Back to Journals » Cancer Management and Research » Volume 11
CIRP regulates BEV-induced cell migration in gliomas
Authors Liu Y ![]() , Zhou JN, Liu K, Fu X, Zhang Z
, Zhou JN, Liu K, Fu X, Zhang Z ![]() , Zhang QH, Yue W
, Zhang QH, Yue W
Received 17 October 2018
Accepted for publication 17 January 2019
Published 11 March 2019 Volume 2019:11 Pages 2015—2025
DOI https://doi.org/10.2147/CMAR.S191249
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Antonella D'Anneo
Yu-Xiao Liu, 1,* Jun-Nian Zhou, 2– 4,* Ke-Hui Liu, 5,* Xiang-pin Fu, 1 Zhi-Wen Zhang, 1 Qin-Hong Zhang, 1 Wen Yue 2
1Department of Neurosurgery, The Fourth Medical Centre of Chinese PLA General Hospital, Beijing 100048, China; 2Stem Cell and Regenerative Medicine Lab, Institute of Health Service and Transfusion Medicine, Beijing 100850, China; 3Experimental Hematology and Biochemistry Lab, Beijing Institute of Radiation Medicine, Beijing 100850, China; 4South China Research Center for Stem Cell & Regenerative Medicine, SCIB, Guangzhou 510005, China; 5State Key Laboratory of Membrane Biology, Institute of Zoology, Chinese Academy of Sciences, Beijing 100101, China
*These authors contributed equally to this work
Purpose: A better understanding of the underlying molecular mechanisms in treatment failure of bevacizumab (BEV) for malignant glioma would contribute to overcome therapeutic resistance.
Methods: Here, we used a quantitative proteomic method to identify molecular signatures of glioblastoma cell after BEV treatment by two-dimensional liquid chromatography-tandem mass spectrometry analysis and 6-plex iTRAQ quantification. Next, the function of cold-inducible RNA-binding protein (CIRP), one of the most significantly affected proteins by drug treatment, was evaluated in drug resistance of glioma cells by invasion assays and animal xenograft assays. Target molecules bound by CIRP were determined using RNA-binding protein immunoprecipitation and microarray analysis. Then, these mRNAs were identified by quantitative real-time PCR.
Results: Eighty-seven proteins were identified with significant fold changes. The biological functional analysis indicated that most of the proteins were involved in the process of cellular signal transduction, cell adhesion, and protein transport. The expression of CIRP greatly decreased after BEV treatment, and ectopic expression of CIRP abolished cell migration in BEV-treated glioma cells. In addition, CIRP could bind mRNA of CXCL12 and inhibit BEV-induced increase of CXCL12 in glioma cells.
Conclusion: These data suggested that CIRP may take part in BEV-induced migration of gliomas by binding of migration-relative RNAs.
Keywords: therapeutic resistance, proteomics, RNA binding, CXCL12
Corrigendum for this paper has been published
Introduction
Glioblastoma multiforme (GBM) was an aggressive and lethal brain cancer. A series of studies pointed out that angiogenesis was the typical hallmark of GBM tumors, and vascular endothelial growth factor (VEGF) was the most critical molecule involved in controlling the complex process of angiogenesis in GBM.1–3 So, bevacizumab (BEV), a recombinant humanized monoclonal antibody to VEGF, was regarded as a successful treatment for recurrent GBM.4–6
However, it showed that the benefits of angiogenesis inhibitors were typically transient and the tumors eventually became resistant to the therapy. Kunkel et al demonstrated that glioma xenografts adopt a more infiltrative and invasive growth pattern after treatment with anti-VEGF or anti-VEGFR antibodies.7 Lucio-Eterovic et al reported that GBM tumors escaped from antiangiogenic treatment through upregulation of other proangiogenic factors, especially the matrix metalloproteinase family members.8 However, the exact mechanism and the relative mediators of tumor invasion were currently unknown. Thus, it was an urgent need for the exploration of underlying mechanisms of the drug resistance.
Proteomic technology was a useful tool to discover the new function of protein in specific pathological activity. Recently, proteomic methods were used for the analysis of variety of central nervous system diseases, including Alzheimer’s disease, Parkinson’s disease, and glioma.9–11 In this study, we used a quantitative proteomic analysis to comprehensively analyze the protein profiling of BEV-resistant GBM cells. Protein changes were measured in glioma cell lines after anti-VEGF treatment. Cold-inducible RNA-binding protein (CIRP), a significantly changed protein, was selected for further analysis using invasion assays, animal xenograft assays, and RNA-binding protein immunoprecipitation (RIP) assays. These results first proved that CIRP was an important mediator in BEV-induced resistance of GBM by binding some migration-relative RNAs.
Methods
Cell culture and treatment
Human GBM cell line U87 and U251 cells were purchased from the Cell Bank of the Chinese Academy of Sciences (Beijing, People’s Republic of China) and maintained in Dulbecco’s modified Eagle’s medium containing 10% fetal bovine serum, at 37°C in 5% CO2 atmosphere. For cell treatment, BEV was added at the concentrations indicated.
LC-MS/MS analysis
After treatment with BEV (2.5 mg/mL) for 48 hours, untreated or BEV-treated U251 cells were collected. A filter-aided sample preparation method was used to digest the proteins in samples. For MS analysis, the peptides were resuspended in 0.1% formic acid and analyzed by an LTQ Orbitrap Elite Mass Spectrometer (Thermo Scientific, Waltham, MA, USA) coupled online to an Easy-nLC 1000 in the data-dependent mode. All MS measurements were performed in the positive ion mode and acquired across the mass range of 300–1,800 m/z. The 15 most intense ions from each MS scan were isolated and fragmented by high-energy collisional dissociation. Raw mass spectrometric files were analyzed using the software MaxQuant (version 1.5.3.28).
Western blot analysis
Untreated or BEV-treated U251 and U87 cells were collected at different time point after BEV treatment. Cells were lysed directly in lysis buffer to collect whole-cell extracts. Protein samples were separated on polyacrylamide gels, transferred onto nitrocellulose membrane by iBlot (Invitrogen), and detected using horseradish-peroxidase-conjugated secondary antibodies and chemiluminescence (Santa Cruz) exposure of BioMax film (Kodak). The following antibodies were used: anti-CIRP (Santa Cruz) and anti-β-actin (Santa Cruz).
Plasmid construct and cell transfections
Human CIRP cDNA was subcloned from U251 or U87 cells and inserted into the lentiviral vector, which carried GFP and/or luciferase. Subsequently, lentiviral particles were produced to transfect the cells.
Mouse tumor model
All animal experiments were approved by the Ethical Committee of Chinese PLA General Hospital (Beijing, China). The procedures in this study were conducted in accordance with the guidelines for the use of experimental animals from the National Institutes of Health.
Human glioma cells (1×106 cells in 5 µL PBS) were implanted into the brains of anesthetized athymic nu/nu mice: a median incision of ~1 cm was made, a burr hole was drilled into the skull, and cells were injected into the right striatum.
Immunofluorescence staining of CIRP in vitro
U251 cells and U87 cells were implanted into the brains of anesthetized athymic nu/nu mice. Six weeks after BEV treatment, tumors from different groups were removed and fixed with formaldehyde, embedded in paraffin wax, and sectioned. Sections were incubated with mouse anti-CIRP monoclonal antibody (Santa Cruz) at 4°C overnight, and then incubated with antimouse IgG secondary antibody for 1 hour. Images were acquired using a microscope. Positive cells per field were calculated in 10 high power microscopic fields.
Wound healing and transwell invasion assays
U251 or U87 cells were divided into three groups: 1) untreated group: cells were not treated with BEV; 2) BEV-treated group: cells were treated with BEV for 48 hours; 3) BEV-treated + CIRP-overexpressed group: cells that overexpressed CIRP were treated with BEV for 48 hours.
For wound healing assay, cells from different groups were trypsinized and plated into 6-well plates at high densities. As the cells reached a high confluency (>90%), a wound was made across the cells with a micropipette tip. The photographs were taken immediately (time zero) and 36 hours after wounding. The distance migrated by the cell monolayer to close the wounded area during this time was measured by ImageJ software.
For transwell assay, cells from different groups were plated into transwell chambers with or without matrix gel (Corning Incorporated, Corning, NY, USA), and 5% FBS was added in the lower chamber as the chemoattractant. After incubation for 24 hours, the cells that attached to the lower surface were fixed in 100% methanol for 2 minutes and stained with DAPI (Invitrogen). The number of invasion cells on the lower surface of the membrane was calculated in microscopic fields. Images were acquired using a microscope.
Bioluminescence imaging (BLI) in vivo
U251 cells with the expression vector of CIRP-luciferase or control vector were implanted into the brains of anesthetized athymic nu/nu mice. Tumors in mice of different groups were measured by BLI signals.
BLI was performed in vivo using NightOWL LB 983 In Vivo Imaging System to assess tumor growth and calculate tumor volumes. The image with the peak BLI intensity was used for quantification in units of photon counts.
Histology and immunohistochemistry staining
U251 cells with the expression vector of CIRP or control vector were implanted into the brains of anesthetized athymic nu/nu mice. Tumors from different groups were removed and fixed with formaldehyde, embedded in paraffin wax, and sectioned. The sections were stained with hematoxylin and eosin or incubated with mouse anti-CXCL12 monoclonal antibody (Santa Cruz) at 4°C overnight, and then incubated with antimouse IgG secondary antibody for 1 hour. Images were acquired using a microscope. Positive cells per field were calculated in 10 high power microscopic fields.
RIP assay
U251 cells were teased and resuspended in complete RIP lysis buffer. Anti-CIRP antibody and normal rabbit IgG (Millipore, Billerica, MA, USA) were used in RIP assays and defined as RIP-CIRP group and RIP-IgG group (negative control). The antibodies were incubated with Protein A/G Magnetic Beads for 30 minutes, and the beads–antibody complexes were incubated with complete RIP Lysis Buffer at 4°C for 10 hours. Fifty microliters each out of 500 µL of the bead suspension during the last wash were removed to test the efficiency of immunoprecipitation by Western blotting. Target RNAs were purified with procedures of proteinase K digestion and phenol–chloroform–isoamyl alcohol (Sigma-Aldrich) extraction, then precipitated in ethanol, and resuspended in 10 µL of RNase-free water.
Microarray assay analysis
Target RNAs bound by CIRP in U251 cells were isolated by RIP, then microarray hybridization was performed according to the standard procedure by CapitalBio Corporation (Beijing, China) using Agilent human lncRNA + mRNA Array version 4.0.
The lncRNA + mRNA array data summarization, normalization, and quality control were analyzed by using the GeneSpring software version 12.0 (Agilent Technologies).
Real-time PCR
Target mRNAs isolated from RIP assay using CIRP antibody (CIRP antibody group), IgG (IgG group), or water (input group) were identified by real-time PCR. RNAs were reverse-transcribed to cDNA and detected according to manufacturer’s protocol (Takara, Otsu, Shiga, Japan). They were examined with real-time PCR using the SYBR Green PCR Master Mix. The forward and reverse primers used are described in Table 1.
 | Table 1 The primer sequences for PCR |
Statistical analysis
Statistical analyses were performed using SPSS software 13.0. All data were presented with mean ± SD. Statistical significance was determined by using Student’s t-test. Results were considered significant at P<0.05.
Results
Identification of proteins with significant fold changes in U251 cells after BEV treatment
For the quantitative proteomics, three biological replicates of untreated and BEV-treated U251 cells were trypsinized and labeled with 6-plex TMT reagents. An average CV of 0.37 was employed to filter out data with poor linearity, corresponding to coverage of >80% of quantified proteins in two groups to maintain a low false-positive rate of comparative analysis (Figure 1A). Eighty-seven proteins with significant fold changes were obtained for further analysis by comparing the total proteomes of cells from untreated or BEV-treated groups (a threshold of ≥2-fold and P<0.05). Among these proteins, 53 proteins were upregulated and 34 were downregulated in treated cells compared with untreated controls (Figure 1B). Most of the proteins changed after BEV treatment were classified as receptors, protein-binding molecules, and cell adhesion molecules (Figure 1C and D), which implied that drug treatment might alter the interaction between cancer cells and their microenvironment. Most of these proteins were also categorized as components of cytoskeleton and lysosome or presented as cytoplasmic and nuclear as shown in Figure 1E, which suggested that BEV treatment could significantly affect the structure of glioma cells.
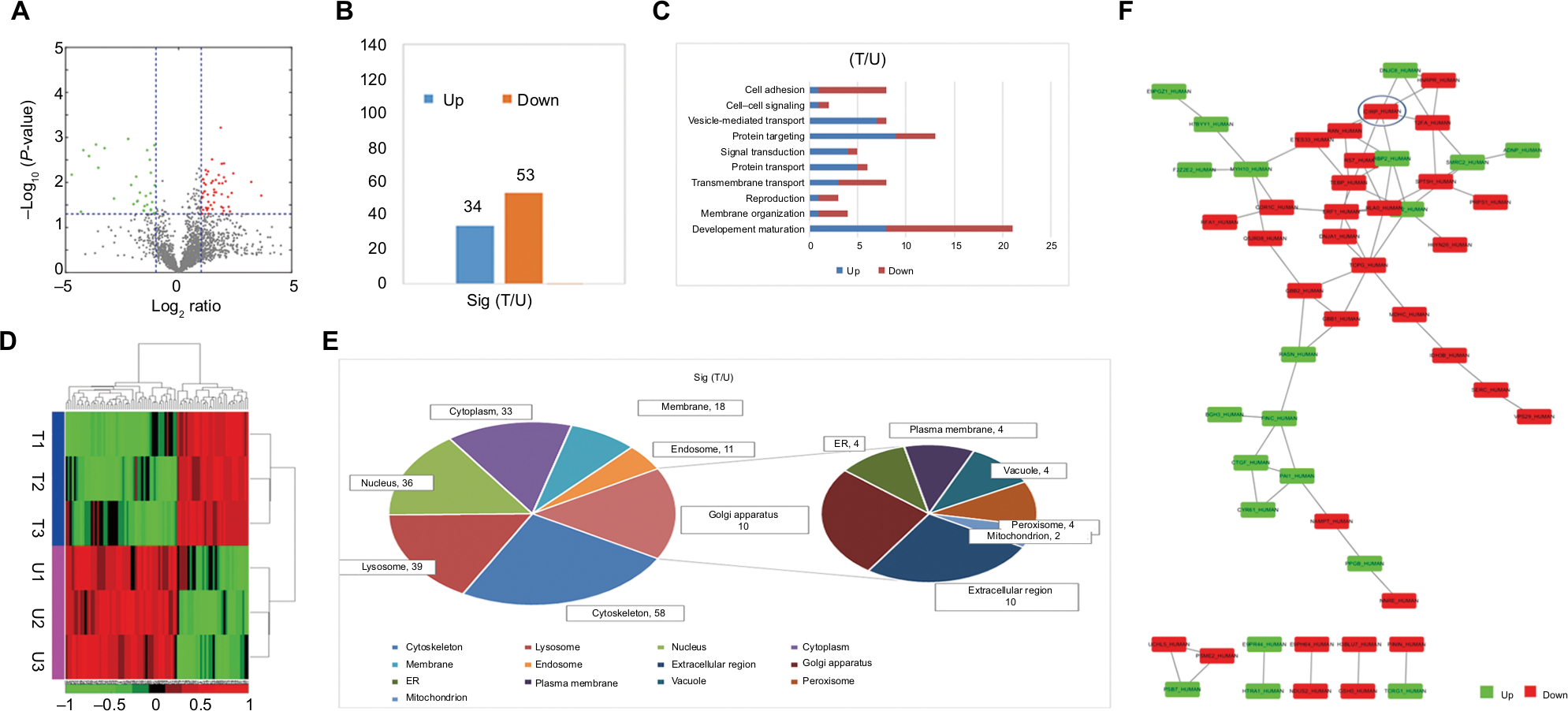 | Figure 1 Functional classification of changed proteins. Notes: (A) Volcano plots of identified proteins changed after BEV treatment in U251 cells. (B) Eighty-seven proteins with significant changes after BEV treatment were identified. (C) Enriched biological functions in which changed proteins are involved. (D) Heatmap showing protein expression profiles in U251 cells 48 hours after BEV treatment (n=3). (E) Subcellular location analysis of proteins identified after treatment in U251 cells. (F) Protein–protein interaction networks were identified in proteins with significant changes after treatment and CIRP was screened by IPA. Abbreviations: BEV, bevacizumab; CIRP, cold-inducible RNA-binding protein; ER, endoplasmic reticulum; IPA, Ingenuity Pathway Analysis; T, BEV treatment group; U, untreated group. |
The results from proteomic measurements also showed that the protein expression of CIRP in U251 cells was 3.85-fold that in BEV-treated U251 cells. The results of Ingenuity Pathway Analysis indicated that the protein synthesis network, cell cycle, and mRNA stabilization were mostly affected. More importantly, CIRP acted as one of the keypoint proteins and had interacted with several factors such as T2FA, HNRPR, and DNJC8 (Figure 1F).
CIRP was downregulated after BEV treatment in vitro and in vivo
Therefore, CIRP was selected for further functional analysis. Two glioma cell lines (U87 and U251 glioma cells, which both release high levels of VEGF) were used for BEV treatment. We measured the expression levels of CIRP in these cells after BEV treatment by real-time PCR and Western blotting, respectively. Our results showed that CIRP was downregulated in both GBM cell lines after BEV treatment at the mRNA level and protein level in vitro (Figure 2A and B).
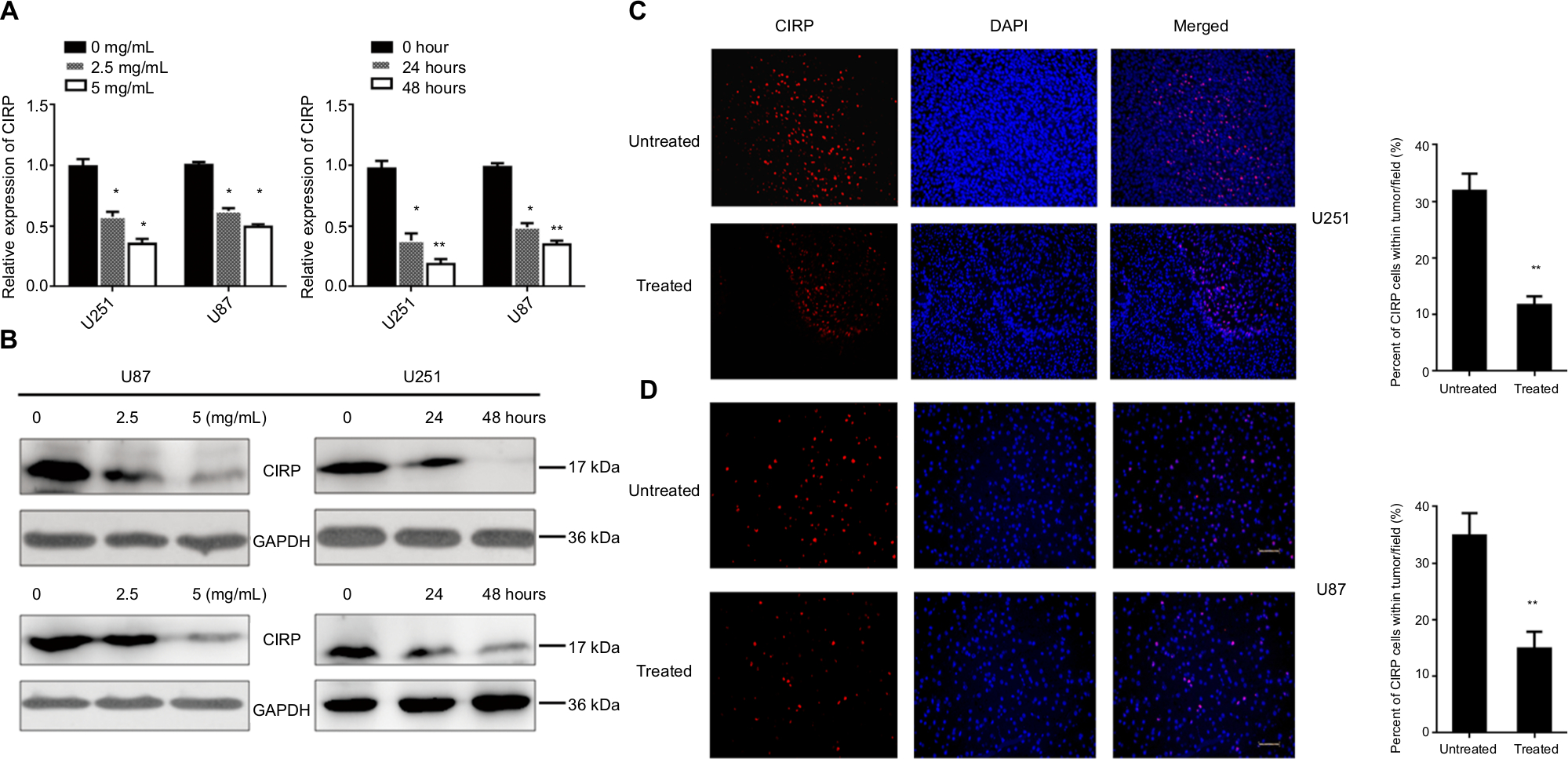 | Figure 2 CIRP was downregulated after BEV treatment in vitro and in vivo. Notes: (A, B) The mRNA and protein expressions of CIRP in U251 cells and U87 cells before and after BEV treatment were analyzed by real-time PCR and Western blot. (C, D) Representative images (left) and quantification (right) of protein levels of CIRP in tumors. U251 cells and U87 cells were implanted into the brains of anesthetized athymic nu/nu mice. Protein levels of CIRP in tumors from the mice were analyzed by immunofluorescence staining, 6 weeks after drug treatment. Untreated: without treated with BEV; treated: treated with BEV. Data represent mean ± SD. *P<0.05, **P<0.01, compared with untreated group. Abbreviations: BEV, bevacizumab; CIRP, cold-inducible RNA-binding protein. |
Next, glioma xenograft mouse models were established using U87 and U251 cells to determine the effect of BEV treatment on the expression of CIRP in vivo. The results confirmed that the expression of CIRP decreased after drug treatment in vivo (Figure 2C and D).
CIRP could inhibit BEV-induced migration in vitro and in vivo
Wound healing assays were conducted to investigate whether CIRP was involved in BEV-induced migration. After treated with BEV (2.5 mg/mL) for 48 hours, both U87 cells and U251 cells immediately closed the scratch wounds compared with the untreated control cells. However, CIRP overexpression could significantly inhibit BEV-induced migration of glioma cells (Figure 3A and B). Furthermore, we confirmed that ectopic expression of CIRP significantly suppressed BEV-induced migration and invasion of malignant glioma cells by transwell assays (Figure 4A and B).
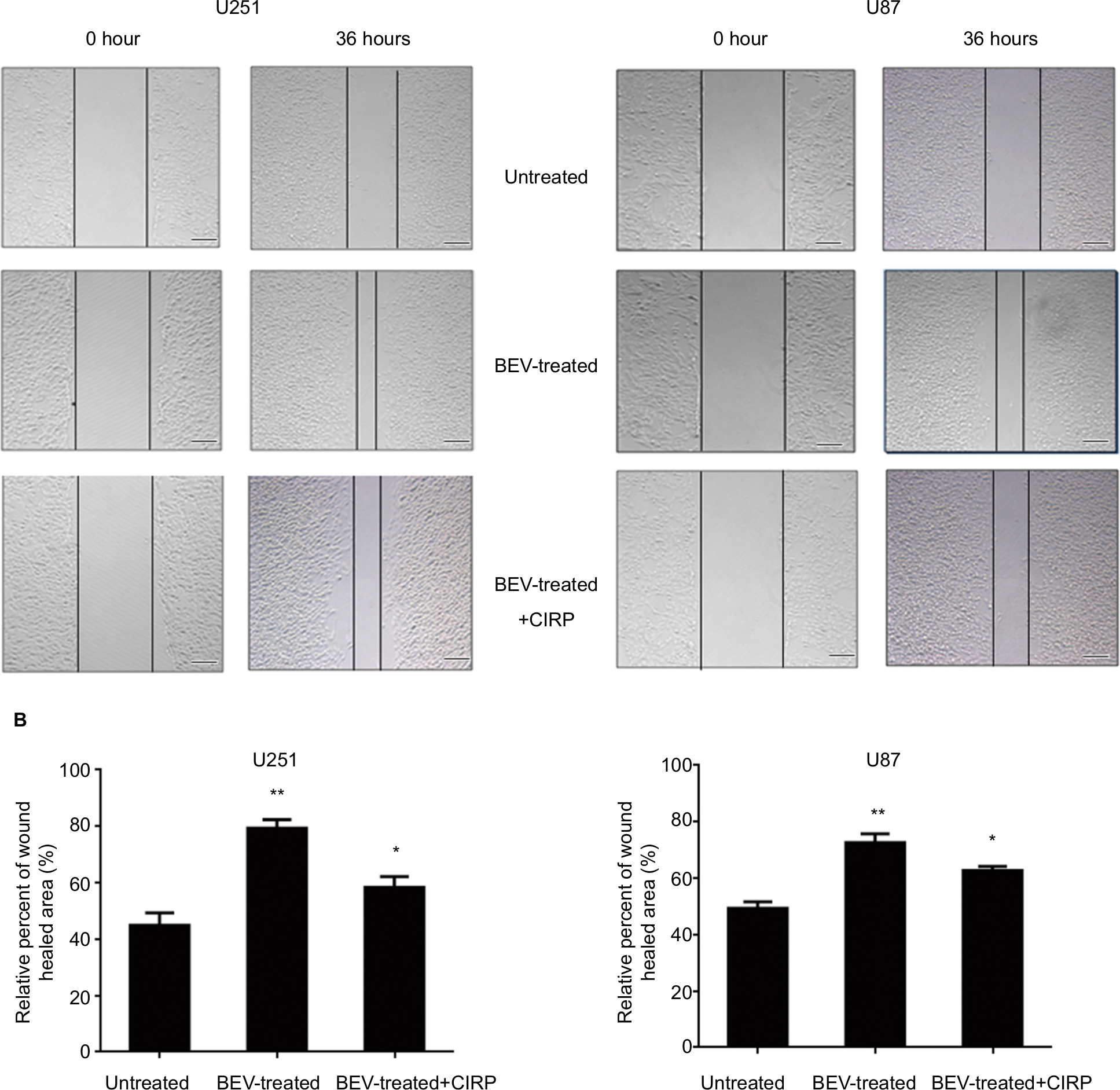 | Figure 3 Cell scratch assays of human glioma cells. Notes: (A) Typical bright field images of wound closures. (B) The statistical wound closure results. The migration capacity of U87 cells or U251 cells from different groups (untreated, BEV-treated, and BEV-treated + CIRP) was tested by scratch wound healing assays. Cell migration was quantified as a percentage of wound healed area. Data represent mean ± SD. *P<0.05, **P<0.01, compared with untreated group. Abbreviations: BEV, bevacizumab; CIRP, cold-inducible RNA-binding protein. |
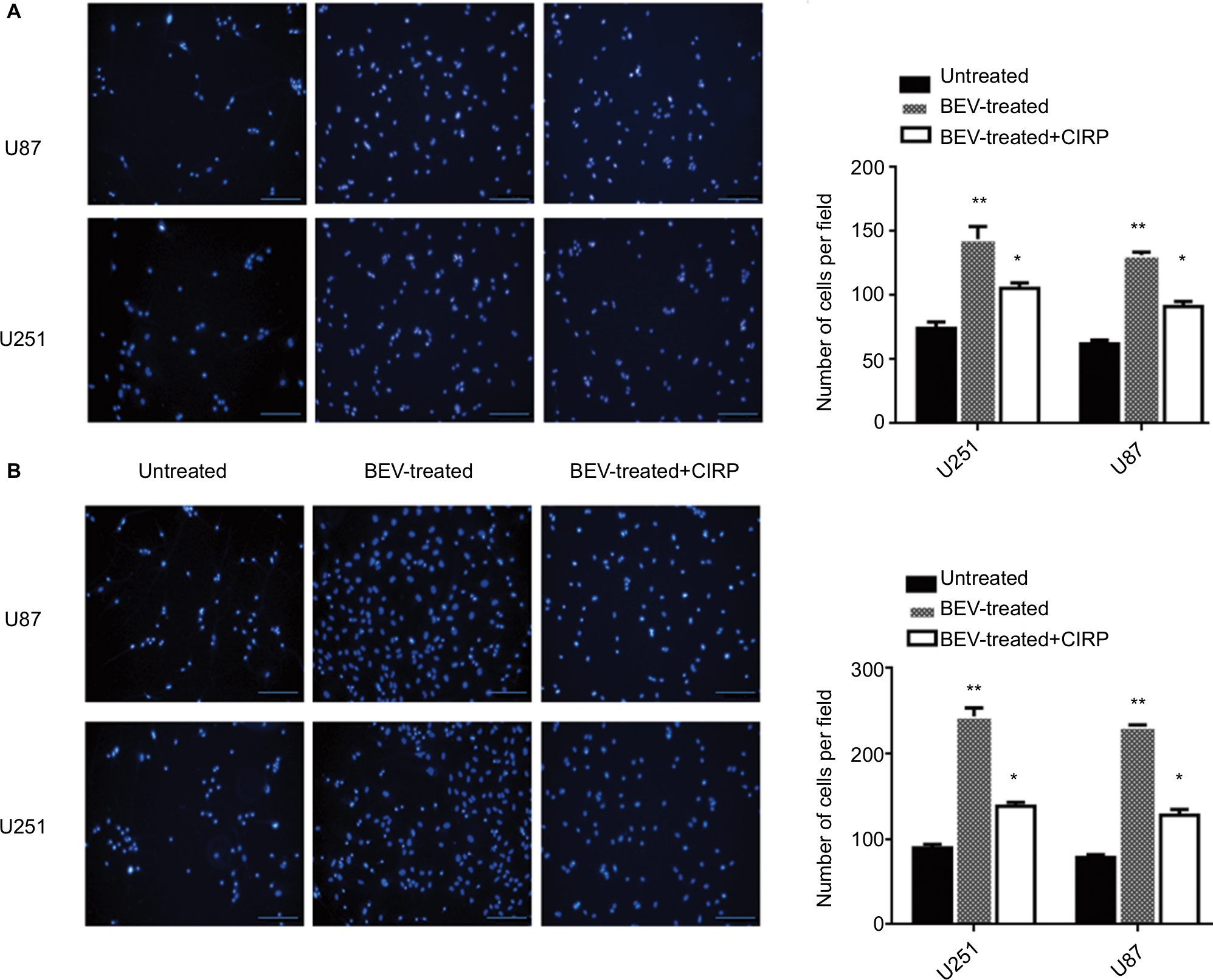 | Figure 4 Transwell invasion assays of human glioma cells. Notes: (A) Representative images (left) and quantification (right) of DAPI-stained invasive cells in transwell assay (with matrix gel). (B) Representative images (left) and quantification (right) of DAPI-stained migratory cells in transwell assay (without matrix gel). Average number of migratory invasive cells/field was analyzed. Data represent mean ± SD, n=10. *P<0.05, **P<0.01, compared with untreated group. Abbreviations: BEV, bevacizumab; CIRP, cold-inducible RNA-binding protein. |
Then, we established glioma xenograft mouse models using CIRP-luciferase or control-luciferase U251 cells to examine the effects of CIRP on BEV-induced migration in vivo. The results showed an increased number of cells undergoing migration in BEV-treated tumors 6 weeks after drug treatment. As expected, BEV-induced migration could be inhibited by CIRP overexpression (Figure 5A–C). These results indicated that overexpression of CIRP could inhibit BEV-induced migration of glioma cells in vitro and in vivo.
 | Figure 5 Overexpression of CIRP inhibits BEV-induced migration in vivo. Notes: (A) Tumors from the mice in different groups were measured by BLI signals. The red circles represented the intensity of BLI signal. (B) Tumors of mice in different groups were observed by H&E staining. The blue arrows indicated the tumors in brain. (C) Statistical analysis for the BLI signal intensities of U251 cell-derived tumors. U251 cells with the expression vector of CIRP-luciferase or control vector of luciferase were implanted into the brains of anesthetized athymic nu/nu mice. The tumors in different groups were analyzed 6 weeks after BEV treatment. Data represent mean ± SD, n=10. *P<0.05, compared with untreated group. Abbreviations: BEV, bevacizumab; BLI, bioluminescence imaging; CIRP, cold-inducible RNA-binding protein. |
Target mRNAs of CIRP in U251 cells
To further elucidate the underlying mechanism of CIRP in glioma cells, CIRP-binding mRNAs in U251 cells were isolated by RIP assay and analyzed by microarray assay. We obtained about a total of 1,745 positive mRNAs on the chip (Figure 6A). Most of the RNAs were located in nucleus, complex macromolecules, and membrane cytoplasm as shown in Figure 6B. These RNAs were mainly involved in metabolic progress, the response to stimulus, and cell growth (Figure 6C). Pathway annotation for the top ten abundant RNAs revealed that most of them were associated with spliceosome, ubiquitin-mediated proteolysis, and regulatory RNA surveillance pathway (Figure 6D). The results also showed that some targeted RNAs bound by CIRP were in adherence and migration-relative pathways (Figure 6D). The top five migration-related mRNAs were presented in Figure 6E. Moreover, the interactions between these mRNAs and CIRP were verified by RIP assays and quantitative real-time PCR (qRT-PCR) (Figure 6F). These results suggested that CIRP may be associated with migration by binding of migration-relative mRNA.
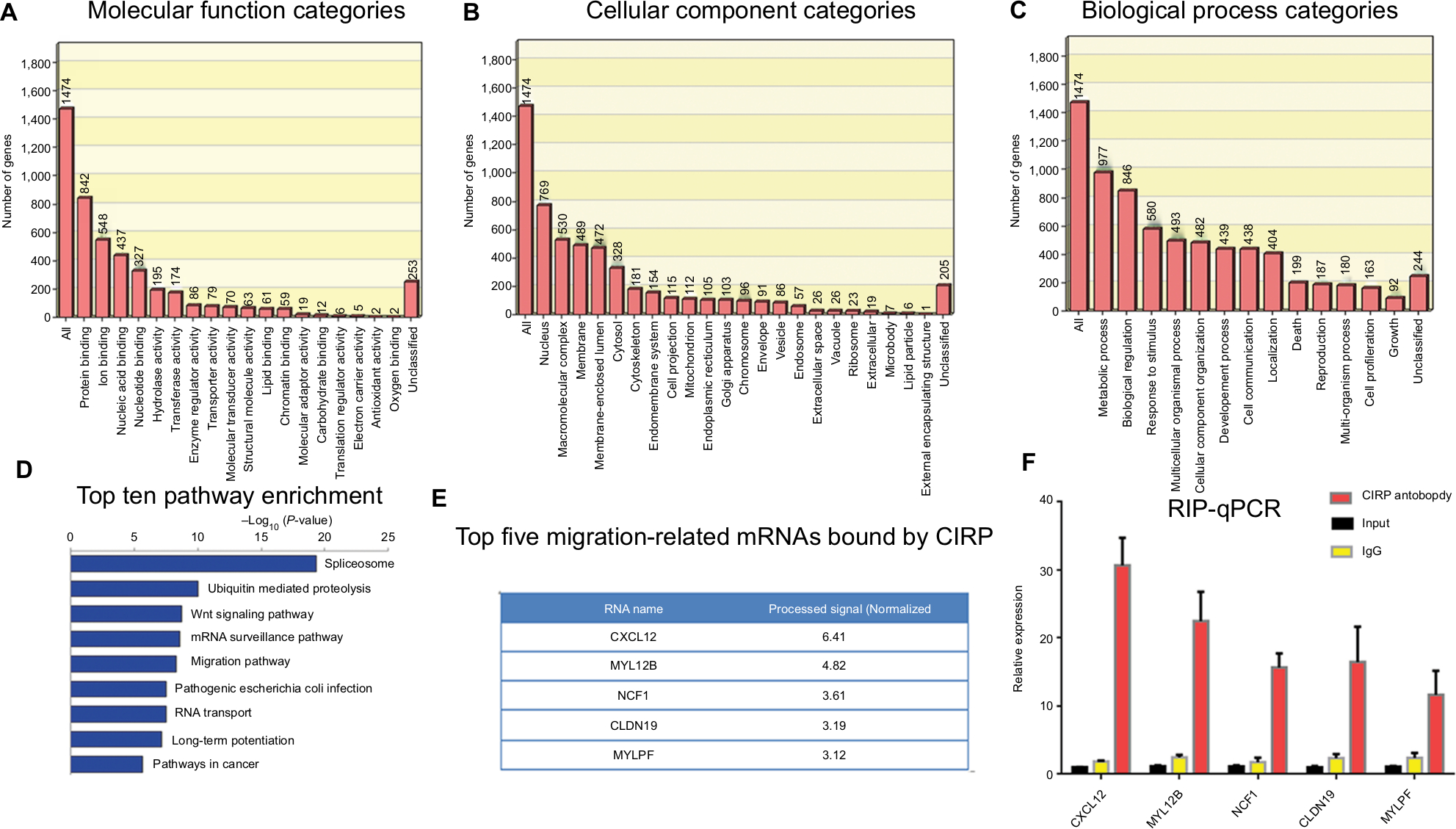 | Figure 6 Analysis of target mRNAs of CIRP in U251 cells. Notes: There were 1,745 mRNAs presented in microarray data. The top 500 abundant mRNAs identified in this study were classified according to their (A) molecular function categories, (B) cellular function categories, (C) biological functions, (D) top ten enriched pathways, and (E) top five migration-related mRNAs bound by CIRP. (F) Target mRNAs were verified by RIP assays and real-time PCR. Abbreviations: CIRP, cold-inducible RNA-binding protein; RIP, RNA-binding protein. |
CIRP inhibits BEV-induced expression of CXCL12
Furthermore, we analyzed the effects of CIRP on migration-relative factor CXCL12 by qRT-PCR. The results showed that the mRNA level of CXCL12 significantly decreased in overexpression glioma cells when compared with control cells and vice versa (Figure 7A and B).
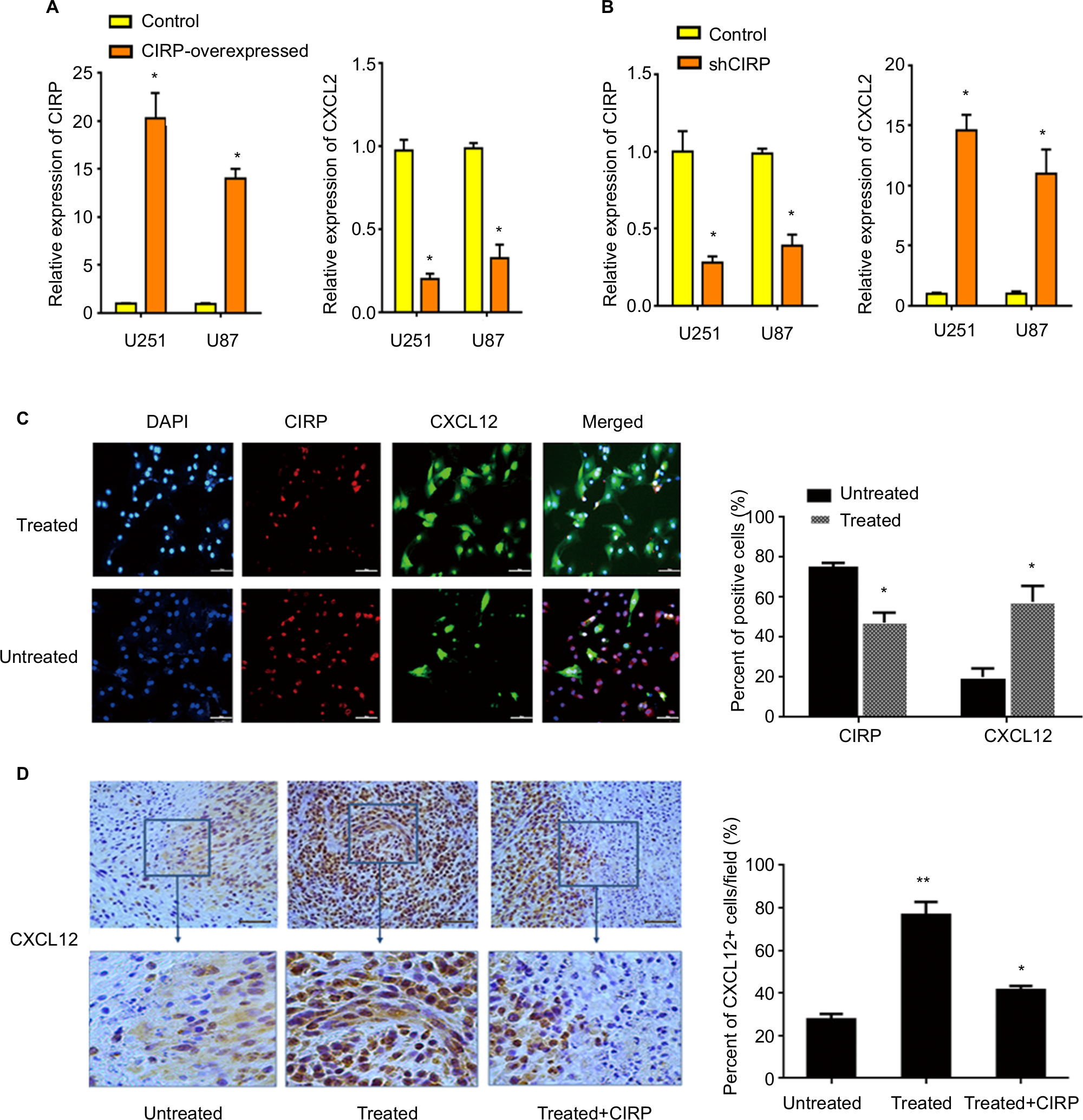 | Figure 7 CIRP inhibits BEV-induced expression of CXCL12 in vitro and in vivo. Notes: (A, B) The effects of CIRP on CXCL12 were analyzed in CIRP overexpressed or knockdown U251 cell lines by real-time PCR. β-actin served as an internal control of real-time PCR. Each point represented the mean of three independent experiments, n=3. *P<0.05, **P<0.01, when compared with the control group. All data were expressed as mean ± SD. (C) Representative images (left) and quantification (right) of the expression of CXCL12 and CIRP in untreated or BEV-treated U251 cells. The results were calculated as ratio of number of positive cells/number of total cells per field. Data were represented as mean ± SD, n=10. *P<0.05. (D) Representative images (left) and quantification (right) of the expression of CXCL12. U251 cells with the expression vector of CIRP or control vector were implanted into the brains of anesthetized athymic nu/nu mice. The percentage of CXCL12+ cells in the tumors was analyzed 6 weeks after BEV treatment. The results were calculated as ratio of CXCL12+ cells/total cells per field. Data represent mean ± SD, n=10. *P<0.05, **P<0.01, compared with untreated group. Abbreviations: BEV, bevacizumab; CIRP, cold-inducible RNA-binding protein. |
Finally, the expression levels of CIRP and CXCL12 after BEV treatment were determined by immunohistochemical staining. The results confirmed that BEV treatment could promote the expression of CXCL12 and inhibit the expression of CIRP (Figure 7C). But the increasing expression of CXCL12 induced by BEV-treated could be abolished by CIRP overexpression (Figure 7D).
Discussion
Currently, the mechanisms by which the resistance to angiogenesis inhibitors occurs were not well understood.12–14 By proteomic and biological functional analysis, we first identified the protein changes of gliomas after BEV treatment. The results revealed that the signal and protein transduction network of glioma cell were broadly affected by BEV. For example, proteins that played roles in RNA post-transcriptional and post-translational modification changed significantly after BEV treatment. Therefore, CIRP, one of the candidate proteins, was selected for further study. CIRP was a highly conserved glycine-rich RNA-binding protein (RBP), which contained an amino-terminal consensus sequence RNA-binding domain and a carboxyl-terminal glycine-rich domain.15,16 Although the exact functions were unknown, CIRP was reported to modulate translation of RNA and affect general protein synthesis.17,18
Zeng et al showed a significant decreased expression of CIRP in human prostate cancer cells. Silencing CIRP by small hairpin RNA could dramatically enhance the therapeutic response of prostate cancer cells to chemotherapy.19 However, whether CIRP played important roles in GBM resistance to BEV therapy remain largely unknown.
Here, we screened and identified CIRP as key regulatory protein in GBM cells after BEV treatment using proteomic techniques for the first time. And we confirmed that BEV treatment decreased CIRP expression of glioma cell lines at a dose-dependent and time-dependent manner in vitro (Figure 2A and B). We also proved that expression of CIRP was inhibited after drug treatment in vivo (Figure 2C and D).
Then we generated CIRP overexpression stable glioma cell lines and proved that ectopic expression of CIRP significantly suppressed BEV-induced migration and invasion of malignant glioma cells in vitro (Figures 3 and 4). We also observed that overexpression of CIRP in glioma cells could inhibit BEV-mediated migration in glioma xenografts (Figure 5).
RBPs could regulate the temporal, spatial, and functional dynamics of RNAs.20 As a vital member of RBPs, CIRP was regarded as RNA chaperones, which played important roles in post-transcriptional regulation of gene expression.21 Xia et al reported that downregulation of CIRP reduced survival time of the target mRNAs.22 So we speculated that binding to CIRP may alter stability of cell migration relative mRNAs. By RIP-Chip, we found that some mRNAs, such as CXCL12, MYL12B, NCF1, CLDN19, and MYLPF which were reported to affect cell migration,23–26 were the target mRNAs bound by CIRP (Figure 6E). And in these mRNAs, CXCL12 had highest processed signal value. Previous studies showed that CXCL12 played pivotal roles in the cell migration, angiogenesis, proliferation, and recurrence of GBM. Here, we found that the expression of CIRP paralleled with a decrease of CXCL12 in glioma cells (Figure 7A and B). Moreover, CIRP overexpression could inhibit the expression of CXCL12 in glioma, which was coincidental with the decrease of BEV-mediated migration in CIRP-overexpressed tumors. Previous study suggested that the recurrence pattern was significantly related to the expression level of CXCL12. However, CXCL12 was not independent prognostic factors for progression-free survival or overall survival in glioma patients.27 In this study, the relationship between the survival of patients and the expression level of CIRP or CXCL12 was not explored, which should be studied in future. These results implied that CIRP may decrease the stability of some mRNAs and affected the migration of glioma cells after BEV treatment. Our findings provide new proofs for the role of CIRP in post-transcriptional regulation of gene expression in gliomas migration after BEV treatment. However, the detailed networks downstream of CIRP that exert migration-inhibiting effects in brain tumor should be further investigated.
Finally, although some studies suggested an increased risk for recurrence or progression of malignant glioma patients treated with BEV, other evidence also proved that BEV did not increase the risk of remote relapse in malignant glioma.28 We speculated that individual differences in patients might cause the differences in experimental data, and more clinical samples were needed. In addition, induced BEV-resistant glioma cell lines should be established because they were more ideal and convincing cell models to study the chemoresistance of GBM.
Conclusion
Our data represented a clear indication that BEV treatment could alter the expression level of CIRP in GBM cells; in turn, the expression level of CIRP could influence BEV-induced migration in GBM cells. These data improve the understanding of the mechanisms by which GBM became resistant to BEV and shed light on the novel drug combination strategies.
Acknowledgments
This study was funded by grant from Special Scientific Research on Health Development of the Capital (grant number: 2014-2-5021) and the National Natural Sciences Foundation of China (grant number: 81472372).
Disclosure
The authors report no conflicts of interest in this work.
References
Jain RK, di Tomaso E, Duda DG, Loeffler JS, Sorensen AG, Batchelor TT. Angiogenesis in brain tumours. Nat Rev Neurosci. 2007;8(8):610–622. | ||
Plate KH, Breier G, Weich HA, Risau W. Vascular endothelial growth factor is a potential tumour angiogenesis factor in human gliomas in vivo. Nature. 1992;359(6398):845–848. | ||
Plate KH, Scholz A, Dumont DJ. Tumor angiogenesis and anti-angiogenic therapy in malignant gliomas revisited. Acta Neuropathol. 2012;124(6):763–775. | ||
Gutin PH, Iwamoto FM, Beal K, et al. Safety and efficacy of bevacizumab with hypofractionated stereotactic irradiation for recurrent malignant gliomas. Int J Radiat Oncol Biol Phys. 2009;75(1):156–163. | ||
Lai A, Filka E, Mcgibbon B, et al. Phase II pilot study of bevacizumab in combination with temozolomide and regional radiation therapy for up-front treatment of patients with newly diagnosed glioblastoma multiforme: interim analysis of safety and tolerability. Int J Radiat Oncol Biol Phys. 2008;71(5):1372–1380. | ||
Narayana A, Golfinos JG, Fischer I, et al. Feasibility of using bevacizumab with radiation therapy and temozolomide in newly diagnosed high-grade glioma. Int J Radiat Oncol Biol Phys. 2008;72(2):383–389. | ||
Kunkel P, Ulbricht U, Bohlen P, et al. Inhibition of glioma angiogenesis and growth in vivo by systemic treatment with a monoclonal antibody against vascular endothelial growth factor receptor-2. Cancer Res. 2001;61(18):6624–6628. | ||
Lucio-Eterovic AK, Piao Y, de Groot JF. Mediators of glioblastoma resistance and invasion during antivascular endothelial growth factor therapy. Clin Cancer Res. 2009;15(14):4589–4599. | ||
Lehnert S, Jesse S, Rist W, et al. iTRAQ and multiple reaction monitoring as proteomic tools for biomarker search in cerebrospinal fluid of patients with Parkinson’s disease dementia. Exp Neurol. 2012;234(2):499–505. | ||
Mustafa DA, Dekker LJ, Stingl C, et al. A proteome comparison between physiological angiogenesis and angiogenesis in glioblastoma. Mol Cell Proteomics. 2012;11(6):M111.008466. | ||
Vafadar-Isfahani B, Ball G, Coveney C, et al. Identification of SPARC-like 1 protein as part of a biomarker panel for Alzheimer’s disease in cerebrospinal fluid. J Alzheimers Dis. 2012;28(3):625–636. | ||
Mitamura T, Pradeep S, Mcguire M, et al. Induction of anti-VEGF therapy resistance by upregulated expression of microseminoprotein (MSMP). Oncogene. 2018;37(6):722–731. | ||
Piao Y, Liang J, Holmes L, et al. Acquired resistance to anti-VEGF therapy in glioblastoma is associated with a mesenchymal transition. Clin Cancer Res. 2013;19(16):4392–4403. | ||
Zarrin B, Zarifi F, Vaseghi G, Javanmard SH. Acquired tumor resistance to antiangiogenic therapy: mechanisms at a glance. J Res Med Sci. 2017;22:117. | ||
Bergeron D, Beauseigle D, Bellemare G. Sequence and expression of a gene encoding a protein with RNA-binding and glycine-rich domains in Brassica napus. Biochim Biophys Acta. 1993;1216(1):123–125. | ||
Burd CG, Dreyfuss G. Conserved structures and diversity of functions of RNA-binding proteins. Science. 1994;265(5172):615–621. | ||
de Leeuw F, Zhang T, Wauquier C, et al. The cold-inducible RNA-binding protein migrates from the nucleus to cytoplasmic stress granules by a methylation-dependent mechanism and acts as a translational repressor. Exp Cell Res. 2007;313(20):4130–4144. | ||
Fujita J. Cold shock response in mammalian cells. J Mol Microbiol Biotechnol. 1999;1(2):243–255. | ||
Zeng Y, Kulkarni P, Inoue T, Getzenberg RH. Down-regulating cold shock protein genes impairs cancer cell survival and enhances chemosensitivity. J Cell Biochem. 2009;107(1):179–188. | ||
Chou HL, Tian L, Kumamaru T, Hamada S, Okita TW. Multifunctional RNA binding protein OsTudor-SN in storage protein mRNA transport and localization. Plant Physiol. 2017;175(4):1608–1623. | ||
Al-Fageeh MB, Smales CM. Cold-inducible RNA binding protein (Cirp) expression is modulated by alternative mRNAs. RNA. 2009;15(6):1164–1176. | ||
Xia Z, Zheng X, Zheng H, et al. Cold-inducible RNA-binding protein (Cirp) regulates target mRNA stabilization in the mouse testis. FEBS Lett. 2012;586(19):3299–3308. | ||
Guo Q, Gao BL, Zhang XJ, et al. CXCL12-CXCR4 axis promotes proliferation, migration, invasion, and metastasis of ovarian cancer. Oncol Res. 2014;22(5-6):247–258. | ||
Kowalewski JM, Shafqat-Abbasi H, Jafari-Mamaghani M, et al. Disentangling membrane dynamics and cell migration; differential influences of F-actin and cell-matrix adhesions. Plos One. 2015;10(8):e0135204. | ||
Peng S, Wang SB, Singh D, et al. Claudin-3 and claudin-19 partially restore native phenotype to ARPE-19 cells via effects on tight junctions and gene expression. Exp Eye Res. 2016;151:179–189. | ||
Prasad R, Kappes JC, Katiyar SK. Inhibition of NADPH oxidase 1 activity and blocking the binding of cytosolic and membrane-bound proteins by honokiol inhibit migratory potential of melanoma cells. Oncotarget. 2016;7(7):7899–7912. | ||
Tang W, Wang X, Chen Y, et al. CXCL12 and CXCR4 as predictive biomarkers of glioma recurrence pattern after total resection. Pathol Biol. 2015;63(4–5):190–198. | ||
Wick A, Dörner N, Schäfer N, et al. Bevacizumab does not increase the risk of remote relapse in malignant glioma. Ann Neurol. 2011;69(3):586–592. |
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.