Back to Journals » Clinical, Cosmetic and Investigational Dermatology » Volume 14
Clinical Approach to Linear Hyperpigmentation: A Review Article
Authors Alkhowailed MS ![]() , Otayf M, Albasseet A, Almousa A, Alajlan Z, Altalhab S
, Otayf M, Albasseet A, Almousa A, Alajlan Z, Altalhab S
Received 7 September 2020
Accepted for publication 24 November 2020
Published 8 January 2021 Volume 2021:14 Pages 23—35
DOI https://doi.org/10.2147/CCID.S280819
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Jeffrey Weinberg
Mohammad S Alkhowailed,1 Mojahed Otayf,2 Abdulrahman Albasseet,2 Abdullah Almousa,2 Ziyad Alajlan,2 Saad Altalhab3
1Department of Dermatology, College of Medicine, Qassim University, Buraydah, Qassim, Saudi Arabia; 2College of Medicine, King Saud University, Riyadh, Saudi Arabia; 3Department of Dermatology, College of Medicine, Al-Imam Mohammad Ibn Saud Islamic University, Riyadh, Saudi Arabia
Correspondence: Mohammad S Alkhowailed
Department of Dermatology College of Medicine, Qassim University, Buraidah 52571, Qassim, Saudi Arabia
Email [email protected]
Abstract: Linear hyperpigmentation is an unusual anatomical configuration in clinical dermatology. Owing to its rarity, consensus on the most effective method of classification is lacking. While linear hyperpigmentation generally follows Blaschko’s lines, this is not universal. Clinical findings such as adherence to Blaschko’s lines, associated morphological findings (including other cutaneous lesions), and systemic manifestations can be used to further characterize and diagnose variants of the disorder. Early detection of any underlying disease is vital, especially in cases with effective management, because the disorder may make it difficult to manage hyperpigmentation. Herein, we introduce a logical clinical diagnostic approach that represents a useful tool for dermatologists to efficiently evaluate patients presenting with linear hyperpigmentation. A simplified systematic and evidence-based approach is useful for this clinical condition owing to the heterogeneous causes and lack of specific diagnostic tools.
Keywords: approach, blaschkitis, Blaschko’s lines, hyperpigmentation, linear, pigmentary mosaicism
Key Points
Linear hyperpigmentation is a rare anatomical configuration in clinical dermatology. Reaching the diagnosis in such presentation is relatively difficult as there is a wide range of differential diagnoses. Creating a classification of linear hyperpigmentation will help dermatologists to reach the diagnosis and recognize and avoid potential systemic complications.
Introduction
Cutaneous hyperpigmentation is a common complaint in dermatologic practice. Clinically, it is classified into diffuse, circumscribed, linear, or reticulated variants. The configuration of the lesion is important to reach the correct diagnosis. Linear hyperpigmentation often has a diagnostic challenge in clinical practice. Various causes including post-inflammatory changes, drug reactions, and mosaic genetic alterations result in hyperpigmentation along Blaschko’s lines. Pigmentary demarcation lines represent the normal dorsal–ventral skin patterning. Blaschko’s lines are attributed to the migration lines of epidermal cells during embryogenesis and are characterized by a V-shaped pattern on the posterior midline, S-shape on the abdomen, and spiral on the posterior scalp.1,2 To the best of our knowledge, there is no existing clinical approach for linear hyperpigmentation diagnosis. We provide a simplified approach for the diagnosis of linear hyperpigmentation through two steps:1 To determine whether hyperpigmentation follows Blaschko’s lines;2 to determine the presence or absence of associated systemic manifestations in addition to other cutaneous findings (Figures 1 and 2).
 |
Figure 1 Clinical approach for linear hyperpigmentation. Abbreviations: XL-HED, X-linked reticulate pigmentary disorder; CALM, café au lait macule; MAS, McCune–Albright syndrome; LWNH, linear whorled nevoid hypermelanosis; FDH, focal dermal hypoplasia; XLRPD, X-linked reticulate pigmentary disorder; CHHS, Conradi–Hünermann–Happle syndrome. |
 |
Figure 2 Cutaneous clues associated with linear hyperpigmentation. Abbreviations: LWNH, linear whorled nevoid hypermelanosis; LS, lichen striatus; XLRPD, X-linked reticulate pigmentary disorder; XL-HED, X-linked reticulate pigmentary disorder. |
Linear Hyperpigmentation Following Blaschko’s Lines Without Systemic Findings
We describe the most common dermatological disorders that may present with linear hyperpigmentation following Blaschko’s lines without associated systemic findings (Table 1).
 |
Table 1 Clinical Clues for Linear Hyperpigmentation That Follow Blaschko’s Lines Without Associated Systemic Manifestations |
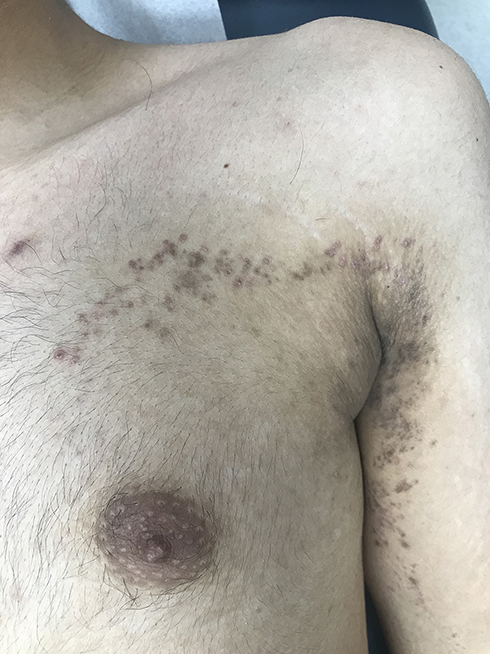 |
Figure 3 Blaschkoid lichen planus. Hyperpigmented macules and patches arranged in a linear pattern following Blaschko’s lines over the arm and extending to the upper chest. |
Blaschkoid Lichen Planus
Blaschkoid lichen planus (LP) is rare, manifesting as hyperpigmented inflammatory macules and patches arranged in a linear, S-shaped, or whorled pattern following Blaschko’s lines (blaschkitis), without koebnerization. It differs from linear LP and accounts for approximately 0.5% of LP cases (Figure 3).3 Mostly, it present as an isolated finding. However, Sil et al recently reported blaschkoid LP in the setting of concurrent childhood‐onset systemic lupus erythematosus.4
Lichen Planus Pigmentosus
Lichen planus pigmentosus (LPP) is another rare form of LP that mostly affects adults with skin phototypes III–V. It presents as dark brown macules and patches that follow Blaschko’s lines and predominately affects sun-exposed areas (especially the forehead, temples, and neck) and flexor folds. Patients with this condition are usually asymptomatic, but some patients report mild pruritus or burning sensation. Unlike classical LP, the palms, soles, nails, and oral mucosa are not involved.5 In one uncontrolled study, lightening of the pigmentation occurred in 54% (7/13) patients treated with topical tacrolimus for 12–16 weeks.6–8
Linear Lichen Planopilaris
Lichen planopilaris (LPP) is a primary diffuse, scaly patch, or plaque associated with cicatricial alopecia that rarely presents in a linear distribution. It may present as a linear patch or plaque associated with scaling alopecia, followed by scarring over the scalp or face. It is characterized by lymphocytic inflammation directed toward the infundibulum and isthmus portions of the hair follicle, leading to follicular scarring and irreversible hair loss.9,10
There are many treatment modalities for LPP, but the current first-line therapy includes high-potency topical and intralesional corticosteroids. Other modalities with varying reports of success include systemic corticosteroids, cyclosporine, mycophenolate mofetil, oral tetracyclines, oral retinoids, thalidomide, and hydroxychloroquine, although relapses after discontinuation of treatment have been reported.10
Linear Atrophoderma of Moulin
Linear atrophoderma of Moulin (LAM) is an acquired dermatological disease primarily affecting children and adolescents. It appears as unilateral round or oval hyperpigmented macules and atrophic band-like skin lesions following Blaschko’s lines over the trunk or limbs. Truncal lesions typically form an S-shaped pattern.11,12 It is typically a non-inflammatory disease although an inflammatory phase was reported. While most patients develop non-sclerotic hyperpigmentation only, atypical presentations can present with bilateral telangiectasias, leukonychia, and lentiginous skin melanoma.12
There is no proven effective treatment for LAM, and both topical steroids and high-dose penicillin have been reported to be ineffective. Few cases have shown stabilization of lesions with oral potassium aminobenzoate 12 g/day. Other studies have reported partial responses to topical calcipotriol and intralesional platelet-rich plasma therapy, while Zaouak et al reported a case successfully treated with methotrexate.13–15
Lichen Striatus and Blaschkitis
Lichen striatus (LS) and blaschkitis are inflammatory conditions following Blaschko lines. LS is primarily affecting children, while blaschkitis is mainly affecting adults. They can be differentiated from other diseases in this category by being inflammatory rather than pigmentary. In dark-skinned individuals, LS may present as a hypopigmented lesion. In blaschkitis, it commonly presents as inflammatory papules and vesicles on adults trunk.16
Other Rare Conditions
There are other few conditions that may present as linear hyperpigmentation following Blaschko’s lines without systemic findings. They include progressive cribriform and zosteriform hyperpigmentation (PCZH), unilateral nevoid acanthosis nigricans (UNAN), and linear macular amyloidosis (LMA). PCZH is a rare condition that presents at birth with Blaschkoid hyperpigmented lesions with no systemic symptoms.17 UNAN is a rare variant of acanthosis nigricans that usually tend to be unilateral and following lines of Blascko without any internal involvement.18 Macular amyloidosis is a depositional disease caused by amyloid deposition in the dermis. In a minority of cases, it presents in a linear configuration and needs skin biopsy to confirm the diagnosis.19
Linear Hyperpigmentation Following Blaschko’s Lines That May Have Systemic Findings
We describe the most common dermatological disorders that may present with linear hyperpigmentation following Blaschko’s lines, and their associated systemic findings (Table 2).
 |
Table 2 Clinical Clues for Linear Hyperpigmentation That Follow Blaschko’s Lines That May Have Systemic Findings |
 |
Figure 4 Linear morphea (limb variant). A unilateral linear band of sclerotic, atrophic, and hyperpigmented plaques affecting the left forearm of an eight years old boy. |
Focal Dermal Hypoplasia
Focal dermal hypoplasia (FDH) or Goltz syndrome is a rare disease associated with various cutaneous and extracutaneous anomalies. It primarily affects 85–90% females due to the X-linked dominant inheritance pattern.20,21
Clinically, it presents with asymmetrical linear atrophic hyperpigmented streaks on the face, trunk, and extremities following Blaschko’s lines, multiple mucosal papillomas, and fatty tissue hernias. Extracutaneous manifestations include mental retardation, osteopathia striata, hypoplastic teeth, microphthalmia, strabismus, ectopia lentis, and coloboma of the iris and choroid. Patients may present with digital anomalies including absent or dystrophic nails, syndactyly, polydactyly, and camptodactyly.21
Linear and Whorled Nevoid Hypermelanosis
Linear and whorled nevoid hypermelanosis (LWNH) is an uncommon sporadic disease thought to occur secondary to mosaicism or cytogenetic aberrations and can present with associated extracutaneous abnormalities. Hyperpigmentation generally becomes apparent at birth or shortly after, and it persists throughout life.22–24 It clinically presents as asymmetrical linear or whorled hyperpigmented macules adhering to Blaschko’s lines on the trunk and thighs while sparing the mucosal surfaces, palms, and soles. Lesions occur without atrophy or preceding inflammation. LWNH is rarely complicated by inflammatory linear verrucous epidermal nevus or ichthyosis vulgaris.23,24
Approximately 10–25% with LWNH present with extracutaneous findings, which typically become apparent during infancy; neurological conditions such as mental retardation, autism, epilepsy, microcephaly, arhinencephaly, developmental delay, hemiatrophy, psychomotor delay, and deafness; musculoskeletal conditions such as brachydactyly; or (less often) cardiac conditions such as congenital heart diseases.24,25 LWNH is usually diagnosed clinically, although chromosomal analysis of skin fibroblasts from hyperpigmented macules can be diagnostic.
X-Linked Reticulate Pigmentary Disorder
X-linked reticulate pigmentary disorder (XLRPD) (also known as familial cutaneous amyloidosis or X-linked cutaneous amyloidosis) is a rare congenital disorder that is inherited as a recessive X-linked trait. In males, it presents as generalized hyperpigmentation with small hypopigmented macules. Distinctive facial characteristics include an upswept frontal hairline and arched eyebrows. This disease is limited to cutaneous expression in females, while males usually suffer from multiple systemic conditions, including pulmonary, ocular, neurological, and gastrointestinal manifestations. Females present with patchy linear hyperpigmentation that adheres to Blaschko’s lines, and it is morphologically similar to stage III incontinentia pigmenti. Histopathologically, the deposition of amyloid-like material in the skin is apparent in adults.26–28
X-Linked Hypohidrotic Ectodermal Dysplasia
Hypohidrotic ectodermal dysplasia (HED) refers to a group of disorders that share the following features: sparse or absent hair, missing or peg-shaped teeth, and decreased ability to sweat. The most common form is X-linked, caused by a mutation in (EDA) codes for a soluble ligand, ectodysplasin A.29
Female patients with the X-linked form of HED present with one of the three categories: (1) Carriers with no identifiable clinical symptoms; (2) limited involvement with findings such as reduced hair density, one or more peg-shaped or missing teeth, a patchy distribution of sweat glands along Blaschko’s lines, and relative hyperpigmentation of the skin that lacks adnexa (most evident on the back); (3) full-blown characteristics of the disorder. This variability results from the random nature of X–inactivation.
Affected individuals typically have an altered facial appearance characterized by a saddle nose, full everted lips, and frontal bossing, thick and viscous nasal secretions and cerumen, recurrent respiratory tract infections, hoarse or raspy voice, gastroesophageal reflux, and feeding problems.30
Stage III Incontinentia Pigmenti
Incontinentia pigmenti (IP) (Bloch-Sulzberger syndrome) is a rare X-linked genetic syndrome that affects multiple organ systems. It primarily affects women and is caused by a mutation in the NF-kappa essential modulator (NEMO) gene or inhibitor of nuclear factor Kappa-B kinase subunit gamma (IKK-γ).31,32
The syndrome progresses through four stages. At the third stage, occurring between the age of 3 and 6 months, lesions develop into hyperpigmented whorled lesions that adhere to Blaschko’s lines.31,32 Extra-cutaneous manifestations include multiple ocular, skull, dental, and palatal defects as well as neurological abnormalities such as epilepsy, microcephaly, mental retardation, and slow motor development.32
Diagnostic criteria for IP were initially established in 1993 and were revised in 2014.31,33 Major clinical criteria are the typical stages of skin eruptions distributed along Blaschko’s lines, while minor criteria include dental anomalies, central nervous system abnormalities, alopecia or abnormal hair, nail dystrophy, palate anomalies, history of multiple male miscarriages in the mother, and typical histopathologic findings in a skin biopsy.33
Early-Stage or Subtle Epidermal Nevus
Epidermal nevus develops sporadically with typical onset within the first year of life, with no gender predilection. The term “nevus” refers to a hamartoma of the epidermis and papillary dermis. It occurs secondary to mosaicism that leads to a mutation in KRT1 or KRT10 versus ATP2A2.34–36 ATP2A2 mutations in epidermal nevi are usually seen in acantholytic epidermal nevi and probably a mosaic form of Darier disease.37
Epidermal nevi most commonly occur as a solitary linear lesion, but they are often seen as multiple unilateral or bilateral linear plaques. Most lesions are well-circumscribed, hyperpigmented (rarely hypopigmented), papillomatous papules, or plaques that are usually asymptomatic. Once developed, the nevi may thicken and become more verrucous, especially over joints and in flexural areas such as the neck. Lesions occur most commonly on the trunk, extremities, or neck.36 Moreover, the nevi follow Blaschko’s lines and may have an abrupt midline demarcation.
Epidermal nevus syndrome is a term used when epidermal nevi occur in combination with other developmental anomalies; the central nervous or musculoskeletal systems are most commonly involved. Neoplasms such as squamous cell carcinoma, basal cell carcinoma, and keratoacanthoma may rarely develop in association with epidermal nevi.38
Conradi–Hünermann–Happle Syndrome
Conradi–Hünermann–Happle (CHH) syndrome is a rare X-linked dominant disorder that occurs primarily in females. This occurs due to a defect in cholesterol biosynthesis caused by mutations in EBP, which encodes the emopamil-binding protein (EBP).39,40
At birth, most CHH syndrome patients have generalized erythema with thick, adherent, feathery scale, streaks, and whorls of hyperkeratosis following Blaschko’s lines. The erythroderma resolves substantially, if not completely, within the first weeks or months of life. In older children, hyperkeratosis is largely replaced by linear or patchy follicular atrophoderma with dilated follicular openings, ice-pick-like scars, and mild residual scaling. The atrophoderma is most pronounced on the forearms and dorsal hands, whereas the palms and soles are usually spared. Hyper- or hypopigmentation along Blaschko’s lines may coexist separately from atrophodermic areas.39 Patchy scarring alopecia, onychoschizia, and flattening of the nail plate can be seen, while the teeth are normal.
Skeletal anomalies are usually asymmetric. They include frontal bossing, malar hypoplasia, a flat nasal bridge, a short neck, rhizomelic shortening of the limbs, and scoliosis. Widespread premature calcifications manifest as stippling (chondrodysplasia punctata) of the long bone epiphyses, tracheal cartilage, and vertebrae. Ocular abnormalities can be seen, most commonly unilateral cataracts, and may be accompanied by microphthalmia or microcornea. Occasionally, other features may be associated, and include congenital heart defects, sensorineural deafness, CNS malformations, and congenital renal anomalies. Intellect is usually not impaired, and life expectancy is normal.30
Linear Morphea
Linear morphea is thought to be the most common subtype of morphea in children; it is one of the five subtypes of morphea: circumscribed (with superficial and deep variants), linear (with trunk/limb variant and head variant), generalized, pansclerotic, and mixed.41 Genetic mosaicism has been suggested as a contributing pathogenic factor, given the strong association between linear morphea and Blaschko’s lines.42
En coup de sabre (ECDS), progressive hemifacial atrophy (Parry-Romberg syndrome), and linear limb involvement are the three most frequently identified forms of linear morphea. These three forms are usually accompanied by underlying tissue atrophy. ECDS usually occurs on the paramedian forehead and is associated with underlying ocular and central nervous system involvement.43 Progressive hemifacial atrophy causes atrophy of the subcutaneous tissues with limited overlaying cutaneous changes. The mean age of onset in patients with ECDS morphea and PRS is 13.6 years, with a female predominance of approximately 2:1. Some authors consider ECDS and PRS as clinical variants of the same disease.44 Linear morphea also mostly affects the limbs, causing muscle atrophy, limb length discrepancies, and joint contractures (Figure 4).45
Extracutaneous manifestations of morphea are usually seen in patients with generalized morphea and include myalgia, arthralgia, and fatigue.45–47 Approximately 4% pediatric morphea patients have symptoms of neurological disease including seizures, headaches, peripheral neuropathy, vascular malformations, and central nervous system vasculitis. Neurologic manifestations are much more frequent in children with ECDS or progressive facial hemiatrophy.47 Therefore, children with head and neck morphea should undergo regular ophthalmologic examinations to monitor for asymptomatic involvement that may lead to irreversible damage. The most common MRI scan findings accompanying morphea are T2 hyperintensities in the subcortical white matter and corpus callosum, focal tissue atrophy, and calcifications.48,49
Becker Nevus
Becker nevus is an acquired hyperpigmentation and hypertrichosis on the upper trunk or proximal upper limb. Androgens likely play a key role, as the majority of cases appear in late childhood and adolescence. Although there are different morphological presentations, most lesions have a geographic or block-like configuration, and rarely present in a linear pattern; however, it is unclear whether Becker nevus adheres to Blaschko’s lines. Although Jackson listed Becker’s nevus as one of the nevoid skin diseases that follow Blaschko’s lines,50,51 this fact was contradicted by Bolognia et al, who insisted that among the lesions listed by Jackson, Becker’s nevus and linear scleroderma do not follow Blaschko’s lines.1
Other cutaneous anomalies can coexist with Becker nevus, such as hypohidrosis; granuloma annulare; and malignancies such as basal cell carcinoma, malignant melanoma, lymphangioma, and osteoma cutis.
Becker nevus is rarely associated with systemic disorders such as spina bifida, scoliosis, pectus carinatum, congenital adrenal hyperplasia, quadriparesis, and an accessory scrotum. Ipsilateral developmental abnormalities such as breast hypoplasia, supernumerary nipples, aplasia of the pectoralis major muscle, limb reduction, segmental odontomaxillary dysplasia, and lipoatrophy have also been reported.52
Café-au-Lait Spots in McCune–Albright Syndrome
McCune–Albright syndrome (MAS) is classically present with café-au-lait spots (CALM) along with other systemic symptoms, including bone deformity and endocrinopathy. It has been reported that CALM may present in a linear configuration.53
Linear Hyperpigmentation Not Following Blaschko’s Lines
We describe the most common dermatological disorders likely to present with linear hyperpigmentation that does not adhere to Blaschko’s lines (Table 3).
 |
Table 3 Clinical Clues for Linear Hyperpigmentation That Does Not Follow Blaschko’s Lines with Associated Cutaneous and Systemic Manifestations |
Linear Postinflammatory Hyperpigmentation
PIH is the most common cause of acquired hyperpigmentation and is more common in darkly pigmented individuals. The circumscribed variant is observed more often than diffuse or linear forms. The presence of linear PIH should raise the possibility of external skin injuries, especially contact dermatitis like jellyfish or Rhus dermatitis. In these conditions, the patient usually presents with inflammatory lesions followed by linear hyperpigmentation.54,55 PIH slowly fades over time, provided that further injury to the skin does not occur. However, resolution may not occur for years, especially in more darkly pigmented individuals and when the melanin is primarily dermal. Disorders that commonly lead to epidermal PIH include acne, insect bites, pyodermas, atopic dermatitis, psoriasis, and pityriasis rosea. However, dermal PIH is associated with dermatoses characterized by degeneration of the basal layer of the epidermis and inflammation at the dermal–epidermal junction, such as lichen planus, lichenoid drug reactions, lupus erythematosus, and fixed drug eruptions.30 In some conditions, like lichen planus, the linear hyperpigmentation may result from koebnerization, which presents as linear hyperpigmentation that is not following Blaschko lines.
Phototoxic Dermatitis
Phototoxic dermatitis is a result of an interaction of solar UVA and exogenous agent. In this reaction, epidermal cell damage leads to inhibition of cell proliferation and DNA synthesis, with subsequent edema, erythema, and excess melanin production presenting as linear hyperpigmentation.
Among these phototoxic agents, psoralen or bergamot, which may lead to dermatitis called berloque dermatitis. Patients usually notice blisters and erythema, which then progress to linear hyperpigmentation that ultimately fades weeks to months after the offending agent is removed.56 Another related condition is phytophotodermatitis, where the patient develops phototoxic reactions caused by sun exposure after contact with furocoumarins-containing plants such as lemon, grapefruit, parsley, and others.57
Linear Lichen Planus
Lichen planus (LP) is an immunological disorder with histologic changes known as lichenoid tissue reactions.7 LP is uncommon in children, constituting only 1–4% of total LP cases but tends to be more severe. There are multiple clinical variants including classical, linear, hypertrophic, ulcerative, and, lastly, the pigmentosus subtype, which is the rarest form.3,7 LP is generally a self-limiting disease that typically causes post-inflammatory hyperpigmentation (PIH).
Clinically, the presentation of LP varies depending on the subtype. Classic LP presents with polygonal, pruritic, violaceous, and flat-topped papules with Wickham’s striae and predominantly affects the flexor aspects of the limbs.58 Linear or zosteriform LP is an uncommon variant that follows the Koebner phenomenon, does not adhere to Blaschko’s lines, and rarely follows a dermatomal arrangement.59 On the other hand, blaschkoid LP is differentiated from linear LP, as it adheres to Blaschko’s lines. Inherited factors in the affected skin, rather than inflammatory cells, cause the linear manifestations of these inflammatory conditions.3
Linear Fixed-Drug Eruption
The diagnosis of fixed drug eruption is the basis of the historical, clinical, and histological findings.60 Linear erythematous and hyperpigmented macular lesions following a neural distribution have been reported secondary to an intramuscular cephazolin injection.61
Flagellate Hyperpigmentation
Flagellate hyperpigmentation can result from bleomycin, shiitake mushroom dermatitis, dermatomyositis, and persistent plaques of Still disease. The lesion resembles the whip-like appendages of bacterial flagella. While there in no consensus on etiology, some researchers suggest that melanocyte accumulation, direct toxicity, and post-inflammatory changes may be causative.30
Bleomycin is an effective chemotherapeutic drug for lymphoma, squamous cell carcinoma, and germ cell tumors.62 Flagellate hyperpigmentation occur in 10–20% patients receiving systemic bleomycin.62,63 Clinical features typically begin as an itchy linear pigmentation arranged in a flagellated pattern on the back and chest. Clinical onset varies between 1 day and 9 weeks after bleomycin administration. High bleomycin doses (accumulative dose: 100–300 mg) are associated with the disease.62,64 However, it also occurs with low doses (15–30 mg), including intralesional injections of warts or vascular malformations.65,66 There is no effective treatment for bleomycin-induced flagellate dermatitis. One study indicated that discontinuation of the drug would lead to gradual resolution of the symptoms within 3–4 months. Topical or systemic corticosteroids and antihistamines may also be useful. However, even without therapy, the cutaneous manifestations are self-limiting and disappear within 6–8 months.64
Shiitake mushroom dermatitis occurs in individuals who have ingested raw or undercooked shiitake mushrooms (Lentinula edodes).67 Lentinan, a thermolabile toxin in shiitake mushrooms, is probably responsible for this dermatosis which has a typically negative result in prick and patch tests.
In shiitake mushroom dermatitis, pruritic erythematous papules, vesicles, and edema develop on the face, scalp, trunk, and extremities 1–2 days after mushroom ingestion. Scratching leads to long, flagellate streaks with erythematous papules and/or petechiae on the extremities and trunk, with sparing of the difficult-to-reach mid-upper back. This can be followed by a linear pattern of either discoloration due to hemosiderin or PIH. Diffuse erythema and edema in photoexposed areas have been observed in patients with shiitake mushroom dermatitis, and fever and malaise are occasionally associated findings.67 Topical corticosteroids and oral antihistamines lead to resolution of symptoms within 1–8 weeks if the mushrooms are avoided. Complete cooking of shiitake mushrooms prevents this disorder.30
Serpentine Supravenous Hyperpigmentation
Serpentine supravenous hyperpigmentation (SSH) is a rare condition that presents with linear hyperpigmentation over superficial veins caused by chemotherapy infusion. It is classically linked to 5-FU, but it has also been reported with other chemotherapy like docetaxel, fotemustine, and vinorelbine. It usually resolves within one to three months.68
Pigmentary Demarcation Lines
Pigmentary demarcation lines (PDL) are symmetric, bilateral, and are present from infancy to adulthood as physiological abrupt transition lines from heavily pigmented areas of the skin to more lightly pigmented areas. PDL is a common finding in dark-skinned women. Approximately 79% of women of African descent have at least one such demarcation line. While the pathogenesis of PDL is uncertain, they may occur secondary to pigmentary mosaicism or the axial-neural theory, as the lines adhere to the cutaneous nerve distribution. Eight types of PDL (A to H) have been reported.69,70
Pigmentary Demarcation Lines in Newborns
Pigmentary demarcation lines in newborns present as a rare congenital pigmentation that follows the horizontal abdominal creases. Hyperpigmentation can be congenital or acquired and may be either localized or diffuse. This hyperpigmentation is classified as a transient benign cutaneous lesion that can present at birth or a few weeks to months later. Hyperpigmentation lines may also affect the back and extremities. However, it may spontaneously regress between the ages 2 and 6 months.71
Linea Nigra
Linea nigra (LN) is a linear hyperpigmentation that appears from the symphysis pubis to the umbilicus on the abdomen. While it is commonly associated with pregnancy (linea gravidarum), it can also be found in males and non-pregnant females, especially in younger age groups. A study conducted in Nigeria revealed that 48% of men with prostatic hyperplasia were affected by LN. As such, researchers suggested that estrogen or lack of sensitivity of androgen receptors could be an etiological factor.72
Sock-Line Hyperpigmentation
Sock line (or mitten line) hyperpigmentation is an infantile skin disorder generally caused by elastic bands that constrict the skin, causing dermal and subcutaneous fat inflammation, which ultimately leads to hyperpigmentation. It usually appears two or more hours after constriction and is characterized by curvilinear patches or plaques surrounded by erythema on the affected extremities.73–75 It should be differentiated from amniotic band syndrome, which has a similar presentation but occurs on the lower abdomen. It is also important to rule out child abuse in such cases.76
Cryofibrinogenemia-Induced Cutaneous Ulcers
Cryofibrinogenemia is an infrequent cause of non-healing dermal ulcers. This small-vessel occlusive vascular disorder is caused by accumulation and precipitation of cryofibrinogens in plasma. It typically manifests as microlivedo, an exquisite form of livedo reticularis that presents as focal violaceous patchy hyperpigmented streaks. These streaks are caused by dermal blood vessels occlusion rather than an increase in melanin content. Diagnosis is challenging, as there are no definitive or agreed-upon criteria.77
Conclusion
Linear hyperpigmentation is a rare presentation of different dermatological diseases. It can be isolated or associated with systemic symptoms. So, early detection of systemic findings will help decrease disease burden in these patients. Using a classification system that segregates linear hyperpigmentation according to adherence to Blaschko’s lines, associated morphological findings (including other cutaneous lesions), and systemic manifestations are important to improve diagnosis.
Disclosure
The authors report no conflicts of interest for this work.
References
1. Bolognia JL, Orlow SJ, Glick SA. Lines of Blaschko. J Am Acad Dermatol. 1994;31(2):157–190.
2. Happle R, Assim A. The lines of Blaschko on the head and neck. J Am Acad Dermatol. 2001;44(4):612–615.
3. Lade NR, Saoji V, Singh AI. Blaschkoid lichen planus: not a koebner phenomenon. Dermatol Online J. 2013;19(4):17.
4. Sil A, Chakraborty S, Panigrahi A, Mondal S. Blaschkoid lichen planus occurring in childhood systemic lupus erythematosus. Pediatr Dermatol. 2020;37(3):579–581.
5. Kanwar AJ, Dogra S, Handa S, Parsad D, Radotra BD. A study of 124 Indian patients with lichen planus pigmentosus. Clin Exp Dermatol. 2003;28(5):481–485.
6. Al-Mutairi N, El-Khalawany M. Clinicopathological characteristics of lichen planus pigmentosus and its response to tacrolimus ointment: an open label, non-randomized, prospective study. J Eur Acad Dermatol Venereol. 2010;24(5):535–540.
7. Akagi A, Ohnishi Y, Tajima S, Ishibashi A. Linear hyperpigmentation with extensive epidermal apoptosis: a variant of linear lichen planus pigmentosus? J Am Acad Dermatol. 2004;50(5):78–80.
8. Hong S, Shin JH, Kang HY. Two cases of lichen planus pigmentosus presenting with a linear pattern. J Korean Med Sci. 2004;19(1):152–154.
9. Ochoa BE, King LE, Price VH. Lichen planopilaris: annual incidence in four hair referral centers in the United States. J Am Acad Dermatol. 2008;58(2):352–353.
10. Assouly P, Reygagne P. Lichen Planopilaris: update on diagnosis and treatment. Semin Cutan Med Surg. 2009;28(1):3–10. doi:10.1016/j.sder.2008.12.006
11. Villani AP, Amini-Adlé M, Wagschal D, Balme B, Thomas L. Linear atrophoderma of Moulin: report of 4 cases and 20th anniversary case review. Dermatology. 2013;227(1):5–9.
12. Atasoy M, Aliagaoglu C, Sahin O, Ikbal M, Gursan N. Linear atrophoderma of Moulin together with leuconychia: a case report. J Eur Acad Dermatol Venereol. 2006;20(3):337–340.
13. Wongkietkachorn K, Intarasupht J, Srisuttiyakorn C, Aunhachoke K, Nakakes A, Niumpradit N. Linear atrophoderma of moulin:a case report and review of the literature. Case Rep Dermatol. 2013;5(1):11–14. doi:10.1159/000346747
14. Kharkar V, Abak B, Mahajan S. Linear atrophoderma of moulin: a rare entity. Indian J Dermatol Venereol Leprol. 2018;84(5):591.
15. Zaouak A, Ghorbel HH, Benmously-Mlika R, et al. A case of linear atrophoderma of Moulin successfully treated with methotrexate. Dermatol Ther. 2014;27(3):153–155.
16. Müller CSL, Schmaltz R, Vogt T, Pföhler C. Lichen striatus and blaschkitis: reappraisal of the concept of blaschkolinear dermatoses. Br J Dermatol. 2011;164(2):257–262. doi:10.1111/j.1365-2133.2010.10053.x
17. Gutte R. Progressive cribriform and zosteriform hyperpigmentation. Indian Dermatol Online J. 2014;5(1):38.
18. Jeong JS, Lee JY, Yoon TY. Unilateral nevoid acanthosis nigricans with a submammary location. Ann Dermatol. 2011;23(1):95. doi:10.5021/ad.2011.23.1.95
19. Abbas O, Ugent S, Borirak K, Bhawan J. Linear macular amyloidosis. J Eur Acad Dermatol Venereol. 2009;23(12):1446–1448.
20. Landa N, Oleaga JM, Ratón JA, Gardeazabal J, Díaz-Pérez JL. Focal dermal hypoplasia (Goltz syndrome): an adult case with multisystemic involvement. J Am Acad Dermatol. 1993;28(1):86–89.
21. Kanitakis J, Souillet A-L, Butnaru C, Claudy A. Melanocyte stimulation in focal dermal hypoplasia with unusual pigmented skin lesions: a histologic and immunohistochemical study. Pediatr Dermatol. 2003;20(3):249–253.
22. Lal K, Di Lernia V. Linear and whorled naevoid hypermelanosis in a patient with trisomy 4 mosaicism. Clin Exp Dermatol. 2015;40(1):45–47.
23. Lu Y, Zhu W-Y. Linear and whorled nevoid hypermelanosis complicated with inflammatory linear verrucous epidermal nevus and ichthyosis vulgaris. J Dermatol. 2007;34(11):765–768.
24. Ertam I, Turk BG, Urkmez A, Kazandi A, Ozdemir F. Linear and whorled nevoid hypermelanosis: dermatoscopic features. J Am Acad Dermatol. 2009;60(2):328–331.
25. Nehal KS. Analysis of 54 cases of hypopigmentation and hyperpigmentation along the lines of Blaschko. Arch Dermatol. 1996;132(10):1167–1170.
26. Pezzani L, Brena M, Callea M, Colombi M, Tadini G. X-linked reticulate pigmentary disorder with systemic manifestations: a new family and review of the literature. Am J Med Genet Part A. 2013;161(6):1414–1420.
27. Anderson RC, Zinn AR, Kim J, Carder KR. X-linked reticulate pigmentary disorder with systemic manifestations: report of a third family and literature review. Pediatr Dermatol. 2005;22(2):122–126.
28. Kim BS, Seo S-H, Jung HD, Kwon K-S, Kim M-B. X-linked reticulate pigmentary disorder in a female patient. Int J Dermatol. 2010;49(4):421–425.
29. Mikkola ML. Molecular aspects of hypohidrotic ectodermal dysplasia. Am J Med Genet Part A. 2009;149A(9):2031–2036.
30. Bolognia J, Schaffer JCL. Dermatology.
31. Landy SJ, Donnai D. Incontinentia pigmenti (Bloch-Sulzberger syndrome). J Med Genet. 1993;30(1):53–59.
32. Cammarata-Scalisi F, Fusco F, Ursini MV. Incontinencia pigmenti. Actas Dermosifiliogr. 2019;110(4):273–278.
33. Minić S, Trpinac D, Obradović M. Incontinentia pigmenti diagnostic criteria update. Clin Genet. 2014;85(6):536–542.
34. Meschia JF, Junkins E, Hofman KJ. Familial systematized epidermal nevus syndrome. Am J Med Genet. 1992;44(5):664–667.
35. Goldman K, Don PC. Adult onset of inflammatory linear verrucous epidermal nevus in a mother and her daughter. Dermatology. 1994;189(2):170–172.
36. Rogers M, McCrossin I, Commens C. Epidermal nevi and the epidermal nevus syndrome. J Am Acad Dermatol. 1989;20(3):476–488.
37. Garcias-Ladaria J, Cuadrado Rosón M, Pascual-López M. Epidermal nevi and related syndromes — part 1: keratinocytic nevi. Actas Dermosifiliogr. 2018;109(8):677–686.
38. Solomon LM, Esterly NB. Epidermal and other congenital organoid nevi. Curr Probl Pediatr. 1975;6(1):3–56.
39. Happle R. X-linked dominant chondrodysplasia punctata. Hum Genet. 1979;53(1):65–73.
40. Braverman N, Lin P, Moebius FF, et al. Mutations in the gene encoding 3β- hydroxysteroid-Δ8,Δ7- isomerase cause X-linked dominant Conradi-Hünermann syndrome. Nat Genet. 1999;22(3):291–294.
41. Laxer RM, Zulian F. Localized scleroderma. Curr Opin Rheumatol. 2006;18(6):606–613.
42. Weibel L, Harper JI. Linear morphoea follows Blaschko’s lines. Br J Dermatol. 2008;159(1):175–181.
43. Tollefson MM, Witman PM. En coup de sabre morphea and Parry-Romberg syndrome: a retrospective review of 54 patients. J Am Acad Dermatol. 2007;56(2):257–263.
44. Blaszczyk M, Królicki L, Krasu M, Glinska O, Jablonska S. Progressive facial hemiatrophy: central nervous system involvement and relationship with scleroderma en coup de sabre. J Rheumatol. 2003;30(9):1997–2004.
45. Christen-Zaech S, Hakim MD, Afsar FS, Paller AS. Pediatric morphea (localized scleroderma): review of 136 patients. J Am Acad Dermatol. 2008;59(3):385–396.
46. Leitenberger JJ, Cayce RL, Haley RW, Adams-Huet B, Bergstresser PR, Jacobe HT. Distinct autoimmune syndromes in morphea: a review of 245 adult and pediatric cases. Arch Dermatol. 2009;145(5):545–550.
47. Zulian F, Athreya BH, Laxer R, et al. Juvenile localized scleroderma: clinical and epidemiological features in 750 children. An international study. Rheumatology. 2006;45(5):614–620.
48. Kister I, Inglese M, Laxer RM, Herbert J. Neurologic manifestations of localized scleroderma: a case report and literature review. Neurology. 2008;71(19):1538–1545.
49. Fett N, Werth VP. Update on morphea. J Am Acad Dermatol. 2011;64(2):231–242.
50. Jackson R. The lines of Blaschko: a review and reconsideration: observations of the cause of certain unusual linear conditions of the skin. Br J Dermatol. 1976;95(4):349–360.
51. Relhan V, Bansal S, Garg V, Goel K. Acquired linear Becker′s nevus on lower limb in blaschkoid pattern. Indian J Dermatol Venereol Leprol. 2015;81(3):328.
52. Glinick SE, Alper JC, Bogaars H, Brown JA. Becker’s melanosis: associated abnormalities. J Am Acad Dermatol. 1983;9(4):509–514.
53. Jung A-J, Soskin S, Paris F, Lipsker D. Syndrome de McCune-Albright révélé par des taches café-au-lait blaschko-linéaires du dos. Ann Dermatol Venereol. 2016;143(1):21–26.
54. Menahem S, Shvartzman P. Recurrent dermatitis from jellyfish envenomation. Can Fam Physician. 1994;40(DEC):2116–2118.
55. Belsito DV. A Sherlockian approach to contact dermatitis. Dermatol Clin. 1999;17(3):705–713.
56. Gruson LM, Chang MW. Berloque dermatitis mimicking child abuse. Arch Pediatr Adolesc Med. 2002;156(11):1091.
57. Bowen AG. Phytophotodermatitis. Am J Contact Dermat. 1999;10(2):89–93.
58. Pandhi D, Singal A, Bhattacharya SN. Lichen planus in childhood: a series of 316 patients. Pediatr Dermatol. 2014;31(1):59–67.
59. Miljković J, Belič M, Godić A, Klemenc P, Marin J. Zosteriform Lichen Planus-Like Eruption. Vol. 15. Acta Dermatovenerologica Alpina, Pannonica et Adriatica; 2006:94–97.
60. Sehgal VN, Gangwani OP. Fixed drug eruption. Int J Dermatol. 1987;26(2):67–74.
61. Sigal-Nahum M, Konqui A, Gaulier A, Sigal S. Linear fixed drug eruption. Br J Dermatol. 1988;118(6):849–851.
62. Yaris N, Çakir M, Kalyoncu M, Aysenur O. Bleomycin induced hyperpigmentation with yolk sac tumor. Indian J Pediatr. 2007;74(5):505–506.
63. Mahmoud BH, Eide MJ. Bendamustine-induced “flagellate dermatitis”. Dermatol Online J. 2012;18(11):12.
64. Grynszpan R, Niemeyer-Corbellini JP, Lopes MSS, Ramos-e-Silva M. Bleomycin-induced flagellate dermatitis. Case Rep. 2013;2013.
65. Cortina P, Garrido JA, Tomas JF, Unamuno P, Armijo M. ‘Flagellate’ erythema from bleomycin. With histopathological findings suggestive of inflammatory oncotaxis. Dermatology. 1990;180(2):106–109.
66. Abess A, Keel DM, Graham BS. Flagellate hyperpigmentation following intralesional bleomycin treatment of verruca plantaris. Arch Dermatol. 2003;139(3):337–339.
67. Hanada K, Hashimoto I. Flagellate mushroom (Shiitake) dermatitis and photosensitivity. Dermatology. 1998;197(3):255–257.
68. Lancman G, Steinberg A, Gabrilove J. Serpentine supravenous hyperpigmentation in an HIV patient receiving R-CHOP for DLBCL. Int J Hematol Stem Cell Res. 2018;12(1):1–3.
69. Al Mohizea S, Bin-Saif G, Al-Balbeesi A, Al-Samary A. Pigmentary demarcation lines on the face in Saudi women. Indian J Dermatol Venereol Leprol. 2010;76(4):378.
70. Somani VK, Razvi F, Sita VNVL. Pigmentary demarcation lines over the face. Indian J Dermatol Venereol Leprol. 2004;70(6):336–341.
71. Fosse N, Itin P. Pigmentary lines of the newborn: a case report and review of the literature. Dermatology. 2014;228(3):198–201.
72. Okeke LI, George AO, Ogunbiyi AO, Wachtel M. Prevalence of linea nigra in patients with benign prostatic hyperplasia and prostate carcinoma. Int J Dermatol. 2012;51(SUPPL.1):41–43.
73. Ciliberto H, Berk D, Salphale P, Bayliss S. Heel-line hyperpigmentation: a variant of sock-line hyperpigmentation after the use of heel-length socks. Pediatr Dermatol. 2013;30(4):473–475.
74. Berk DR. Sock-line hyperpigmentation: case series and literature review. Arch Dermatol. 2007;143(3):423.
75. Berk DR, Bayliss SJ. Mitten-line hyperpigmentation: a reactive process analogous to sock-line hyperpigmentation. Pediatr Dermatol. 2010;27(4):401–402.
76. Dyer JA, Chamlin S. Acquired raised bands of infancy: association with amniotic bands. Pediatr Dermatol. 2005;22(4):346–349.
77. Grada A, Falanga V. Cryofibrinogenemia-induced cutaneous ulcers: a review and diagnostic criteria. Am J Clin Dermatol. 2017;18(1):97–104.
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.