery. J Clin Endocrinol Metab. 2009;94(1):223–230. | ||
Godbout A, Manavela M, Danilowicz K, Beauregard H, Bruno OD, Lacroix A. Cabergoline monotherapy in the long-term treatment of Cushing’s disease. Eur J Endocrinol. 2010;163(5):709–716. | ||
Pecori Giraldi F, Scaroni C, Arvat E, et al. Effect of protracted treatment with rosiglitazone, a PPARgamma agonist, in patients with Cushing’s disease. Clin Endocrinol (Oxf). 2006;64(2):219–224. | ||
Occhi G, Albiger N, Berlucchi S, et al. Peroxisome proliferator-activated receptor gamma in the human pituitary gland: expression and splicing pattern in adenomas versus normal pituitary. J Neuroendocrinol. 2007;19(7):552–559. | ||
Dang CN, Trainer P. Pharmacological management of Cushing’s syndrome: an update. Arq Bras Endocrinol Metabol. 2007;51(8):1339–1348. | ||
Pecori Giraldi F, Ambrogio AG, Andrioli M, et al. Potential role for retinoic acid in patients with Cushing’s disease. J Clin Endocrinol Metab. 2012;97(10):3577–3583. | ||
Occhi G, Regazzo D, Albiger NM, et al. Activation of the dopamine receptor type-2 (DRD2) promoter by 9-cis retinoic acid in a cellular model of Cushing’s disease mediates the inhibition of cell proliferation and ACTH secretion without a complete corticotroph-to-melanotroph transdifferentiation. Endocrinology. 2014;155(9):3538–3549. | ||
Boscaro M, Ludlam WH, Atkinson B, et al. Treatment of pituitary-dependent Cushing’s disease with the multireceptor ligand somatostatin analog pasireotide (SOM230): a multicenter, phase II trial. J Clin Endocrinol Metab. 2009;94(1):115–122. | ||
Bruns C, Lewis I, Briner U, Meno-Tetang G, Weckbecker G. SOM230: a novel somatostatin peptidomimetic with broad somatotropin release inhibiting factor (SRIF) receptor binding and a unique antisecretory profile. Eur J Endocrinol. 2002;146(5):707–716. | ||
Cuevas-Ramos D, Fleseriu M. Somatostatin receptor ligands and resistance to treatment in pituitary adenomas. J Mol Endocrinol. 2014;52(3):R223–R240. | ||
Giustina A, Chanson P, Kleinberg D, et al. Expert consensus document: A consensus on the medical treatment of acromegaly. Nat Rev Endocrinol. 2014;10(4):243–248. | ||
Wolin EM, Hu K, Hughes G, Bouillaud E, Giannone V, Resendiz KH. Safety, tolerability, pharmacokinetics, and pharmacodynamics of a long-acting release (LAR) formulation of pasireotide (SOM230) in patients with gastroenteropancreatic neuroendocrine tumors: results from a randomized, multicenter, open-label, phase I study. Cancer Chemother Pharmacol. 2013;72(2):387–395. | ||
van der Hoek J, Waaijers M, van Koetsveld PM, et al. Distinct functional properties of native somatostatin receptor subtype 5 compared with subtype 2 in the regulation of ACTH release by corticotroph tumor cells. Am J Physiol Endocrinol Metab. 2005;289(2):E278–E287. | ||
Golor G, Hu K, Ruffin M, et al. A first-in-man study to evaluate the safety, tolerability, and pharmacokinetics of pasireotide (SOM230), a multireceptor-targeted somatostatin analog, in healthy volunteers. Drug Des Devel Ther. 2012;6:71–79. | ||
Mackenzie Feder J, Bourdeau I, Vallette S, Beauregard H, Ste-Marie LG, Lacroix A. Pasireotide monotherapy in Cushing’s disease: a single-centre experience with 5-year extension of phase III Trial. Pituitary. Epub 2013 Nov 28. | ||
Trementino L, Cardinaletti M, Concettoni C, Marcelli G, Boscaro M, Arnaldi G. Up-to 5-year efficacy of pasireotide in a patient with Cushing’s disease and pre-existing diabetes: literature review and clinical practice considerations. Pituitary. Epub 2014 Jun 21. | ||
Trementino L, Cardinaletti M, Concettoni C, et al. Salivary cortisol is a useful tool to assess the early response to pasireotide in patients with Cushing’s disease. Pituitary. Epub 2014 Jan 31. | ||
Pivonello R, Petersenn S, Newell-Price J, et al. Pasireotide B2305 Study Group. Pasireotide treatment significantly improves clinical signs and symptoms in patients with Cushing’s disease: results from a Phase III study. Clin Endocrinol (Oxf). 2014;81(3):408–417. | ||
Badia X, Valassi E, Roset M, Webb SM. Disease-specific quality of life evaluation and its determinants in Cushing’s syndrome: what have we learnt? Pituitary. 2014;17(2):187–195. | ||
Webb SM, Badia X, Barahona MJ, et al. Evaluation of health-related quality of life in patients with Cushing’s syndrome with a new questionnaire. Eur J Endocrinol. 2008;158(5):623–630. | ||
Webb SM, Ware JE, Forsythe A, et al. Treatment effectiveness of pasireotide on health-related quality of life in patients with Cushing’s disease. Eur J Endocrinol. 2014;171(1):89–98. | ||
Feelders RA, de Bruin C, Pereira AM, et al. Pasireotide alone or with cabergoline and ketoconazole in Cushing’s disease. N Engl J Med. 2010;362(19):1846–1848. | ||
Mazziotti G, Gazzaruso C, Giustina A. Diabetes in Cushing syndrome: basic and clinical aspects. Trends Endocrinol Metab. 2011;22(12):499–506. | ||
Breitschaft A, Hu K, Hermosillo Reséndiz K, Darstein C, Golor G. Management of hyperglycemia associated with pasireotide (SOM230): healthy volunteer study. Diabetes Res Clin Pract. 2014;103(3):458–465. | ||
Inzucchi SE, Bergenstal RM, Buse JB, et al; American Diabetes Association (ADA); European Association for the Study of Diabetes (EASD). Management of hyperglycemia in type 2 diabetes: a patient-centered approach: position statement of the American Diabetes Association (ADA) and the European Association for the Study of Diabetes (EASD). Diabetes Care. 2012;35(6):1364–1379. | ||
Petersenn S, Bollerslev J, Arafat AM, et al. Pharmacokinetics, pharmacodynamics, and safety of pasireotide LAR in patients with acromegaly: A randomized, multicenter, open-label, phase I study. J Clin Pharmacol. 2014;54(11):1308–1317. | ||
Dietrich H, Hu K, Ruffin M, et al. Safety, tolerability, and pharmacokinetics of a single dose of pasireotide long-acting release in healthy volunteers: a single-center Phase I study. Eur J Endocrinol. 2012;166(5):821–828. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.