Back to Journals » Therapeutics and Clinical Risk Management » Volume 13
Clinical and genetic characteristics in a group of 45 patients with Turner syndrome (monocentric study)
Authors Bucerzan S, Miclea D, Popp R, Alkhzouz C, Lazea C ![]() , Pop IV, Grigorescu-Sido P
, Pop IV, Grigorescu-Sido P
Received 4 November 2016
Accepted for publication 20 March 2017
Published 4 May 2017 Volume 2017:13 Pages 613—622
DOI https://doi.org/10.2147/TCRM.S126301
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Garry Walsh
Simona Bucerzan,1 Diana Miclea,1,2 Radu Popp,2 Camelia Alkhzouz,1 Cecilia Lazea,1 Ioan Victor Pop,2 Paula Grigorescu-Sido1
1Emergency Hospital for Children, “Iuliu Hatieganu” University of Medicine and Pharmacy, Cluj-Napoca, Romania; 2Department of Molecular Sciences, “Iuliu Hatieganu” University of Medicine and Pharmacy, Cluj-Napoca, Romania
Introduction: Recent years have seen a shift in perspective on Turner syndrome, as it is no longer considered a significant disability due to therapeutic advances. The delay of diagnosis and the underdiagnosis are common in Turner syndrome, especially because of the great phenotypic variability and lack of firm diagnostic criteria.
Aim: Our first aim was to assess the clinical and the cytogenetic characteristics and growth rate in growth hormone (GH)-treated patients as compared to those with spontaneous growth. The second aim was to analyze the Y chromosomal sequences.
Materials and methods: We analyzed 45 patients diagnosed with Turner syndrome in Genetic Pathology Centre of Cluj Emergency Children’s Hospital. We carried out a study of the clinical features, the correlations between the karyotype and the phenotype, and we also made a research of Y chromosome sequences.
Results: The average age at diagnosis was 8.9±5.4 years. A significant association was observed between the number of external phenotypical abnormalities and internal malformations (r=0.45), particularly the cardiovascular ones (r=0.44). Patients treated with GH showed improvement in growth rate, with final stature significantly better than in untreated patients; benefits following treatment were greater if diagnosis was made before the age of 5 years. Thirteen percent of patients experienced spontaneous and complete puberty, whereas 30% experienced incomplete puberty. Patients with the 45,X genotype had a greater stature deficit and a higher incidence of cardiac malformations, compared with patients with 45,X/46,XX mosaic karyotype. Y chromosome sequences were found in only one patient, who subsequently underwent gonadectomy.
Conclusion: The importance of this study resides, to the best of our knowledge, in the fact that the largest group of patients in Romania was analyzed and assessed. To draw firm conclusions on the most valuable clinical indicators for Turner syndrome diagnosis in clinical practice, studies on large groups of patients should be conducted.
Keywords: Turner syndrome, diagnosis, phenotype, karyotype, GH treatment, Y chromosome sequences
Introduction
Recent years have seen a shift in perspective on Turner syndrome pathology, as it is no longer considered a significant disability due to therapeutic advances. At present, appropriate treatment with growth hormone (GH), initiated as early as 2 years of age, can lead to normal stature in adulthood. At the same time, the consequences of gonadal dysgenesis are mitigated by improved pharmacological feminization and by the possibility of procreating using either oocytes from a healthy donor or, as a more recent development, one’s own oocytes that have been cryopreserved at an early age.1–4 These effects and the implicitly higher quality of life that come with them are, however, dependent on an early diagnosis and the initiation of treatment at the optimal age.
Unfortunately, late diagnosis and underdiagnosis are common with Turner syndrome, the main reason for this being its considerable clinical variability. Less than 10% of patients present with the classical clinical picture, while the others have only a few signs or isolated short stature.5 Thus, it is well known that over 50% of patients with Turner syndrome remain undiagnosed.6
Although age at diagnosis has decreased from 15 to 16 years (2 decades ago) to 10 years,5,6 in order to obtain an optimal response to current therapeutic possibilities, the age at diagnosis should drop to 2–4 years.7–9 Use of the current diagnosis guidelines could bring improvements in this respect, although they still do not provide definitive diagnostic criteria.10,11
Another important aspect in the management of Turner syndrome is the need to evaluate the patient’s Y sequences using molecular techniques. Research has shown that about 5%–10% (or even 25%) of patients with Turner syndrome have fragments of the Y chromosome, often in mosaic, and usually not found following a standard karyotype.12 The Y chromosomal fragments are associated with a 30% risk of developing gonadoblastoma; consequently, prophylactic gonadectomy is mandatory.13,14 Although some studies support the need for research into the Y chromosome sequences of each patient with Turner syndrome, there is still no clear consensus regarding routine testing.12,15–17 If present, virilization may be an indication of the presence of Y chromosomal sequences, but most often the clinical picture does not suggest this.12
Given the above data, a retrospective study has been conducted on patients with Turner syndrome who were under observation at the Cluj Centre of Genetic Pathology. Our aim was to further assess the clinical characteristics and their prevalence in patients diagnosed at various ages, along with the cytogenetic characteristics, age when diagnosis was made, and growth rate in GH-treated patients as compared to those with only spontaneous growth. The retrospective study was completed with the assessment of the Y chromosomal sequences.
Materials and methods
This monocentric study evaluated 45 patients with Turner syndrome aged 1 month–16 years, who were diagnosed and followed-up at the Genetic Pathology Center of Cluj Emergency Children’s Hospital, Romania over a period of 20 years (1991–2010). The patients came from 14 regions, predominantly from the northern, western and central regions of Romania (Figure 1). The patients’ charts were completed by the attending physicians when the diagnosis was made and during case evolution, at every clinical evaluation. The charts contained data on auxological parameters (from birth to the last check-up), the clinical phenotype, imaging data (hand radiography, ultrasound, etc.), laboratory investigations (hormonal, biochemical and hematological), cytogenetic analysis and clinical data on GH treatment.
 | Figure 1 Patients’ distribution according to their geographic area of origin in Romania. |
Cytogenetic analysis
For cytogenetic analysis, peripheral blood was used as biological material. The culture was made using Lymphochrome culture medium (Lonza), while to obtain and band the chromosomes, previously described protocol stages were followed.18
Y chromosome analysis
Analysis of Y chromosome sequences was carried out in all patients, except those with Y chromosome found in the karyotype. The molecular analysis consisted of polymerase chain reaction (PCR) using four sets of primers corresponding to the SRY, PABY, DYS1 and DYZ3 regions. For each of the four sets of primers, PCR was performed in a total volume of 25 μL, containing 100 ng of genomic DNA, 1.5 mmol/L of MgCl2, 0.2 mmol/L of each dNTP, 20 pM of each primer and 0.5 U of Taq DNA polymerase.19 The amplification consisted of initial denaturation at 94°C (10′), 30 cycles of denaturation at 94°C (30′′), annealing at 60°C (30′′) and extension at 72°C (2′′). The electrophoresis of the PCR products was performed on 1% agarose gel, stained with ethidium bromide and the gel was then observed under UV light.
Statistical study and ethics
For the descriptive study of clinical features, we calculated the mean and standard deviation of the quantitative variables, while for the qualitative variables, we calculated the incidence. The correlation coefficient (r) was used to assess the correlation between clinical abnormalities, while the chi-squared test and Student’s t-test were used to assess the phenotype–karyotype associations. To analyze the role of age at therapeutic initiation on GH treatment effects, the patients were classified into three categories according to their age at diagnosis: 0–5, 6–10 and 11–16 years. For each patient, informed consent was obtained and the study was approved by the ethics committee of “Iuliu Hatieganu” University of Medicine and Pharmacy, Cluj-Napoca. The parents or legal guardians of the patients provided written informed consent. Parents of the children in the images provided written informed consent for the images to be published.
Results
Clinical characteristic in patients with Turner syndrome
All of the patients (100% of the study group) presented with a dysmorphic syndrome, whose characteristics are described in Figure 2.
 | Figure 2 The main craniofacial dysmorphic signs observed in our patients. |
Forty-one patients (91% of the group) presented with a malformation syndrome, consisting mainly of skeletal, cardiovascular and renal malformations. Their overall number and frequency, as well as the types of malformation within the three above-mentioned categories, are shown in Figure 3A–D. Skeletal abnormalities were found to be predominant (91%) as compared to cardiovascular (33%) and renourinary (29%) abnormalities.
 | Figure 3 (A) Frequency of the main malformations (n=45); (B) skeletal abnormalities; (C) cardiovascular malformations; (D) renal malformations. |
Diagnosis in patients with Turner syndrome
Mean age at diagnosis was 8.9±5.4 years. The main clinical characteristics that led to diagnosis for the age groups of <1, 1–10 and >10 years are shown in Figure 4.
 | Figure 4 The main clinical signs leading to diagnosis at different ages. |
When diagnosis was made from newborn period to the age of 1 year (n=10), the most frequent clinical sign was lymphedema (9 out of 10 patients). In four patients, it was isolated, whereas in another four it was associated with pterygium colli and in one patient it was associated with pterygium colli and coarctation of the aorta. The next most frequent sign was pterygium colli, which was found in six patients (isolated in only one case). This was followed by coarctation of the aorta, which was found in one patient.
In patients for whom the diagnosis was made between the ages of 1 and 10 years (n=14), the most common clinical sign was short stature (13 out of 14 patients); this was isolated or associated with the dysmorphic syndrome in four and nine patients, respectively.
Short stature was found in all the patients diagnosed after the age of 10 years (n=21), less commonly as isolated sign (3 patients), and frequently associated with delayed puberty, craniofacial dysmorphism or both (7, 7 and 4 patients, respectively).
Of the 45 patients of the study group, 23 (51%) had two clinical signs specific to Turner syndrome that are obvious in the newborn: pterygium colli and lymphedema located at the distal extremities of one or more limbs, which would have allowed a clinical diagnosis on the first day of life. Nevertheless, the diagnosis in these patients was made between 3.5 and 16 years (mean =10.6±3.5 years).
In the remaining 22 patients, the most suggestive clinical sign was short stature. However, the time interval between the moment when short stature became obvious (stature below −2 SD for age and sex) and the point when Turner syndrome was diagnosed was 7.7±4.5 years.
Auxological data
Auxological data are shown in Table 1, detailing: 1) the length and weight within normal range in newborns; 2) delayed growth at the diagnosis (SD =−2.79±2.15), with discordant predictable final height compared with genetic potential (SD =−1.92±1.28 vs −0.17±0.89) and 3) weight gain toward the upper limit for age and sex (SD =2.27±2.34).
 | Table 1 Auxology in patients with Turner syndrome |
Figure 5 shows height at diagnosis (expressed as SD) versus age at diagnosis for each of the 45 patients. The following findings are shown: a bimodal distribution of age at diagnosis, with a first peak in the first year of life (10 out of 45 patients =22%) and a second peak between 9 and 16 years of age (29 out of 45 patients =64%). Also, SD for stature was negative in 42 patients and positive in only three patients, all of whom were diagnosed in the first 3 months of life; short stature worsened as age advanced. In 23 out of 45 patients, SD for stature was less than −3 at diagnosis.
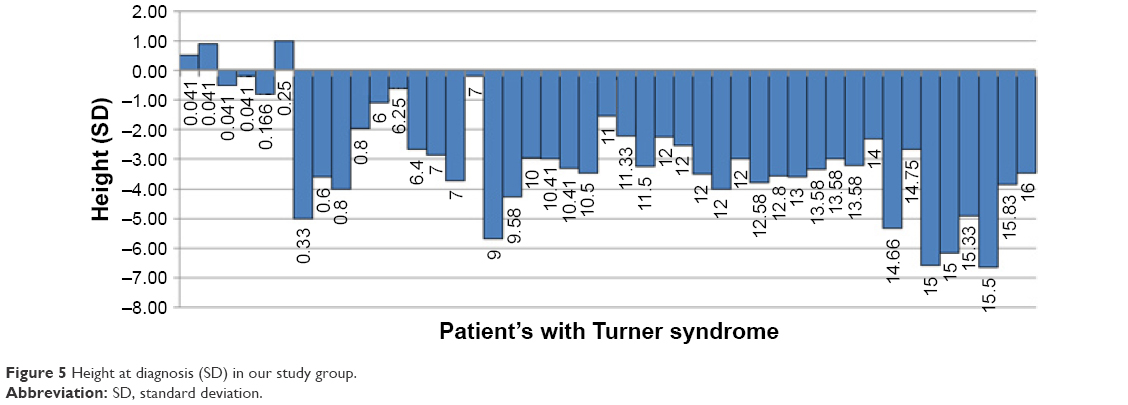 | Figure 5 Height at diagnosis (SD) in our study group. |
Correlations between clinical signs in patients with Turner syndrome
A significant association between the number of external phenotypical abnormalities and the internal malformations (r=0.45), particularly cardiovascular ones (r=0.44), was found. A larger number of external phenotypical abnormalities were also found to correlate with younger age at diagnosis (r=−0.35). The correlation was even more significant when pterygium colli and limbs lymphedema (r=−0.55) constituted the external phenotypical sign. Pterygium colli and lymphedema were found to be more frequently associated with cardiovascular malformations (r=0.50), while short stature (DS) correlated with younger age at diagnosis (r=−0.68).
Figure 6 (a 10-year-old patient) shows the classical phenotype of Turner syndrome. Pterigium colli, low posterior hairline, increased biacromial diameter, cubitus valgus, bilateral short 4th metatarsals (bilateral 4th brachymetatarsia) and hypostature were present but thelarche was not.
 | Figure 6 Classical phenotype of a patient (10 years old) with Turner syndrome. |
Effect of GH treatment
Treatment with GH was prescribed in 26 out of 45 patients (58% of the group). The treatment was initiated at various ages (between 4.7 and 15 years; mean =10.1±3.2 years), depending on the age at diagnosis. When assessing this group, GH treatment duration ranged between 0.3 and 4 years (mean =2.1±1.1 years). Height for patients who received GH treatment versus the rest of patients is shown in Figure 7. An almost equal value of height SD was found in the two categories when GH treatment was initiated (−3.2 vs −3.05; P=0.28); also, at the last assessment, an obvious improvement in the height SD was noticed in treated patients (after 2.1±1.1 years of treatment) versus its worsening in untreated patients (−2.9 vs −3.64; P=0.028) (Figure 7A). In addition, an almost double annual growth rate was found in treated patients (6.3±3.1 vs 3.6±1.9 cm/year; P=0.0007) (Figure 7B).
 | Figure 7 Stature growth in patients with Turner syndrome with/without GH treatment. |
Our analysis of the response to therapy, depending on the age at which GH treatment was initiated and the age at diagnosis, showed that only patients diagnosed in the first 5 years of life had a good response to therapy, such that their SD for stature fell within the normal range (−1.8±1.4 SD).
Puberty development in patients with Turner syndrome
When evaluating the 30 patients aged >12 years (the maximum age for the onset of puberty in girls) in terms of puberty retardation, the following was found (Figure 8): four patients with spontaneous and complete puberty (4 out of 30, 13%), two patients with a karyotype 45,X and another two with a karyotype 45,X/46,XX. The average age of menarche onset was 13.5 years. Of nine patients (9 out of 30, 30%), five had karyotype 45,X/46,XX and four had karyotype 45,X; they all showed incomplete spontaneous puberty, limited to Tanner II and III stages. These nine patients, along with those who showed no signs of puberty (26 patients altogether), required estroprogestatives and hormone replacement therapy.
 | Figure 8 Secondary sexual characteristics in patients with Turner syndrome, aged ≥12 years (n=30). |
Correlations karyotype–phenotype in patients with Turner syndrome
Thirty-three out of 45 patients (73%) had a 45,X karyotype and mosaicism 45,X/46,XX was found in 9 out of 45 patients (20%). The other three cases of mosaicism that we found (45,X/47,XXX; 45,X/46,Xiso (Xq); 45,X/46,XY) were present in one patient each (2%). We compared the influence of karyotypes 45,X versus 45,X/46,XX on phenotypical traits. No significant difference was found between patients with 45,X and those with 45,X/46,XX in terms of the majority of phenotypical signs. Significant differences were only seen for height at the time of diagnosis, namely −3.79±1.23 45 DS in patients with 45,X as compared to −2.23±1.9 DS in those with mosaicism 45,X/46,XX (P=0.002) and for cardiac malformations, which were present in 45% of cases with homogeneous monosomy X (15 out of 33 patients), but absent in patients with mosaicism 45,X/46,XX (P=0.03).
Y chromosome sequences in patients with Turner syndrome
Our research using PCR technology on the Y chromosome sequences of patients with Turner syndrome revealed only one positive patient (1 out of 44 patients, 2%). She was diagnosed with Turner syndrome at the age of 3 years due to the presence of a classical Turner phenotype, but with the absence of virilization or developmental abnormalities of the external genitalia; gonadectomy was performed at the age of 12 years, as recommended after the fragments of Y chromosome were found.
Discussion
Our study included patients with Turner syndrome from the central and north-western regions of the country, the study group being the largest one described in the national literature to date.
The age at diagnosis for the patients in this study (8.9±5.4 years) was similar to the value reported in literature and was consistent with the trend of a decrease in the age at diagnosis observed over the last few years.5 The lack of previous data on age at diagnosis in Romania prevented us from evaluating any probable progress, as was done in other recent studies. Considering all the benefits of early diagnosis, age at diagnosis could be improved. Studies have demonstrated that classical dysmorphic signs, which are most useful for diagnosis, are observed in 40% of cases with Turner syndrome at most.2,20–23 This is the main reason why underdiagnosis of Turner syndrome is an important issue, both in Romania and worldwide.6
External phenotypical abnormalities (Figure 2) were more commonly found in the chest, neck and limbs in our study, which was also consistent with the work of other authors.20–22 Occurring as a consequence of haploinsufficiency of the lymphogenic gene, pterygium colli or lymphedema were reported as having similar incidence in other studies, usually being found in 22%–36% of patients 20–22 versus 33% in our study. When these signs are present in a newborn with external female genitalia, diagnosis should be made relatively quickly, as these signs are believed to represent diagnostic markers that always involve a karyotype analysis. Regarding the craniofacial dysmorphic syndrome (Figure 2), the low occipital hairline was the most common sign found in our study, with 69% versus 42% in Sybert publication;20 it should, therefore, be routinely assessed in the clinical examination of a female patient, particularly if she shows hypostature.
Ninety-one percent of patients presented with a malformation syndrome (Figure 3), the most common being skeletal abnormality. Mention must be made that, in comparison to other studies, we found increased incidence of the broad shield-like chest deformity 91% versus 80%,23 and of short 4th and 5th metacarpals (bradymetacarpia), with 67% versus 37%.21 The proportion of patients with heart abnormalities (Figure 3C) (33%), especially those with a bicuspid aortic valve, was somewhat similar to that reported by other authors (40%),20,23–25 but the coarctation of the aorta was more rare. Among renal malformations noted in 29% of the cases investigated by ultrasound (Figure 3D), pyeloureteral abnormalities (pyeloureteral duplications) were found quite frequently (13%). This is not surprising, considering that they are known as the most common abnormalities in Turner syndrome.10,21,22 The horseshoe kidney malformation was less commonly found (4%), as compared to the studies of other authors (~10%).10,21
Regarding a possible association pattern of the various phenotypic abnormalities, our study found an association between pterygium colli and cardiac malformations, consistent with other publications.10,26 This observation can be explained by lymphatic abnormalities in patients with Turner syndrome, which underlie changes observed in both the neck and heart.27 The clinical characteristics discussed above are considered diagnostic criteria for Turner syndrome and are included in the current diagnostic guidelines. The purpose of analyzing them in this study was to assess their diagnostic weight. The guidelines proposed by Savendahl et al for the diagnosis of Turner syndrome recommended karyotype testing of any female with one or more of the following signs: short stature, pterygium colli, lymphedema in the limbs, coarctation of the aorta and delayed puberty.28 Massa et al have recommended cytogenetic analysis for any female presenting with hypostature (stature <−2 SD) or stature below the genetic growth potential, as shown by the target stature.5 The last international consensus on Turner syndrome (Turner Syndrome Consensus Study Group, 2007) recommended that the diagnosis of Turner syndrome be considered in any female with short stature and/or delayed puberty, knowing that these two signs are found in the majority of patients.10 However, just as many girls with Turner syndrome do not present with evident hypostature until later, toward puberty age; in these cases, diagnosis at an early age is often difficult.29
Short stature in patients who received GH treatment was significantly improved in comparison with those who did not receive the treatment (−2.9 SD vs −3.6 SD; P=0.029). An important effect of GH therapy was seen on annual growth rate of 6.3 cm versus 3.6 cm without treatment (P=0.0007), similar effect was also described by other authors.30 Age at diagnosis influenced stature prognosis when GH treatment had been given: postpuberty stature in patients diagnosed within the first 5 years of life became normal under GH treatment (−1.8±1.4 SD). These findings are consistent with those reported in the literature30 and support the need for early diagnosis in order to ensure optimal GH treatment.
Regarding puberty, complete spontaneous puberty was found in 13% of cases, of whom almost all were diagnosed after the age of 10 years. This observation can probably be explained by the clinical phenotype, including the gonadal phenotype that was less expressed and did not allow for an early diagnosis. Although this finding is consistent with other studies,31,32 it should be noted that in many of these patients, secondary amenorrhea will occur. Incomplete puberty was found in 30% of cases, often limited to Tanner stage II. Although the absolute number of patients who had complete or incomplete puberty with monosomy X or mosaicism 45,X/46,XX was approximately equal, the percentage relative to the total number of patients with a homogeneous karyotype or mosaicism in each subgroup showed that the probability for developing spontaneous puberty was greater when the patient had mosaicism 45,X/46,XX.
Some studies have reported correlations between karyotype and phenotype in Turner syndrome, particularly the association between homogeneous monosomy X a more severe phenotype than with other chromosomal formulas.2,6,10,21,22 In our study, these associations were not very obvious, with two exceptions, a more severe short stature at diagnosis and an increased incidence of cardiac malformations in homogeneous monosomy X. Massa et al reported a younger age at diagnosis (at least 1 year less) in patients with karyotype 45,X compared to other karyotypes;5 in our study, we found a slight difference, but it was not statistically significant. The lack of a clear correlation between karyotype and phenotype could be due to cytogenetic techniques that are based on standard methods and do not allow a proper assessment of low-level mosaicisms due to the limited number of cells analyzed.
Detection of Y chromosome sequences is important for the clinical management of Turner syndrome, given the known risk of ~30% for gonadoblastoma development.13,14 It is known that the gonadoblastoma could be developed even at the age of 4–6 years in patients with Turner syndrome and Y sequences13; therefore, an early diagnosis of Turner syndrome and at the same moment the testing for Y sequences are absolutely necessary. In this study, we found Y chromosome sequences in one patient (2%), which is similar to the data reported by other authors.33,34 However, some studies have found fragments of Y chromosome in as many as 60% of patients with Turner syndrome.35 Such differences could be explained by the selection process for the different study groups; those with higher percentages of Y chromosome sequences may include patients with chromosome marker or other cytogenetic abnormalities that may derive from the Y chromosome. The critical region in the development of gonadoblastoma (GBY – gonadoblastoma locus on Y) is thought to be located on the short arm of the Y chromosome.36 Gonadoblastoma is an in situ cancer of germ cells and can lead to invasive cancer, such as disgerminome (in 60% of cases), choriocarcinoma, embryonal carcinoma or yolk sac tumor.37 Thus, the identification of Y chromosome sequences requires a recommendation of gonadectomy and an early diagnosis of Y chromosome sequences is vital for a better prognosis. PCR-based techniques are sensitive and inexpensive methods for detecting Y chromosome sequences and therefore they should be recommended to each patient with Turner syndrome, even in the absence of virilization or of the marker chromosome, as they contribute to improved quality of life and survival.
Limitations
One limitation of our study is that the phenotype information may have been incomplete due to it being a retrospective study. Another limitation is that we evaluated already-diagnosed patients whose clinical pictures could have been more pronounced than those who remain undiagnosed. The importance of this study resides, to the best of our knowledge, in the fact that the largest group of patients in Romania was analyzed and assessed.
Conclusion
Our study described the clinical characteristics of a group of Romanian patients with Turner syndrome and the most important findings indicate that for a better diagnosis, it is important to make a good evaluation of lymphedema, pterigium colli and coarctation of the aorta in newborn because these were the signs which often led to diagnosis in infancy in our study and thus permit an early diagnosis; also the short stature and craniofacial dysmorphism in a girl led more often to diagnosis between 1 and 10 years, and short stature associated with puberty retardation and craniofacial dysmorphism are diagnosis markers after the age of 10 years. Also, we find that pubertal development could be normal in some cases with Turner syndrome, 13% of our patients experienced spontaneous and complete puberty, whereas 30% experienced incomplete puberty. We also show the importance to detect Y chromosome sequences at diagnosis for gonadoblastoma prophylaxis by gonadectomy. To draw firm conclusion on the most valuable clinical indicators for making the Turner syndrome diagnosis in clinical practice, studies on large groups of patients from other regions of the country should be conducted. Initiation of the National Register for Turner Syndrome is also of utmost importance.
Acknowledgments
Diana Miclea would like to thank the “Iuliu Hatieganu” University of Medicine and Pharmacy of Cluj-Napoca (Miclea Diana, research grant 4944/24/08.03.2016) for financial support of this article. Simona Bucerzan and Diana Miclea are co-first authors.
Disclosure
The authors report no conflicts of interest in this work.
References
Davenport ML. Growth hormone therapy in Turner syndrome. Pediatr Endocrinol Rev. 2012;9(2):723–724. | ||
Cabrol S. Syndrome de Turner. Ann Endocrinol. 2007;68:2–9. | ||
Nwosu EC, Brockbank A, Walkinshaw SA. A successful pregnancy in a Turner syndrome with oocyte donation. J Obstet Gynaecol. 2012;32(5):419–421. | ||
Hovatta O. Ovarian function and in vitro fertilization (IVF) in Turner syndrome. Pediatr Endocrinol Rev. 2012;9(2):713–717. | ||
Massa G, Verlinde F, De Schepper J, et al. Trends in age at diagnosis of Turner syndrome. Arch Dis Child. 2005;90(3):267–268. | ||
Stochholm K, Juul S, Juel K, Naeraa RW, Gravholt CH. Prevalence, Incidence, Diagnostic Delay, and Mortality in Turner Syndrome. J Clin Endocrinol Metab. 2006;91(10):3897–3902. | ||
Hughes IP, Choong CS, Harris M, et al. Growth hormone treatment for Turner syndrome in Australia reveals that younger age and increased dose interact to improve response. Clin Endocrinol (Oxt). 2011;74(4):473–480. | ||
Davenport ML, Crowe BJ, Travers SH, et al. Growth hormone treatment of early growth failure in toddlers with Turner syndrome: a randomized, controlled, multicenter trial. J Clin Endocrinol Metab. 2007;92(9):3406–3416. | ||
Backeljauw P. Does growth hormone therapy before 4 years of age enhance the linear growth of girls with Turner’s syndrome? Nat Clin Pract Endocrinol Metab. 2008;4(2):78–79. | ||
Bondy CA; Turner Syndrome Study Group. Care of girls and women with Turner syndrome: a guideline of the Turner Syndrome Study Group. J Clin Endocrinol Metab. 2007;92(1):10–25. | ||
Davenport ML, Punyasavatsut N, Stewart PW, Gunther DF, Sävendahl L, Sybert VP. Growth failure in early life: an important manifestation of Turner syndrome. Horm Res. 2002;57(5–6):157–164. | ||
Miclea DL, Grigorescu-Sido P, Bucerzan S, Pop IV. Y chromosome in Turner syndrome and FISH technique usefulness in cytogenetic diagnosis. Rev Rom Med Lab. 2012;20(4):327–333. | ||
Manuel M, Katayama PK, Jones HWJ. The age of occurrence of gonadal tumors in intersex patients with a Y chromosome. Am J Obstet Gynecol. 1976;124(3):293–300. | ||
Verp MS, Simpson JL. Abnormal sexual differentiation and neoplasia. Cancer Genet Cytogenet. 1987;25(2):191–218. | ||
Reena Rahayu MZ, Sharifah NA, Zubaidah Z, et al. Identification of Y chromosomal material in Turner syndrome by fluorescence in situ hybridisation (FISH). Med Health. 2008;3(1):22–29. | ||
Nazmy NA. Detection of low level sex chromosome mosaicism in Turner syndrome by FISH. Bull Alex Fac Med. 2009;45(1):215–220. | ||
Wiktor AE, Van Dyke DL. Detection of low level sex chromosome mosaicism in Ullrich-Turner syndrome patients. Am J Med Genet A. 2005;138(3):259–261. | ||
Rondón Lagos SM, Rangel Jiménez NE. Cytogenetic analysis of primary cultures and cell lines: generalities, applications and protocols. In: Tirunilai P, editor Recent Trends in Cytogenetic Studies – Methodologies and Applications. Rijeka: Intechopen; 2012:1–24. | ||
Bashamboo A, Rahman MM, Prasad A, Padinjarel Chandy S, Ahmad J, Ali S. Fate of SRY, PABY, DYS1, DYZ3 and DYZ1 loci in Indian patients harbouring sex chromosomal anomalies. Mol Hum Reprod. 2005;11(2):117–127. | ||
Sybert VP, McCauley E. Turner’s syndrome. N Engl J Med. 2004;351(12):1227–1238. | ||
Elsheikh M, Dunger DB, Conway GS, Wass JAH. Turner’s syndrome in adulthood. Endocr Rev. 2002;23(1):120–140. | ||
Gravholt CH. Epidemiological, endocrine and metabolic features in Turner syndrome. Eur J Endocrinol. 2004;151(6):657–687. | ||
Saenger P. Turner’s syndrome. N Engl J Med. 1996;335(23):1749–1754. | ||
Cramer JW, Bartz PJ, Simpson PM, Zangwill SD. The spectrum of congenital heart disease and outcomes after surgical repair among children with Turner syndrome: a single-center review. Pediatr Cardiol. 2014;35(2):253–260. | ||
Gotzsche C, Krag-Olsen B. Prevalence of cardiovascular malformations and association with karyotypes in Turner’s syndrome. Arch Dis Child. 1994;71(5):433–436. | ||
Loscalzo ML, Van PL, Ho VB, et al. Association between fetal lymphedema and congenital cardiovascular defects in Turner syndrome. Pediatrics. 2005;115(3):732–735. | ||
Bondy CA, Cheng C. Monosomy for the X chromosome. Chromosome Res. 2009;17:649–658. | ||
Savendahl L, Davenport ML. Delayed diagnoses of Turner’s syndrome: proposed guidelines for change. J Pediatr. 2000;137(4):455–459. | ||
Even L, Cohen A, Marbach N, et al. Longitudinal analysis of growth over the first 3 years of life in Turner’s syndrome. J Pediatr. 2000;137(4):460–464. | ||
Sas TC, de Muinck Keizer-Schrama SM, Stijnen T, et al. Normalization of height in girls with Turner syndrome after long-term growth hormone treatment: results of a randomized dose-response trial. J Clin Endocrinol Metab. 1999;84(12):4607–4612. | ||
Pasquino AM, Passeri F, Pucarelli I, Segni M, Municchi G. Spontaneous pubertal development in Turner’s syndrome. Italian Study Group for Turner’s Syndrome. J Clin Endocrinol Metab. 1997;82(6):1810–1813. | ||
Mohamed S, Roche EF, Hoey HM. High Prevalence of Spontaneous Puberty in Patients with Turner Syndrome in Tertiary Referral Center in Ireland. Acta Endo (Buc). 2015;11(1):60–63. | ||
Fernandes S, Ventura V, Doria S, Barros A. Y-Chromosome Detection in Turner Syndrome. Human Genet Embryol. 2013;3(3):1–4. | ||
Sallai A, Sólyom J, Dobos M, et al. Y-chromosome markers in Turner syndrome: screening of 130 patients. J Endocrinol Invest. 2010;33(4):222–227. | ||
Bianco B, Lipay M, Guedes A, Oliveira K, Verreschi IT. SRY gene increases the risk of developing gonadoblastoma and/or nontumoral gonadal lesions in turner syndrome. Int J Gynecol Pathol. 2009;28(2):197–202. | ||
Tsuchiya K, Reijo R, Page DC, Disteche CM. Gonadoblastoma: molecular definition of the susceptibility region on the Y chromosome. Am J Hum Genet. 1995;57(6):1400–1407. | ||
Looijenga LH, Stoop H, de Leeuw HP, et al. POU5F1 (OCT3/4) identifies cells with pluripotent potential in human germ cell tumors. Cancer Res. 2003;63(9):2244–2250. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.