Back to Journals » Clinical Ophthalmology » Volume 11
Clinical spectrum and management options in Vogt–Koyanagi–Harada disease
Authors Lodhi SA, Reddy JM ![]() , Peram V
, Peram V
Received 17 February 2017
Accepted for publication 14 June 2017
Published 7 August 2017 Volume 2017:11 Pages 1399—1406
DOI https://doi.org/10.2147/OPTH.S134977
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Scott Fraser
Sikander AK Lodhi, JM Lokabhi Reddy, Venkataratnam Peram
Department of Ophthalmology, Osmania Medical College/Sarojini Devi Eye Hospital, Hyderabad, Telangana, India
Purpose: The aim of this study was to describe the clinical features, treatment options, and visual outcome of Vogt–Koyanagi–Harada (VKH) disease patients over a 9-year period.
Method: A retrospective chart analysis of 32 patients with VKH, from January 2007 to December 2015, at a tertiary care government medical college eye hospital in South India.
Results: A total of 32 patients were diagnosed with VKH. The mean age at diagnosis was 32.03±8.8 years. There were 24 patients (42 eyes) with acute VKH and eight patients (16 eyes) with recurrent/chronic VKH. The mean baseline best-corrected visual acuity on presentation in the acute VKH group was 5/60 (1.114±0.565) and at last follow-up it was 6/9 (0.225±0.157). Intravenous methyl prednisolone (IVMP) was administered for 3 days to all patients with acute and recurrent VKH, followed by posterior subtenon triamcinolone (40 mg/mL) and oral azathioprine.
Conclusion: VKH-related uveitis is more common in the female gender in this South Indian population. Posterior uveitis is the most common initial manifestation. Initial aggressive treatment with IVMP, peribulbar long-acting corticosteroids, and immunosuppressives, avoiding side effects of systemic steroids, gives a good visual outcome without recurrences. Cases of unilateral VKH, seen in six patients, are the initial manifestations in the natural course of the disease, which if managed aggressively at the acute stage prevents recurrence in the other eye.
Keywords: Vogt–Koyanagi–Harada disease, female, posterior uveitis, retinal detachment, triamcinolone, azathioprine
Introduction
Vogt–Koyanagi–Harada (VKH) is a multisystem autoimmune inflammatory disorder with ophthalmic, auditory, dermatologic, and neurologic manifestations.1 The prevalence of this disease varies with ethnicity. VKH characteristically affects pigmented groups, such as Hispanics, Asians, people from the Middle East, and Asian Indians, but not the blacks of sub-Saharan African descent.2 The extraocular manifestations appear in different phases of the disease and show ethnic variations.3–5 VKH disease accounts for 8% of uveitis in Japan, 2.9% in the Middle East, 1.2% in Europe, and 1%–4% in the USA.6,7 In a study from a referral eye care center in South India, the prevalence is reported as 1.4%–3.5%.8 VKH is seen more frequently in an adult population, and women are more frequently affected.9 VKH disease usually manifests as four distinct clinical phases: prodromal, acute uveitic, convalescent, and chronic recurrent.1 The prodromal phase features neurologic and auditory manifestations, including a headache, tinnitus, neck stiffness, and hearing loss. The acute uveitic phase shows diffuse choroiditis with exudative retinal detachment (RD) and optic disc swelling. A variable amount of anterior chamber and vitreous inflammation may be seen. In the acute phase, anterior uveitis seen in some patients usually presents as low-grade non-granulomatous anterior uveitis, while in the chronic recurrent phase, patients present with recurrent granulomatous anterior uveitis. “Sunset glow fundus,” due to depigmentation of the choroid, is the hallmark of the convalescent phase. Recurrent episodes of anterior uveitis are characteristic of the chronic recurrent phase.
In a study by Rao et al, in an ethnically and geographically diverse group of patients, it is revealed that there are two clinical signs which are highly specific to VKH – exudative RD during the acute phase and sunset glow fundus during the chronic phase of the disease.3
Imaging technology has allowed a better visualization and understanding of the disease process in VKH. The first international workshop on VKH disease proposed criteria to make diagnosis.2 According to the revised diagnostic criteria, VKH disease can be complete, incomplete, and probable. All the three categories have the absolute requirement of bilateral eye disease; however, a unilateral or delayed involvement of the other eye can occur in rare cases.10–13 Complete disease represents the involvement of ocular, neurologic/auditory, and integumentary systems. Incomplete disease is an eye disease with either neurological/auditory or integumentary signs. Probable VKH is uveitis consistent with VKH, without any extraocular manifestations.
Multiple treatment regimens have been tried for VKH disease, including intravenous, oral, regional corticosteroid, cyclosporine, antimetabolites, and alkylating agents.14 Improper and inadequate immunosuppressive therapy during initial-onset acute VKH disease will lead to chronic recurrent VKH disease manifesting as granulomatous anterior uveitis with sunset glow fundus. Vision threatening complications like secondary glaucoma and cataract occur in the chronic recurrent phase. Early and high-dose systemic corticosteroids, followed by slow tapering, are mandatory to suppress the inflammation in the acute posterior uveitis stage. Adding immunomodulatory agents can improve the outcome and can reduce the risk of integumentary involvement. High-dose corticosteroids are associated with side effects such as hyperglycemia, hypertension, immunodeficiency, and secondary glaucoma. Posterior subtenon triamcinolone (PST) acetonide injection is a safe and more convenient treatment compared with systemic steroid administration, avoiding the systemic side effects and patient compliance. Most of the recurrences of RD and inflammation are due to noncompliance or a very rapid tapering of systemic corticosteroids.
The aim of this study was to analyze the demographic characteristics, clinical features, efficacy of subtenon triamcinolone acetonide injection with immunomodulatory therapy (ITP), and visual outcome in VKH patients referred to a medical college tertiary eye care center in South India.
Methods
This is a retrospective, nonrandomized, noncomparative interventional study. Patients diagnosed with VKH disease, attending the retina/uvea outpatient department of a medical college tertiary eye hospital, between January 2007 and December 2015, were included in the study. Informed consent was obtained from all the patients included in the study, and the study adhered to the tenets of the Declaration of Helsinki. Institutional ethics committee (Ethics Committee of Osmania Medical College) approval was obtained for a retrospective chart review. Retrospective chart review was performed for 32 patients (58 eyes) diagnosed with VKH for demographic data, clinical signs, investigations, treatment received, complications, and visual outcome (Tables 1 and 2). All patients underwent a comprehensive ophthalmic examination, including measurement of best-corrected visual acuity (BCVA) with Snellen charts, applanation tonometry, slit lamp examination, ophthalmoscopy, and fundus photography. Ultrasound examination of the eye and fundus fluorescein angiography (FFA) were performed in all the cases, and optical coherence tomography (OCT) was performed whenever possible. A general examination was done to look for any integumentary findings. The clinical presentation and its confirmation on FFA formed the basis for the diagnosis of VKH disease. Previous history of any trauma was ruled out in all the cases. Investigations were done to rule out tuberculosis (Mantoux test, chest X-ray) and syphilis (venereal disease research laboratory [VDRL] test). Revised diagnostic criteria for VKH disease by Read et al2 were used for classifying the cases into complete, incomplete, or probable VKH. Cases with unilateral involvement were diagnosed to have VKH when all the characteristic ocular manifestations were present because unilateral or delayed involvement of the other eye can occur in rare cases. A uniform protocol was followed in treating all the cases. All acute cases and recurrent cases on presentation (31 patients) were treated with intravenous methyl prednisolone (IVMP) 1 g for 3 days followed by PST acetonide 40 mg/mL in the affected eyes. Simultaneously, oral azathioprine 1–2 mg/kg was started and continued for 6–9 months. Patients were followed-up at monthly intervals. PST was used in 44 eyes. PST was repeated after 3 months, if required. Continuing inflammation or recurrence was monitored by BCVA, signs of inflammation, and OCT. PST was required to be repeated in 4 eyes. In our earlier cases (not included in this study), we used IVMP followed by oral corticosteroids for 1 year. The compliance with oral steroids was very poor, and therefore, the strategy was modified to use PST combined with the immunosuppressive agent azathioprine. Patients with anterior segment inflammation were treated with topical corticosteroids and cycloplegics in addition to systemic treatment. Any case that had raised intraocular pressure (IOP) was managed with medical therapy. Use of azathioprine was monitored by complete blood count and liver function tests. Complications such as cataract, glaucoma, and choroidal neovascular membrane were noted, and any extraocular manifestations were recorded in addition to chronic stage features such as sunset glow fundus and chorioretinal atrophy patches. For statistical analysis, visual acuity data were converted to logMAR values. The data were entered in an Excel 2007 spreadsheet and statistically analyzed with SPSS software.
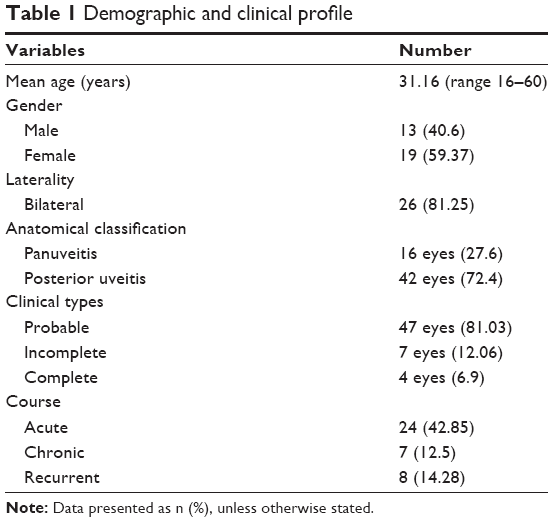 | Table 1 Demographic and clinical profile |
 | Table 2 Comparison of results between acute and chronic recurrent Vogt–Koyanagi–Harada (VKH) |
Results
A total of 32 patients were seen during the study period. The average age of the patients was 31.16 years (range 16–60 years). There were 13 male and 19 female patients (Table 1). The average follow-up was 18 months (range 2–84 months). All patients were from South Indian states of Telangana and Andhra Pradesh. Twenty-six patients had a bilateral presentation and six patients had only unilateral VKH. The type of VKH, according to diagnostic criteria, was classified as complete in 2 patients, incomplete in 5 patients, and probable in 25 patients. We have classified our patients into two groups depending on the stage of the disease at presentation (Table 2). At presentation, there were 24 patients (75%) with acute VKH and 8 patients (25%) with chronic recurrent VKH disease. This amounts to 42 eyes with acute VKH and 16 eyes with chronic recurrent VKH, and all patients with unilateral VKH were in the acute stage. Comparisons between acute and chronic VKH with respect to clinical features at presentation, clinical evaluation, and follow-up are presented in Table 2. Meningism and headache were the most common systemic associations in the acute stage (Table 3). Three patients in chronic VKH had alopecia and poliosis, and three patients had a hearing disturbance. Clinically, 16 eyes presented with panuveitis and 42 eyes presented with posterior uveitis. Slit lamp examination showed keratic precipitates in 14 eyes and flare and cells in 10 eyes. All the 24 cases in the acute stage presented with posterior segment manifestations, and the most common finding was multiple serous RDs in the posterior pole in all the 42 eyes. Vitreous cells were seen in 38 eyes. Disc hyperemia was seen in 55 eyes and disc edema in 26 eyes. Sunset glow fundus was seen in 24 eyes (which includes chronic VKH cases on presentation and acute VKH cases in convalescence on follow-up; Table 3). FFA (Figures 1–3) performed in all patients (except one) showed multiple pinpoint hyperfluorescent leaks in 54 eyes and late pooling in serous RDs in 42 eyes. Late disc staining was seen in 46 eyes. 10 MHz ultrasonography showed a diffuse choroidal thickening in all the acute cases, which was most marked in the juxta papillary region (Figures 1 and 2). OCT in acute cases showed multilobular RDs, subretinal septa, retinal pigment epithelium undulations, and choroidal thickening (Figures 1 and 2).
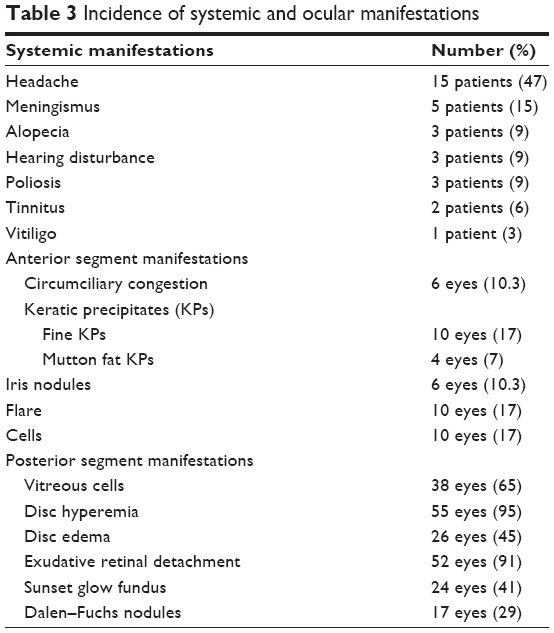 | Table 3 Incidence of systemic and ocular manifestations |
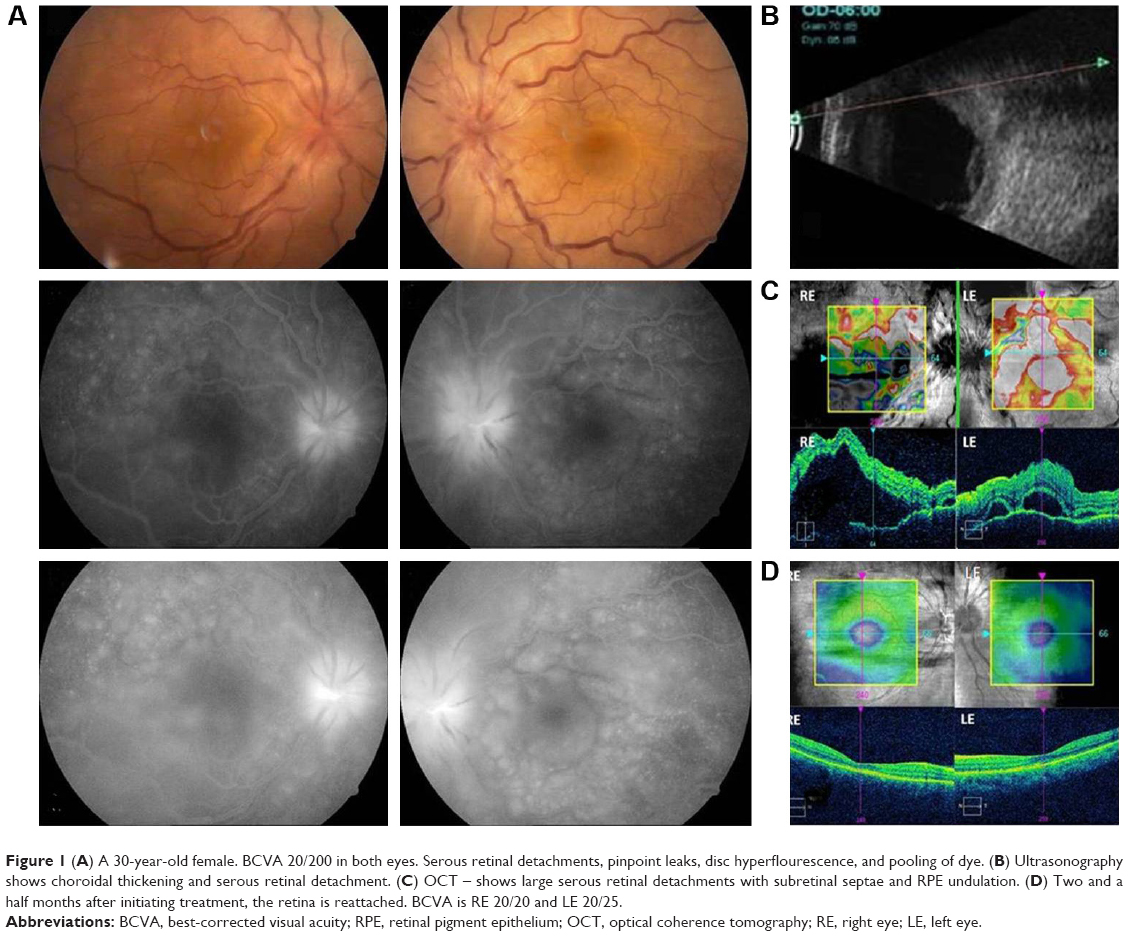 | Figure 1 (A) A 30-year-old female. BCVA 20/200 in both eyes. Serous retinal detachments, pinpoint leaks, disc hyperflourescence, and pooling of dye. (B) Ultrasonography shows choroidal thickening and serous retinal detachment. (C) OCT – shows large serous retinal detachments with subretinal septae and RPE undulation. (D) Two and a half months after initiating treatment, the retina is reattached. BCVA is RE 20/20 and LE 20/25. |
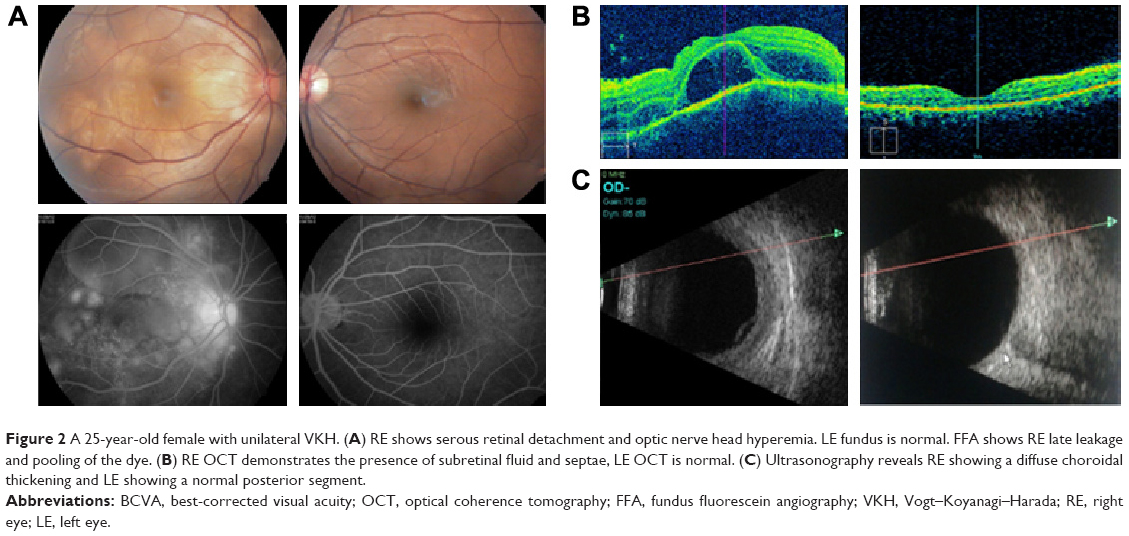 | Figure 2 A 25-year-old female with unilateral VKH. (A) RE shows serous retinal detachment and optic nerve head hyperemia. LE fundus is normal. FFA shows RE late leakage and pooling of the dye. (B) RE OCT demonstrates the presence of subretinal fluid and septae, LE OCT is normal. (C) Ultrasonography reveals RE showing a diffuse choroidal thickening and LE showing a normal posterior segment. |
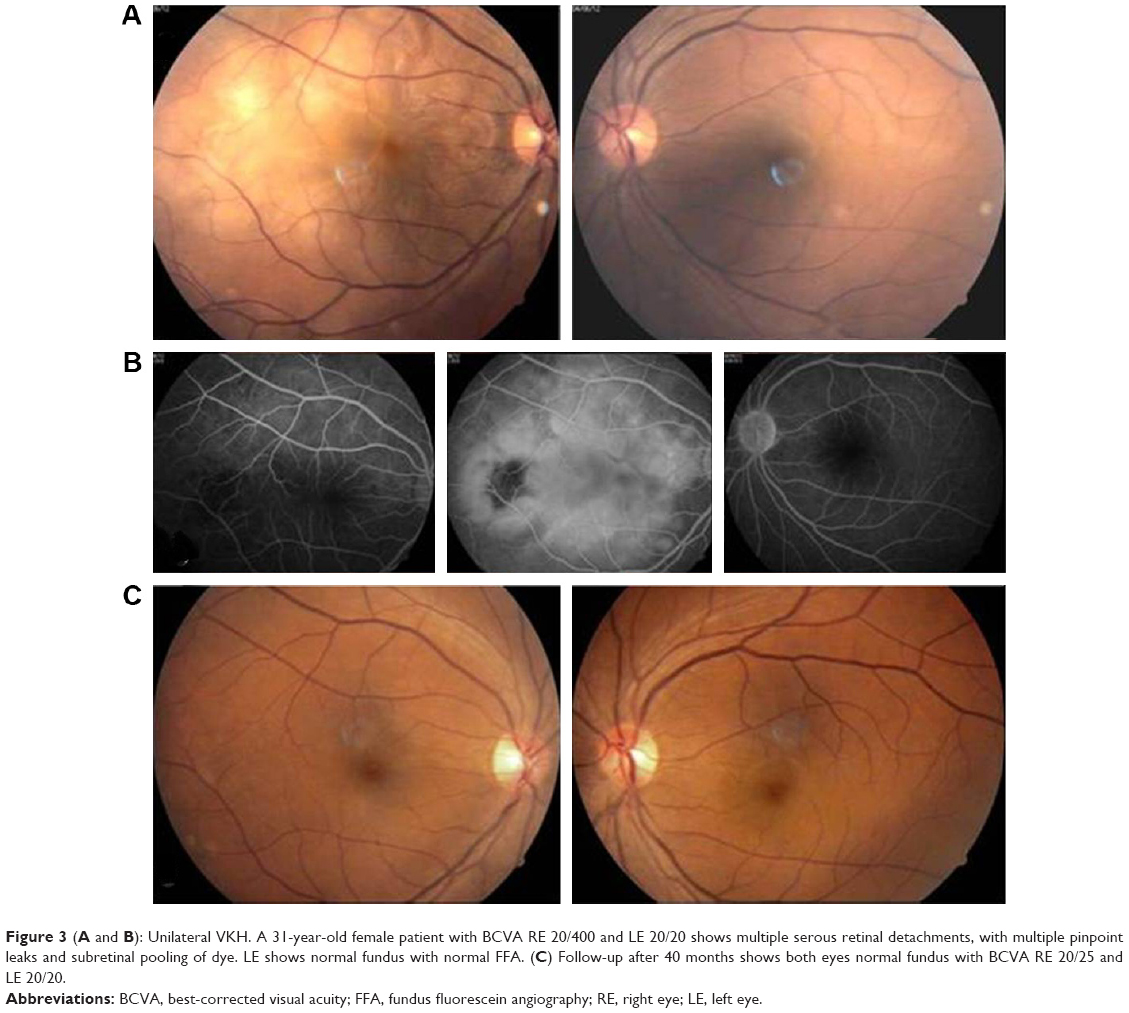 | Figure 3 (A and B): Unilateral VKH. A 31-year-old female patient with BCVA RE 20/400 and LE 20/20 shows multiple serous retinal detachments, with multiple pinpoint leaks and subretinal pooling of dye. LE shows normal fundus with normal FFA. (C) Follow-up after 40 months shows both eyes normal fundus with BCVA RE 20/25 and LE 20/20. |
Twenty-five cases of acute VKH were treated with the above-mentioned strategy. Four cases were lost to follow-up after 2 months. Twenty cases completed follow-up from 6 months to 5 years. One case had multiple recurrences over a follow-up period of 1.5 years and was treated with a combination of immunosuppressives (azathioprine and methotrexate). We found six cases of unilateral VKH in this study. All these cases clinically showed uniocular multiple serous detachments in the posterior pole, with angiographic features of choroidal delay, multiple pinpoint hyperfluorescent leaks, and pooling in the serous elevations by late phases. Ultrasound examination showed choroidal thickening in all these cases. All these cases were treated aggressively with IVMP, PST acetonide, and immunosuppression with azathioprine for 6–9 months. One other case presented with unilateral VKH in the right eye (RE) and treated with IVMP followed by PST was lost to follow-up. After 3 years, she presented with acute VKH in the left eye (LE) with BCVA RE 6/24 and LE 6/60. She was treated with the same protocol; 6 months later, she had BCVA RE 6/24 and LE 6/9 (Figure 2). Once the inflammation has subsided, RDs have resolved, and with no disc hyperemia, the fundus had a near normal appearance in 21 patients and sunset glow fundus in 6 patients of acute VKH and 6 patients with chronic and recurrent uveitis.
In this series, visual acuity of 6/12 or better was maintained in 38 eyes that presented with acute VKH. Of these, 21 eyes had a BCVA better than 6/12 after at least 1-year follow-up without recurrences (Table 2). Chronic and recurrent VKH cases had a poor visual outcome, <6/60. Complicated cataract was seen in 14 eyes, one chronic VKH patient had raised IOP, and eight patients with acute VKH had secondary glaucoma after PST injection. All these cases were managed with medical therapy. The choroidal neovascular membrane was seen in 3 eyes and chronic vitritis in 6 eyes of chronic VKH cases (Table 2).
Discussion
This retrospective study investigated patients with VKH disease over a 9-year period. Demographic features in the present case series are almost similar to those studied in other countries.1,7,8,15–18 Most of the studies show female preponderance. In accordance with the published literature, we also found that 59.37% of the cases were females. In this series, majority of the cases presented with posterior uveitis in the acute stage, and therefore, as per the revised diagnostic criteria, 25 patients (78.12%) are in the probable category. Compared to studies from Japan,19,20 we have a higher proportion of probable VKH. A study by Arevalo et al from Saudi Arabia also shows a higher proportion (58.4%) of probable VKH cases.21 Extraocular signs such as poliosis, alopecia, vitiligo, and hearing loss develop during the course of VKH disease and are seen mainly in the chronic stage of the disease.4 In a multicenter study by Rao et al, in ethnically and geographically diverse study groups, exudative RD was the most statistically significant feature of the acute disease.3 They report that robust positive and negative predictive values and likelihood ratios of various documented clinical findings indicate that in acute uveitis, the presence of bilateral exudative RDs in a patient with bilateral intraocular inflammation strongly suggests acute VKH. In the present study, exudative RD and disc hyperemia were the most common posterior segment findings in the acute cases, and sunset glow fundus was the most common finding in chronic/convalescent cases. Ancillary testing supports the clinical diagnosis of VKH disease. FFA performed in all cases showed multiple hyperfluorescent pinpoint leaks in 56 eyes (96.55%) and late pooling in serous RDs in 54 eyes (93.1%). Rao et al, in their multiethnic diverse study, reported one or more angiographic findings (choroidal delay, multifocal leaks, subretinal pooling of dye) in 83% of VKH cases.3 Another test that supported the diagnosis was ultrasound examination, performed in all cases, which showed choroidal thickening in all acute cases.
To the best of our knowledge, the number of cases of unilateral VKH seen in this study is more than that reported by any other study (Figures 2 and 3). As has been observed by Rao et al,3 from multiple papers proposing or evaluating diagnostic criteria for VKH,2,22,23 a definitive set of criteria has not been universally agreed upon. This is mainly because of the multiphasic nature of VKH, with varying clinical features in different stages of the disease as well as due to ethnic variation in disease manifestations. What we are seeing as unilateral VKH may be the early phase of the disease which if not treated aggressively in the initial stages may occur in the other eye in due course. All the six cases of unilateral VKH did not have a recurrence because of early aggressive and prompt treatment and continuation of immunosuppressive therapy for a sufficient period of time. The one case of unilateral VKH that was lost to follow-up and was not sufficiently treated in the early phase had a recurrence in the other eye after 3 years.
Prompt and aggressive treatment with corticosteroids with slow tapering over a period of 6 months is recommended to avoid recurrence and complications.24,25 Systemic corticosteroids are associated with possible side effects like hyperglycemia, hypertension, gastritis, and rising IOPs. Compliance with the use of systemic corticosteroids is usually poor. Recurrences are usually due to early and abrupt discontinuation of corticosteroids. Rubsamen and Gass reported recurrences in 43% and 52% of their patients occurring in the first 3 and 6 months of the disease, associated in most cases with rapid tapering of corticosteroids.26 Ocular manifestations of VKH can be managed by local administration of steroids.27–29 Moreker and Lodhi, from their study on the use of intravitreal triamcinolone acetonide (IVTA) in VKH, have concluded that IVTA, when used as an adjuvant, can induce remission in the acute stage of VKH and help avoid the long-term use of systemic corticosteroids.27 Hosoda et al demonstrated that isolated PST is a useful primary treatment option for patients with acute VKH without systemic disorders. In 77.8% of patients, they achieved complete resolution of serous RD without recurrence after PST therapy.30 Paredes et al31 reported their outcomes in 13 patients with VKH disease and suggested that immunomodulatory therapy as a first-line therapy for VKH is associated with a superior outcome when compared to steroid as monotherapy or with delayed addition of immunomodulatory drugs.
To overcome the problem of compliance, side effects, and recurrences, we have used a combination of PST acetonide and systemic immunosuppression with azathioprine. Azathioprine is shown to be effective for the treatment of chronic uveitis, usually in combination with corticosteroids.32–34 Azathioprine may show some adverse side effects, such as myelosuppresson, and serious side effects are unusual with the low dose (1–2 mg/kg/day) compared to high dose of azathioprine. A visual acuity of 6/12 or better was maintained in 44 eyes that presented with acute VKH that had no recurrences in the follow-up. Periocular injection of depot steroids likely induces the IOP to rise. In our study, 8 eyes of four patients showed ocular hypertension (IOP >21 mmHg) after the injection. The IOP was controlled by topical medications only, and no additional glaucoma treatment was required. In a recent review by Rao et al it was concluded that, “The initial treatment is with high dose corticosteroids, then steroid-sparing therapy, with a focus on getting the inflammatory response under control to prevent the development of sequelae such as a sunset glow fundus, cataracts, glaucoma, and choroidal neovascularization.”14
Conclusion
This study shows that in acute VKH, initial aggressive corticosteroid treatment with IVMP, followed by peribulbar depot steroids in combination with systemic immunosuppression, is effective in treating the ocular inflammation with rare recurrences.
Disclosure
The authors report no conflicts of interest in this work.
References
Burkholder BM. Vogt–Koyanagi–Harada disease. Curr Opin Ophthalmol. 2015;26(6):506–511. | ||
Read RW, Holland GN, Rao NA, et al. Revised diagnostic criteria for Vogt–Koyanagi–Harada disease: report of an international committee on nomenclature. Am J Ophthalmol. 2001;131:647–652. | ||
Rao NA, Gupta A, Dustin L, et al. Frequency of distinguishing clinical features in Vogt–Koyanagi–Harada disease. Ophthalmology. 2010;117:591–599. | ||
Moorthy RS, Inomata H, Rao NA. Vogt–Koyanagi–Harada syndrome. Surv Ophthalmol. 1995;39:265–292. | ||
Beniz J, Forster DJ, Lean JS, Smith RE, Rao NA. Variations in clinical features of the Vogt–Koyanagi–Harada syndrome. Retina. 1991;11:275–280. | ||
Wakabayashi T, Morimura Y, Miyamoto Y, Okada AA. Changing patterns of intraocular inflammatory disease in Japan. Ocul Immunol Inflamm. 2003;11:277–286. | ||
Nashtaei EM, Soheilian M, Herbort CP, Yaseri M. Patterns of uveitis in the Middle East and Europe. J Ophthalmic Vis Res. 2011;6(4):233–240. | ||
Martin TD, Rathinam SR, Cunningham ET Jr. Prevalence, clinical characteristics, and causes of vision loss in children with Vogt–Koyanagi–Harada disease in South India. Retina. 2010;30(7):1113–1121. | ||
Mota LA, Santos AB. Vogt–Koyanagi–Harada’s syndrome and its multisystem involvement. Rev Assoc Med Bras. 2010;56(5):590–595. | ||
Neves A, Cardoso A, Almeida M, et al. Unilateral Vogt–Koyanagi–Harada Disease: a clinical case report. Case Rep Ophthalmol. 2015;6:361–365. | ||
Forster DJ, Green RL, Rao NA. Unilateral manifestation of Vogt–Koyanagi–Harada syndrome in a 7-year old child. Am J Ophthalmol. 1991;111:380–382. | ||
Usui Y, Goto H, Sakai J, Takeuchi M, Usui M, Rao NA. Presumed Vogt–Koyanagi–Harada disease with unilateral ocular involvement: report of three cases. Graefes Arch Clin Exp Opthalmol. 2009;247:1127–1132. | ||
Agrawal A, Biswas J. Unilateral Vogt–Koyanagi–Harada disease: report of two cases. Middle East Afr J Ophthalmol. 2011;18:82–84. | ||
O’Keefe GA, Rao AN. Vogt–Koyanagi–Harada disease. Surv Ophthalmol. 2017;62(1):1–25. | ||
Al-Mezaine HS, Kangave D, Abu El-Asrar AM. Patterns of uveitis in patients admitted to a university hospital in Riyadh, Saudi Arabia. Ocul Immunol Inflamm. 2010;18(6):424–431. | ||
Khairallah M, Zaouali S, Messaoud R, et al. The spectrum of Vogt–Koyanagi–Harada Disease in Tunisia, North Africa. Int Ophthalmol. 2007;27(2–3):125–130. | ||
Tugal-Tutkun I, Ozyazgan Y, Akova YA, et al. The spectrum of Vogt–Koyanagi–Harada disease in Turkey: VKH in Turkey. Int Ophthalmol. 2007;27(2–3):117–123. | ||
Chee SP, Jap A, Bacsal K. Spectrum of Vogt–Koyanagi–Harada disease in Singapore. Int Ophthalmol. 2007;27(2–3):137–142. | ||
Kitamura M, Takami K, Kitachi N, et al. Comparative study of two sets of criteria for the diagnosis of Vogt–Koyanagi–Harada’s disease. Am J Ophthalmol. 2005;139:1080–1085. | ||
Yamaki K, Hara K, Sakuragi S. Application of revised diagnostic criteria for Vogt–Koyanagi–Harada disease in Japanese patients. Jpn J Ophthalmol. 2005;49:143–148. | ||
Arevalo JF, Lasave AF, Gupta V, et al. Clinical outcomes of patients with Vogt–Koyanagi–Harada disease over 12 years at a tertiary center. Ocul Immunol Inflamm. 2016;24(5):521–529. | ||
Kitamura M, Takami K, Kitachi N, et al. Comparative study of two sets of criteria for the diagnosis of Vogt–Koyanagi–Harada’s disease. Am J Ophthalmol. 2005;139:1080–1085. | ||
Yamaki K, Hara K, Sakuragi S. Application of revised diagnostic criteria for Vogt–Koyanagi–Harada disease in Japanese patients. Jpn J Ophthalmol. 2005;49:143–148. | ||
Read RW, Yu F, Accorinti M, et al. Evaluation of the effect on outcomes of the route of administration of corticosteroids in acute Vogt–Koyanagi–Harada disease. Am J Ophthalmol. 2006;142:119–124. | ||
Bykhovskaya I, Thorne JE, Kempen JH, Dunn JP, Jabs DA. Vogt–Koyanagi–Harada disease: clinical outcomes. Am J Ophthalmol. 2005;140:674–678. | ||
Rubsamen PE, Gass JD. Vogt–Koyanagi–Harada syndrome. Clinical course, therapy, and long-term visual outcome. Arch Ophthalmol. 1991;109:682–687. | ||
Moreker MR, Lodhi SA, Pathengay A. Role of intravitreal triamcinolone as an adjuvant in the management of Vogt–Koyanagi–Harada disease. Indian J Ophthalmol. 2007;55(6):479–480. | ||
Taylor SR, Isa H, Joshi L, Lightman S. New developments in corticosteroid therapy for uveitis. Ophthalmologica. 2010;224:46–53. | ||
Andrade RE, Muccioli C, Farah ME, Nussenblatt RB, Belfort R Jr. Intravitreal triamcinolone in the treatment of serous retinal detachment in Vogt–Koyanagi–Harada syndrome. Am J Ophthalmol. 2004;137:572–574. | ||
Hosoda Y, Hayashi H, Kuriyama S. Posterior subtenon triamcinolone acetonide injection as a primary treatment in eyes with acute Vogt-Koyanagi-Harada disease. Br J Ophthalmol. 2015;99:1211–1214. | ||
Paredes I, Ahmed M, Foster CS. Immuomodulatory therapy for Vogt–Koyanagi–Harada patients as first line therapy. Ocul Immunol Inflamm. 2006;14:87–90. | ||
Jabs DA, Rosenbaum JT, Foster CS, et al. Guidelines for the use of immunosuppressive drugs in patients with ocular inflammatory disorders: recommendations of an expert panel. Am J Ophthalmol. 2000;130:492–513. | ||
Lustig MJ, Cunningham ET Jr. Use of immunosuppressive agents in uveitis. Curr Opin Ophthalmol. 2003;14:399–412. | ||
Okada AA. Immunomodulatory therapy for ocular inflammatory disease: a basic manual and review of the literature. Ocul Immunol Inflamm. 2005;13:335–351. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.