Back to Journals » International Journal of Chronic Obstructive Pulmonary Disease » Volume 11 » Issue 1
Cigarette smoke-induced lung inflammation in COPD mediated via LTB4/BLT1/SOCS1 pathway
Authors Dong R, Xie L ![]() , Zhao K, Zhang Q, Zhou M, He P
, Zhao K, Zhang Q, Zhou M, He P ![]()
Received 15 September 2015
Accepted for publication 7 November 2015
Published 22 December 2015 Volume 2016:11(1) Pages 31—41
DOI https://doi.org/10.2147/COPD.S96412
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Richard Russell
Ran Dong,1,* Liang Xie,1,* Kaishun Zhao,2,* Qiurui Zhang,1 Min Zhou,1 Ping He3
1Department of Respiratory Medicine, Ruijin Hospital, Shanghai Jiao Tong University School of Medicine, 2Department of Respiratory Medicine, Jiading Central Hospital, 3Department of Microbiology and Parasitology, Institutes of Medical Sciences, Shanghai Jiao Tong University School of Medicine, Shanghai, People’s Republic of China
*These authors contributed equally to this work
Background: Evidence suggests that suppressor of cytokine signaling 1 (SOCS1) is crucial for the negative regulation of inflammation. We investigated the relationship between smoking, SOCS1, and leukotriene B4 (LTB4) in vitro and in clinical samples of COPD; besides which we detected the impact of LTB4 receptor 1 (BLT1) antagonist on inflammation.
Methods: SOCS1 expression in bronchial mucosa was determined by immunohistochemistry and real-time polymerase chain reaction. We also detect SOCS1 and BLT1 expression in alveolar macrophages from bronchoalveolar lavage fluid (BALF) by real time-PCR, in addition to measuring the level of cytokines in BALF using enzyme-linked immunosorbent assay. In vitro, we investigated the expression of SOCS1 in cigarette smoke extract-induced mouse macrophage cell line RAW264.7 by real-time polymerase chain reaction and Western blot, and detected the level of cytokines in the supernatant by enzyme-linked immunosorbent assay. Then, we investigated the effects of BLT1 antagonist U-75302 on SOCS1 expression in these cells.
Results: We obtained endobronchial biopsies (15 COPD patients and 12 non-COPD control subjects) and BALF (20 COPD patients and 20 non-COPD control subjects), and our results showed that SOCS1 expression significantly decreased in lung tissues from COPD patients. Inflammatory cytokines in BALF were higher in COPD and these inflammatory cytokines negatively correlate with SOCS1 levels. Further, the BLT1 antagonist restored SOCS1 expression and in turn inhibited inflammatory cytokine secretion in vitro.
Conclusion: Long-term cigarette smoke exposure induced SOCS1 degradation and LTB4 accumulation, which was associated with emphysema and inflammation. A BLT1 antagonist might be a potential therapeutic candidate for the treatment of COPD.
Keywords: treatment, LTB1 antagonist, endobronchial biopsies, BALF, inflammatory cytokines
Introduction
COPD is a chronic inflammatory disease of the airways that is characterized by progressive and irreversible decline in lung function, excessive mucus secretion, and limited airflow.1,2 According to the World Health Organization, the annual mortality due to COPD is ~3 million people worldwide. It causes significant social and economic burden and is a serious public health issue.3 Cigarette smoking (CS) is the main risk factor for COPD and persistent airway inflammation, which contributes to irreversible pathological changes in the lung, such as airway remodeling and pulmonary emphysema.4 However, the mechanisms of pathogenesis are still not fully elucidated. To our knowledge, cigarette smoke triggers immune dysregulation and pulmonary inflammation and activates innate immune cells such as macrophages, which play a crucial role in promoting inflammation and secretion of inflammatory cytokines by triggering pattern recognition receptors, either directly or indirectly via the release of damage-associated molecular patterns from stressed or dying cells.5–7 Inflammation is a complex defense response.8 An uncontrolled and prolonged inflammatory response may cause lung tissue destruction and remodeling, as chronic inflammation continues even after smoking cessation.9,10 Therefore, elimination of inflammation caused by CS is a COPD treatment goal, which is difficult in clinical practice.
The suppressor of cytokine signaling 1 (SOCS1) mediates innate immune regulation with a negative feedback loop, which is important in inflammatory cytokine production.11–13 These studies suggest a novel regulatory mechanism prompting us to speculate that the absence of SOCS1, a negative regulatory protein, may lead to accumulation of proinflammatory cytokines and chronic inflammatory reaction in COPD. Therefore, we investigated the SOCS1 expression and inflammatory cytokine level in COPD patient’s lung tissues and bronchoalveolar lavage (BAL). The results may provide evidence supporting our hypothesis. However, the precise mechanism is still unclear.
The most recent study suggested that leukotriene B4 (LTB4) combined with its receptor, leukotriene B4 receptor 1 (BLT1), accelerates the degradation of SOCS1 mRNA.14,15 LTB4 is a key chemokine in the accumulation of granulocytes and macrophages via BLT1 and BLT2, a G protein-coupled cell surface receptor. It plays an important role in the COPD inflammatory response.16–20 Clinically, LTB4 promotes the survival of neutrophils in exhaled breath condensate (EBC) and sputum of COPD patients.21 The effects of SOCS1 on LTB4/BLT1 pathway in CS-induced airway inflammation are not well known. It is reasonable to hypothesize that accumulation of LTB4 in the CS-exposed lung might suppress SOCS1 transcription and expression, causing the release of a series of downstream proinflammatory cytokines. Thus, we chose the selective BLT1 receptor antagonist U-75302 to investigate if U-75302 can prevent inflammation in vitro. Our results may provide experimental evidence for the underlying mechanisms of COPD pathogenesis and a novel therapeutic approach for COPD via modulation of LTB4/BLT1 pathway.22
Materials and methods
Subjects
We obtained endobronchial biopsies (15 COPD patients and 12 non-COPD control subjects) from the Respiratory Ward at Ruijin Hospital affiliated to the School of Medicine, Shanghai Jiao Tong University. In addition, BAL was performed in 20 COPD patients and 20 non-COPD control subjects in either the middle lobe or the lingula. All the samples are from clinically stable patients and the patients and controls for endobronchial biopsy and BAL were completely different, and the characteristics of subjects are listed in Table 1. All the COPD patients met the criteria proposed by the Global initiative for chronic Obstructive Lung Disease. All COPD patients were in a stable condition at the time of study. All control subjects had normal lung function without any history of asthma or chronic bronchitis and smoking history. No patients were diagnosed with current or recent (8 weeks) respiratory tract infection, or any exacerbations requiring oral steroid or antibiotic treatment. All subjects provided written informed consent to participate in the study, which was reviewed and approved by the Ruijin Hospital Ethics Committee Shanghai Jiao Tong University of Medicine. All measurements and experiments were performed in a blinded fashion.
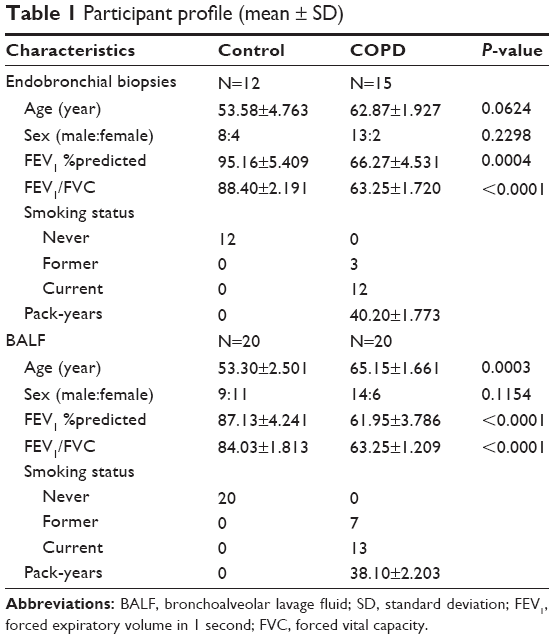 | Table 1 Participant profile (mean ± SD) |
Preparation of alveolar macrophages from BALF
By infusing 50 mL of sterile sodium chloride into either the middle lobe or the lingula, we obtained approximately 30 mL of bronchoalveolar lavage fluid (BALF) from each subject. BALF was centrifuged for 20 minutes at 1,500 rpm and the supernatant was discarded. The precipitate was resuspended in Dulbecco’s Modified Eagle’s Medium (DMEM; Thermo Fisher Scientific, Waltham, MA, USA) containing 10% fetal bovine serum, supplemented with 100 U/mL penicillin and 100 mg/mL streptomycin. Cells were cultured on 3.5 cm plates for 3 hours, and washed three times by phosphate-buffered saline. The cells adhering to the bottom were alveolar macrophages (AMs), which were verified by cellular staining.
Immunohistochemical analysis
Immunohistochemistry was used to analyze the protein levels and distribution of SOCS1 in bronchial tissues of COPD and control subjects. Briefly, we embedded lung tissues (endobronchial biopsies) in paraffin and placed 5 μm-thick sections on the slides, dewaxed in xylene, and rehydrated. Endogenous peroxidases were inhibited with 0.5% hydrogen peroxide in methanol for 10 minutes, followed by overnight incubation at 4°C with a rabbit polyclonal IgG antibody against SOCS1 (Abcam, Cambridge, UK). Immunodetection was performed with diaminobenzidine, biotinylated goat anti-rabbit IgG reagent, and horseradish peroxidase reagent.
RNA extraction and real-time PCR
Total RNA from AMs and bronchial tissues were extracted using an RNeasy kit (Qiagen, Valencia, CA, USA), and total RNA from RAW264.7 cells was extracted using TRIzol reagents (Thermo Fisher Scientific), according to the manufacturer’s instructions. Single-stranded cDNA was synthesized for each sample with oligo (dT) primers using RevertAid™ First Strand cDNA Synthesis Kit (Fermentas, Waltham, MA, USA), according to the manufacturer’s protocol. Total RNA (500 ng) was amplified with a 7500 Fast Real-Time PCR System (Thermo Fisher Scientific) with FastStart Universal SYBR Green (Hoffman-La Roche Ltd., Basel, Switzerland) after cDNA synthesis. The standard curve for quantification was created using a modified procedure as previously described.23 The sequences of all primers are shown in Table 2. Fold change of the gene expression was calculated by 2−ΔΔCt relative to the internal reference gene (glyceraldehyde-3-phosphate dehydrogenase, GAPDH). The results were repeated at least three times, respectively.
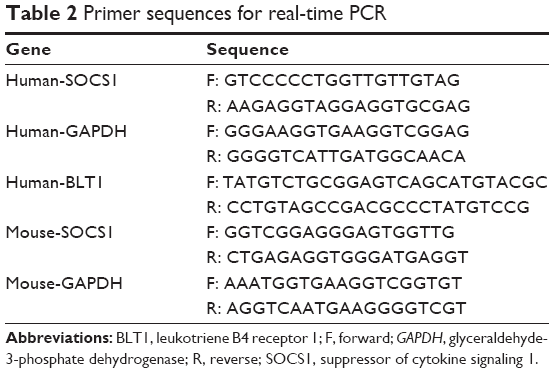 | Table 2 Primer sequences for real-time PCR |
Cell line and CS extract
The RAW264.7 mouse macrophage cell line was obtained from the Cell Collection and Research Center of Chinese Academy of Science. CS was collected and treated with DMEM by continuous suction in a chamber connected to a vacuum. Treatment with 500 mL of DMEM required mainstream smoke from ten cigarettes and 5 minutes for each cigarette. The resultant CS extract (CSE)–DMEM was considered as 100% stock solution and ready for use after adjusting the pH to 7.4 and sterile filtration (0.2 μm). Serial dilutions of 50%, 30%, 20%, and 10% CSE solution were prepared for cell culture. DMEM only was considered as 0% CSE.
Exposure of RAW264.7 culture to CSE and U-75302 treatment
The most appropriate condition was determined using a combination of different concentrations of CSE stimulation at different exposure times of 3, 6, 12, 24, and 48 hours. Basically, 30% CSE was the maximal dilution without excessive cell mortality after 24 hours exposure. For treatment with U-75302, RAW264.7 cells were seeded overnight and treated with 20% CSE with 10 nmol/mL U-75302 (Enzo, Telluride, CO, USA) for 24 hours. Supernatant was then harvested and total cellular RNA and protein were extracted.
Cytokine enzyme-linked immunosorbent assays
The concentrations of interleukin-8 (IL-8) and tumor necrosis factor-α (TNF-α) in the BALF, and the mouse IL-6 and TNF-α levels in the cell culture supernatant were determined using DuoSet ELISA kits (R&D Systems, Minneapolis, MN, USA) according to the manufacturer’s instructions. LTB4 concentration in BALF and cell culture supernatants were measured using a specific enzyme immunoassay with a commercial kit (Cayman Chemical Co, Ann Arbor, MI, USA).
Western blot
Cells were lysed with 100 μL radio immunoprecipitation assay lysis buffer (Sigma-Aldrich Co., St Louis, MO, USA). Protein concentrations were measured with bicinchoninic acid assay kit (Thermo Fisher Scientific) according to the manufacturer’s protocol. Equal amounts of proteins (30 μg) were subjected to a 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis. Gels were run at 80 V for 30 minutes followed by 120 V for 1 hour before transferring onto a polyvinylidene fluoride membrane (EMD Millipore, Billerica, MA, USA). After blocking with phosphate-buffered saline containing 5% nonfat milk for 2 hours at room temperature, we incubated overnight at 4°C with rabbit polyclonal anti-SOCS1 antibody (1:1,000; Abcam), or a β-actin antibody (1:1,000; Cell Signaling, Danvers, MA, USA). The membrane was washed for 5 minutes with 15 mL tris-buffered saline and tween 20 three times, and then incubated with horseradish peroxidase-conjugated goat anti-rabbit IgG antibody (1:2,000; Sigma) for 1 hour at room temperature. After washing, we added 1 mL chemiluminescent substrate (Thermo Fisher Scientific) to the membrane. The signal was detected and quantified with an enhanced chemiluminescence system (ImageQuant LAS-4000 MINI; GE Healthcare Bio-Sciences, Pittsburgh, PA, USA). All samples were normalized to β-actin.
Statistical analysis
Results are presented as mean ± standard error of the mean if not stated otherwise. A two tailed t-test was performed in all results’ statistical analysis for comparisons of baseline characteristic. A linear regression was adopted for association between SOCS1 mRNA levels, LTB4 expression level, and FEV1 (forced expiratory volume in 1 second) %predicted using Spearman’s rank correlation test. Statistical analyses were performed as described in each figure legend, using GraphPad Prism. P-values ≤0.05 were considered statistically significant.
Results
Immunohistochemistry of SOCS1 in bronchial mucosa of COPD patients
Immunohistochemistry staining showed the presence of SOCS1 protein mainly in the airway epithelial cells and AMs (Figure 1). We found that the expression level of SOCS1 in bronchial mucosa was significantly lower in patients with COPD than in control subjects (808.17±697.06 vs 15,944±7,181.53; Figure 1A–D).
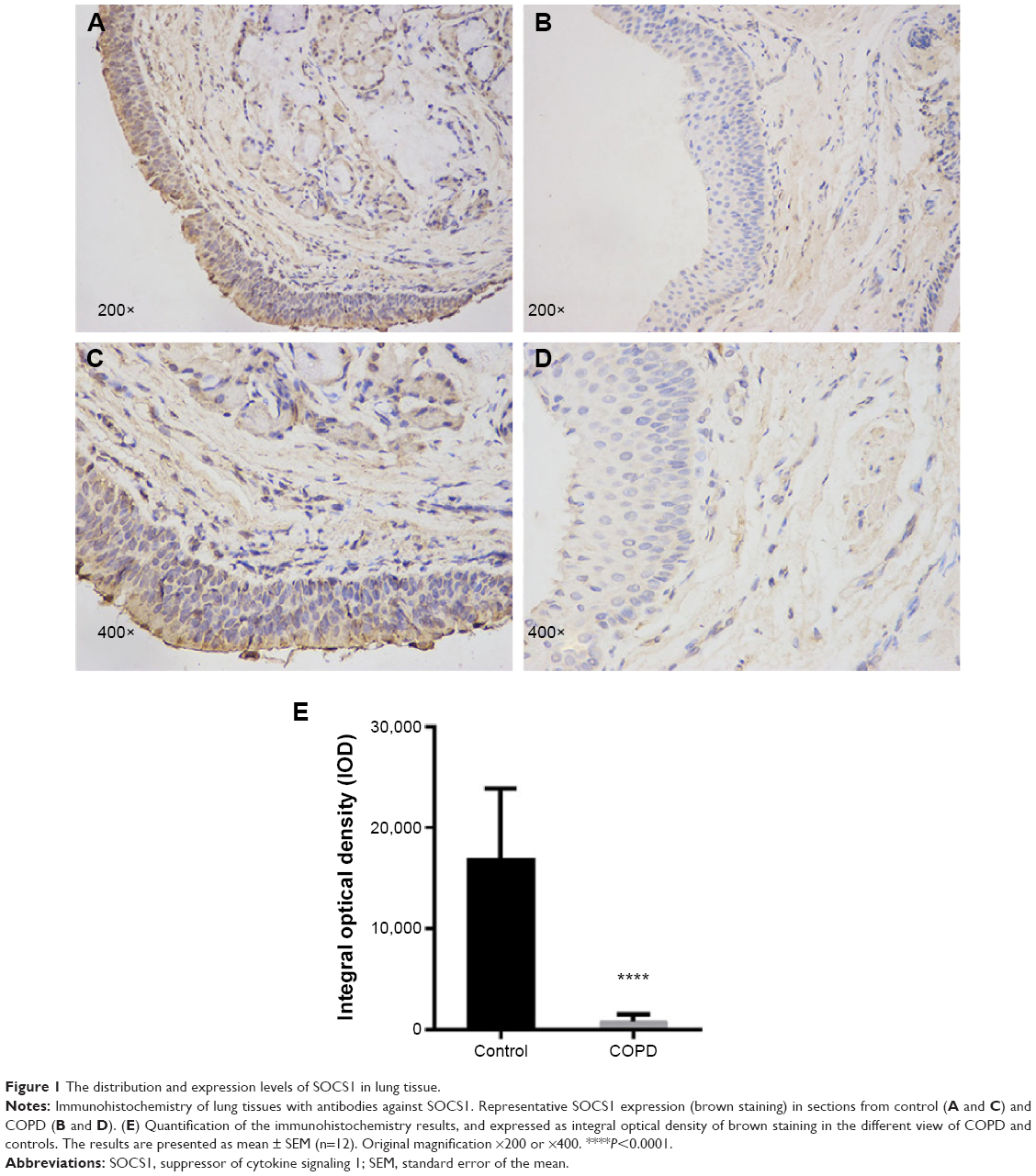 | Figure 1 The distribution and expression levels of SOCS1 in lung tissue. |
SOCS1 mRNA expression in the bronchial mucosa
The SOCS1 mRNA level in bronchial biopsies was compared between 12 non-COPD and 15 COPD subjects by real-time reverse transcription quantitative polymerase chain reaction (RT-qPCR). The SOCS1 expression relative to the housekeeping gene GAPDH was significantly lower in COPD subjects than in control subjects (0.6587±0.1045 vs 2.786±0.4748; shown in Figure 2). These results were consistent with immunohistochemistry results illustrated in Figure 1, indicating that SOCS1 expression was significantly decreased in lung tissues of COPD patients.
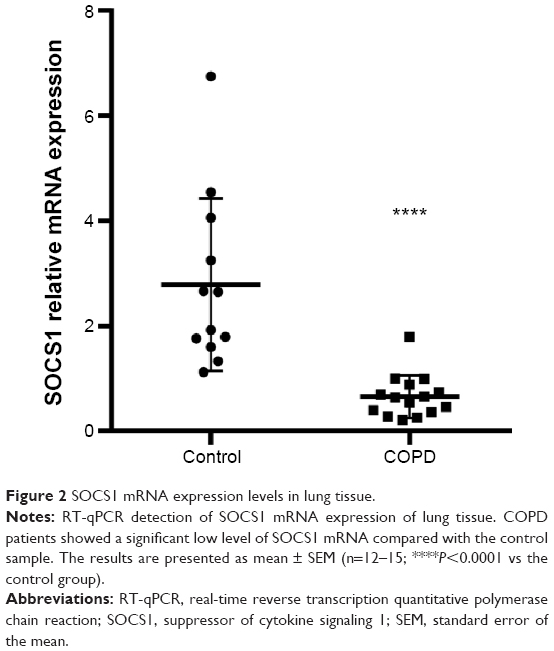 | Figure 2 SOCS1 mRNA expression levels in lung tissue. |
SOCS1 and BLT1 mRNA expression in the AMs
Total RNA of AMs in BALF was isolated and the mRNA of SOCS1 and LTB4 receptor BLT1 was then determined by RT-qPCR. The SOCS1 expression relative to GAPDH was significantly lower in COPD subjects than in control subjects (3.583±0.5430 vs 1.173±0.1867), whereas the BLT1 mRNA expression was higher (0.7000±0.1168 vs 2.670±0.6158) than that of SOCS1 (Figure 3) suggesting increased BLT1 expression after long-term cigarette smoke exposure.
 | Figure 3 mRNA expression of SOCS1 and BLT1 in alveolar macrophages. |
Cytokine levels in alveolar BALF
The IL-8 and TNF-α levels were higher in BALF of COPD patients than in control subjects (Figure 4A–C). These results indicated persistent airway inflammation in COPD patients.
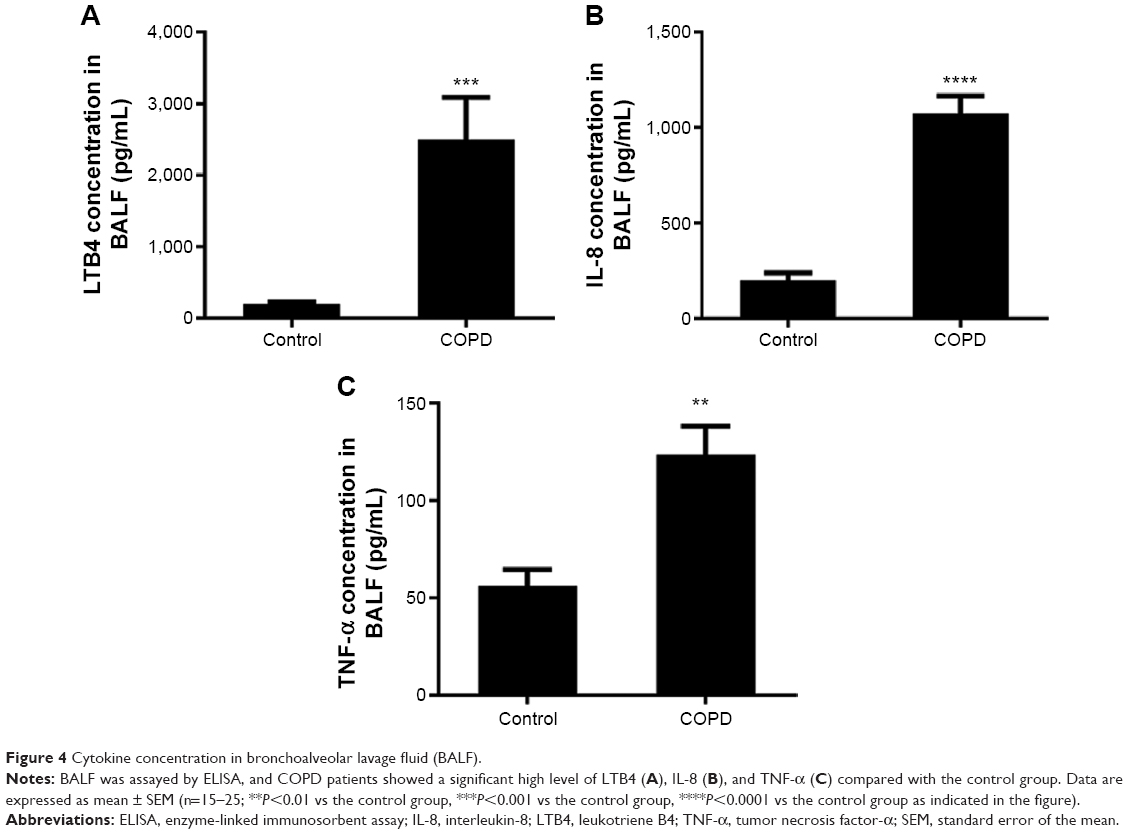 | Figure 4 Cytokine concentration in bronchoalveolar lavage fluid (BALF). |
SOCS1 and LTB4 expression and clinical outcomes
In this study, a negative correlation existed between SOCS1 mRNA level in lung tissue and LTB4 concentration in BALF (Figure 5A). Significant positive correlation was seen between SOCS1 mRNA level in lung tissues and FEV1 %predicted (Figure 5B). These data indicated that SOCS1/LTB4 pathway was crucial for the progression of COPD.
 | Figure 5 Correlations between SOCS1 mRNA levels, LTB4 expression level, and FEV1 %predicted. |
Expression of SOCS1 and cytokines in CSE-induced RAW264.7 cells
To detect the potential effects of CSE on the expression of SOCS1, we used 20% CSE to treat RAW264.7 cells for the indicated period (6, 12, and 24 hours). The cells were harvested, and total RNA was isolated to analyze the mRNA expression by RT-qPCR. In addition, concentrations of various cytokines in supernatants were measured by enzyme-linked immunosorbent assay. The results showed that the level of LTB4 was significantly upregulated in CSE-treated RAW264.7 cells than in control groups. However, the mRNA level of SOCS1 reached its peak 6 hours later, after treatment, and decreased subsequently at 12 and 24 hours (Figure 6A and B). Next, we assessed the proinflammatory cytokines in CSE-stimulated RAW264.7 cells, as shown in Figure 6C and D. Western blot analyses suggest that the expression of SOCS1 protein was increased in the early stages of CSE treatment and remarkably decreased compared with the control group at 24 hours (Figure 6E). Collectively, these data indicated that SOCS1 plays an important role in the process of inflammation.
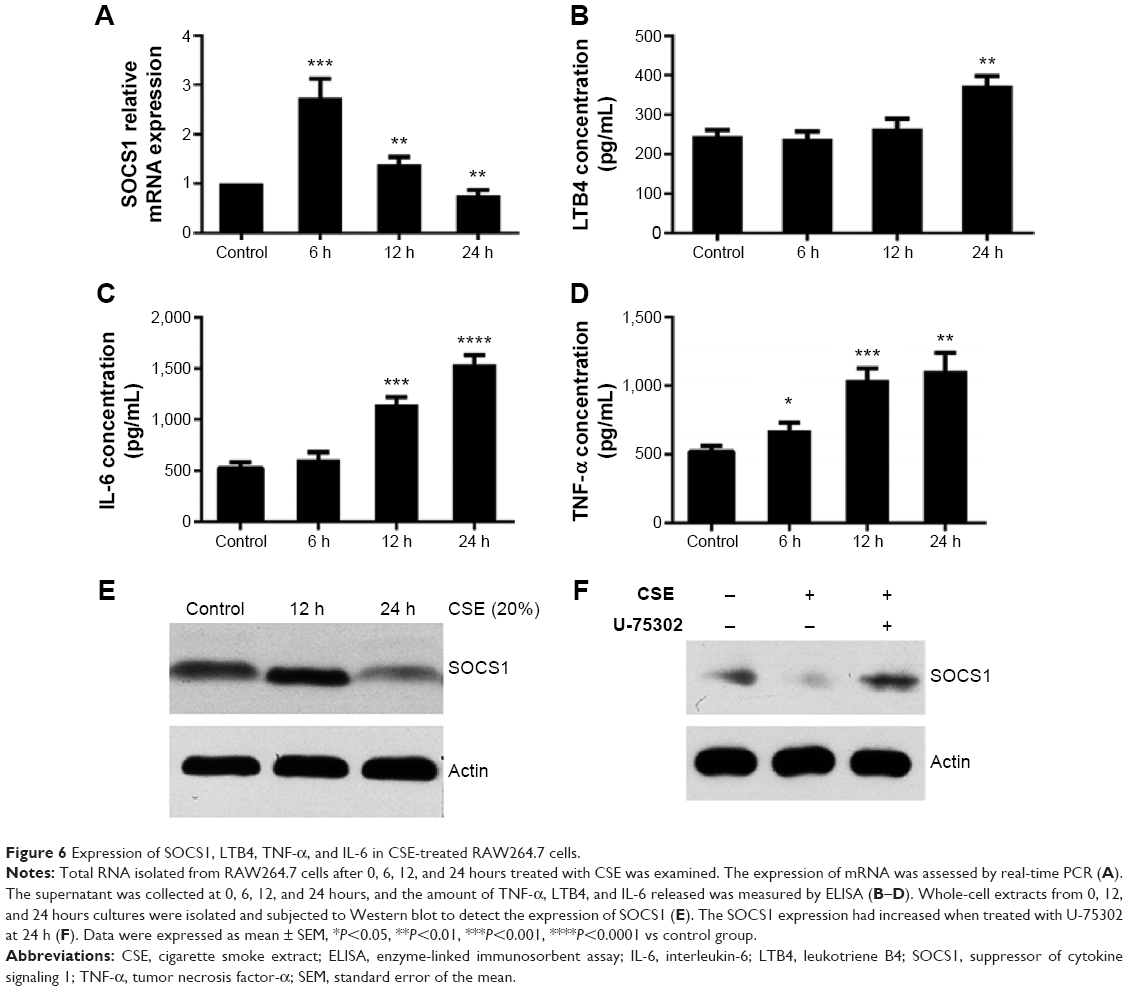 | Figure 6 Expression of SOCS1, LTB4, TNF-α, and IL-6 in CSE-treated RAW264.7 cells. |
Effect of U-75302 on SOCS1 expression in RAW 264.7 cells
In order to analyze the role of LTB4 in the degeneration of SOCS1 expression, U-75302, a BLT1 antagonist, was used to treat CSE-stimulated RAW264.7 cells. As shown in Figure 7A, addition of U-75302 enhanced SOCS1 mRNA and protein expression (Figure 6F). U-75302 also abolished the increase in TNF-α and IL-6 mRNA and protein expression in CSE-stimulated cells (shown in Figure 7B–D). These data supported the hypothesis correlating SOCS1 expression with LTB4 accumulation.
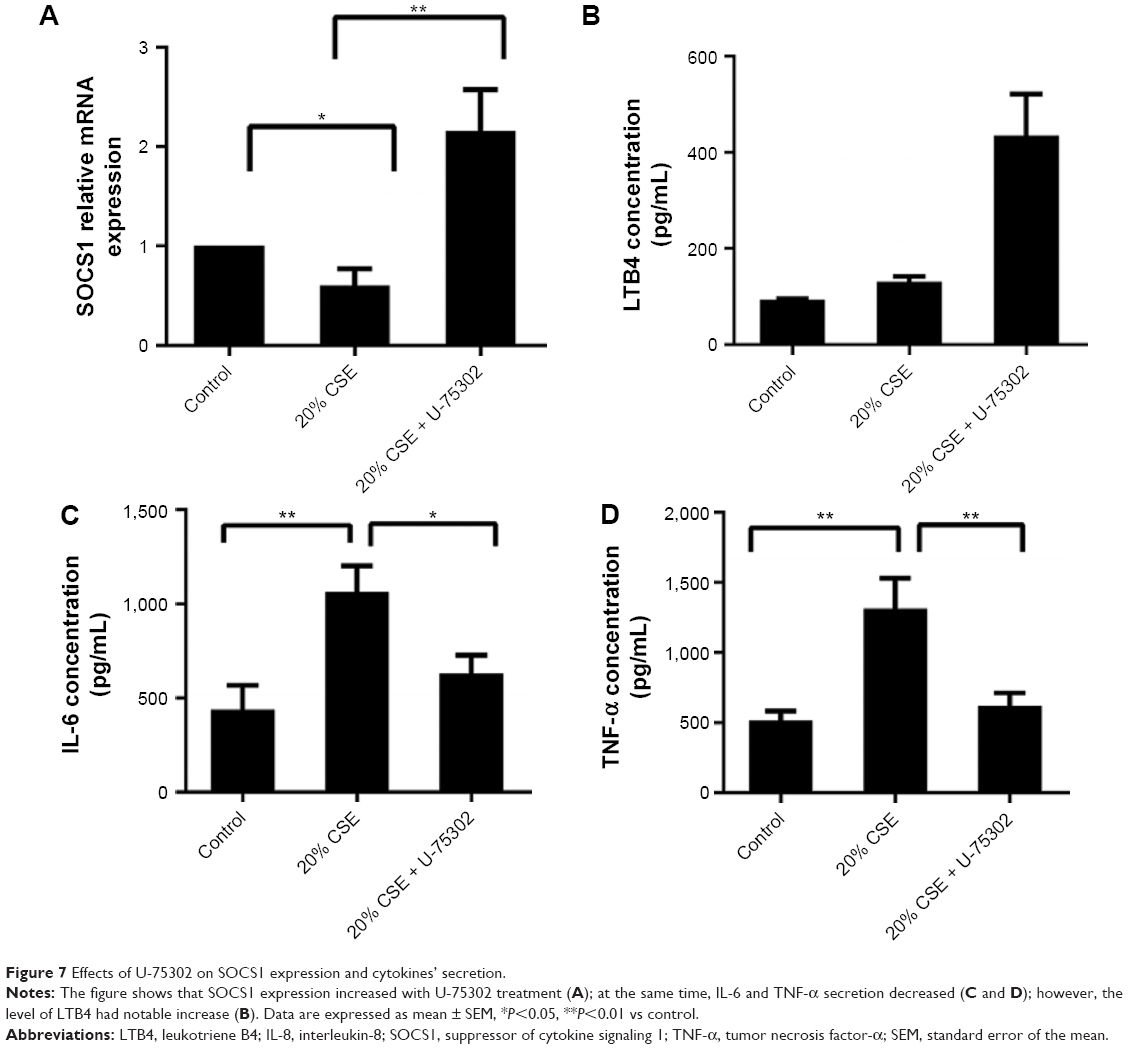 | Figure 7 Effects of U-75302 on SOCS1 expression and cytokines’ secretion. |
Discussion
Our study demonstrated lower expression of SOCS1 and higher levels of LTB4 in COPD patients; importantly, BLT1 antagonist could restore SOCS1 expression and prevent inflammation in vitro, thus, our results revealed a novel therapeutic target in COPD smoke-induced airway inflammation. As the primary mediator of innate immune response, AMs are initially activated by CS. AMs are directly involved in the secretion of cytokines, including IL-8, TNF-α, and LTB4 resulting in chronic inflammatory response and tissue destruction, exacerbating emphysema in COPD patients.24–26 Therefore, inhibition of inflammatory cytokine secretion in macrophages is a potential therapeutic strategy in inflammation and autoimmune diseases.27,28 Similarly, our study showed high levels of IL-8, TNF-α, and LTB4 in AMs, BALF, and lung tissue in COPD patients. For the first time, we showed that the basal level of SOCS1 decreased while the expression of BLT1 increased in bronchial mucosa and AMs from patients with COPD. Our data suggest that the exaggerated inflammatory response may be associated with poor SOCS1 expression.
SOCS1 mediates the negative regulation of inflammatory response. Previous studies reported an increased level of inflammatory cytokines, including TNF-α, IL-6, and IL-8 in SOCS1-deficient macrophages.29,30 Wang et al14 and Serezani et al15 showed that LTB4/BLT1 pathway enhanced degradation of SOCS1 mRNA via miR-155, consistent with other studies.31–34 Some studies showed that LTB4 was involved in the pathogenesis of pulmonary emphysema.35–37 We observed higher levels of LTB4 in the BALF of COPD patients and in the EBC, consistent with previous studies.38,39 LTB4 is a key neutrophil chemotactic factor, causing neutrophil infiltration in COPD patients.21 We found for the first time a significant negative correlation between the LTB4 levels in BALF and SOCS1 expression in bronchial mucosa from patients with COPD, suggesting that the accumulation of LTB4 contributes negatively to inflammation in vivo. We found that membrane injury in airway epithelial and AMs caused by longtime CS exposure exaggerated the LTB4 levels in the BALF of COPD patients and then activated LTB4/BLT1 signaling. Once triggered, the SOCS1 mRNA was degraded resulting in uncontrolled inflammatory response. We, therefore, hypothesized that BLT1 antagonist might prevent these events.
To test this hypothesis, we identified SOCS1 expression in CSE-stimulated macrophages in vitro, for the first time. Interestingly, we found that SOCS1 mRNA level was significantly upregulated in CSE-stimulated RAW264.7 during the early period. To our knowledge, JAK/STAT signal pathway plays a vital role in various conditions, including immune reaction, cell growth, and differentiation.40 Studies showed that CSE induced the release of damage-associated molecular pattern molecules and activated pattern recognition receptors, and downstream signaling pathways that trigger a negative feedback loop and enhance SOCS1 expression to control inflammatory response.13,41–44 Increased inflammatory cytokines such as IL-8, TNF-α, and LTB4 were also secreted in clinical samples and in vitro.45–48 Further, our data showed that the SOCS1 expression significantly decreased with increased LTB4 in the supernatants after 24 hours CSE treatment. For the first time, we showed that BLT1 antagonist U-75302 restored SOCS1 expression induced by LTB4 and reduced the levels of inflammatory cytokines IL-6 and TNF-α in vitro. The results showed that BLT1 antagonist may control chronic airway inflammation and prevent the progress of COPD pathology. Our results were consistent with other findings. Watanabe et al49 found that the pathogenesis of airway remodeling was inhibited by the BLT1 specific antagonist, U-75302. Further, U-75302 reduced neutrophil chemotactic activity in samples of EBC and induced sputum from patients with COPD.21 More recently, animal experiments showed that U-75302 attenuated lung injury and the level of systemic cytokines in polymicrobial sepsis-induced mice.50 However, previous studies showed that LTB4 antagonist did not have any benefit in COPD patients,51–53 and these results may be associated with the uncontrolled clinical trials with novel measures of pharmacodynamic effect, patient selection, end points, and disease indications; furthermore, the LTB4 antagonist in these studies was not specific and dual BLT1 and BLT2 inhibition may exert a lack of effect if the functions of these two receptors are opposing.53 Thus, selective BLT1 inhibition could reduce inflammation,22,54,55 although simply BLT1 antagonist therapeutic may not be effective in COPD patients, because BLTI receptor should have other protective roles in cell survival, etc, however, we might at least discover a target to further study.
There were also some limitations in our study; we utilized murine macrophages for the in vitro study and whether the results can be extrapolated to humans is unknown. However, our study indicated the effect of CS-induced lesions on SOCS1 expression and LTB4/BLT1 pathway. Further investigation of U-75302 in an animal model will be interesting.
In summary, this study demonstrated lower levels of SOCS1 and higher levels of LTB4 in COPD patients when compared with control groups. Importantly, SOCS1 expression was negatively correlated with that of LTB4, which indicated that LTB4/BLT1 pathway might play a critical role in airway inflammation in COPD. Thus, BLT1 antagonist could be a new therapeutic candidate in COPD treatment targeting smoke-induced inflammation.
Acknowledgment
This work was supported by grants from the National Natural Science Foundation of China (No. 81200027 and No. 81570029).
Disclosure
The authors report no conflicts of interest in this work.
References
Decramer M, Janssens W, Miravitlles M. Chronic obstructive pulmonary disease. Lancet. 2012;379(9823):1341–1351. | ||
MacNee W. Pathogenesis of chronic obstructive pulmonary disease. Proc Am Thorac Soc. 2005;2(4):258–266; discussion 290–291. | ||
Diaz-Guzman E, Mannino DM. Epidemiology and prevalence of chronic obstructive pulmonary disease. Clin Chest Med. 2014;35(1):7–16. | ||
Tuder RM, Yun JH. It takes two to tango: cigarette smoke partners with viruses to promote emphysema. J Clin Invest. 2008;118(8):2689–2693. | ||
Brussel GG, Joos GF, Bracke KR. Chronic obstructive pulmonary disease 1 new insights into the immunology of chronic obstructive pulmonary disease. Lancet. 2011;378(9795):1015–1026. | ||
Valledor AF, Comalada M, Santamaria-Babi LF, Lloberas J, Celada A. Macrophage proinflammatory activation and deactivation: a question of balance. Adv Immunol. 2010;108:1–20. | ||
de Geus ED, Vervelde L. Regulation of macrophage and dendritic cell function by pathogens and through immunomodulation in the avian mucosa. Dev Comp Immunol. 2013;41(3):341–351. | ||
Hirahara K, Poholek A, Vahedi G, et al. Mechanisms underlying helper T-cell plasticity: implications for immune-mediated disease. J Allergy Clin Immunol. 2013;131(5):1276–1287. | ||
Scrivo R, Vasile M, Bartosiewicz I, Valesini G. Inflammation as “common soil” of the multifactorial diseases. Autoimmun Rev. 2011;10(7):369–374. | ||
Cowburn AS, Condliffe AM, Farahi N, Summers C, Chilvers ER. Advances in neutrophil biology: clinical implications. Chest. 2008;134(3):606–612. | ||
Lesinski GB, Zimmerer JM, Kreiner M, et al. Modulation of SOCS protein expression influences the interferon responsiveness of human melanoma cells. BMC Cancer. 2010;10:142. | ||
Hashimoto M, Ayada T, Kinjyo I, et al. Silencing of SOCS1 in macrophages suppresses tumor development by enhancing antitumor inflammation. Cancer Sci. 2009;100(4):730–736. | ||
Tajiri K, Imanaka-Yoshida K, Matsubara A, et al. Suppressor of cytokine signaling 1 DNA administration inhibits inflammatory and pathogenic responses in autoimmune myocarditis. J Immunol. 2012;189(4):2043–2053. | ||
Wang Z, Filgueiras LR, Wang S, et al. Leukotriene B4 enhances the generation of proinflammatory microRNAs to promote MyD88-dependent macrophage activation. J Immunol. 2014;192(5):2349–2356. | ||
Serezani CH, Lewis C, Jancar S, Peters-Golden M. Leukotriene B4 amplifies NF-kappaB activation in mouse macrophages by reducing SOCS1 inhibition of MyD88 expression. J Clin Invest. 2011;121(2):671–682. | ||
Yokomizo T, Izumi T, Chang K, Takuwa Y, Shimizu T. A G-protein-coupled receptor for leukotriene B4 that mediates chemotaxis. Nature. 1997;387(6633):620–624. | ||
Kavelaars A, Vroon A, Raatgever RP, et al. Increased acute inflammation, leukotriene B4-induced chemotaxis, and signaling in mice deficient for G protein-coupled receptor kinase 6. J Immunol. 2003;171(11):6128–6134. | ||
Huang WW, Garcia-Zepeda EA, Sauty A, Oettgen HC, Rothenberg ME, Luster AD. Molecular and biological characterization of the murine leukotriene B4 receptor expressed on eosinophils. J Exp Med. 1998;188(6):1063–1074. | ||
Miyahara N, Miyahara S, Takeda K, Gelfand EW. Role of the LTB4/BLT1 pathway in allergen-induced airway hyperresponsiveness and inflammation. Allergol Int. 2006;55(2):91–97. | ||
Serhan CN, Prescott SM. The scent of a phagocyte: Advances on leukotriene b(4) receptors. J Exp Med. 2000;192(3):F5–F8. | ||
Corhay JL, Henket M, Nguyen D, Duysinx B, Sele J, Louis R. Leukotriene B4 contributes to exhaled breath condensate and sputum neutrophil chemotaxis in COPD. Chest. 2009;136(4):1047–1054. | ||
Waseda K, Miyahara N, Kanehiro A, et al. Blocking the leukotriene B4 receptor 1 inhibits late-phase airway responses in established disease. Am J Respir Cell Mol Biol. 2011;45(4):851–857. | ||
Zhang Y, Lou XL, Yang HL, et al. Establishment of a leptospirosis model in guinea pigs using an epicutaneous inoculations route. BMC Infect Dis. 2012;12:20. | ||
Liu G, Yang H. Modulation of macrophage activation and programming in immunity. J Cell Physiol. 2013;228(3):502–512. | ||
Cheng C, Huang C, Ma TT, et al. SOCS1 hypermethylation mediated by DNMT1 is associated with lipopolysaccharide-induced inflammatory cytokines in macrophages. Toxicol Lett. 2014;225(3):488–497. | ||
Cosio MG, Saetta M, Agusti A. Immunologic aspects of chronic obstructive pulmonary disease. N Engl J Med. 2009;360(23):2445–2454. | ||
De Palma M, Lewis CE. Macrophage regulation of tumor responses to anticancer therapies. Cancer Cell. 2013;23(3):277–286. | ||
Wynn TA, Chawla A, Pollard JW. Macrophage biology in development, homeostasis and disease. Nature. 2013;496(7446):445–455. | ||
Lee TL, Yeh J, Van Waes C, Chen Z. Epigenetic modification of SOCS-1 differentially regulates STAT3 activation in response to interleukin-6 receptor and epidermal growth factor receptor signaling through JAK and/or MEK in head and neck squamous cell carcinomas. Mol Cancer Ther. 2006;5(1):8–19. | ||
Guenterberg KD, Lesinski GB, Mundy-Bosse BL, et al. Enhanced anti-tumor activity of interferon-alpha in SOCS1-deficient mice is mediated by CD4(+) and CD8(+) T cells. Cancer Immunol Immunother. 2011;60(9):1281–1288. | ||
Lu LF, Thai TH, Calado DP, et al. Foxp3-dependent microRNA155 confers competitive fitness to regulatory T cells by targeting SOCS1 protein. Immunity. 2009;30(1):80–91. | ||
Sun YX, Cai JJ, Ma F, Lu PP, Huang HF, Zhou J. miR-155 mediates suppressive effect of progesterone on TLR3, TLR4-triggered immune response. Immunol Lett. 2012;146(1–2):25–30. | ||
Wang P, Hou J, Lin L, et al. Inducible microRNA-155 feedback promotes type I IFN signaling in antiviral innate immunity by targeting suppressor of cytokine signaling 1. J Immunol. 2010;185(10):6226–6233. | ||
Rodriguez A, Vigorito E, Clare S, et al. Requirement of bic/microRNA-155 for normal immune function. Science. 2007;316(5824):608–611. | ||
Shim YM, Paige M, Hanna H, Kim SH, Burdick MD, Strieter RM. Role of LTB(4) in the pathogenesis of elastase-induced murine pulmonary emphysema. Am J Physiol Lung Cell Mol Physiol. 2010;299(6):L749–L759. | ||
Dahlen SE, Hedqvist P, Hammarstrom S, Samuelsson B. Leukotrienes are potent constrictors of human bronchi. Nature. 1980;288(5790):484–486. | ||
Wardlaw AJ, Hay H, Cromwell O, Collins JV, Kay AB. Leukotrienes, LTC4 and LTB4, in bronchoalveolar lavage in bronchial asthma and other respiratory diseases. J Allergy Clin Immunol. 1989;84(1):19–26. | ||
Biernacki WA, Kharitonov SA, Barnes PJ. Increased leukotriene B4 and 8-isoprostane in exhaled breath condensate of patients with exacerbations of COPD. Thorax. 2003;58(4):294–298. | ||
Kostikas K, Gaga M, Papatheodorou G, Karamanis T, Orphanidou D, Loukides S. Leukotriene B4 in exhaled breath condensate and sputum supernatant in patients with COPD and asthma. Chest. 2005;127(5):1553–1559. | ||
Harrison DA. The Jak/STAT pathway. Cold Spring Harb Perspect Biol. 2012;4(3):a011205. | ||
Pace E, Ferraro M, Siena L, et al. Carbocysteine regulates innate immune responses and senescence processes in cigarette smoke stimulated bronchial epithelial cells. Toxicol Lett. 2013;223(2):198–204. | ||
Prele CM, Woodward EA, Bisley J, Keith-Magee A, Nicholson SE, Hart PH. SOCS1 regulates the IFN but not NFkappaB pathway in TLR-stimulated human monocytes and macrophages. J Immunol. 2008;181(11):8018–8026. | ||
Oudijk EJ, Lammers JW, Koenderman L. Systemic inflammation in chronic obstructive pulmonary disease. Eur Respir J Suppl. 2003;46:5s–13s. | ||
Heijink IH, Pouwels SD, Leijendekker C, et al. Cigarette smoke induced DAMP release from necrotic neutrophils triggers pro-inflammatory mediator release. Am J Respir Cell Mol Biol. 2015:554–562. | ||
Doz E, Noulin N, Boichbt E, et al. Cigarette smoke-induced pulmonary inflammation is TLR4/MyD88 and IL-1R1/MyD88 signaling dependent. J Immunol. 2008;180(2):1169–1178. | ||
MacRedmond RE, Greene CM, Dorscheid DR, McElvaney NG, O’Neill SJ. Epithelial expression of TLR4 is modulated in COPD and by steroids, salmeterol and cigarette smoke. Respir Res. 2007;8:84. | ||
Pace E, Giarratano A, Ferraro M, et al. TLR4 upregulation underpins airway neutrophilia in smokers with chronic obstructive pulmonary disease and acute respiratory failure. Hum Immunol. 2011;72(1):54–62. | ||
Yin Y, Hou G, Li ER, Wang QY, Kang J. PPAR Gamma agonists regulate tobacco smoke-induced toll like receptor 4 expression in alveolar macrophages. Respir Res. 2014;15:28. | ||
Watanabe S, Yamasaki A, Hashimoto K, et al. Expression of functional leukotriene B4 receptors on human airway smooth muscle cells. J Allergy Clin Immunol. 2009;124(1):59–65.e51–e53. | ||
Li XJ, Fu HY, Yi WJ, et al. Dual role of leukotriene B4 receptor type 1 in experimental sepsis. J Surg Res. 2015;193(2):902–908. | ||
Belvisi MG, Hele DJ, Birrell MA. New anti-inflammatory therapies and targets for asthma and chronic obstructive pulmonary disease. Expert Opin Ther Targets. 2004;8(4):265–285. | ||
Barnes PJ. New anti-inflammatory targets for chronic obstructive pulmonary disease. Nat Rev Drug Discov. 2013;12(7):543–559. | ||
Hicks A, Monkarsh SP, Hoffman AF, Goodnow R Jr. Leukotriene B4 receptor antagonists as therapeutics for inflammatory disease: preclinical and clinical developments. Expert Opin Investig Drugs. 2007;16(12):1909–1920. | ||
de Hoog VC, Bovens SM, de Jager SC, et al. BLT1 antagonist LSN2792613 reduces infarct size in a mouse model of myocardial ischaemia-reperfusion injury. Cardiovasc Res. Epub 2015 Oct 8. | ||
Han CG, Huang HG, Hu M, et al. [Involvement of leukotrine B4 receptors in the inflammatory responses and immunological regulation in vitro]. Zhongguo Ying Yong Sheng Li Xue Za Zhi. 2009;25(2):273–276. Chinese. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.