Back to Journals » Clinical, Cosmetic and Investigational Dermatology » Volume 13
Clinical and Molecular Investigation of Familial Multiple Lipomatosis: Variants in the HMGA2 Gene
Authors Mejía Granados DM, de Baptista MB ![]() , Bonadia LC
, Bonadia LC ![]() , Bertuzzo CS, Steiner CE
, Bertuzzo CS, Steiner CE
Received 27 April 2019
Accepted for publication 24 September 2019
Published 7 January 2020 Volume 2020:13 Pages 1—10
DOI https://doi.org/10.2147/CCID.S213139
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Jeffrey Weinberg
Diana Marcela Mejía Granados, Marcella Bergamini de Baptista, Luciana Cardoso Bonadia, Carmen Silvia Bertuzzo, Carlos Eduardo Steiner
Department of Medical Genetics and Genomic Medicine, School of Medical Sciences, University of Campinas, Campinas, São Paulo, Brazil
Correspondence: Diana Marcela Mejía Granados
Department of Medical Genetics and Genomic Medicine, School of Medical Sciences, University of Campinas, Rua Tessália Vieira De Camargo 126, Cidade Universitária, Campinas, São Paulo 13083-887, Brazil
Tel +55 19 9 7151-1695
Email [email protected]
Background: Familial multiple lipomatosis (FML) is an autosomal dominant disorder characterized by the slow growth of encapsulated nodules spread across the trunk and limbs. Currently, there is no specific etiology; therefore, its molecular and biological bases need to be better understood. High-throughput sequencing technologies appear to be a cost-effective tool and have a pivotal role in elucidating different genodermatoses.
Objective: This study aimed to perform a clinical and molecular characterization of constitutional DNA of seven individuals belonging to five unrelated families diagnosed with FML.
Patients and methods: Clinical aspects were obtained from medical records and physical examination. HMGA2 gene was investigated using Sanger sequencing method. Mutational analysis of other genes associated with syndromic lipomatosis AKT1, APC, PIK3CA, MEN-1, and PTEN was performed through next-generation sequencing.
Results: In this series, FML was predominant among women who were overweight and reaching the age of thirty and was associated with gastrointestinal comorbidity. Histopathological diagnosis of biopsies revealed typical features of both lipoma and angiolipoma. We identified two identical novel variants with unknown significance in exon 5 of the HMGA2 gene in two participants of different families. There were no additional changes in exons 1 to 4 of the HMGA2 gene. Multi-gene panel was normal in all cases.
Conclusion: Variants found in exon 5 of the HMGA2 gene have not been described and have an uncertain significance in the genesis of FML. Further studies, including a more significant number of affected individuals and functional analysis of the novel variants of HGMA2 gene, should be undertaken to better understand its biological role in FML.
Keywords: HMGA2 gene, next-generation sequencing, familial lipomatosis
Introduction
Familial Multiple Lipomatosis (FML, OMIM% 151,900) is a rare autosomal dominant disorder of hypodermis characterized by the development of well-encapsulated subcutaneous nodules on the extremities and trunk.1 First reports of multiple adipocytic tumors were made since 1846 by Sir Benjamin Brodie.2 Later results from Blaschko (1891), and Alsberg (1892) demonstrated a familial recurrence of the lesions and observed an increased incidence among males.1,3,4 In subsequent years, many other authors published families with characteristics highly suggestive of FML,1,5,6 and finally in 1970 Das Gupta classified lipomatous tumors in three categories: solitary or sporadic, FML and Multiple Symmetric Lipomatosis (MSL).7,8
Solitary lipomas are considered the most common benign neoplasm of soft tissues in adults, but multiple lipomas occur in 5% of individuals.9,10 The exact prevalence of FML is unknown, but it has been estimated to be 0.002%.8,11,12 Age-onset of nodules is between 30 and 40 years, reaching the maximum peak at the 50s.6,8,13,14 It has been widely held that this disorder exhibit a preference for males, however, a large number of works demonstrate that both sexes are equally affected.15–17 Although it is considered a benign disease, cosmetic concerns may appear in some individuals, impairing their quality of life. Moreover, it is also prevalent in obese individuals.9
Up to now, available research in FML has been explicitly focused on clinical aspects, and molecular issues are usually not investigated. The literature suggests that at least 70% of sporadic lipomas result from cytogenetic rearrangements involving the 12q13-15 band, which could lead to a deregulated expression of the HMGA2 (High Mobility Group AT-hook 2) gene. This gene encodes non-histones chromatin proteins responsible for DNA conformational changes, aberrant cell proliferation, and development of benign mesenchymal tumors.18–20
Recognizing the molecular base of genetic conditions and the implementation of high-throughput sequencing technologies has permitted to explore and elucidate other mechanisms involved in the etiology of several genodermatosis. Therefore, this study aimed to perform a clinical and molecular characterization of seven individuals with FML to detect variants in six candidate genes related to lipomatosis using Sanger and next-generation sequencing (NGS) techniques.
Materials and Methods
Patients and Ethical Aspects
This research was conducted in compliance with the principles of the Declaration of Helsinki. Approval was obtained from the Institutional Review Board before the study (The Research Ethics Committee at FCM-UNICAMP, file N° 1.313.164) and all participants gave written informed consent. We conducted the study in the outpatient clinic of medical genetics service at the Clinical Hospital of the University of Campinas (HC-UNICAMP) in São Paulo, Brazil. A total of seven individuals belonging to five unrelated families (named from A to E) were eligible. Primary inclusion criteria for selecting the subjects were as follows: i) age above 18 years, ii) clinical and histopathological diagnosis of lipoma or angiolipoma and iii) familial recurrence suggesting autosomal dominant inheritance. Main clinical aspects were gathered from medical records using a structured form. Physical assessment involved the participation of both dermatologist and geneticist specialties. The Pathology Department at HC-UNICAMP provided the histopathologic specimens. Clinical findings are detailed in Table 1.
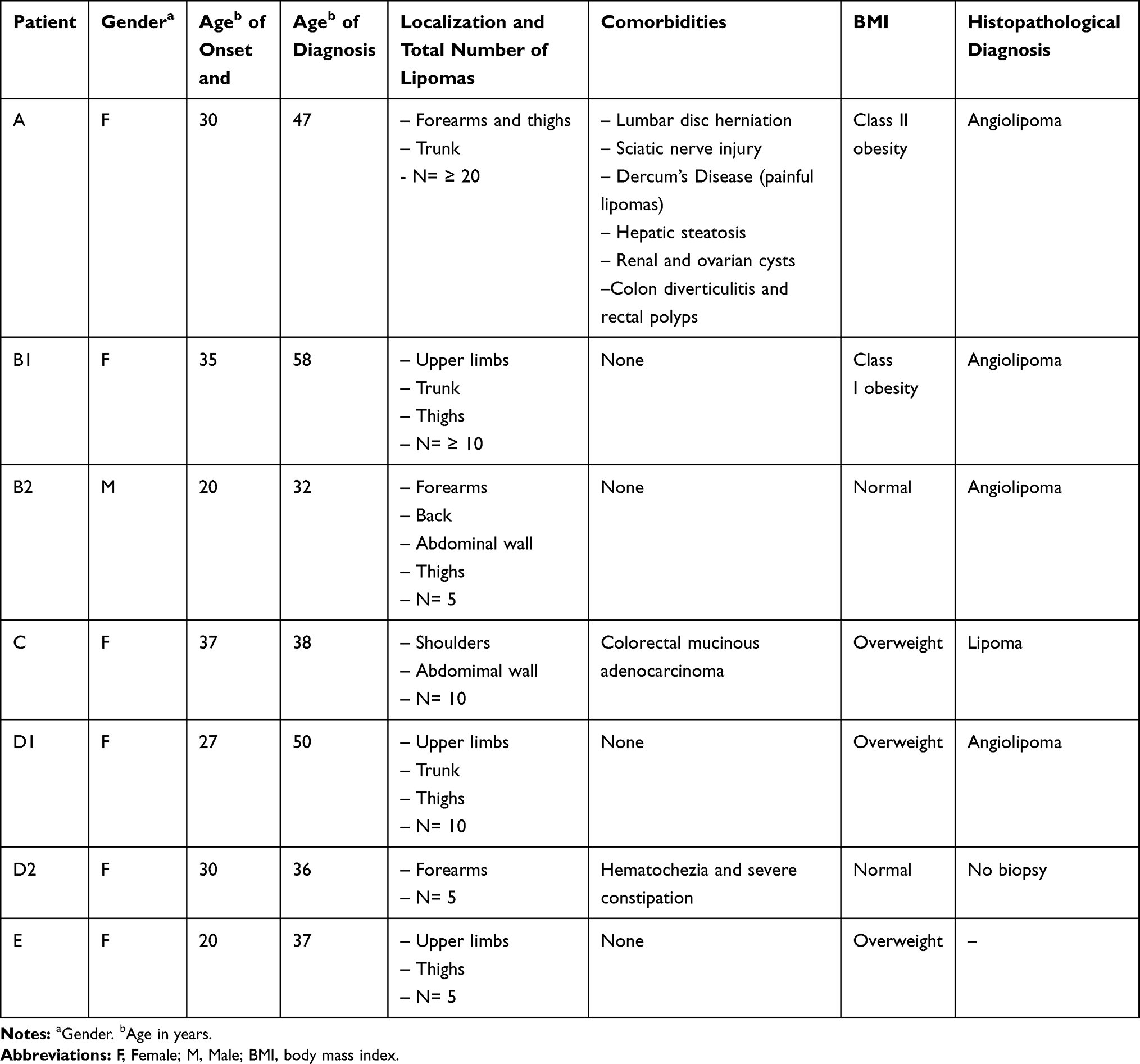 |
Table 1 Clinical Summary of Patients with FML |
Sanger Sequencing of the HMGA2 Gene
Genomic DNA was extracted from peripheral EDTA blood samples of seven patients according to the conventional phenol-chloroform procedure used by Sambrook et al21
A polymerase chain reaction (PCR) was carried out to amplify five coding exons and intron-exon junctions regions of the HMGA2 gene. Using two online bioinformatic tools, Primer3 (https://primer3plus.com/primer3web/primer3web_input.htm), and OligoAnalyer 3.1 (https://www.idtdna.com/calc/analyzer), we designed the specific pair of primers, as summarized in Table 2.
 |
Table 2 Specific Primers of the HMGA2 Gene for PCR |
PCR products were verified on a 12% polyacrylamide gel and then purified directly with Illustra ExoStar™ (GE Healthcare Life Sciences). All selected exons were sequenced on an automatic capillary system, ABI3500xL DNA analyzer (BigDye® Terminator Cycle Sequencing kit v3.1, Life Technologies®) following the manufacturer’s instructions. Chromatograms were examined using Chromas v.2.6.5. Results were compared with HMGA2 Ensembl transcript number ENST00000403681 (https://grch37.ensembl.org/index.html). We checked the novel variants in three public datasets: i) The Global Alliance for Genomics and Health (GA4GH) Beacon network (https://beacon-network.org/#/), ii) The Exome Aggregation Consortium (http://exac.broadinstitute.org/) and, iii) Online Archive of Brazilian Mutations (http://abraom.ib.usp.br/). To evaluate the pathogenicity scores and damage effects of novel variants in candidate HMGA2 gene, we used different in silico prediction models like VEP (Variant Effect Predictor),22 FATHMM (Functional Analysis Through Hidden Markov Models),23 UMD-predictor,24 SIFT (Sorting Intolerant from Tolerant),25 PROVEAN (Protein Variation Effect Analyzer),26 and Mutation Taster.27
Target Multi-Gene Panel
A custom panel was performed to assess the genes involved in syndromic lipomatosis. To perform this technique, we obtained genomic DNA from saliva specimens of five patients (A, B, C, D, and E) using the Oragene® DNA self-collection kit (DNA Genotek, Inc., Ottawa, Ontario, Canada). Samples were then enriched and hybridized against target-specific sequences. The capture was done using next-generation Illumina® MiSeq (TruSeq Capture) sequencer in 150bp paired-end mode according to the standard protocol for this platform. Depth of coverage of target regions was higher than 50x. Enrichment and analysis were focused on the coding sequences of five transcripts (AKT1 NM_005163.2, APC NM_000038.5, MEN1 NM_130799.2, PIK3CA NM_006218.2, and PTEN NM_000314.4), 10bp of intronic sequences and other regions presumably causative of disease. Exonic deletions, duplications and splice-site variations were also considered during the investigation. Reads were aligned and compared with human genome reference (GRCh37). Multiplex Ligation-dependent Probe Amplification (MLPA) was chosen to validate variants as mentioned by Invitae® Corporation (1400 16th Street, San Francisco, CA 94103, #05D2040778).
Data Availability
Sequences were submitted to GenBank (accession number: BankIt2217069 Seq1 -MK875826). Datasets related to this article are available at the European Bioinformatics Institute EMBL-EBI through ENA repository with study accession number PRJEB28960 (https://www.ebi.ac.uk/ena).
Results
Clinical Analysis
Seven individuals from five unrelated families (A, B1, B2, C, D1, D2, and E) were included, as depicted in Figure 1. We did not identify parental consanguinity between the participants. Ethnical origin was self-reported as mixed by each proband. In a general overview, families A and B were observed to have three successive or more generations involved. Male to female sex ratio was estimated in 1:6. Mean age at inclusion was 42 years (SD 11.8), while the mean age of lesions onset was 28 years (SD 11.8). Females reported a peak incidence of nodules after their pregnancies. Classic clinical features of FML included soft nodules highly variable in number and size, located in subcutaneous fat and confined to the trunk and extremities, as depicted in Figure 2. Discomfort and marked tenderness to palpation were noticed only in individual A. Comorbid gastrointestinal disorders were present in patients A, C, and D2. They reported diverticulitis and polyps, severe chronic constipation, and colorectal cancer respectively. Four patients had histological evaluation of subcutaneous lesions. The presence of mature adipocytes rounded by numerous capillaries confirmed angiolipoma diagnostic, as referenced by Bancroft and Fletcher et al10,28 (Figure 2). Other relevant findings are described in Table 1.
 |
Figure 1 Overview of the pedigrees of families studied (A–E). Notice how females and males are equally involved and the autosomal dominant inheritance pattern with almost two successive generations affected. Family A had the highest number of generations developing multiple lipomas (a). The proband A (III 2) was the only within her family group and in the casuistry in referring pain when was examined (a). Arrows indicate the probands. |
 |
Figure 2 Clinical characteristics of FML and histopathologic aspects of lipomas (HE stain (10×). Primary findings include soft, mobile, and circumscribe painless nodules, located in subcutaneous fat. The most common topographical distribution comprises the upper limbs (A, B and D), anterior abdominal wall, back and thighs (C). Lipoma usually presents a lobulated pattern of white adipocytes with uniform nuclei and a thin fibrous capsule (E). Angiolipomas are composed of mature fat in association with numerous small blood vessels, that are predominantly capillaries. Fibrin thrombi (arrows) is very common (F). Images courtesy of The Department of Pathology and Medical Genetics, School of Medical Sciences, UNICAMP. |
Molecular
Sanger sequencing revealed two novel variants in exon 5 of the HMGA2 gene. Both variants were identified in patient A and B1. The first one was a synonymous heterozygous variant c.327C>T p.(Asp=) considered as low impact or neutral, based on several prediction tools (VEP, FATHMM, UMD-predictor, SIFT, PROVEAN). In contrast, the second alteration was a nonsense change heterozygous variant c.328T>C (p.*110Glnext*16) which causes a stop codon substitution by glutamine amino acid and the subsequent extension of final protein (https://www.hgvs.org/mutnomen/recs-prot.html). Figure 3 schematizes the structure of HMGA2 gene, the protein binding domains, and the novel variants.
 |
Figure 3 Structure of the human HMGA2 gene. (A) Located on 12q14.3, HMGA2 gene spans more than 140 kb and encompasses five exons. Exon 1, 2 and 3 (blue boxes) encode for an AT-hook domain (blue circles). The fourth exon (lilac) encodes for a spacer region. Exon 5 (pink box)encodes the C-terminal domain of the protein of 109 amino acids. (B) Electropherogram from patients A, B1, and reference. Two identical novel variants were found in these patients. A synonymous heterozygous variant c.327C>T (p.Asp=) and a no-stop change heterozygous variant c.328T>C (p.*110Glnext*16). |
The variant c.328T>C (p.*110Glnext*16) was defined as 100% pathogenic or high impact according to mentioned in silico algorithms. However, Mutation Taster prediction system demonstrated a discrepancy between the two variants described, as displayed in Table 3. The targeted multi-gene panel of AKT1, APC, MEN-1, PIK3CA, and PTEN in families A, B, C, D, and E, did not detect deleterious changes in sequences evaluated.
 |
Table 3 Variants Found in Individuals A and B1 and Comparison Among Prediction Tools |
Discussion
The current study set out to identify the molecular bases of FML through the integration of modern sequencing techniques such as Sanger sequencing and NGS combined with traditional clinical approaches.
Following previous studies, the present research shows some divergences regarding epidemiological data. We observed that there is a common misconception about the classification and diagnosis of different types of lipomatosis.7,10,15 Several authors emphasize that men have a high tendency to present multiple lipomas,11,29,30 however, our results show an apparent high frequency of FML in females. It is possible that this finding merely reflects a selection bias due to small sample size, or may also be explained by the fact that women are more likely to seek health medical care because of cosmetic concerns.31 Nevertheless, the pedigrees indicate that the proportion of individuals affected in both sexes was similar, leading to a sex ratio close to 1:1 (10 men vs 13 females) which may underestimate the true prevalence of FML.6,8,16 Interestingly, the total number of lipomas noted in women was higher compared to the unique male examined.1,14
Factors such as overweight and exacerbated growth of nodules during and after pregnancies suggest that exogenous factors (diet), metabolic changes (dyslipidemia or alterations in fatty acid desaturation) and hormonal mechanisms may be involved in adipocyte hyperplasia.32,33 Yee et al compared differences in enzymatic desaturation of stearoyl-CoA between individuals with rare adipose disorders (RAD) and obese control group and concluded that individuals with FML and obesity showed the highest rates of desaturation, thus increasing the lipogenesis process.32,34
One unexpected finding was that patient A was the only within the casuistic and her family group to manifest pain, daily weakness, fatigue, and psychiatric comorbidity (chronic depression and anxiety disorder). These symptoms are strongly associated with Dercum’s Disease, which is considered a differential diagnosis or a variant of FML.35 Based on the new Dercum’s Disease classification proposed by Hansson et al,36 we identified in this patient the generalized nodular form, characterized by intense pain on the surface of the fatty tissue and around lipomas. Another study that supports this hypothesis was published by Campen et al37 They described a family of nine members diagnosed with multiple lipomas who displayed variable symptoms ranged from total disability to asymptomatic nodules. It can, therefore, be assumed that the segregation follows an autosomal dominant pattern with variable expressivity and possibly represents an extreme manifestation of FML.
Concerning the histopathological diagnosis, we evidenced two main types: lipoma and angiolipoma. Microscopically, the most common noticeable feature of angiolipoma is the proliferation of fine blood vessels which contain fibrin microthrombi in association with mature adipocytes.10,28 Angiolipomas appear to have a high incidence in young adult men who occasionally manifest mild pain or tenderness. This type of tumor is also related to the history of local trauma or the use of steroid therapy.13,38 Although many authors recognize both diagnoses as different pathological entities, some case reports do not make this distinction due to very subtle differences between them.39 In this context, our analysis confirms that it is possible to have angiolipomas with familial recurrence and this diagnoses could be part of the broad spectrum of FML.40–43 Furthermore, multiple spindle cell lipomas, another type of lipoma, has also been reported in several families.44
Although there are several studies focused on expression and direct cytogenetic analysis on sporadic lipomas, few studies have been concerned about constitutional causes of FML.2 Extensive lipoma series in adults confirm multiple structural rearrangements with frequencies ranging from 50 to 75%.10,28,45 Among those balanced chromosomal alterations, translocation t(3; 12)(q27-28; q13-15) is observed in nearly 25% of tumors.9 In most cases, breakpoint affects the HMGA2 gene which is composed of 5 exons and generates a protein of 109 amino acids (Figure 3).18,46,47 Ligon et al reported the case of an eight-year-old boy with a constitutional rearrangement affecting the band 12q14.3 (de novo pericentric inversion) thus, involving HMGA2 gene. However, the clinical phenotype characterized by somatic overgrowth, the advancement of bone and dental age, cerebellar tumor, and multiple lipomas,48 differs from our casuistic.
We analyzed the heterozygous variants c.327C>T p.(Asp=) and c.328T>C (p.*110Glnext*16) of exon 5 of the HMGA2 gene in diverse prediction programs. The synonymous or silent alteration was interpreted as benign, whereas the change in the translation stop codon was considered as 100% pathogenic or high impact. In this way, it might indicate that changes involving the 3ʹ UTR (untranslated) region would trigger the neoplastic transformation of the HMGA2 gene.49 Fusco et al highlighted the crucial role of the HMGA2 gene in mesenchymal tumors. They proposed that in the HMGA2 gene, the 3ʹ UTR region is generally silenced by Let-7 miRNA and any alteration at this level, either truncation or fusion with ectopic sequences, drives to its up-regulation and consequently to an abnormal replication of cells.46
It is somewhat surprising that in family B we had detected both novel variants only in the mother (B1). It could indicate that they do not segregate with FML phenotype, being discarded at least in this family as an etiological cause. On the other hand, patient A presented the same two novel variants but demonstrated a phenotype highly suggestive of Dercum’s disease. Despite these promising findings, they are somewhat difficult to interpret because it was not possible to assess other relatives affected with FML.
Focal fatty infiltration and multiple lipomas may be present in several syndromes. Proteus syndrome and CLOVES (Congenital Lipomatous asymmetric Overgrowth, Vascular malformations, Epidermal nevi, Skeletal, and spinal anomalies) are examples of somatic mutations where lipomatosis is present.50,51 On the other hand, Bannayan-Riley-Rubacalva Syndrome, Multiple Endocrine Neoplasia type 1, Cowden syndrome and Gardner syndrome are conditions resulting from germline mutations that have concomitant lipomatosis.52–55 Even some mutations involving mitochondrial function are related to abnormal adipose tissue growth mostly in the upper trunk and neck and the best example is the Madelung Disease.56 However, the negative panel of AKT1, APC, MEN-1, PIK3CA, and PTEN excludes that they are responsible for the FML phenotype, at least in this research.
Conclusion
FML is a rare and heterogeneous disease that may be overlapping with other dermatological syndromes such as Dercum’s Disease. The natural history of FML in this series revealed that the development of lesions was progressive and reached its highest incidence in middle-aged individuals. FML affected both sexes and presented a topographical distribution with predominance in extremities and trunk. The sex ratio among evaluated patients showed a female prevalence, but pedigree data displayed a more balanced distribution. The diagnosis of Dercum’s Disease was also recognized in one individual (patient A).
The molecular approach has provided a more in-depth insight into the genetic basis of FML and has attempted to thoroughly investigate the largest number of candidate genes linked to it. We highlighted the utility of NGS in addition to Sanger method in the comprehension of the full spectrum of FML, due to it might offer an alternative for the etiology identification and characterization of rare genetic conditions in clinical settings. Variants in exon 5 of the HMGA2 gene have not been described, and until now, they have an uncertain significance in the genesis of FML. Further studies, including a more significant number of affected individuals and functional analysis of the novel variants of HGMA2 gene, should be undertaken to determine its biological function in FML better.
Abbreviations
FML, Familial Multiple Lipomatosis; HMGA2, High Mobility Group AT-hook 2 gene; NGS, next-generation sequencing; BMI, body mass index.; Tm, Melting temperature; bp, base pair.
Acknowledgments
The authors thank patients who provided clinical data and samples. The authors also acknowledge the Laboratory of Molecular Genetics and Genomic Medicine at the School of Medical Sciences of the University of Campinas, São Paulo, Brazil, and Genia® Laboratory – Blvr Artigas 922, Montevideo, Uruguay, for their technical assistance. This research was supported by The National Council for the Improvement of Higher Education (CAPES) grant No. 01P3368/2017.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Leffell DJ, Braverman IM. Familial multiple lipomatosis. Report of a case and a review of the literature. J Am Acad Dermatol. 1986;15(2 Pt 1):275–279. doi:10.1016/S0190-9622(86)70166-7
2. Lee C-H, Spence RAJ, Upadhyaya M, Morrison PJ. Familial multiple lipomatosis with clear autosomal dominant inheritance and onset in early adolescence. BMJ Case Rep. 2011;2011:2010–2012. doi:10.1136/bcr.10.2010.3395
3. BLASCHKO H. Eine seltene erbliche Lipombildung. Vircbows Arch Pathol Anat. 1891;124:175. doi:10.1007/BF01984919
4. Humphrey AA, Kingsley PC. Familial multiple lipomas: report on a family. Arch Derm Syphilol. 1938;37(1):30–34. doi:10.1001/archderm.1938.01480070033004
5. Kurzweg FT, Spencer R. Familial multiple lipomatosis. Am J Surg. 1951. doi:10.1056/NEJMicm1316241
6. Shanks JA, Paranchych W, Tuba J. Short communications: familial multiple lipomatosis. Can Med Assoc J. 1957;77:881–884.
7. Das Gupta T. Tumors and tumor-like conditions of the adipose tissue. Curr Probl Surg. 1970;7(3):1–60. doi:10.1016/S0011-3840(70)80010-7
8. Gologorsky D, Gologorsky Y, Yarygina AS, Surti U, Zirwas MJ. Familial multiple lipomatosis: report of a new family. Cutis. 2007;79(3):227–232.
9. Sandberg AA. Updates on the cytogenetics and molecular genetics of bone and soft tissue tumors: lipoma. Cancer Genet Cytogenet. 2004;150(2):93–115. doi:10.1016/j.cancergencyto.2003.12.018
10. Bancroft LW, Kransdorf MJ, Peterson JJ, O’Connor MI. Benign fatty tumors: classification, clinical course, imaging appearance, and treatment. Skeletal Radiol. 2006;35(10):719–733. doi:10.1007/s00256-006-0189-y
11. Ware R, Mane A, Saini S, Saini N. Familial multiple lipomatosis - a rare syndrome diagnosed on FNAC. Int J Med Sci Public Heal. 2016;5(2):367–369. doi:10.5455/ijmsph.2016.1007201547
12. Ronan SJ, Broderick T. Minimally invasive approach to familial multiple lipomatosis. Plast Reconstr Surg. 2000;106(4):878–880. doi:10.1097/00006534-200009020-00021
13. Weedon D. Section 7: tumors. In: Weedon’s Skin Pathology.
14. Medina CR, Schneider S, Mitra A, Spears J. Giant submental lipoma: case report and review of the literature. Can J Plast Surg. 2007;15(4):219–222. doi:10.1177/229255030701500405
15. Tadisina KK, Mlynek KS, Hwang LK, Riazi H, Papay FA, Zins JE. Syndromic lipomatosis of the head and neck: a review of the literature. Aesthetic Plast Surg. 2015;39(3):440–448. doi:10.1007/s00266-015-0478-8
16. Keskin D, Ezirmik N, Çelik H. Familial multiple lipomatosis. Isr Med Assoc J. 2002;4(12):1121–1123. doi:10.1056/NEJMicm1316241
17. Chirilã D, Gligor D. Familial multiple lipomatosis. Human & veterinary medicine. Int J Bioflux Soc. 2014;6:66–69. doi:10.1056/NEJMicm1316241
18. Cleynen I, Van de Ven WJM. The HMGA proteins: a myriad of functions (Review). Int J Oncol. 2008;32(2):289–305. doi:10.3892/ijo.32.2.289
19. Fedele M, Battista S, Manfioletti G, Croce CM, Giancotti V, Fusco A. Role of the high mobility group A proteins in human lipomas. Carcinogenesis. 2001;22(10):1583–1591. doi:10.1093/CARCIN/22.10.1583
20. Ozturk N, Singh I, Mehta A, Braun T, Barreto G. HMGA proteins as modulators of chromatin structure during transcriptional activation. Front Cell Dev Biol. 2014;2:1–9. doi:10.3389/fcell.2014.00005
21. Sambrook J, Fritsch EF, Maniatis T. Molecular Cloning: A Laboratory Manual. Cold Spring Harbor Laboratory Press; 1989.
22. McLaren W, Gil L, Hunt SE, et al. The ensembl variant effect predictor. Genome Biol. 2016;17(1):1–14. doi:10.1186/s13059-016-0974-4
23. Shihab HA, Gough J, Cooper DN, et al. Predicting the functional, molecular, and phenotypic consequences of amino acid substitutions using hidden markov models. Hum Mutat. 2013;34(1):57–65. doi:10.1002/humu.22225
24. Salgado D, Desvignes JP, Rai G, et al. UMD-predictor: a high-throughput sequencing compliant system for pathogenicity prediction of any human cDNA substitution. Hum Mutat. 2016;37(5):439–446. doi:10.1002/humu.22965
25. Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc. 2009;4(7):1073–1082. doi:10.1038/nprot.2009.86
26. Choi Y, Chan AP. PROVEAN web server: a tool to predict the functional effect of amino acid substitutions and indels. Bioinformatics. 2015;31(16):2745–2747. doi:10.1093/bioinformatics/btv195
27. Schwarz JM, Cooper DN, Schuelke M, Seelow D. Mutationtaster2: mutation prediction for the deep-sequencing age. Nat Methods. 2014;11(4):361–362. doi:10.1038/nmeth.2890
28. Fletcher CDM, Unni K, Mertens F. World Health Organization Classification of Tumours Pathology and Genetics of Tumours of Soft Tissue and Bone. Fletcher CDM, Unni KK, Mertens F, eds. Lyon, France: IARC Press International Agency for Research on Cancer (IARC) 69008 Lyon, France; 2002. doi:10.1016/j.suronc.2004.03.001
29. Paneru DP, Naik VA, Nilgar BR. Multiple lipomas diagnosed on cytology-A. Asian J Med Clin Sci. 2014;3(1):36–39.
30. Imerci A, Kaya A, Ilyas G, Surer L. Familial Multiple Lipomatosis: Report of Two Cases. Fırat Tıp Dergisi. 2012;17(4-1):17–20.
31. Frederick DA, Lever J, Peplau LA. Interest in cosmetic surgery and body image: views of men and women across the lifespan. Plast Reconstr Surg. 2007;120(5):1407–1415. doi:10.1097/01.prs.0000279375.26157.64
32. RUBINSTEIN A, GOOR Y, GAZIT E, CABILI S. Non‐symmetric subcutaneous lipomatosis associated with familial combined hyperlipidaemia. Br J Dermatol. 1989;120(5):689–694. doi:10.1111/j.1365-2133.1989.tb01357.x
33. Herbst KL. Rare adipose disorders (RADs) masquerading as obesity. Acta Pharmacol Sin. 2012;33(2):155–172. doi:10.1038/aps.2011.153
34. Yee JK, Phillips SA, Allamehzadeh K, Herbst KL. Subcutaneous adipose tissue fatty acid desaturation in adults with and without rare adipose disorders. Lipids Health Dis. 2012;11(19):11. doi:10.1186/1476-511X-11-19
35. Campen RB, Sang CN, Duncan LM. Case 25-2006 : a 41-year-old woman with painful subcutaneous nodules. N Engl J Med. 2006;355(7):714–722. doi:10.1056/NEJMcpc069018
36. Hansson E, Svensson H, Brorson H. Review of Dercum’s disease and proposal of diagnostic criteria, diagnostic methods, classification and management. Orphanet J Rare Dis. 2012;7(23):1–15. doi:10.1186/1750-1172-7-23
37. Campen R, Mankin H, Louis DN, Hirano M, MacCollin M. Familial occurrence of adiposis dolorosa. J Am Acad Dermatol. 2001;44(1):132–136. doi:10.1067/mjd.2001.110872
38. Sesthapongvanich K, Puangpet P. Familial multiple angiolipomatosis : a case report and review of the literature. 2011;27(3):193–197.
39. Kumar R, Pereira BJG, Sakhuja V, Chugh KS. Autosomal dominant inheritance in familial angiolipomatosis. Clin Genet. 1989;35(3):202–204. doi:10.1111/j.1399-0004.1989.tb02928.x
40. Cina SJ, Radentz SS, Smialek JE. A case of familial angiolipomatosis with Lisch nodules. Arch Pathol Lab Med. 1999;123(10):946–948. doi:10.1043/0003-9985(1999)123<0946:ACOFAW>2.0.CO;2
41. Hapnes SA, Boman H, Skeie SO. Familial angiolipomatosis. Clin Genet. 1980;17(3):202–208. doi:10.1111/j.1399-0004.1980.tb00133.x
42. Kanter WR, Wolfort FG. Multiple familial angiolipomatosis: treatment of liposuction. Ann Plast Surg. 1988;20(3):277–279. doi:10.1097/00000637-198803000-00018
43. Abbasi NR, Brownell I, Fangman W. Familial multiple angiolipomatosis. Dermatol Online J. 2007;13(1):12–13.
44. Fanburg-Smith J, Devaney K, Miettinen M, Weiss S. Multiple spindle cell lipomas: a report of 7 familial and 11 nonfamilial cases. Am J Surg Pathol. 1998;22(1):40–48. doi:10.1016/B978-1-4377-1422-7.00102-5
45. van de RM, Fletcher J. Genetics of soft tissue tumors. Annu Rev Pathol Mech Dis. 2006;1(1):435–466. doi:10.1146/annurev.pathol.1.110304.100052
46. Fusco A, Fedele M. Roles of HMGA proteins in cancer. Nat Rev Cancer. 2007;7(12):899–910. doi:10.1038/nrc2271
47. Kubo T, Matsui Y, Naka N, et al. Specificity of fusion genes in adipocytic tumors. Anticancer Res. 2010;30(2):661–664.
48. Ligon AH, Moore SDP, Parisi MA, et al. Constitutional rearrangement of the architectural factor HMGA2: a novel human phenotype including overgrowth and lipomas. Am J Hum Genet. 2005;76(2):340–348. doi:10.1086/427565
49. Bartuma H, Panagopoulos I, Collin A, et al. Expression levels of HMGA2 in adipocytic tumors correlate with morphologic and cytogenetic subgroups. Mol Cancer. 2009;8:1–11. doi:10.1186/1476-4598-8-36
50. Biesecker LG, Sapp JC. Proteus Syndrome. In: Adam MPA, Pagon HH, Wallace RA, Bean SE, Stephens LJH, Karen Amemiya AA, editors, Geneviews® [Internet]. Seattle:University of Washington, Seattle; 1993-2019. Available from: https://www.ncbi.nlm.nih.gov/books/NBK99495/. Accessed 13 December 2019.
51. Gopal B, Keshava SN, Selvaraj D. A rare newly described overgrowth syndrome with vascular malformations-Cloves syndrome. Indian J Radiol Imaging. 2015;25(1):71–73. doi:10.4103/0971-3026.150166
52. Pilarski R, Burt R, Kohlman W, Pho L, Shannon KM, Swisher E. Cowden syndrome and the PTEN hamartoma tumor syndrome: systematic review and revised diagnostic criteria. J Natl Cancer Inst. 2013;105(21):1607–1616. doi:10.1093/jnci/djt277
53. Eng C. PTEN hamartoma tumor syndrome. In: Adam MP, Ardinger HH, Pagon RA, et al. editors. GeneReviews® [Internet]. Seattle: University of Washington, Seattle; 1993-2019. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1488/. Accessed 13 December 2019.
54. Giusti F, Marini F, Brandi ML. Multiple endocrine neoplasia type 1. In: Adam MP, Ardinger HH, Pagon RA, et al. editors. GeneReviews® [Internet]. Seattle: University of Washington, Seattle; 1993–2019. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1538/. Accessed 13 December 2019.
55. Aihara H, Kumar N, Thompson CC. Diagnosis, surveillance, and treatment strategies for familial adenomatous polyposis: rationale and update. Eur J Gastroenterol Hepatol. 2014;26(3):255–262. doi:10.1097/MEG.0000000000000010
56. Enzi G, Busetto L, Cescin E, Coin A, Digito M, Pigozzo S. Multiple symmetric lipomatosis: clinical aspects and outcome in a long-term longitudinal study. Int J Obes. 2002;26(2):253–261. doi:10.1038/sj/ijo/0801867
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.