Back to Journals » Open Access Journal of Clinical Trials » Volume 6
Clinical trial design in the era of comparative effectiveness research
Received 16 April 2014
Accepted for publication 23 May 2014
Published 3 October 2014 Volume 2014:6 Pages 101—110
DOI https://doi.org/10.2147/OAJCT.S39758
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Anke C Winter, Graham A Colditz
Division of Public Health Sciences, Department of Surgery, Washington University School of Medicine, St Louis, MO, USA
Abstract: Clinical trials are one of the key study designs in the evolving field of comparative effectiveness research. Evaluating the effectiveness of interventions in real-world settings is complex and demands a rethinking of the traditional clinical trial approach as well as transformation of the clinical trial landscape. Novel strategies and refinement of existing approaches have been proposed to generate evidence that can guide health care stakeholders in their decision process. The purpose of this review is to discuss clinical trial design approaches in the era of comparative effectiveness research. We will focus on aspects relevant to the type of clinical trial, study population and recruitment, randomization process, outcome measures, and data collection.
Keywords: review, clinical trial, comparative effectiveness research
Introduction
Comparative effectiveness research (CER) aims to provide health care stakeholders, including patients, clinicians, and policymakers, with evidence necessary to make informed health care decisions.1 One important aspect of CER is the generation of evidence that is applicable to a broad patient population and reflects real-world circumstances, allowing efficient translation and implementation of findings into patient care. Clinical trials are one of the key study designs in CER and can be utilized to evaluate the effectiveness of a broad spectrum of health care interventions such as treatments, behavioral interventions, clinical evaluation strategies, health care delivery methods, and policy interventions.2 However, conducting a clinical trial in a real-world setting is complex and demands a shift in the traditional clinical trial paradigm.3
The investment of over $1 billion in CER through the American Recovery and Reinvestment Act of 2009 has resulted in a growing demand and interest in CER in the research community in the USA. Funding agencies including the Agency for Healthcare Research and Quality and the Patient-Centered Outcomes Research Institute have issued proposal requests to develop CER infrastructure and conduct CER studies including pragmatic clinical trials.4 The governmental commitment and investment in CER provides the research community with exciting opportunities to address important CER questions. However, many health researchers and decision makers may not yet be familiar with clinical trial design features and concepts in the rapidly evolving CER field. The purpose of this review is to provide a broad overview of clinical trial design concepts in the context of CER and to discuss some aspects relevant to the design and interpretation of clinical trials. Within the scope of this review, we will focus on the definition of trial type, study population and recruitment, randomization process, outcomes measures, and data collection. We will discuss some methodological points to consider when designing a clinical trial, acknowledging that we are unable to cover every aspect that has been proposed in this evolving field.
Type of trial
Effectiveness or pragmatic trials have been proposed as one key trial design in CER to generate evidence that can be efficiently translated into patient care. An effectiveness or pragmatic trial seeks to answer the question whether an intervention works under usual conditions. An efficacy or explanatory trial is designed to evaluate whether an intervention works under ideal circumstances.5,6 These distinctions also have implications for the design and interpretation of the trial. A pragmatic/effectiveness trial is designed to determine the effectiveness of an intervention in a real-world setting and will include a broad spectrum of patients. The trial will be embedded in routine care or reflect real-world circumstances of patient care. The intervention will be compared with an alternative intervention or usual care. The trial design will allow a certain degree of flexibility in administering the intervention and in following up patients without compromising the internal validity of the trial. In contrast, to determine the efficacy of an intervention, an efficacy/explanatory trial will enroll a selective patient population, likely to be highly responsive to the intervention. The intervention will be compared with placebo or a well controlled alternative intervention. The trial will be performed in a tightly controlled study setting with little flexibility and patients will be closely monitored and followed.6
Both efficacy and effectiveness trials add valuable findings to the whole body of evidence and the choice of trial should be guided by the underlying research question. Understanding the purpose and design features of the trial are important for the interpretation of the trial and the generalizability of trial results. Results of an efficacy trial indicating a beneficial effect do not allow the conclusion that the intervention will always work in usual practice, whereas a “negative” efficacy trial strongly suggests that the intervention would not work under usual conditions. An intervention that has been demonstrated to be effective under usual conditions will probably show similar results under ideal circumstances, whereas a “negative” effectiveness trial does not prove that its intervention cannot work under other circumstances.
However, labels such as pragmatic or explanatory are an oversimplification and imply a dichotomy. In reality, a trial is rarely completely pragmatic or explanatory and will be on a continuum between these two extremes. To provide guidance for trial design and to support trialists in the evaluation of the degree of pragmatism, a pragmatic-explanatory continuum indicator summary (PRECIS) tool has been developed by an international group of trialists.6 The PRECIS instrument describes ten domains that affect the degree to which a trial is pragmatic or explanatory (Table 1). The graphical representation of the ten domains is a useful instrument to identify those domains that are not as pragmatic or explanatory as the trial designer desires (Figure 1). The PRECIS instrument has been primarily developed to guide the trial design at the planning stage but may also have an application in peer reviews from study reports.
 | Figure 1 Pragmatic-explanatory continuum indicator summary (PRECIS) tool examples. |
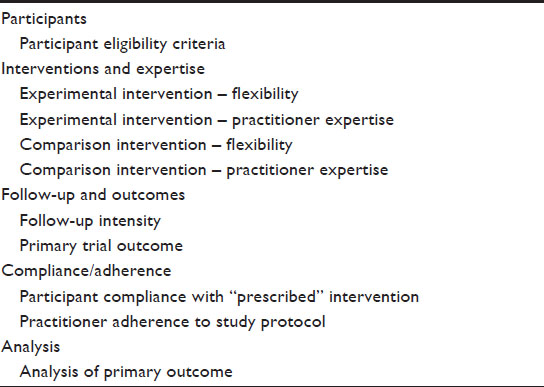 | Table 1 Ten domains of the PRECIS model |
In a recently published study, the PRECIS criteria were applied to the POWER (Practice-Based Opportunities for Weight Reduction) trials.7 The POWER trials were three individual studies designed to test the effectiveness of interventions for obesity treatment in primary care settings. As part of a common National Institutes of Health funding mechanism, all trials shared certain design features. Trial-specific elements included different types of interventions and secondary outcome measures. Two raters from each trial and three independent raters were asked to rate the three studies on the ten PRECIS domains, using a 0–4 point scale (0, completely explanatory; 4, completely pragmatic). In Figure 1, the PRECIS diagram of the “POWER Hopkins” study7 is presented. Overall, all trials were rated in a moderate range on the PRECIS scale, with mean scores ranging from 1.82 to 2.36. The inter-rater reliability on the composite PRECIS score was high (r=0.88) and there was moderate agreement on the individual level. Despite the small sample size, the study is an important first step to evaluate the applicability of the PRECIS criteria in post hoc reviews. In addition, the authors introduced a scoring system to more objectively quantify the degree of pragmatism. It is unclear to date how the degree of pragmatism of a trial will impact the adoption and implementation of findings in patient care. Hopefully, future studies in the field will help to define how the choices made at the design stage can affect the translation into care.
Another important concept to further define and to classify clinical trials is according to the underlying research hypothesis.
In the context of CER, noninferiority trials are important since they can be used to guide the decision process between two interventions that have similar therapeutic effects but differ in terms of other aspects relevant to stakeholders, such as costs, adverse event profile, and/or route of administration.8 The noninferiority trial aims to show that the difference between the treatment of interest and the reference treatment (active control) is less than the prespecified noninferiority margin.9 Figure 2 displays a schematic presentation of the possible scenarios of observed treatment differences in noninferiority trials.
 | Figure 2 Possible scenarios of observed treatment differences in non-inferiority trials. |
The design and quality of a noninferiority trial depends on the proper choice of the noninferiority margin. Defining the noninferiority margin can be complex and quiet challenging. Factors that can provide guidance in the development of noninferiority are evidence from previous studies, preliminary data, and/or clinical judgment. Sufficient evidence from previous efficacy studies or preliminary data is very helpful in allowing the trialists to make reasonable assumptions about an anticipated effect of the reference treatment. However, some points should be considered when utilizing previous evidence for the definition of the noninferiority margin. First, patient populations enrolled in previous efficacy trials may be highly selective and not representative of the targeted population in the noninferiority trial. Second, trials demonstrating a beneficial effect of the reference treatment must have been conducted recently enough to ensure that no substantial changes in medical practice and important medical advances occurred. Third, the chosen endpoint in the planned trial must be sensitive to the proposed effect in both the intervention and reference group to demonstrate a true difference.10
Many trials have to enroll a chronic disease population in order to address important CER hypotheses that are pertinent to real-world patient care. Designing a noninferiority trial in a patient population with chronic disease is particularly challenging as previous evidence about possible anticipated effect sizes may be lacking for this specific patient population, cointerventions may occur, and patients may change their treatment regime throughout the trial. In addition, efficacy studies can fail to distinguish between treatment and placebo effect, or the effect can vary according to the type of placebo used in some chronic conditions. This makes a noninferiority trial difficult to design.11 Some strategies, such as stratification, are available at the design stage to control for anticipated or known cointerventions. However, stratification of multiple factors complicates the trial design and it may be impossible to anticipate any possible cointervention upfront. Although the randomization process ideally balances the possible cointerventions between the groups, the possibility that the effect will be diluted and results will be biased toward the null cannot be ruled out. In the context of a noninferiority trial, a bias toward the null has special implications as it can lead to the false conclusion of noninferiority.
Study population and recruitment
Recruitment of a large representative study population in a timely and cost-efficient manner is one of the major challenges in the CER field. To assure generalizability of results, an effectiveness trial aims to include a broad and representative study population. In particular, the inclusion of populations that have been traditionally underrepresented in clinical trials such as the elderly, minorities, and underserved populations is an important aspect of CER. Some comparative effectiveness trials addressing important gaps in the field require a large sample size to demonstrate effectiveness of interventions and may therefore not be feasible to conduct. A long recruitment process is not desirable because it increases costs and unnecessarily delays the translation of evidence into patient care. Utilization of existing health care infrastructures, such as registries, health insurances, and primary care networks, for trial recruitment is a promising approach to overcome some of these challenges.
TASTE (Thrombus Aspiration in ST-Elevation Myocardial Infarction in Scandinavia) is an example of a recently published clinical trial that utilized the infrastructure of a population-based national registry, SCAAR (the Swedish Coronary Angiography and Angioplasty Registry), to establish feasibility and to facilitate patient enrollment and data collection.12 This government-funded registry included data from all 29 Swedish and one Icelandic coronary intervention centers. The patients were randomized using an online randomization tool within the SCAAR database and the intervention was embedded in routine care. Using the existing registry infrastructure, the investigators were able to recruit and randomize over 6,000 patients between June 2000 and September 2012 at an incremental cost of $50 per patient.13
MI FREE (Post-Myocardial Infarction Free Rx Event and Economic Evaluation Trial) is an example of a cluster randomized trial that was conducted within a large insurance system (Aetna) in the USA. The aim of the trial was to compare the effectiveness of full prescription drug coverage for statins, beta-blockers, angiotensin-converting enzyme inhibitors, and angiotensin II receptor blockers versus usual prescription coverage in the secondary prevention of myocardial infarction. Hospital discharge claims were evaluated by the insurance provider to identify eligible patients with a discharge diagnosis of new acute myocardial infarction. Randomized assignments to the two insurance benefits groups occurred at the level of the plan sponsor. During the total study period of 34 months, 5,855 patients were included in the trial and followed for a minimum of 1 year. Outcome information has been ascertained through Aetna’s health care utilization databases.14
Although both the registry-based and insurance-based designs are promising and novel concepts of efficient and cost-effective recruitment of a large number of trial participants, there are some limitations to these designs worth considering. Despite the broad inclusion criteria of the TASTE trial, approximately 40% of registry patients did not enter the trial, mainly because they were unable to provide informed consent. These patients had a higher 30-day mortality rate when compared with the enrolled patient population, which limits the generalizability of the study results.12 Choudry and Shrank shared their experience with designing the MI FREE trial in an insurance setting and discussed several challenges.15 Noteworthy in the context of patient recruitment and study population characteristics, the potential inaccuracy of claims-based identification methods, the impact of claims lag on the timely enrollment of subjects, and the reluctance of patients to participate in insurance-based interventions were described as challenges the trialists faced throughout the trial. In addition, the trial included neither patients over 65 years old as they receive health benefits through Medicare nor those patients who received health benefits through other mechanisms.
Recruitment of patients through primary care practices and community-based health care providers is another important strategy for assembling a study population that reflects real-world patient care. In particular, primary care practices may give access to multimorbid and elderly patients, a population typically underrepresented in clinical trials.16 Practice-based research networks have been developed and initiated worldwide, and provide an infrastructure for conducting research in primary care settings.17 Recruitment of participants in primary care settings may be associated with some unique challenges. When clinicians and/or practice staff are involved in the screening and enrolling process, barriers such as lack of time and resources, concerns with the study protocol, and the possible negative impact on the patient-clinician relationship can affect the success of recruitment. In addition, some primary care practices lack the infrastructure necessary to recruit and conduct research.18 Proposed strategies to overcome some of these recruitment difficulties in primary care include identification of eligible patients through electronic health records and minimizing the impact on general practice operations, but it is unclear to date whether these strategies can be applied to and adopted by the majority of primary care settings or whether individually tailored strategies are necessary.19–21
Utilization of existing health care infrastructures for patient recruitment may not assure the participation of ethnic minorities and other underrepresented populations in clinical trials. Factors impacting minority clinical trial enrollment range from individual to policy level factors, so strategies to enhance minority recruitment possibly require interventions at multiple levels. A framework to develop and implement an institutional strategy to increase minority recruitment in therapeutic cancer trials at a US academic institution has been published recently.22 Within 5 years after implementation of structural changes on four different levels, minority accrual to therapeutic trials increased from 12% to 14%. Another strategy that has been proposed to enhance minority participation in clinical research is engagement of community members in research activities through community-based participatory research.23 Both implementing changes at the institutional level and community-based participatory research are promising approaches to address the underrepresentation of minority groups in clinical trials. However, these approaches require long-term commitment and support from institutions and researchers to implement structural changes at the institutional level as well as to build and sustain community partnerships.
Randomization process
Many research questions in the CER field do not allow randomization at the individual level. They may require a cluster randomized trial design because interventions are delivered at the system level or because individual allocation of the intervention creates the possibility of contamination between those who receive the intervention and those who do not, either through the patients or the provider who delivers the intervention. In a cluster randomized trial, the intervention is randomly assigned to a group (ie, cluster) of patients and each patient within a cluster receives the same intervention. James et al provide an example of a cluster randomized trial designed to test system interventions to promote colon cancer screening among underinsured and uninsured patients.24 In this pragmatic clinical trial, community health centers will be randomly assigned to evidence-based implementation strategies for increasing colorectal cancer screening. The primary outcome, colon cancer screening rates, will be assessed at the patient level. Implementation outcomes, defined according to the RE-AIM (Reach, Efficacy/Effectiveness, Adoption, Implementation, and Maintenance) conceptual framework,25 will be collected at the patient, provider, and practice levels.
Compared with an individual randomized trial, the cluster trial is more complex to design and execute, and poses some methodological challenges.26,27 Allocating interventions to a cluster of patients has important implications for both the sample size calculations and the analyses approach. Patients within a cluster may share certain similarities and cannot be considered as independent observations. Independence is one important assumption of standard statistical tests used for sample size calculations, and the trial designer needs to account for possible correlations of patient characteristics including the outcome of interest within a cluster. The intraclass correlation coefficient, defined as the ratio of the variance between clusters divided by the sum of the variance between clusters plus the variance among patients within a cluster, quantifies the amount of agreement in a characteristic (ie, the primary endpoint of a study) between individuals of the same cluster.26 Estimation of the intraclass correlation coefficient is one key component of the trial design as it informs the calculation of the design effect, an inflation factor used to adjust standard sample size calculations. The sample size for a cluster randomized trial is commonly estimated by calculating the number of participants for an individual randomized trial with the same effect size, significance level, and power, and then multiplying the sample size by the design effect.28 Failure to incorporate the design effect into the sample size calculations results in a possible underestimation of the sample size necessary to detect the anticipated outcome difference between the intervention groups. Estimation of a design effect in the planning stages of a cluster randomized trial is challenging, particularly when preliminary data are not available to make reasonable assumptions.
Although accounting for correlations in sample size calculations and in the analyses approach is an important aspect in the design of cluster randomized trials, many cluster randomized trials still use inappropriate statistical methods or fail to report important methodological aspects. In a systematic review of 73 cluster randomized trials in residential facilities, only 27% reported accounting for clustering in sample size calculations and 74% in the analyses approach.29 There is some evidence that the quality of reporting cluster randomized trials has improved in a few aspects since the introduction of the extended Consolidated Standards of Reporting Trials (CONSORT) statement. However, no improvements were observed in reporting essential methodological features.28,30 In a recently published systematic review, journal endorsement of the CONSORT statement was not associated with trial quality, but trials with support from statisticians and/or epidemiologists were more likely to account for clustering in sample size calculations and analyses.29
One unique challenge of cluster randomized trials is that successful randomization at the system level, resulting in balanced characteristics between clusters, does not guarantee that characteristics are balanced at the individual level. Imbalance on the individual level is a threat to the internal validity of study results, and strategies to address possible imbalances should be considered at the design stage of the trial. Simple randomization techniques may pose a higher risk for covariate imbalance in cluster randomized trials, and more complex allocation techniques such as restricted randomization including matching, stratification, and minimization, as well as covariate-constrained randomization techniques, have been proposed to minimize the risk of imbalances.31
Outcome measures
Traditionally, primary and secondary endpoints in clinical trials have been chosen to be well defined clinically relevant outcome measures, such as mortality and disease-free survival, and measures indicating physiological and disease status changes. Although outcomes such as mortality and disease-free survival are certainly important to multiple stakeholders and should be part of the decision process, these endpoints do not reflect the patient’s experience and perspectives about the benefits and harms of an intervention. The value of incorporating patient-reported outcomes that allow conclusions about the effect of an intervention on patient’s symptoms, functional status, and quality of life has been extensively discussed and broadly accepted by the CER community as an important strategy to generate evidence that matters to patients and helps engage them in the clinical decision process.
Given the variety of instruments available to assess patient-reported outcomes, one of the challenges at the trial planning stage is to choose the appropriate outcome measure. A patient-reported outcome can be defined as a self-reported measure of patient health status, such as health-related quality of life, functional status, and patient satisfaction. Several patient-reported outcome measures including health-related quality of life have their roots in the social sciences using different conceptual frameworks as a basis for the instrument development.32 Instrument development is a complex process that involves patient input in qualitative assessments of the instrument, validation of the scoring system, and possibly different translations, and quantitative assessment of how to interpret score differences and establishment of meaningful thresholds.33,34 Clinical researchers may not yet be familiar with a meaningful interpretation of the mostly multidimensional instruments, and it can be challenging to choose an instrument that best suits the specific objectives of the trial. Criteria to evaluate the appropriateness of an instrument include evidence for its reliability and validity in relation to the study population of the trial and its responsiveness to change.35 Patient-reported outcomes are often derived from multi-item instruments and summarized in scores, and may be less intuitive to interpret compared with outcomes such as mortality and disease-free survival. Some patient-reported outcome instruments allow derivation and definition of multiple endpoints (ie, overall score at the end of the study, mean change of score during follow-up, percent change of baseline score) and a careful decision about the endpoint definition and anticipated magnitude of the effect size should be taken at the designing stage to avoid selective reporting of results and to assure appropriate power and sample size estimates.36–38
Several initiatives have been established to develop and standardize patient-reported outcome measures. The Patient Reported Outcomes Measurement Information System (PROMIS) is a US National Institutes of Health-funded network of outcomes researchers with the overarching goal of developing a framework for patient-reported outcomes.39 Following the World Health Organization definition of health, PROMIS distinguishes between three areas of health (physical, mental, and social) and further defines subdomains, including physical function, fatigue, pain, emotional distress, social function, and global health. PROMIS measures were developed using data from general population samples across multiple chronic conditions. One advantage is that these universally relevant measures with a common metric can be compared across diseases and conditions. However, universally relevant measures may not be as sensitive as disease-specific instruments to assess the health status and to detect changes over time in certain disease populations. Controversy currently exists about the utilization of universally relevant measures versus disease-specific measures, and future studies are warranted to better understand the relationship between these two types of measures and their application in different disease populations.40,41
Heterogeneity of outcomes measures makes it challenging to synthesize existing evidence through meta-analyses and systematic reviews. Achieving consensus about endpoints including patient-reported outcomes in clinical trials is crucial. Working groups worldwide have been launched to define and standardize disease-specific core outcome sets that are comparable across trials.42,43
Data collection and follow-up
Integration of electronic medical record information into clinical trials through automated processes is an emerging concept in the clinical trial enterprise.44,45 Effectiveness trials embedded in primary care and clinical settings can utilize patient data that have been routinely collected in clinical care through electronic medical records. This approach has advantages, as it allows collection of baseline and long-term follow-up data for large and highly representable trial populations in a timely and cost-effective manner. Patients eligible for the trial can be identified automatically and utilization of electronic record information allows comparisons of enrolled trial patients with those not enrolled to monitor the representativeness of the trial population. Linkages to other data sources, including national death registers, hospital records, and registries, allow capture of important outcome information and reduce the amount of loss to follow-up. Self-reported patient information collected through electronic devices can be linked to the trial database, enriching the trial data and allowing incorporation of patient-reported outcomes.46
Data collection and patient follow-up through utilization and linkage of electronic health records provides the CER enterprise with exciting opportunities. However, the electronic health record has not been primarily designed for research purposes, which poses some major challenges. The data captured in an electronic health record may reflect the interactions of the patient with the health care system rather than a well defined disease status. Strategies to accurately “phenotype” patients according to the available electronic health record information have been developed, and efforts are underway to standardize and validate procedures across electronic medical record systems and institutions.47 Data quality is another issue that has been broadly discussed in the context of use of electronic medical records for research purposes, including data completeness and accuracy. Missing data, erroneous data, inconsistencies among providers, across institutions, and over time, as well as data stored in noncoded text notes, are some of the data challenges identified.48
Complete, valid, and reliable baseline, follow-up, and endpoint data are crucial to assure high internal validity of trial results. A recently published study from Scotland compared cardiovascular endpoint detection through record linkage of death and hospitalization records with events that were reported through a standard clinical trial mechanism in the West of Scotland Coronary Prevention study.49 The study showed excellent matching between record linkage and endpoints assessed through standard trial mechanisms for unambiguous endpoints such as mortality, but complex diagnoses such as transient ischemic attack/stroke and identification of subsequent events was associated with imperfect matching of events. Important to note, the study was conducted in Scotland, a country with a unified health care system that facilitates patient follow-up. In a more scattered health care system with frequent insurance coverage transitions like the USA, patient follow-up through electronic health record linkage may be limited and prone to missing information. Missing information can introduce bias and lead to false conclusions of intervention effectiveness. Although there are analytical strategies to handle missing information, the best approach is to prevent occurrence of missing information.50 Further studies will hopefully help to identify the best approach for electronic medical records’ utilization in clinical trials without compromising data quality and accuracy.
Conclusion
The evolving field of CER requires a shift in the traditional clinical trial paradigm and will continue to challenge and change the clinical trials’ landscape. Careful study design and consistent use of terminology and standards in reporting of trial results will facilitate meaningful interpretation and translation of findings. Incorporation of patient-reported outcomes into clinical trials will provide stakeholders with important information on patient’s experiences and perspectives. Identification of gaps in the field will foster development of novel methodological approaches. Key to a successful transition of the clinical research enterprise is investment in sustainable research infrastructures, further development and refinement of methodological approaches, and continuing training of the research community in relevant CER methods.
Acknowledgments
The authors thank Jennifer Tappenden for providing assistance with manuscript writing. Drs Colditz and Winter are supported by funds from the Washington University School of Medicine, the Barnes-Jewish Hospital Foundation, and the Siteman Cancer Center.
Disclosure
The authors report no conflicts of interest in this work.
References
Institute of Medicine. Initial National Priorities for Comparative Effectivene ss Research. Washington, DC, USA: National Academies Press; 2009. | |
Hlatky MA, Douglas PS, Cook NL, et al. Future directions for cardiovascular disease comparative effectiveness research: report of a workshop sponsored by the National Heart, Lung, and Blood Institute. J Am Coll Cardiol. 2012;60(7):569–580. | |
Luce BR, Kramer JM, Goodman SN, et al. Rethinking randomized clinical trials for comparative effectiveness research: the need for transformational change. Ann Intern Med. 2009;151(3):206–209. | |
Sox HC. Comparative effectiveness research: a progress report. Ann Intern Med. 2010;153(7):469–472. | |
Schwartz D, Lellouch J. Explanatory and pragmatic attitudes in therapeutical trials. J Chronic Dis. 1967;20(8):637–648. | |
Thorpe KE, Zwarenstein M, Oxman AD, et al. A pragmatic-explanatory continuum indicator summary (PRECIS): a tool to help trial designers. J Clin Epidemiol. 2009;62(5):464–475. | |
Glasgow RE, Gaglio B, Bennett G, et al. Applying the PRECIS criteria to describe three effectiveness trials of weight loss in obese patients with comorbid conditions. Health Serv Res. 2012;47(3 Pt 1):1051–1067. | |
Piaggio G, Elbourne DR, Altman DG, Pocock SJ, Evans SJ; CONSORT Group. Reporting of noninferiority and equivalence randomized trials: an extension of the CONSORT statement. JAMA. 2006;295(10):1152–1160. | |
Christensen E. Methodology of superiority vs equivalence trials and noninferiority trials. J Hepatol. 2007;46(5):947–954. | |
Friedman LM, Furberg CD, DeMets DL. Fundamentals of Clinical Trials. 4th ed. New York, NY, USA: Springer; 2010. | |
Meissner K, Fassler M, Rucker G, et al. Differential effectiveness of placebo treatments: a systematic review of migraine prophylaxis. JAMA Intern Med. 2013;173(21):1941–1951. | |
Frobert O, Lagerqvist B, Olivecrona GK, et al. Thrombus aspiration during ST-segment elevation myocardial infarction. N Engl J Med. 2013;369(17):1587–1597. | |
Lauer MS, D’Agostino RB Sr. The randomized registry trial – the next disruptive technology in clinical research? N Engl J Med. 2013;369(17):1579–1581. | |
Choudhry NK, Avorn J, Glynn RJ, et al. Full coverage for preventive medications after myocardial infarction. N Engl J Med. 2011;365(22):2088–2097. | |
Choudhry NK, Shrank WH. Implementing randomized effectiveness trials in large insurance systems. J Clin Epidemiol. 2013;66(Suppl 8):S5–S11. | |
McMurdo ME, Roberts H, Parker S, et al. Improving recruitment of older people to research through good practice. Age Ageing. 2011;40(6):659–665. | |
Green LA, Hickner J. A short history of primary care practice-based research networks: from concept to essential research laboratories. J Am Board Fam Med. 2006;19(1):1–10. | |
Daly JM, Xu Y, Ely JW, Levy BT. A randomized colorectal cancer screening intervention trial in the Iowa Research Network (IRENE): study recruitment methods and baseline results. J Am Board Fam Med. 2012;25(1):63–72. | |
Love MM, Pearce KA, Williamson MA, Barron MA, Shelton BJ. Patients, practices, and relationships: challenges and lessons learned from the Kentucky Ambulatory Network (KAN) CaRESS clinical trial. J Am Board Fam Med. 2006;19(1):75–84. | |
Reed RL, Barton CA, Isherwood LM, Baxter JM, Roeger L. Recruitment for a clinical trial of chronic disease self-management for older adults with multimorbidity: a successful approach within general practice. BMC Fam Pract. 2013;14:125. | |
Stuardi T, Cox H, Torgerson DJ. Database recruitment: a solution to poor recruitment in randomized trials? Fam Pract. 2011;28(3):329–333. | |
Anwuri VV, Hall LE, Mathews K, et al. An institutional strategy to increase minority recruitment to therapeutic trials. Cancer Causes Control. 2013;24(10):1797–1809. | |
De las Nueces D, Hacker K, DiGirolamo A, Hicks LS. A systematic review of community-based participatory research to enhance clinical trials in racial and ethnic minority groups. Health Serv Res. 2012;47(3 Pt 2):1363–1386. | |
James AS, Richardson V, Wang JS, Proctor EK, Colditz GA. Systems intervention to promote colon cancer screening in safety net settings: protocol for a community-based participatory randomized controlled trial. Implement Sci. 2013;8:58. | |
Glasgow RE, Vogt TM, Boles SM. Evaluating the public health impact of health promotion interventions: the RE-AIM framework. Am J Public Health. 1999;89(9):1322–1327. | |
Glynn RJ, Brookhart MA, Stedman M, Avorn J, Solomon DH. Design of cluster-randomized trials of quality improvement interventions aimed at medical care providers. Med Care. 2007;45(10 Suppl 2):S38–S43. | |
Puffer S, Torgerson DJ, Watson J. Cluster randomized controlled trials. J Eval Clin Pract. 2005;11(5):479–483. | |
Campbell MK, Piaggio G, Elbourne DR, Altman DG. Consort 2010 statement: extension to cluster randomised trials. BMJ. 2012;345:e5661. | |
Diaz-Ordaz K, Froud R, Sheehan B, Eldridge S. A systematic review of cluster randomised trials in residential facilities for older people suggests how to improve quality. BMC Med Res Methodol. 2013;13:127. | |
Ivers NM, Taljaard M, Dixon S, et al. Impact of CONSORT extension for cluster randomised trials on quality of reporting and study methodology: review of random sample of 300 trials, 2000–2008. BMJ. 2011;343:d5886. | |
Ivers NM, Halperin IJ, Barnsley J, et al. Allocation techniques for balance at baseline in cluster randomized trials: a methodological review. Trials. 2012;13:120. | |
Bakas T, McLennon SM, Carpenter JS, et al. Systematic review of health-related quality of life models. Health Qual Life Outcomes. 2012;10:134. | |
Izem R, Kammerman LA, Komo S. Statistical challenges in drug approval trials that use patient-reported outcomes. Stat Methods Med Res. Epub February 21, 2013. | |
Rothman M, Burke L, Erickson P, Leidy NK, Patrick DL, Petrie CD. Use of existing patient-reported outcome (PRO) instruments and their modification: the ISPOR Good Research Practices for Evaluating and Documenting Content Validity for the Use of Existing Instruments and Their Modification PRO Task Force Report. Value Health. 2009;12(8):1075–1083. | |
Calvert M, Brundage M, Jacobsen PB, Schunemann HJ, Efficace F. The CONSORT Patient-Reported Outcome (PRO) extension: implications for clinical trials and practice. Health Qual Life Outcomes. 2013;11:184. | |
Al-Marzouki S, Roberts I, Evans S, Marshall T. Selective reporting in clinical trials: analysis of trial protocols accepted by The Lancet. Lancet. 2008;372(9634):201. | |
Cappelleri JC, Bushmakin AG. Interpretation of patient-reported outcomes. Stat Methods Med Res. Epub February 19, 2013. | |
Julious SA, Walters SJ. Estimating effect sizes for health related quality of life outcomes. Stat Methods Med Res. Epub February 19, 2013. | |
Cella D, Riley W, Stone A, et al. The Patient-Reported Outcomes Measurement Information System (PROMIS) developed and tested its first wave of adult self-reported health outcome item banks: 2005–2008. J Clin Epidemiol. 2010;63(11):1179–1194. | |
The Patient-Reported Outcomes Measurement Information System Statistical Center Working Group. The Patient-Reported Outcomes Measurement Information System (PROMIS) perspective on: universally-relevant vs disease-attributed scales. Available from: http://www.nihpromis.org/(X(1)S(nhwsekmzio2cx4ct0u3ko0gn))/Documents/Universally-Relevant_vs_Disease-Attributed_2014-2-12_final508.pdf. Accessed February 15, 2014. | |
Coon CD, McLeod LD. Patient-reported outcomes: current perspectives and future directions. Clin Ther. 2013;35(4):399–401. | |
Idzerda L, Rader T, Tugwell P, Boers M. Can we decide which outcomes should be measured in every clinical trial? A scoping review of the existing conceptual frameworks and processes to develop core outcome sets. J Rheumatol. 2014;41(5):986–993. | |
Core Outcome Measures in Effectiveness Trials (COMET) Initiative. Available from: http://www.comet-initiative.org/. Accessed February 15, 2014. | |
Goodman K, Krueger J, Crowley J. The automatic clinical trial: leveraging the electronic medical record in multisite cancer clinical trials. Curr Oncol Rep. 2012;14(6):502–508. | |
Yamamoto K, Yamanaka K, Hatano E, et al. An eClinical trial system for cancer that integrates with clinical pathways and electronic medical records. Clin Trials. 2012;9(4):408–417. | |
Staa TP, Goldacre B, Gulliford M, et al. Pragmatic randomised trials using routine electronic health records: putting them to the test. BMJ. 2012;344:e55. | |
Richesson RL, Hammond WE, Nahm M, et al. Electronic health records based phenotyping in next-generation clinical trials: a perspective from the NIH Health Care Systems Collaboratory. J Am Med Inform Assoc. 2013;20(e2):e226–e231. | |
Bayley KB, Belnap T, Savitz L, Masica AL, Shah N, Fleming NS. Challenges in using electronic health record data for CER: experience of 4 learning organizations and solutions applied. Med Care. 2013;51(8 Suppl 3):S80–S86. | |
Barry SJ, Dinnett E, Kean S, Gaw A, Ford I. Are routinely collected NHS administrative records suitable for endpoint identification in clinical trials? Evidence from the West of Scotland Coronary Prevention Study. PLoS One. 2013;8(9):e75379. | |
Li T, Hutfless S, Scharfstein DO, et al. Standards should be applied in the prevention and handling of missing data for patient-centered outcomes research: a systematic review and expert consensus. J Clin Epidemiol. 2014;67(1):15–32. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.