Back to Journals » Clinical Ophthalmology » Volume 11
Clinical presentation and management of congenital ptosis
Authors Marenco M ![]() , Macchi I, Macchi I, Galassi E, Massaro-Giordano M, Lambiase A
, Macchi I, Macchi I, Galassi E, Massaro-Giordano M, Lambiase A ![]()
Received 22 April 2016
Accepted for publication 19 July 2016
Published 27 February 2017 Volume 2017:11 Pages 453—463
DOI https://doi.org/10.2147/OPTH.S111118
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Scott Fraser
Marco Marenco,1,* Ilaria Macchi,2,* Iacopo Macchi,3 Emilio Galassi,4 Mina Massaro-Giordano,5 Alessandro Lambiase1
1Department of Sense Organs, University of Rome “Sapienza”, 2Department of Ophthalmology, Campus Bio-Medico University of Rome, Rome, 3Department of Ophthalmology, University of Catania, Catania, 4Ophthalmic Clinic, Department of Ophthalmology, University of L’Aquila, L’Aquila, Italy; 5Department of Ophthalmology, Scheie Eye Institute, University of Pennsylvania, Philadelphia, PA, USA
*These authors contributed equally to this work
Abstract: Congenital ptosis is a rare condition characterized by lower positioning of the upper eyelid that is present at birth and is a clinical condition that is persistent if not treated. It may be unilateral or bilateral and may be associated with other ocular disorders or systemic conditions, including Marcus Gunn, Horner, and Duane syndromes. It is a benign condition but causes functional, cosmetic, and psychological problems in children. However, not all patients need to undergo surgery, and usually only patients at risk of amblyopia need a prompt surgical correction, while in other cases, surgery can be postponed. The grade of ptosis, the eyelid function, and the amblyopic risk are the parameters that affect the ophthalmologist’s decision on timing of surgery and the surgical technique to be used. In fact, there are several types of surgical techniques to correct a congenital ptosis, although very often more than one is needed to obtain an acceptable result. This paper reviews the causes of congenital ptosis and associated diseases. Particular emphasis is given to surgical management and different procedures available to correct the upper eyelid anomaly and avoid permanent damage to visual function.
Keywords: ptosis, extraocular muscle development, neurologic dysfunction, surgical approach
Introduction
Congenital ptosis is a rare condition characterized by an abnormal drooping of the upper eyelid that is present since birth or occurs within the first year of life, resulting in the reduction of the vertical palpebral fissure. Although isolated congenital ptosis can have an autosomal, either dominant or recessive, inheritance, it may be part of a larger spectrum of birth defects combined with other ocular or systemic conditions. It usually presents with a variable loosening of the upper eyelids due to a loss of muscular or nerve function that can be unilateral or bilateral.1–3
The superior branch of the third cranial nerve (CN III) is involved in the contraction of the levator palpebrae superioris muscle and the superior rectus muscle that are responsible for the elevation of the eyelids. The impairment of this function leads to abnormal visual development, resulting in a long-lasting uncorrected astigmatism or deprivational amblyopia. This form of amblyopia is defined as a disruption in the normal image-forming ability of the eye early in life caused by diminished performance of the visual system and severe reduction of the visual acuity due to an obscured visual field.4 The incidence of amblyopia in the overall population has been assessed at ~3%. However, a recent paper published by Willshaw5 provides information of an overall rate of amblyopia of up to 26.45% in a group of pediatric patients with blepharoptosis of different etiologies. Among them, 18.7% have visually significant refractive errors, while 14.19% are present with squint.
Patients with congenital ptosis have to be closely monitored in order to preserve their visual function and establish timely and appropriate medical or surgical treatment if needed.6
This review focuses on the genetics, main causes, and management of congenital ptosis, with a particular focus on the timing of surgical intervention and the options that are available.
Prevalence and incidence
The incidence rate of congenital ptosis worldwide has not been officially reported. However, according to a recent study over a 40-year time period on the incidence and demographics of childhood ptosis published by Griepentrog et al,7 ~90% of known cases were of congenital onset, one out of 842 births. Only 3% were bilateral and the left side was consistently more affected (68%) compared to the right.
These data have been recently confirmed by a retrospective study conducted on 336 children with ptosis where 69% were congenital. Left ptosis was again clearly predominant (74%) and unilateral in 65% of cases.8
Berry-Brincat and Willshaw5 reviewed all cases of childhood ptosis over a 9-year time period at the Birmingham Children’s Hospital. They estimated the incidence of congenital ptosis at 41% (76 children out of 186) while in the remaining patients, ptosis was an associated sign of a systemic syndrome.
These results apparently contradict the report of one of the largest studies conducted on >700,000 people from multiple Chinese provinces by Hu in 1987.9 In fact, the prevalence of congenital ptosis in that study was 0.18%, with a predominance of sporadic onset. Although these findings are not suitable to extrapolate to other ethnic groups, it is important to note that there was an autosomal recessive pattern of inheritance in 14.5% of patients and an autosomal dominant pattern in 18.4% of patients of the same group.
Genetics
Many theories have been proposed regarding the pathogenesis of congenital ptosis. Among them, only a few authors underlined the high prevalence of chromosomal alterations detected by genomic hybridization or karyotyping such as chromosomal deletion, chromosomal pericentric inversion, chromosomal micro-replication, or mosaic gain. The history of ptosis associated with developmental delay or a systemic disorder, should prompt an early genetic consultation and chromosomal testing in order to plan timely treatment and avoid visual loss. A recent study conducted by Stein et al10 demonstrated the relationship between congenital ptosis and the underlying chromosomal alterations, genetic syndromes, or neurological disorders. The first genetic locus identified for isolated congenital ptosis was (PTOS1) in the short arm of human chromosome 1 (1p32–34.1). According to Engle11 the critical region for the PTOS1 disease gene is 3 cM, an autosomal dominant inherited gene with incomplete penetrance pattern, defined by the polymorphic markers D1S447/D1S2733 and D1S1616. A few reports by McMullan et al12 suggested a clearly distinct pattern of inheritance and defined a new condition of X-linked dominant inheritance in a family affected by isolated, congenital bilateral ptosis where no male-to-male transmission was observed. More accurate and advanced molecular and genetic analysis led to definition of a critical region between Xq24 and Xq27.1. Further studies conducted by the same group of McMullan et al13 identified two chromosome breakpoints and a de novo balanced translocation of chromosomes 10 and 8 affecting the ZFH4 gene (8q21.1). The modified gene, which codes for a zinc-finger home domain protein and acts as a transcription factor, leads to an impaired development of cranial nerve tissue and muscles. Depending on the condition that exacerbates the ptosis, a variety of genes with new loci involved in this process can be identified. A group of systemic syndromes that commonly present with congenital ptosis because of abnormal extraocular muscle innervation are reviewed.
- Isolated Duane syndrome. This disorder recognizes cytogenetic abnormalities both on the q arm of chromosome 2 (2q31) and the q arm of chromosome 8. In this condition, the lateral rectus muscle acquires an aberrant innervation from the CN III as a result of a deficient function of the sixth cranial nerve. Both abduction and adduction may be affected as it presents with one of the three types of diseases known.14–18
- Autosomal-dominant blepharophimosis-ptosis-epicanthus inversus syndrome (BPES). An Italian study published in Nature19 identified a winged helix/forkhead transcription factor gene, FOXL2, which is mutated in BPES. This mutation produces altered proteins, which are truncated in type I BPES and larger in type II BPES. The modified proteins are expressed both in the mesenchyme of developing eyelids and the ovary. A heterozygous mutation in the UBE3B gene (12q23) is the underlying cause of the blepharophimosis-ptosis-intellectual disability syndrome, which is responsible for the intellectual infirmity in patients with BPES.
- Lymphedema-distichiasis-syndrome. Although it is not specifically one of the main causes of congenital ptosis, it is caused by a mutation of the FOX2 gene, a member of the same forkhead/winged-helix gene family responsible for BPES. It is an autosomal dominant condition typically characterized by distichiasis of the upper and lower lids and lymphedema of the extremities, with ptosis present in 30% of cases.20,21
- Congenital fibrosis of the extraocular muscles (CFEOM) types 1, 2, and 3 are autosomal dominant (types 1 and 3) and recessive (type 2) disorders belonging to the congenital cranial dysinnervation disorder family. CFEOM1 is an autosomal dominant condition that results from heterozygous mutations in KIF21A (12q12). Genetic analysis conducted on a group of people affected by CFEOM3 reveals linkage to markers on 16q24.2q24 inherited in an autosomal dominant pattern with incomplete penetrance throughout the family.22,23
- Congenital myasthenic syndrome is a heterogeneous group of autosomal recessive disorders characterized by altered neuromuscular transmission. Mild ptosis is the most common sign at first inspection and usually does not require surgical intervention. Congenital myasthenic syndrome may be associated to other ocular or bulbar signs or to a decreased pulmonary function. The majority of all cases of congenital myasthenic syndrome are characterized by an altered neuromuscular transmission, mainly affecting the postsynaptic region, due to genetic mutations of the proteins involved in this signaling. Gene defects may otherwise affect presynaptic and synaptic structures. Recent discoveries focus on the muscle-specific protein kinase (MUSK, 9q31.3–q32) gene and the RAPSN gene (11p11.2–p11.1), which play crucial roles, respectively, in synaptic differentiation and clustering of acetylcholine receptors.24
Causes
Congenital ptosis can be associated with both anomalies of extraocular muscle development and of innervation.
Muscle
Histologically, the levator muscle and the aponeurotic tissues of patients with congenital ptosis are replaced by fibrous and adipose tissue to a grade of severity, which causes, in worse cases, a complete replacement of the striated muscle.25
Congenital fibrosis of the extraocular muscles
CFEOM types 1, 2, and 3 are a group of conditions that share common features such as paralytic strabismus or other restricted eye movements, ophthalmoplegia and ptosis.22,23,26–29 CFEOM1 is an autosomal dominant disorder with severe restriction of elevation of the eye above the midline and a typical chin-up position of the head. Heterozygous mutations in KIF21A (12q12), a kinase protein involved in the process of neuronal development and axonal signal conduction, have been individuated. Inappropriate division of the oculomotor cranial nerve due to the lack of such signals causes severe atrophy and impaired function of the levator palpebrae superioris and superior rectus muscle. CFEOM2 is an autosomal recessive disease due to mutations in PHOX2A/ARIX (11q13) characterized by congenital bilateral ptosis with exotropic ophthalmoplegia. It may be associated with pupillary defects, in particular, miosis. Guo et al30 suggested that mutation in PHOX2A/ARIX transcription factor leads to congenital ptosis in CFEOM2 as a result of hypoplasia of the oculomotor and trochlear cranial nerve nuclei derived from incomplete development. CFEOM3 is an autosomal dominant condition with an incomplete penetrance pattern characterized by a mutation either to chromosome 16q24.2–q24.3 or in KIF21A. Although patients present with a heterogeneous phenotype, they all manifest with ptosis and ophthalmoplegia. Cognitive impairment, facial dysmorphisms, and/or digital anomalies, are also common features. Doherty et al31 proposed to consider CFEOM3 as a variant form of CFEOM that differed from the first type both genotypically and phenotypically.
Currently, the term congenital cranial dysinnervation disorder32 is preferred to define these syndromes. This updated definition, in fact, reflects the underlying cause of cranial nerve/brainstem deficient development and aberrant innervation of the extraocular musculature.
This group of disorders may also include Duane’s syndrome and Marcus Gunn jaw winking syndrome.
Nerves
Congenital ptosis can also be the result of a neurologic dysfunction or a neuromuscular junction failure of the levator muscle.33 The superior branch of the CN III innervates the levator palpebra superioris muscle and the superior rectus muscle. CN III is located in the midbrain and is composed of multiple subnuclei. The oculomotor nucleus, ventrally located, controls the levator muscle and the extraocular muscles, with the exception of the lateral rectus and the superior oblique. The inferior oblique subnucleus located above, serves the ipsilateral muscle. The sphincter pupillae muscle and the ciliary muscle are otherwise controlled by parasympathetic fibers originating from the Edinger–Westphal nucleus through the ciliary ganglion.
Duane retraction syndrome
Duane retraction syndrome is a congenital eye movement pathological condition that belongs to the family of incomitant strabismus, characterized by an incomplete development of the abducens nerve and corresponding midbrain nucleus. As a result, the lateral rectus is not properly innervated by the sixth cranial nerve and acquires an aberrant innervation by a branch of the CN III. The most common features include contraction of the palpebral fissure and reduction or total absence of either adduction or abduction.15 Although the synkinesis produced does not involve lid innervation, retraction of the globe and ptosis may result on attempted adduction.34
If not diagnosed early in children, Duane retraction syndrome can lead to amblyopia, a permanent uncorrectable loss of vision.
Duane syndrome is often clinically subdivided into three types 1–3 with a broad spectrum of severity and phenotypic segregation as described by Chung et al35 within the same family. Type 1 is a marked limitation of abduction combined with contraction of the lateral and the medial rectus muscles when adduction is struggled. Adduction is not altered in this case. Type 2 is a limitation of adduction in the affected eye while abduction is usually preserved. Huber (1974) explains this condition as a result of the concomitant action of both lateral and medial rectus muscles while attempting an adduction.36 Type 3 refers to a marked limitation of both adduction and abduction. Chung et al, Evans et al, and Al-Baradie et al described the concomitant presence of more than one type of Duane syndrome within a single pedigree.16,36,37 Duane syndrome occurs in >65% of cases as an isolated finding that can be associated with further ocular, neural, skeletal or auricular, malformations, as well as with other well-defined syndromes such as morning glory syndrome and Goldenhar syndrome.
Marcus Gunn syndrome
One of the more common congenital oculofacial synkineses, the Marcus Gunn syndrome, accounts for almost 5% of all congenital ptosis. Described by Gunn in 1883, the synkinetic winking motion of the eyelid on the movement of the jaw bears his name. Typically, the infant affected presents with variable degree of severity of blepharoptosis that may be either bilateral or, more frequently, unilateral. The wink reflex is characterized by a momentary upper eyelid retraction upon stimulation of the ipsilateral pterygoid muscle, elicited by chewing, laughing, or by usual movements of the jaw. The underlying cause of this reflex may be referred to an anomalous link between the motor fibers that innervate the levator superioris muscle, which belongs to the oculomotor nerve, and those belonging to the motor branches of the trigeminal nerve that innervate the external pterygoid muscle. The synkinetic winking motion rapidly ends with a quick return to a lower position.38–40
BPES
BPES is a rare disorder with an autosomal dominant pattern of inheritance characterized by blepharophimosis, blepharoptosis, epicanthus inversus, and telecanthus. The main findings are the horizontal narrowing of the eyelids (blepharophimosis), the loosening of the upper eyelids (blepharoptosis) followed by a skinfold that goes vertically from the lower eyelid to one side of the nose (epicanthus inversus). Because of these anatomical malpositions, it is usually associated with lacrimal duct anomalies. Additional signs include a broad nasal bridge, short philtrum, or low set ears. Zlotogora et al41 identified two types of BPES. In type 1 BPES, palpebral anomalies are associated with premature ovarian failure, early menopause, or complete infertility inherited as an autosomal dominant sex-limited trait. Type 2 presents with the same facial features without premature ovarian failure. Diagnosis of BPES is based on presence at birth of the four major eyelids features and when identified later in life, is often associated with premature ovarian failure (amenorrhea of >6 months, age <40 years, and follicle stimulating hormone concentration >40 IU/L); this is type 1 BPES. The clinical suspicion is confirmed by the identification of a genetic mutation in the FOXL2 gene (3q23). FOXL2 mutations have been identified in both types of BPES.42,43
Sympathetic nervous system
Ptosis can also be seen in association with dysfunction of the sympathetic nervous system, as in Horner syndrome, or with other forms of strabismus, including congenital esotropia or exotropia.
Horner syndrome
Horner syndrome, also known as Horner–Bernard syndrome or oculosympathetic palsy, is a combination of signs and symptoms caused by the disruption in the sympathetic nervous system of a nerve pathway from the brain to the face and eye on one side of the body. Horner syndrome may develop from lesions at any point along the sympathetic pathway. Central lesions are those between the hypothalamus and the fibers that exit from the spinal cord [C8 to T2], while peripheral lesions are mainly located at the superior cervical ganglion or in the cervical sympathetic chain. The classic triad of ipsilateral miosis, anhidrosis, and ptosis due to dysfunction of the sympathetic innervation to the Muller’s muscle is the pathognomonic sign of the disorder. Additional signs and symptoms in children with Horner syndrome may include lighter iris color in the affected eye because of an incomplete development of the iris melanocyte due to a deficient sympathetic innervation and a reduction of redness (flushing) on the affected side of the face that would normally appear from heat, physical exertion, or emotional reactions.44,45
Timing
Appropriate and timely treatment is the result of an accurate diagnosis. Correct management of congenital ptosis starts with determining the etiology of the ptosis, whether it is a genetic condition or if there are systemic syndromes associated and considering how the vision is affected by the eyelid position. These ultimately determine if and when surgical management should be undertaken. Congenital mild ptosis without serious refractive errors, strabismus, amblyopia, or abnormal torticollis should only be considered for cosmetic reasons focusing on the psychological impact of the ptosis on the child. Patients should be closely monitored for an increasing astigmatism due to the compressive force applied by the droopy eyelid on the eye or deprivational amblyopia that represents the ultimate clinical complication of the disorder.4,46,47 Before approaching a surgical intervention, avenues of targeted medical treatments are available that match the underlying cause of congenital ptosis. A new prospective is represented by the use of gene therapy to provide a healthy copy of the mutated genes. However, when the upper eyelid interferes with the visual axis (Figure 1) causing a reduction of the visual field or induces amblyogenic astigmatism or abnormal head position, surgical intervention is mandatory and has to be performed as soon as possible.47–49 In other cases, surgery may be postponed; some authors recommend waiting until 4 years of age to make the most accurate preoperative examination.46
 | Figure 1 Upper eyelid interferes with the visual axis causing stimulus deprivation or induces amblyogenic astigmatism. |
Surgical approach
An accurate preoperative examination includes evaluation of the degree of ptosis (measured as the marginal reflex distance under the primary gaze), the levator function (eyelid excursion from maximal downgaze to maximal upgaze with the frontalis muscle immobilized), the position and extent of the skin crease and the coexistence of Bell’s phenomenon or other associated signs (pupillary changes, myasthenic signs, and fundus abnormality). The amount of ptosis and the levator function are the two main parameters that should be considered before surgery. Among several procedures described, the most common and widely used are the frontal sling, levator resection, Fasanella–Servat procedure, Muller’s muscle resection, and Whitnall ligament sling.
This review focuses on the plethora of surgical approaches available for congenital ptosis repair and the surgical indication depending on the clinical presentation and the underlying cause of ptosis. Furthermore, the surgeon’s comfort level and experience are important factors that should be considered. Careful preoperative evaluation, planning, and counseling are necessary to obtain satisfactory surgical results, leading to happy parents and patients. Complications and indications are summarized in Table 1.50
 | Table 1 Choice of surgical technique according to the grade of ptosis and levator function |
Procedures
There are several options available with regard to procedures. The autogenous fascia lata is still considered the best long-term result material, especially because of a low risk of infection or breakage that may occur with foreign materials. However, its use in very young patients is limited by age and size as for the need of harvesting and additional surgery because of reabsorption. The fascia lata is a versatile source of autograft material that is useful in a variety of surgical techniques because of its great dimension, and the relatively low morbidity and complications for the donor site.51 In facial nerve reanimation surgery, the fascia lata is used for static, ancillary procedures such as the labial commissure or lower lid suspension.52 Several techniques have been described for its harvest, many of which may require endoscopes, strippers, or fasciotomes. In case of upper lid suspension, only a little amount of fascia lata is needed and the harvesting technique is quite easy to perform. The leg is medially rotated to expose and easily access the lateral surface of the thigh. Under general anesthesia, a skin incision is made 3–4 cm under an ideal line between the lateral condyle of the tibia and the anterior superior iliac crest spine, 6–8 cm from the knee, and extends upwards 4–5 cm. After the skin incision, the underlying fat tissue is dissected to reach the fascia. Once the fascia lata is exposed, the graft is tailored with a number 15 blade and released from its muscular connections by using the Metzenbaum scissors. A piece of fascia 5–8 cm long is collected from this site. Few absorbable stitches are placed between the two cutting edges before donor site closure in order to avoid muscular postoperative herniation. Banked fascia lata is an alternative option that solves the problem of harvesting and looking for new operative sites but may elicit an immune reaction or recurrent inflammation. Nonautogenous/synthetic materials are preferred to autogenous in very young patients who are anatomically too small for fascia lata to be harvested. The nonautogenous suspensory materials are elastic and allow easier replacement or adjustment when necessary but may be subjected to immune reaction or rejection. Among the nonautogenous materials, the most commonly used are silicone, nylon, polyester, gut chromic, polypropylene, collagen, silk, and polytetrafluoroethylene. Balacco et al53 described the use of Mersilene mesh slings developed in order to overcome the problems of failure, slippage, or extrusion within the surgical procedure.
Frontalis sling: Crawford technique and Fox pentagon
The frontalis sling is the most common surgical technique for congenital severe ptosis with high risk of amblyopia with <4 mm of levator function. The frontalis muscle elevates the eyebrow and partially the eyelid. This technique consists of forming a direct connection between the tarsus and the frontalis muscle by creating a double triangle suspension from the eyelid to the superior eyebrow area. Several variations to the original frontalis sling technique have been proposed. We may classify the surgical approaches to the frontalis sling by considering the connection surface/area and the type of material employed into two major groups: Crawford frontalis sling technique (using prevalently autogenous fascia lata) and Fox pentagon technique (using nonautogenous materials)54 (Figure 2).
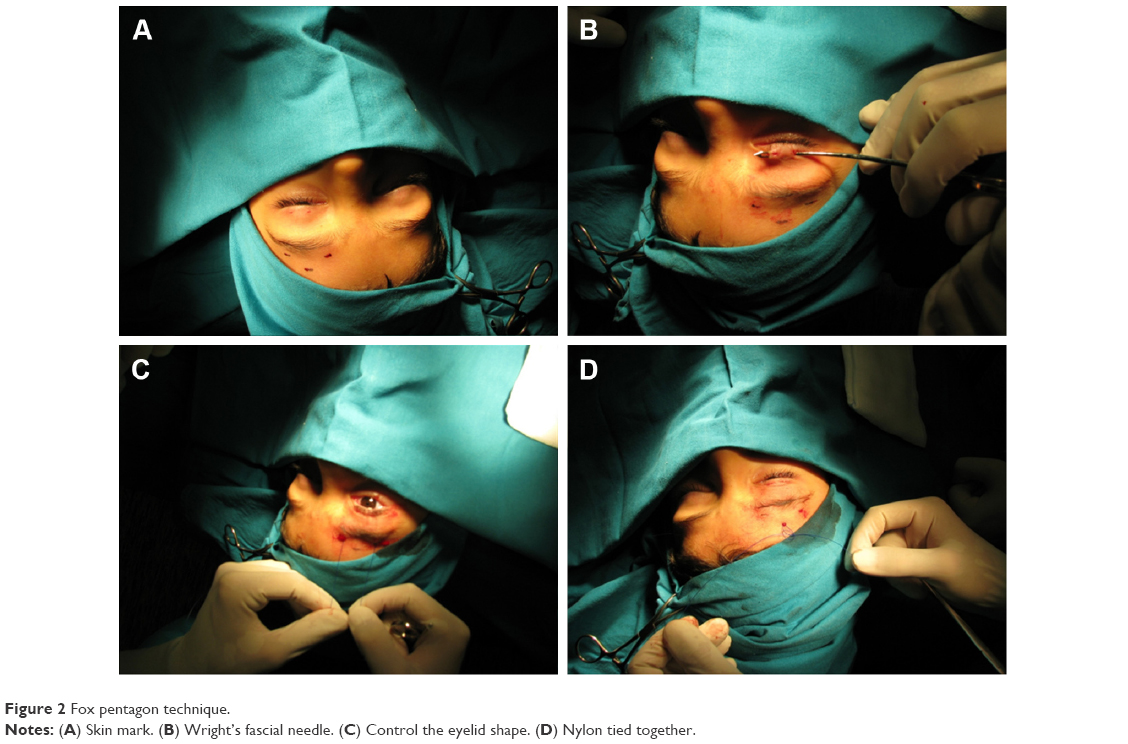 | Figure 2 Fox pentagon technique. |
Crawford technique
Before approaching the surgical procedure on the eyelid area, four strips of autogenous fascia lata 10–12 cm long have to be collected from the patient’s leg, along a line between the caput fibulae and the anterior superior spina iliaca, or received from a tissue bank. The skin crease is individuated by a lateral, medial, and central skin mark approximately 3 mm from the lash line. Two more 3 mm skin marks are made vertically above on the eyebrow, one slightly laterally and the other slightly medially from the eyelid marks created before, while a third forehead mark, 5 mm above and between the two eyebrow marks, draws an ideal isosceles triangle. A Wright fascial needle is used to loop the strips through the lid. At this point of the surgery the surgeon has to be careful not to catch/incarcerate the periostium, as it will not allow the frontalis muscle to lift the lids. Finally, both ends of the strips are tied together while a 6-0 absorbable suture reinforces the knot.
Fox pentagon
This surgical procedure is an alternative technique to Crawford’s original frontalis sling. The main difference between the two is the use of one single strip made of nonautogenous material to connect the lid to the forehead muscles. The surgical technique consists of only two incisions on the eyelids instead of the three expected in the Crawford technique, connected to the three higher incisions, two on the eyebrow and one on the central forehead as for the Crawford frontalis sling (Figure 2A). Pulling the strip through the incisions will draw a pentagon rather than two separate triangles (Figure 2B–D). Complications such as failure of the surgical procedure, extrusion, or slippage can be avoided by the use of a nonabsorbable, synthetic material such as Mersilene.53
There is some controversy about whether to perform bilateral suspension in patients with unilateral ptosis. A recent paper by Nemet55 demonstrates that Hering’s law, which refers to the compensatory retraction of the contralateral upper eyelid in case of unilateral ptosis, does not manifest in congenital ptosis. A bilateral procedure may, however, improve the symmetry between the eyes.
The frontalis sling is a simple, well-known procedure (Figure 3), that can be repeated or revised if needed. However, the use of autogenous materials limits the time and chances available to be removed as they easily incorporate into the tissue. Nonautogenous materials can otherwise be removed at any time and do not exacerbate the inflammatory granulomatous reactions, although they might be exposed to rejection. Other limits to the use of the frontalis sling surgical technique are the moderate immobilization of the lids and the increase in postsurgical lagophthalmos during sleep or in the extreme downgaze because of the concomitant orbicularis muscle detention.
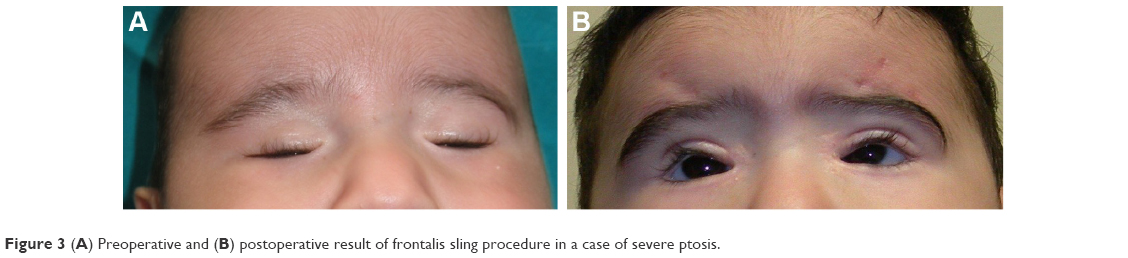 | Figure 3 (A) Preoperative and (B) postoperative result of frontalis sling procedure in a case of severe ptosis. |
Levator’s resection and aponeurosis surgery
This technique is indicated in the correction of ptosis in patients with >5 mm of levator function. It enhances the elevator function by shortening the elevator complex (muscle and aponeurosis) (Figure 4).
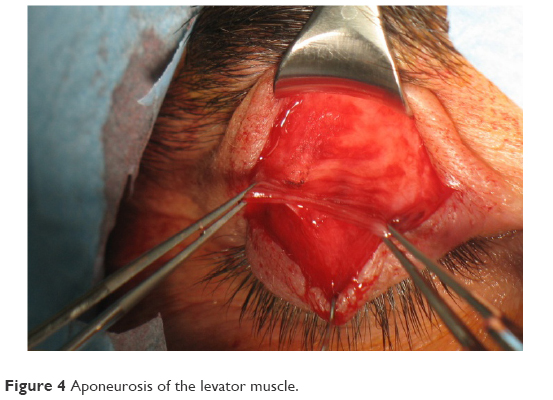 | Figure 4 Aponeurosis of the levator muscle. |
In order to obtain an acceptable level of eyelid suspension, Collins50 suggests to work on the following amount of resection:
Levator function 8–10 mm: 14–18 mm resection
Levator function 6–7 mm: 18–22 mm resection
Levator function 4–5 mm: 22–26 mm resection.
There are two possible approaches described. The anterior approach consists of exposure of the levator aponeurosis using a superior eyelid crease incision and the advancement of the levator aponeurosis by folding or excising the muscle. The aponeurosis is reattached to the anterior surface of the tarsus with nonabsorbable sutures. The transconjunctival approach or the small skin incision (8–13 mm) represents an alternative to the anterior procedure, leading to a reduced distortion of the tissue and a better cosmetic result. The main disadvantage of the small incision approach is otherwise the limited view of the surgical field. The benefits obtained by this type of surgery are the preservation of the anatomical planes and the elevating structures of the eyelids, including Muller’s muscle and Whitnall’s ligament. Furthermore, it allows the surgeon to customize the amount of eyelid elevation intraoperatively. Although the levator resection outcomes are not completely predictable, a 10-year retrospective study conducted by Abrishami et al56 on a group of patients where congenital ptosis accounted for an 88.2% reports an overall success rate of 78.7%, which increased after the second and/or third procedures. This is in line with a previous study conducted by Cates and Tyers57 who reported an overall 76% success rate, falling to 74% at 6 months postoperatively. Scuderi et al report the success of this technique associated with Muller’s muscle technique.58
Muller’s muscle resection
Muller’s muscle resection was first described by Putternam in 1985.59 Muller’s muscle is responsible for eyelid elevation up to 2–3 mm. The indication for this type of surgery is the resolution of ptosis after one drop of phenylephrine associated with a good function of levator muscle. A conjunctival approach was originally proposed to pursue resection of the Muller’s muscle and the conjunctiva, without involving the tarsus. An open sky approach was later introduced to save the healthy conjunctiva and avoid an incorrect and limited muscle resection.60 Baldwin et al obtained successful results from the Muller muscle-conjunctiva resection in phenylephrine test-negative patients with ptosis.61 The potential complication is the corneal abrasion because of the exposed sutures, risk of upper fornix alteration, damage to accessory tear glands and upper lid entropion.62
Whitnall ligament sling
The Whitnall ligament sling procedure is indicated in cases of mild-to-severe ptosis with a levator function >3 mm.63,64 The role and anatomical position of the Whitnall’s ligament were first described by Anderson et al.65 It is an orbital fascial condensation that crosses the anterosuperior orbit between the trochlea and the lacrimal gland fascia, and lies near the junction of the muscular and aponeurotic levator. Because of its hidden position, it might be intraoperatively confused with other structures of the levator complex or mistaken as if frequently happens with the transverse ligament. If these mistakes happen, the elevation will be insufficient and the surgical outcome will be poor. The Whitnall sling procedure allows improving the effort of the levator muscle by adding a sustainment structure to the eyelid. The levator aponeurosis is cut up to the point of the maximal levator resection, and both the levator muscle and the overlying Whitnall’s ligament are sutured to the superior portion of the tarsal plate.64
Fasanella–Servat
The Fasanella–Servat procedure was first described in 1961.66 The technique is generally used for those patients who present with a good levator function and ptosis <3 mm. The Fasanella–Servat procedure might also be used in congenital ptosis with poor levator function because of the surgeon’s choice not to remove the healthy tarsus for its functional role in eyelid stability. Fasanella–Servat reduces ptosis by removing conjunctival tissue, Muller muscle, and part of the tarsus using a posterior internal eyelid approach. Many studies have been carried out and dozens of adjustments to the original technique have been proposed by oculoplastic surgeons to achieve betters results.67 In the original Fasanella–Servat procedure, the eyelid is gently everted, and the upper border of the tarsus is clamped to the inferior side of the Muller’s muscle and conjunctiva. After clamping, the same needle is used to create a running suture with an absorbable suture through the lid margin. The sutures stay in place for a few days while the artery forceps are removed. Both arms of the suture are then pulled through the conjunctival wound and tied. The use of softer suture has consistently reduced one of the main complications represented by the corneal irritation due to scratching. Other common complications include faulty placement of the clamps, postoperative keratitis, suture granuloma, and postoperative hemorrhages. A damage to the lacrimal glands may worsen the superficial keratopathy. As a result of the loss of conjunctival tissue from the superior fornix, an entropion may form. Many authors have studied the success rate of the Fasanella–Servat procedure in congenital ptosis. Pang et al performed 169 cases of ptosis repair using Fasanella–Servat. In this study, only 18 cases were classified as congenital ptosis. The success rate was 89.5%, but congenital cases had the lowest success rate of all studied subgroups (76.4%). In a larger study of 155 congenital ptosis, Berry-Brincat et al performed 15 Fasanella–Servat procedures.The overall success rate was 71%, with a 20% reintervention rate.5
Discussion
The most common and resolutive approach to the treatment of congenital ptosis and its phenotypical presentation is surgical intervention. The recommendations can be either functional or aesthetic. Accurate preoperative evaluation is undoubtedly the most important thing to consider in order to plan a timely, valuable surgery and to choose the best surgical technique that adapts to the clinical presentation and the underlying cause of ptosis. Above all, the levator’s function and the degree of ptosis in primary position represent the parameters that should guide this choice. The optimal time for intervention is related to the visual impairment and the psychological impact on the young patient. When the upper eyelid interferes with the visual axis, leading to deprivation or developing a compressive astigmatism, the risk of amblyopia is high. The surgical correction of blepharoptosis induces anterior corneal surface modification, restoring corneal symmetry and regular corneal astigmatism.68 However, recent papers demonstrate that both blepharoplasty and levator resection may induce significant changes of corneal curvature (eg, central corneal power and corneal astigmatism) by repositioning of the upper eyelid.69 Deprivational amblyopia4,46,47 represents the only emergency that requires immediate surgical intervention. In this case, the frontalis sling is the most commonly used and recommended procedure. A limit to this type of technique in very young patients might be the insufficient length of the leg for harvesting the fascia lata. The Fox pentagon technique overcomes this problem by using nonautogenous, synthetic materials that can be adapted to the growth of the patient. Several studies have shown that this material does not integrate with the fabrics and offers the possibility of reintervention.
The most common complications described are under- or overcorrection and lagophthalmos, leading to superficial punctate keratitis or even corneal ulcer, depending on the degree of exposure. The surgical outcome is therefore considered very poor as these conditions are associated with high ocular discomfort or pain. Other surgical techniques that require a conjunctival approach such as the Fasanella–Servat and the Muller’s muscle resection, may present with the same pattern of superficial complications like corneal abrasions due to the exposure of conjunctival sutures.
Complications that belong to the material used for the frontal sling are widely described by Simon.70 Although autogenous materials, such as fascia lata, allow an optimal execution of the surgical intervention, they require a proper length of the leg to be harvested. The very young age and the limited physical development of patients (usually under 3 years old) who undergo this procedure is a limit to the use of autogenous materials.71 Another limitation to the use of fascia lata is the high incidence of severe infections due to its exposure. Further reinterventions are also not allowed as the autogenous materials rapidly integrate with the tissues. Alternatively, nonautogenous materials like Mersilene present a very low risk of reintervention (~12.5%) but few soft tissue complications as reported by Mehta et al.72 The nylon monofilament is extremely easy to use but is affected by a high recurrence rate.71
Conclusion
In literature, several different options to correct congenital ptosis are described. The treatment of eyelid ptosis associated with a rare disease does not differ from the management of congenital ptosis. Frequently, more than one surgical intervention is required to pursue a satisfactory surgical outcome. The choice of the surgical technique adopted depends on the experience of the surgeon but mainly on preoperative considerations. The grade of ptosis, the levator eyelid function, and the risk of amblyopia are the parameters that will guide the timing and the choice of the surgical technique.
Disclosure
The authors report no conflicts of interest in this work.
References
Sakol PJ, Mannor G, Massaro BM. Congenital and acquired blepharoptosis. Curr Opin Ophthalmol. 1999;10(5):335–339. | ||
SooHoo JR, Davies BW, Allard FD, Durairaj VD. Congenital ptosis. Surv Ophthalmol. 2014;59(5):483–492. | ||
Vestal KP, Seiff SR, Lahey JM. Congenital ptosis in monozygotic twins. Ophthal Plast Reconstr Surg. 1990;6(4):265–268. | ||
Von Noorden GK, Maumenee E. Clinical observations on stimulus-deprivation amblyopia (amblyopia ex anopsia). Am J Ophthalmol. 1968;65(2):220–224. | ||
Berry-Brincat A, Willshaw H. Paediatric blepharoptosis: a 10-year review. Eye (Lond). 2009;23(7):1554–1559. | ||
Lee V, Konrad H, Bunce C, Nelson C, Collin JR. Aetiology and surgical treatment of childhood blepharoptosis. Br J Ophthalmol. 2002;86(11):1282–1286. | ||
Griepentrog GJ, Diehl NN, Mohney BG. Incidence and demographics of childhood ptosis. Ophthalmology. 2011;118(6):1180–1183. | ||
El Essawy R1, Elsada MA. Clinical and demographic characteristics of ptosis in children: a national tertiary hospital study. Eur J Ophthalmol. 2013;23(3):356–360. | ||
Hu DN. Prevalence and mode of inheritance of major genetic eye diseases in China. J Med Genet. 1987;24(10):584–588. | ||
Stein A, Kelly JP, Weiss AH. Congenital eyelid ptosis: onset and prevalence of amblyopia, associations with systemic disorders, and treatment outcomes. J Pediatr. 2014;165(4):820–824.e2. | ||
Engle EC. Applications of molecular genetics to the understanding of congenital ocular motility disorders. Ann NY Acad Sci. 2002;956:55–63. | ||
McMullan TF, Collins AR, Tyers AG, Robinson DO. A novel X-linked dominant condition: X-linked congenital isolated ptosis. Am J Hum Genet. 2000;66(4):1455–1460. | ||
McMullan TW, Crolla JA, Gregory SG, et al. A candidate gene for congenital bilateral isolated ptosis identified by molecular analysis of a de novo balanced translocation. Hum Genet. 2002;110(3):244–250. | ||
DeRespinis PA, Caputo AR, Wagner RS, Guo S. Duane’s retraction syndrome. Surv Ophthalmol. 1993;38(3):257–288. | ||
Hotchkiss MG, Miller NR, Clark AW, Green WR. Bilateral Duane’s retraction syndrome. A clinical-pathologic case report. Arch Ophthalmol. 1980;98(5):870–874. | ||
Evans JC, Frayling TM, Ellard S, Gutowski NJ. Confirmation of linkage of Duane’s syndrome and refinement of the disease locus to an 8.8-cM interval on chromosome 2q31. Hum Genet. 2000;106(6):636–638. | ||
Murillo-Correa CE, Kon-Jara V, Engle EC, Zenteno JC. Clinical features associated with an I126M alpha2-chimaerin mutation in a family with autosomal-dominant Duane retraction syndrome. J AAPOS. 2009;13(3):245–248. | ||
Miller NR, Kiel SM, Green WR, Clark AW. Unilateral Duane’s retraction syndrome (Type 1). Arch Ophthalmol. 1982;100(9):1468–1472. | ||
Crisponi L, Deiana M, Loi A, et al. The putative forkhead transcription factor FOXL2 is mutated in blepharophimosis/ptosis/epicanthus inversus syndrome. Nat Genet. 2001;27(2):159–166. | ||
Tanpaiboon P, Kantaputra P, Wejathikul K, Piyamongkol W. c. 595–596 insC of FOXC2 underlies lymphedema, distichiasis, ptosis, ankyloglossia, and Robin sequence in a Thai patient. Am J Med Genet A. 2010;152A(3):737–740. | ||
Dellinger MT, Thome K, Bernas MJ, Erickson RP, Witte MH. Novel FOXC2 missense mutation identified in patient with lymphedema-distichiasis syndrome and review. Lymphology. 2008;41(3):98–102. | ||
Mackey DA, Chan WM, Chan C, et al. Congenital fibrosis of the vertically acting extraocular muscles maps to the FEOM3 locus. Hum Genet. 2002;110(5):510–512. | ||
Marszalek JR, Weiner JA, Farlow SJ, Chun J, Goldstein LS. Novel dendritic kinesin sorting identified by different process targeting of two related kinesins: KIF21A and KIF21B. J Cell Biol. 1999;145(3):469–479. | ||
Allen RC. Genetic diseases affecting the eyelids: what should a clinician know? Curr Opin Ophthalmol. 2013;24(5):463–477. | ||
Hoşal BM, Ayer NG, Zilelioğlu G, Elhan AH. Ultrasound biomicroscopy of the levator aponeurosis in congenital and aponeurotic blepharoptosis. Ophthal Plast Reconstr Surg. 2004;20(4):308–311. | ||
Heidary G, Engle EC, Hunter DG. Congenital fibrosis of the extraocular muscles. Semin Ophthalmol. 2008;23(1):3–8. | ||
Wang SM, Zwaan J, Mullaney PB, et al. Congenital fibrosis of the extraocular muscles type 2, an inherited exotropic strabismus fixus, maps to distal 11q13. Am J Hum Genet. 1998;63(2):517–525. | ||
Yazdani A, Chung DC, Abbaszadegan MR, et al. A novel PHOX2A/ARIX mutation in an Iranian family with congenital fibrosis of extraocular muscles type 2 (CFEOM2). Am J Ophthalmol. 2003;136(5):861–865. | ||
Nakano M, Yamada K, Fain J, et al. Homozygous mutations in ARIX(PHOX2A) result in congenital fibrosis of the extraocular muscles type 2. Nat Genet. 2001;29(3):315–320. | ||
Guo S, Brush J, Teraoka H, et al. Development of noradrenergic neurons in the zebrafish hindbrain requires BMP, FGF8, and the homeodomain protein soulless/Phox2a. Neuron. 1999;24(3):555–566. | ||
Doherty EJ, Macy ME, Wang SM, Dykeman CP, Melanson MT, Engle EC. CFEOM3: a new extraocular congenital fibrosis syndrome that maps to 16q24.2-q24.3. Invest Ophthalmol Vis Sci. 1999;40(8):1687–1694. | ||
Gutowski NJ, Bosley TM, Engle EC. 110th ENMC International Workshop: the congenital cranial dysinnervation disorders (CCDDs). Naarden, the Netherlands, 25e27 October 2002. Neuromuscul Disord. 2003;13(7–8):573–578. | ||
Traboulsi EI. Congenital abnormalities of cranial nerve development: overview, molecular mechanisms, and further evidence of heterogeneity and complexity of syndromes with congenital limitation of eye movements. Trans Am Ophthalmol Soc. 2004;102:373–389. | ||
Appukuttan B, Gillanders E, Juo SH, et al. Localization of a gene for Duane retraction syndrome to chromosome 2q31. Am J Hum Genet. 1999;65(6):1639–1646. | ||
Chung M, Stout JT, Borchert MS. Clinical diversity of hereditary Duane’s retraction syndrome. Ophthalmology. 2000;107(3):500–503. | ||
Huber A. Electrophysiology of the retraction syndromes. Br J Ophthalmol. 1974;58(3):293–300. | ||
Al-Baradie R, Yamada K, St Hilaire C, et al. Duane radial ray syndrome (Okihiro syndrome) maps to 20q13 and results from mutations in SALL4, a new member of the SAL family. Am J Hum Genet. 2002;71(5):1195–1199. | ||
Bowyer JD, Sullivan TJ. Management of Marcus Gunn jaw winking synkinesis. Ophthal Plast Reconstr Surg. 2004;20(2):92–98. | ||
Davis G, Chen C, Selva D. Marcus Gunn syndrome. Eye (Lond). 2004;18(1):88–90. | ||
Pratt SG, Beyer CK, Johnson CC. The Marcus Gunn phenomenon. A review of 71 cases. Ophthalmology. 1984;91(1):27–30. | ||
Zlotogora J, Sagi M, Cohen T. The blepharophimosis, ptosis, and epicanthus inversus syndrome: delineation of two types. Am J Hum Genet. 1983;35(5):1020–1027. | ||
Bell R, Murday VA, Patton MA, Jeffery S. Two families with blepharophimosis/ptosis/epicanthus inversus syndrome have mutations in the putative forkhead transcription factor FOXL2. Genet Test. 2001;5(4):335–338. | ||
Gulati R, Verdin H, Halanaik D, Bhat BV, De Baere E. Co-occurrence of congenital hydronephrosis and FOXL2-associated blepharophimosis, ptosis, epicanthus inversus syndrome (BPES). Eur J Med Genet. 2014;57(10):576–578. | ||
Zafeiriou DI, Economou M, Koliouskas D, Triantafyllou P, Kardaras P, Gombakis N. Congenital Horner’s syndrome associated with cervical neuroblastoma. Eur J Paediatric Neurol. 2006;10(2):90–92. | ||
Mirzai H, Baser EF. Congenital Horner’s syndrome and the usefulness of the apraclonidine test in its diagnosis. Indian J Ophthalmol. 2006;54(3):197–199. | ||
Harrad RA, Graham CM, Collin JR. Amblyopia and strabismus in congenital ptosis. Eye (Lond). 1988;2(Pt 6):625–627. | ||
Oral Y, Ozgur OR, Akcay L, Ozbas M, Dogan OK. Congenital ptosis and amblyopia. J Pediatr Ophthalmol Strabismus. 2010;47(2):101–104. | ||
Beckingsale PS, Sullivan TJ, Wong VA, Oley C. Blepharophimosis: a recommendation for early surgery in patients with severe ptosis. Clin Experiment Ophthalmol. 2003;31(2):138–142. | ||
Avgitidou G, Koch KR, Cursiefen C, Heindl LM. Current aspects of eyelid, lacrimal and orbital surgery in childhood. Ophthalmologe. 2015;112(2):102–109. | ||
Collins JRO. A Manual of Systematic Eyelid Surgery, 3rd ed. London: Butterworth-Heinemann; 2006. | ||
Wheatcroft SM, Vardy SJ, Tyers AG. Complications of fascia lata harvesting for ptosis surgery. Br J Ophthalmol. 1997;81(7):581–583. | ||
Drever JM. A simple method for obtaining fascia lata grafts. Plast Reconstr Surg. 1972;50(2):196–197. | ||
Balacco GC, Recupero SM, Contestabile MT, Pacella E, Abdolrahimzadeh S. Fox’s modified technique using the Mersilene mesh sling in the management of blepharoptosis. Ophthalmic Surg Lasers. 1996;27(11):924–928. | ||
DeMartelaere SL, Blaydon SM, Cruz AA, Amato MM, Shore JW. Broad fascia fixation enhances frontalis suspension. Ophthal Plast Reconstr Surg. 2007;23(4):279–284. | ||
Nemet AY. Is Hering’s law as important in congenital blepharoptosis as in acquired ptosis? Aesthet Surg J. 2013;33(8):1110–1115. | ||
Abrishami A, Bagheri A, Salour H, Aletaha M, Yazdani S. Outcomes of levator resection at tertiary eye care center in Iran: a 10-year experience. Korean J Ophthalmol. 2012;26(1):1–5. | ||
Cates CA, Tyers AG. Outcomes of anterior levator resection in congenital blepharoptosis. Eye (Lond). 2001;15(Pt 6):770–773. | ||
Scuderi N, Chiummariello S, De Gado F, Alfano C, Scuderi G, Recupero SM. Surgical correction of blepharoptosis using the levator aponeurosis-Müller’s muscle complex readaptation technique: a 15-year experience. Plast Reconstr Surg. 2008;121(1):71–78. | ||
Putterman AM. Müllers muscle-conjunctival resection ptosis procedure. Aust NZ J Ophthalmol. 1985;13(2):179–183. | ||
Lake S, Mohammad-Ali FH, Khooshabeh R. Open sky Müller’s muscle-conjunctiva resection for ptosis surgery. Eye (Lond). 2003;17(9):1008–1012. | ||
Baldwin HC, Bhagey J, Khooshabeh R. Open sky Muller muscle-conjunctival resection in phenylephrine test-negative blepharoptosis patients. Ophthal Plast Reconstr Surg. 2005;21(4):276–280. | ||
Peter NM, Khooshabeh R. Open-sky isolated subtotal Muller’s muscle resection for ptosis surgery: a review of over 300 cases and assessment of long-term outcome. Eye (Lond). 2013;27(4):519–524. | ||
Anderson RL, Nowinski TS. The five-flap technique for blepharophimosis. Arch Ophthalmol. 1989;107(3):448–452. | ||
Anderson RL, Jordan DR, Dutton JJ. Whitnall’s sling for poor function ptosis. Arch Ophthalmol. 1990;108(11):1628–1632. | ||
Anderson RL, Dixon RS. The role of Whitnall’s ligament in ptosis surgery. Arch Ophthalmol. 1979;97(4):705–707. | ||
Fasanella Rm, Servat J. Levator resection for minimal ptosis: another simplified operation. Arch Ophthalmol. 1961;65:493–496. | ||
Pang NK, Newsom RW, Oestreicher JH, Chung HT, Harvey JT. Fasanella-Servat procedure: indications, efficacy, and complications. Can J Ophthalmol. 2008;43(1):84–88. | ||
Savino G, Battendieri R, Riso M, et al. Corneal topographic changes after eyelid ptosis surgery. Cornea. 2016;35(4):501–505. | ||
Kim YK, In JH, Jang SY. Changes in corneal curvature after upper eyelid surgery measured by corneal topography. J Craniofac Surg. 2016;27(3):e235–e238. | ||
Ben Simon GJ, Macedo AA, Schwarcz RM, Wang DY, McCann JD, Goldberg RA. Frontalis suspension for upper eyelid ptosis: evaluation of different surgical designs and suture material. Am J Ophthalmol. 2005;140(5):877–885. | ||
Wasserman BN, Sprunger DT, Helveston EM. Comparison of materials used in frontalis suspension. Arch Ophthalmol. 2001;119(5):687–691. | ||
Mehta P, Patel P, Olver JM. Functional results and complications of Mersilene mesh use for frontalis suspension ptosis surgery. Br J Ophthalmol. 2004;88(3):361–364. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.