Back to Journals » Drug Design, Development and Therapy » Volume 13
Chronic myelogenous leukemia, a still unsolved problem: pitfalls and new therapeutic possibilities
Authors Flis S ![]() , Chojnacki T
, Chojnacki T
Received 22 October 2018
Accepted for publication 9 January 2019
Published 8 March 2019 Volume 2019:13 Pages 825—843
DOI https://doi.org/10.2147/DDDT.S191303
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Jianbo Sun
Sylwia Flis,1 Tomasz Chojnacki2
1Department of Pharmacology, National Medicines Institute, 00-725 Warsaw, Poland; 2Department of Hematology, Military Institute of Medicine, 04-141 Warsaw, Poland
Abstract: Chronic myeloid leukemia (CML) is a clonal myeloproliferative disorder of hematopoietic stem cells. At the molecular level, the disorder results from t(9;22)(q34;q11) reciprocal translocation between chromosomes, which leads to the formation of an oncogenic BCR–ABL gene fusion. Instead of progress in the understanding of the molecular etiology of CML and the development of novel therapeutic strategies, clinicians still face many challenges in the effective treatment of patients. In this review, we discuss the pathways of diagnosis and treatment of patients, as well as the problems appearing in the course of disease development. We also briefly refer to several aspects regarding the current knowledge on the molecular basis of CML and new potential therapeutic targets.
Keywords: chronic myeloid leukemia, CML, BCR–ABL, tyrosine kinase inhibitors, TKI, TKI withdrawal, stem cells, autophagy
Introduction
Chronic myeloid leukemia (CML) is a widely described malignant disorder of hematopoietic stem cells (HSCs) that accounts for 15%–20% of all cases of leukemia in adults.1 The main history of CML begins in 1960 when Peter Nowel and David Hungerford discovered an abnormally small G-group chromosome – now called the Philadelphia (Ph) chromosome. This was the first proof that the disease results in some changes to DNA. In 1973, Janet Rowley recognized that the Ph chromosome was the product of a t(9;22)(q34;q11) reciprocal translocation between chromosomes, and then later in the 1980s, Nora Heisterkamp discovered that this translocation generates the BCR–ABL fusion oncogene.2 In the ABL gene, the break point is generally located upstream of the second exon (a2), whereas in the BCR gene the breakage takes place usually in one of the three regions called major (M-bcr), minor (m-bcr), and micro-bcr (μ-bcr) break point regions. Depending on the location of the chromosome breakage in the BCR gene, three different types of BCR–ABL proteins, differing in mass as well as biological properties, can be formed (Figure 1). The majority of CML patients have a p210 BCR–ABL gene (M-bcr), in which the fusion is located downstream of the 14 or 13 exons of the BCR gene resulting in the creation of mRNA transcripts, which have an e14 and/or an e13 junction, and thus a 210 kDa chimeric protein is produced from this mRNA. The smallest of the fusion proteins, p190 BCR–ABL, is developed as a result of the minor break point region (m-bcr) of the BCR gene leading to the transcript e1a2. The p190 BCR–ABL form is mainly associated with Ph-positive acute lymphoblastic leukemia and rarely appears in patients with CML and might correlate with an aggressive course of the disease. A third break point in the region of the BCR gene called μ-bcr results in the transcription of an e19/a2 mRNA that codes a 230 kDa BCR–ABL protein. This form of fusion protein is associated with the rare Ph-positive chronic neutrophilic leukemia3 (Figure 1). Although other atypical BCR–ABL transcripts may occur, the form of p210 BCR–ABL is found in over 90% of CML patients. Therefore, it is accepted that acquisition of the BCR–ABL oncogene (especially the p210 BCR–ABL form) is the initiating step in the development of CML. The acquisition of the BCR–ABL gene initially occurs in a single pluripotent HSC that gains a proliferative advantage and/or aberrant differentiation capacity over its normal counterparts, giving rise to the expanded myeloid compartment.4 This process is possible because the BCR–ABL oncoprotein is constitutively active tyrosine kinase as a result of oligomerization via the coiled-coil region of BCR and a deletion of the inhibitory SH3 domain of ABL. This results in autophosphorylation of p210 BCR–ABL on the Y177 tyrosine residue and leads to phosphorylation of many downstream targets.5 Activation of various signaling pathways such as Ras/mitogen-activated protein kinase (MAPK), phosphoinositide 3-kinase (PI3K), or signal transducer and activator of transcription 5 (STAT5) by BCR–ABL kinase leads to tumor transformation in conjunction with dysfunction of underlying cellular processes associated with the control of proliferation, differentiation, and survival. BCR–ABL-positive cells become independent of the presence of growth factors in the environment; these cells are characterized by increased proliferation, apoptosis resistance, and genetic instability leading to CML progression, as well as impaired cell adhesion leading to their spread and abnormal release of immature cells to the peripheral blood.6
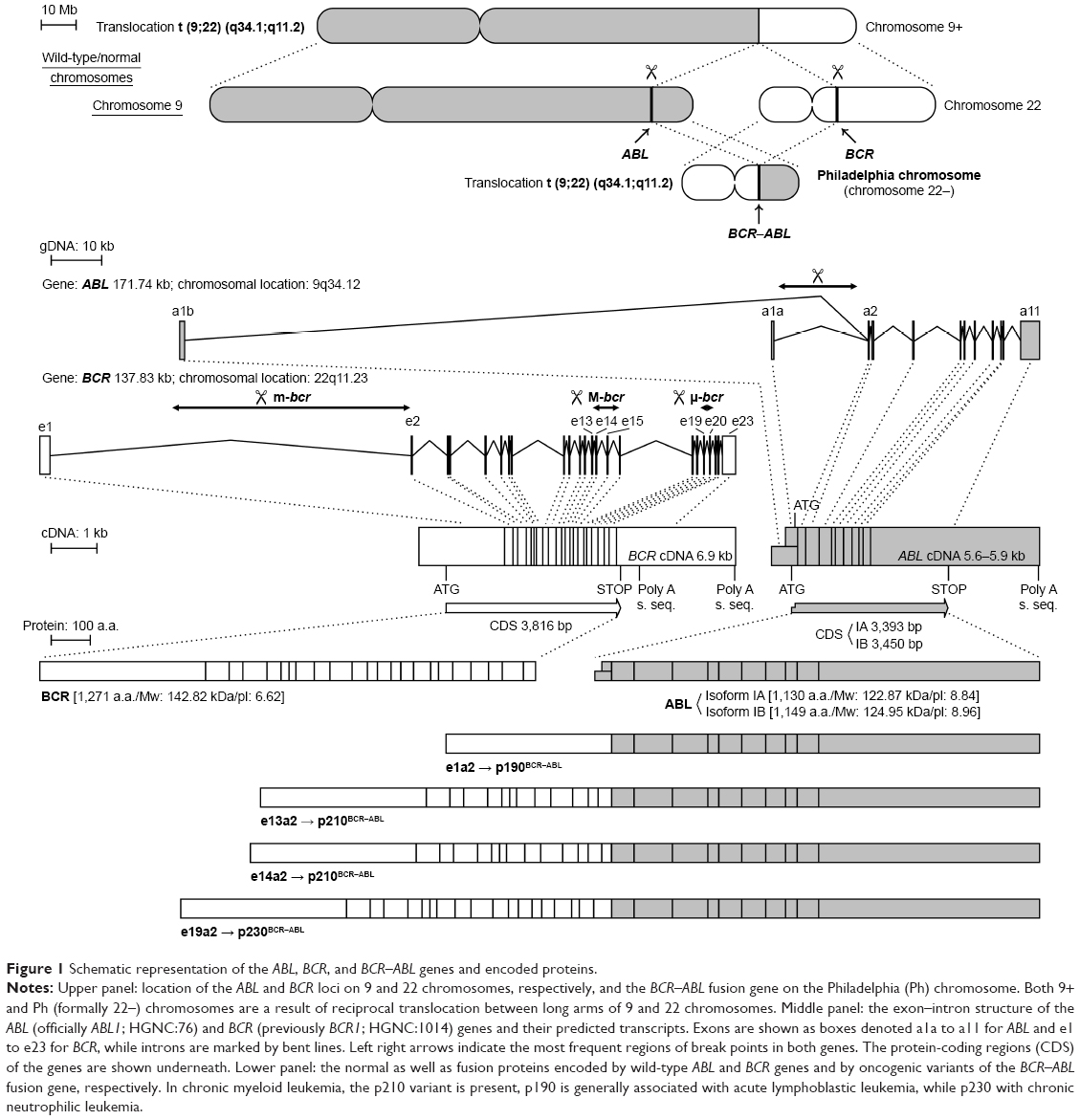 | Figure 1 Schematic representation of the ABL, BCR, and BCR–ABL genes and encoded proteins. |
CML is a triphasic myeloproliferative disorder that begins from a latent phase called a chronic phase (CP). Generally, CML-CP is a leukemia stem cell (LSC)-derived disease, in which deregulated growth of LSC-derived leukemia progenitor cells (LPCs) leads to the manifestation of disease symptoms.7 Untreated CML-CP progresses spontaneously to a more advanced accelerated phase (CML-AP) of the disease and subsequently to its very aggressive blast crisis phase (CML-BP).8 During disease progression, further molecular and biological alterations in the population of HSC cells arise. In consequence, patients become unresponsive to therapy.
Diagnostic standard
In many cases, CML is accidentally detected in routine blood tests. The patient who was diagnosed with leukocytosis often has no symptoms of the disease. More frequently, however, an in-depth medical history reveals a deterioration of well-being, impaired exercise tolerance, loss of appetite, feeling of fullness in the abdomen, weight loss, and increased sweating. Much less often, patients with significant leukocytosis manifest the symptoms of excessive blood viscosity – priapism, visual disturbances, tinnitus, and consciousness disturbances. There are isolated cases in which the diagnosis is preceded by pain in the left subcostal area caused by infarction, rupture of the spleen, or inflammation of the spleen capsule. Physical examination usually demonstrates splenomegaly.
The results of laboratory tests immediately raise the suspicion of CML. Leukocytosis usually shows the features of microscopic granulocytes at all stages of maturation: metamyelocytes, myelocytes, promyelocytes, and sometimes myeloblasts. This is accompanied by thrombocythemia, basophilia, and/or eosinophilia. Low granulocytic activity of phagocytosis (FAG) is typical of CML and differentiates the condition from other chronic myeloproliferative disorders.
Bone marrow biopsy is the next stage of diagnosis. During this process, material is collected for cytological, cytogenetic, and molecular examinations. Sometimes, immunophenotyping of myeloid cells (to assess the number and characteristics of blasts) and histopathological examination of the bone marrow (optional) are also useful. The myelogram most often reveals a rich-cell bone marrow with an increased percentage of granulopoietic cells at all stages of development.
To make a diagnosis, it is necessary to demonstrate the presence of the Ph chromosome in the karyotype examination or the BCR–ABL transcript by polymerase chain reaction (PCR) (or optionally fluorescence in situ hybridization).
The next stage of diagnosis is to determine the phase of the disease. Most patients at the point of diagnosis are in a CP. However, there are patients who are diagnosed with an acceleration phase (AP) or a BP. Determination of the phase of the disease is important due to the necessity to choose the appropriate treatment. The AP diagnosis criteria were revised in the WHO classification of myeloproliferative neoplasms in 2016.9 Despite these changes, the criteria are still not harmonized and differ significantly, eg, from the European LeukemiaNet (ELN) guidelines, International Bone Marrow Transplant Registry, and MD Anderson Cancer Center (Table 1).10 According to WHO, the AP should be diagnosed in the presence of blasts in the bone marrow or peripheral blood at a percentage of 10%–19%, while according to ELN this range is higher and amounts to 15%–29%. In the majority of studies concerning blast crisis, the criterion for diagnosing this phase is the value of at least 30% of blasts in peripheral blood or bone marrow. According to the WHO criteria, 20% of blasts in the bone marrow or peripheral blood is sufficient to diagnose a blast crisis. The value of 30% of blastic cells, however, seems to better reflect the nature of this phase because the prognosis of patients with a blast percentage between 20% and 29% is better than those with a percentage ≥30%.11 The wide use of tyrosine kinase inhibitors (TKIs) may require a shifting of the boundaries between CP, AP, and BP and to some extent a modification of the classic division of CML into three phases, but the data collected are not yet sufficient to change this definition. However, in comparison with the previous editions of the WHO classification, new parameters have appeared, the presence of which justifies the diagnosis of AP. These include, among others, persistent leukocytosis (>10×109/L) not responding to treatment, persistent splenomegaly not responding to treatment, and additional clonal chromosomal aberrations (so-called major route) in Ph+ cells present at the point of diagnosis. Criteria of persistent leukocytosis and persistent splenomegaly are new for 2017 WHO classification. In WHO 2008 classification, there were: “increasing WBC count not responding to treatment” and “increasing spleen size”. In 2017 there are: “persistent or increasing WBC count (>10×109/L) not responding to treatment” and “persistent or increasing splenomegaly not responding to treatment”. Provisional criteria for the response to the TKI therapy have also been developed. These include hematological resistance to TKI administered in the first-line treatment or the absence of complete hematological response (CHR) during the first-line treatment with TKI; hematological, cytogenetic, or molecular resistance to treatment with another TKI; and the presence of two or more BCR–ABL mutations during the TKI therapy. These changes have meant that the AP should be recognized much more frequently than, eg, that based on the ELN criteria. This is important in particular for patients already being treated with TKI, as it increases the proportion of individuals with indications for allogeneic hematopoietic stem cell transplantation (allo-HSCT). This contrasts with everyday practice and the tendency to marginalize the role of hematopoietic cell transplantation in this disease in the TKI era. The definition of BP is slightly simpler. It is diagnosed when the percentage of blasts in the peripheral blood or bone marrow is ≥30% according to ELN (≥20% according to WHO) and in the case of extramedullary blast proliferation.
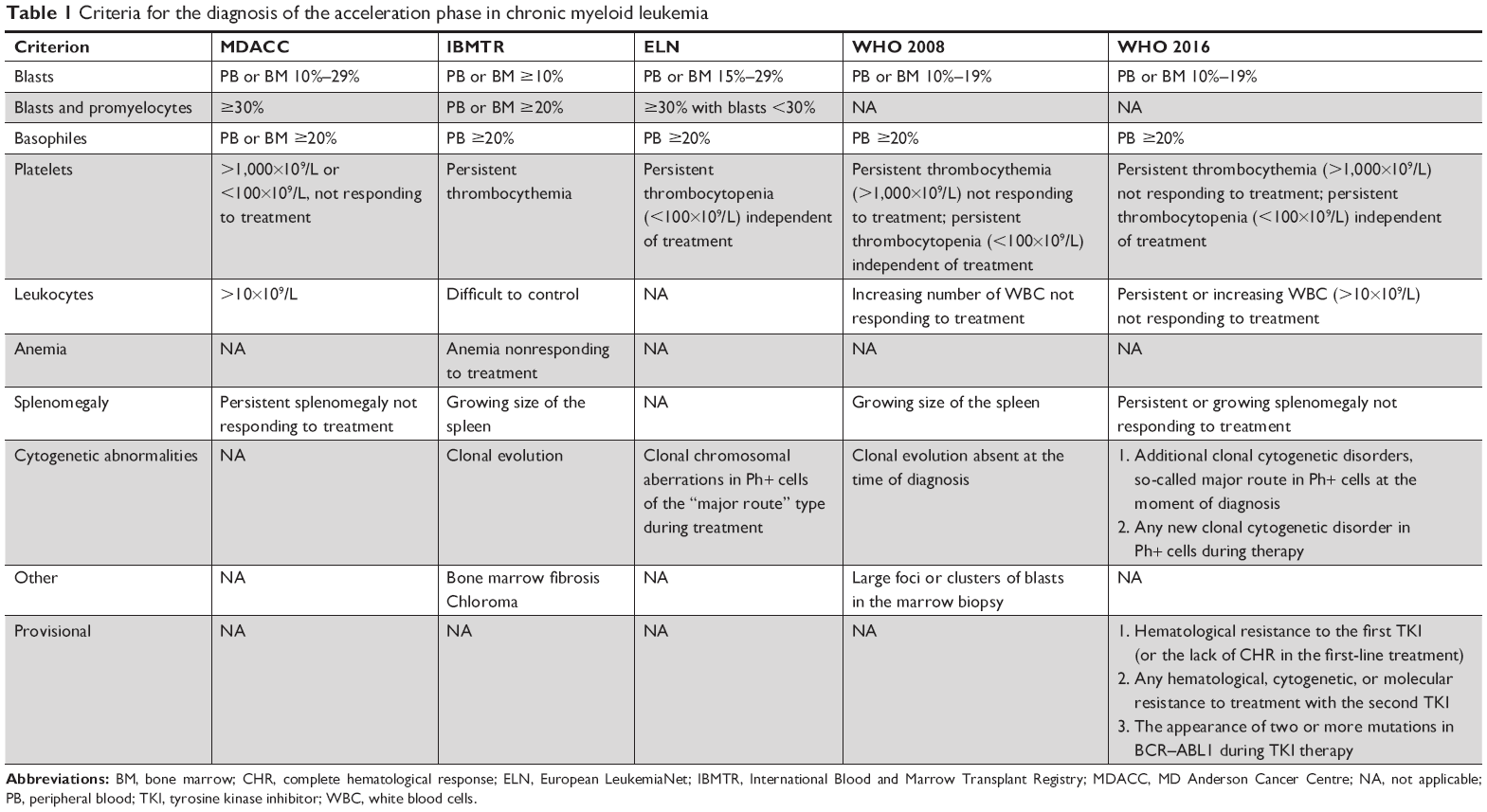 | Table 1 Criteria for the diagnosis of the acceleration phase in chronic myeloid leukemia |
In relation to the diagnostic standard, another important element is the evaluation of prognosis. The most important prognostic factors are of course the phase of the disease at the point of diagnosis and the response to TKI at each stage of treatment. Three prognostic scales are currently administered to determine prognosis: Sokal, Hasford, and EUTOS, with the latter being the most useful and the easiest to administer in clinical practice. The EUTOS scale assesses the probability of 5-year progression-free survival (PFS), stratifying patients into low- and high-risk groups based on two parameters – the size of the spleen and the number of basophiles.12
The size of the spleen is determined by the number of centimeters to which the spleen protrudes from under the left costal arch. The number of basophils is determined by measuring the percentage of these cells in a peripheral blood smear. Finally, the following formula is administered to calculate the risk: spleen size × 4 + basophiles × 7. A value >87 qualifies the patient to a high-risk group. This entails the necessity to take further actions:
- According to ELN 2013, a patient already at the beginning of the treatment should be qualified for the “warning” group (this will be discussed later).
- It is advisable to look for a family donor. This does not mean, of course, that at this stage we should plan for allo-HSCT.13
- According to some researchers, this is an indication for the administration of second-generation TKI as the first-line treatment.14
The newer risk factors evaluated in the further stages of treatment include response to TKI treatment, additional clonal cytogenetic aberrations (CCAs) in Ph+ cells (Ph trisomy, 8, 19, iso17), and genetic markers, eg, CIP2A, transcript type (e14a2 vs e13a2), gene expression profile, genetic polymorphism, mutations within the BCR–ABL gene coding domain, the BCR–ABL transcript level, MDR, and OCT1. Many of these are subjects of current scientific research, and their role is not fully documented.
Some studies based on large cohorts of CML patients treated with TKIs show that some minor-route additional cytogenetic aberrations (ACAs) have significant adverse prognostic value.15,16 This question is still unresolved and requires further research.
On the other hand, it is important to note that older prognostic tools described earlier, such as scales of Sokal, Hasford and EUTOS, are still in use, but their prognostic value appears to be limited in TKI era. Jabbour et al reported in 2012 that the EUTOS score was not predictive for outcome in CML.17 But as for now it is just a single-center experience.
Taken all together the treatment for CML should be started as soon as possible after diagnosis and completion of risk assessment.
Treatment standard
The goals of CML treatment are the prevention of progression of the disease to more advanced stages, reducing the risk of death, extending the patient’s life to the length typical of the general population, and improving the quality of life. These goals are achieved by reducing the number of Ph+ cells as much as possible. The therapy scheme depends on the phase of the disease at the point of diagnosis.
There are three TKIs to choose from for the treatment of patients in the CP: imatinib (IM) (first-generation TKI), and dasatinib and nilotinib (NIL) (second-generation TKIs). IM is administered at a dose of 400 mg/day, dasatinib at a dose of 100 mg/day, and NIL at a dose of 2×300 mg/day. There are no specific recommendations indicating which TKI to choose. In practice, we have to consider several aspects. First, second-generation TKIs are more effective and provide faster and deeper response to treatment.18,19 Therefore, they should be administered in the first-line treatment of patients for whom a deep response is particularly important, eg, young patients. The second group in which the administration of second-generation TKI seems justified is high-risk patients, as defined according to EUTOS. Second, when choosing TKI, we should take into consideration the side effect profile for each of the drugs. The safest of these three is IM. This should be recommended for older patients with comorbidities, for whom improvement in the quality of life is relevant. Dasatinib and NIL are significantly different in terms of their major side effects. During treatment with dasatinib, pleural effusion, congestive heart failure, and pulmonary hypertension are more common. NIL, on the contrary, causes a worsening of pancreatic and hepatic capacity, glucose metabolism disorders, and progression of atherosclerosis, which increases the risk of cardiovascular diseases. Therefore, the metabolic risk should be determined when choosing a second-generation TKI, eg, based on the Pol-SCORE 2015 (or EURO-SCORE) scale. When choosing a TKI, the metabolic risk should be determined and drugs known to have no negative effects on glucose and lipid metabolisms ought to be recommended.20 Third, when choosing a TKI, the expected compliance of the patient with the rules of taking regular medication should be considered. The recommendation to take NIL entails the need for the daily administration of four quite large (21.2 mm long) capsules and the need to abstain from eating for 2 hours before and 1 hour after taking the drug. On the contrary, dasatinib has the form of an oval tablet (15 mm in diameter) administered once, at the same time of the day, regardless of meals.
The diagnosis of the AP or BP creates the need to radically change the treatment strategy. According to ELN 2013, this management is different in newly diagnosed patients and those previously treated with TKI. Among the group of newly diagnosed patients, we should distinguish those with the BCR–ABL coding domain T315I mutation, with the subsequent administration of ponatinib and allo-HSCT, preferably after reaching the CP. These patients may be considered for polychemotherapy including ponatinib. The remaining patients ought to receive IM at a dose of 2×400 mg/day or dasatinib 140 mg/day or 2×70 mg/day (in the blast phase). Allo-HSCT is initially recommended only for patients in BP and those with no optimal response. In this group of patients, polychemotherapy may also be required to achieve control over the disease and to appropriately prepare for allo-HSCT. In patients previously treated with TKI, the progression to AP or BP entails the need to change TKI to any drug that was not administered before the progression (ponatinib only in the presence of the T315I mutation). In this group, according to ELN 2013, allo-HSCT is recommended in all patients, preferably after reaching the CP. Polychemotherapy may be necessary to prepare the patient for transplantation.
The European Society for Blood and Marrow Transplantation guidelines recommend allo-HSCT in the following cases:21
- In patients with suboptimal response or failure of the first-line therapy in the following cases:
- An EBMT risk score of 0–1 (the inclusion of second-generation TKI and transplantation are recommended after obtaining the optimal response);
- An EBMT risk score of 0–4 in cases of a loss of response to IM (hematological or cytogenetic).
- In patients with no hematological response to second-generation TKI, regardless of the EBMT risk score (however, considering mutations within the BCR–ABL coding domain and previous allo-HSCT, these patients may benefit from treatment with third-generation TKI);
- In patients with failure to respond to IM therapies and second-generation TKI treatment-resistant BCR–ABL mutations as well as an EBMT risk score of 0–4;
- In patients in AP or BP after previous preparation with TKI or TKI in combination with polychemotherapy. Transplantation should be performed as soon as possible after reaching the second CP, without the need to achieve a deep cytogenetic or molecular response.
The use of the NCCN guidelines on the progression of the disease to the AP during first-line treatment with TKI has identical consequences. Second-line therapy with another TKI is considered a “bridging” treatment, which makes it possible to achieve an optimal response before HSCT. According to these guidelines, the application of omacetaxine is an alternative.22,23
Experts from Hammersmith Hospital in London presented a similar, somewhat more intuitive approach to the topic of qualifying patients to HSCT after failure of first-line therapy (Table 2).24
 | Table 2 Indications for allo-HSCT in chronic myeloid leukemia – the stance of experts from Hammersmith Hospital |
This brief analysis of treatment strategies in patients with advanced stages of the disease (AP and BP) indicates that this significantly differs from the treatment regimen of CP patients. More important is the unification of the AP and BP diagnostic criteria. The second important conclusion from this analysis relates to the effectiveness of AP and BP treatment with TKI. When IM is administered in the blast phase at a dose of 800 mg/day, complete hematological remission is achieved only in 25% of patients and 3-year PFS in 10%. In patients in the myeloid blast crisis (MBP) treated with dasatinib at 140 mg/day in one dose, a major cytogenetic response (MCyR) is achieved by 25% of individuals and by 28% of those receiving dasatinib in two doses (2×70 mg/day). In patients in lymphoblastic crisis (LBP), MCyR is reached by 50% and 40%. Twenty-four-month overall survival (OS) is achieved by 24% and 28% of patients with MBP and 21% and 16% of those with LBP, respectively.25 These data have encouraged researchers to seek new molecular-oriented drugs that are an alternative to TKI.
Treatment of the second and subsequent lines is administered when the first-line therapy fails or the patient develops intolerance. The choice of the subsequent method of treatment is governed by additional rules:
- In the event of failure of first-line treatment with IM or its intolerance, we can administer dasatinib 100 mg/day, NIL 2×400 mg/day, or bosutinib 500 mg/day.
- In the event of failure of first-line treatment with dasatinib or its intolerance, we can administer NIL at a dose of 2×400 mg/day or bosutinib at a dose of 500 mg/day.
- In the event of failure of first-line treatment with NIL or its intolerance, we can administer dasatinib 100 mg/day or bosutinib 500 mg/day.
- Allo-HSCT is recommended only if first-line treatment with all available TKIs fails and the disease progresses to AP or BP, as well as in patients with the T315I mutation. This point does not administer to TKI intolerance. In subjects eligible for allo-HSCT, an attempt should be made to treat them with second-generation TKI and continue the treatment if: i) a minimal cytogenetic response (percentage of Ph+ cells <95%) is achieved after 3 months; ii) complete cytogenetic response (CCyR) is achieved after 6 months; and iii) major molecular response (MMR) is achieved after 1 year.
If this treatment is unsuccessful, allo-HSCT should be performed. - In the case of failure of second-line treatment with dasatinib or its intolerance, third-line treatment can be administered with NIL at a dose of 2×400 mg/day, bosutinib at a dose of 500 mg/day, or ponatinib at a dose of 45 mg/day.
- In the case of failure of second-line treatment with NIL or its intolerance, in the third line we can administer dasatinib at a dose of 100 mg/day, bosutinib at a dose of 500 mg/day, or ponatinib at a dose of 45 mg/day.
- In the case of failure of second-line treatment with bosutinib or its intolerance, in the third-line therapy we can administer NIL 2×400 mg/day, dasatinib 100 mg/day, or ponatinib 45 mg/day.
- Allo-HSCT is recommended for all patients after failure of second-line therapy. This point does not apply to cases of intolerance to TKI.
- An attempt to treat with IM at a dose of 600–800 mg/day can be made in patients who are IM-resistant at a dose of 400 mg/day, have multiple comorbidities, are at high risk of second-generation TKI toxicity, and have good tolerance of IM.
Assessment of responses to treatment
The evaluation of the degree of remission at individual stages of treatment is important for the implementation of therapeutic goals. Treatment effects should be monitored every 3 months using a cytogenetic test (especially relevant at the beginning of treatment) and/or quantification of the BCR–ABL transcript. According to ELN (the guidelines from 2013), patients are qualified to three groups depending on the degree of remission at specific time points. These groups are “optimal,” “warning,” and “failure.” The “optimal” group includes patients responding well to treatment, ie, achieving CHR (WBC <10 G/L, PLT <450 G/L, no immature granulocytes in peripheral blood smear, no splenomegaly in the palpation examination, and basophiles <5% in peripheral blood smear) and with a reduction of the BCR–ABL transcript ≤10% in International Units (IS) and/or with a percentage of Ph+ cells ≤35% of all metaphases after 3 months of treatment, a reduction of BCR–ABL transcript amount <1% IS and/or CCyR after 6 months of treatment, and a decrease in the BCR–ABL transcript ≤0.1% IS (MMR) after 12 months of treatment.26 The condition that is necessary to qualify the patient to the “optimal” group at the later stages of treatment is the maintenance of MMR. The lack of CHR and/or the percentage of Ph+ cells >95% after 3 months of therapy, the presence of BCR–ABL transcript >10% IS and/or the percentage of Ph+ cells >35% after half a year, and the failure to reach CCyR and/or the amount of BCR–ABL transcript >1% after 1 year of therapy indicate the failure of treatment and the need to change the management program. At the later stages of treatment, therapeutic failure should be recognized in cases of CHR and/or CCyR loss, which are confirmed in two successive MMR (including one BCR–ABL test ≥1% IS) and the appearance of additional CCAs in Ph+ cells (CCA/Ph+). In all intermediate cases, which do not qualify the patient to the “optimal” group, but where at the same time there are no reports of treatment failure at any stage, patients are classified as “warning.” This group also includes high-risk cases at the point of diagnosis (as previously described), patients with the presence of CCA/Ph+ type “major route” [+8, +19, additional Ph chromosome (1der(22) t(9;22) (q34;q11)), isochromosome 17 (i(17) (q10)), and 1der(22) (q10) t(9;22) (q34;q11)] before the beginning of treatment and the appearance of monosomy 7 or del7q at later stages of treatment. The appearance of −7 or del7q in Ph− cells may indicate an increased risk of myelodysplasia and blastic transformation, and this is associated with the need to monitor bone marrow cytology. CCA, which appears in Ph− (CCA/Ph−) cells in 5%–10% of patients, is not considered an adverse prognostic factor.
In recent years, particular attention has been paid to the so-called early molecular response (EMR), defined as the amount of BCR–ABL transcript ≤10% IS after 3 months of treatment. The lack of EMR is associated with a lower rate of event-free survival, PFS, OS, as well as with an increased risk of transformation to AP and BP.27 A reduction of the amount of transcript ≤1% IS after 3 months of therapy gives a greater chance to achieve MMR and deep molecular response. This decrease is significantly more common after first-line treatment with second-generation TKI. According to NCCN, the lack of EMR is an indication of the need to test for the presence of BCR–ABL coding domain mutations. It is recommended to repeat the test preferably after 1 month to ensure that an increase in the amount of transcript expresses the actual trend. It can be assumed that the change of the drug to second-generation TKI in the absence of EMR is crucial for the success of second-line therapy. The definitions of treatment responses are the same for patients in CP, AP, and BP for first-line treatment and second-line therapy, but only if the former was changed due to drug intolerance.
Similar rules as in first-line treatment (“optimal,” “warning,” and “failure” groups) are administered to assess the response to second-line treatment in the case of IM treatment failure. However, these criteria are slightly less restrictive.
In CP, a deeper response beyond CCyR/MMR confers no survival benefit but may be one step closer to TKI discontinuation. But in BP of CML, it is probably different. In the study based on large cohort of CML patients in BP, the authors showed that there are significant differences in OS between groups of patients with different depth of response. Patients who achieved molecular undetectable leukemia status had a better survival than those who achieved MMR. The results are similar for patients with myeloid BP and for patients with lymphoid BP.28
TKI withdrawal
Many clinical trials to evaluate the impact of TKI withdrawal in specific groups of patients on long-term treatment are currently underway. Withdrawal of TKI is a particularly interesting option, eg, for women planning pregnancy. It is also justified from an economic point of view. Such an attempt can be made only if a prolonged (at least 1 year) and deep molecular response (at ≥MR4) is obtained in patients treated for at least 3 years. The results of these tests are encouraging.12 Studies on IM withdrawal have shown that ~40% of patients maintain the same depth of molecular response in long-term observations. Importantly, almost all patients who relapsed after the TKI withdrawal achieved equally deep molecular response after returning to IM therapy.29 However, all major global guidelines allow for TKI withdrawal only as part of a clinical trial. The initial results of clinical trials focused on the withdrawal of second-generation TKI are similar and equally encouraging.30
A British group has recently proposed another de-escalation strategy. The TKI dose was reduced by half in subjects with a stable deep molecular response. Among 174 subjects, only 12 had a relapse, and all of these 12 achieved a deep molecular response after returning to the standard treatment.31
Clinical trials conducted or being carried out for CML
There are many clinical trials on CML in the world. Few of them apply to the first-line treatment, where TKIs have an established position. Most of them focus on treatment-free remission or TKI holiday for improved tolerability. Another group of clinical trials in CML is research on optimizing treatment after relapse. They concern mainly bosutinib and ponatinib. The effectiveness of bosutinib is also studied in specific groups of patients, eg, in pediatrics or older patients. In France, there is ongoing study on the combination of TKI and sequential therapy with TKIs in patients with CML-CP. However, there are a lot of studies on the use of new drugs in combination with TKIs or alone. As an example, let us use research on PF-114 (in Russia), ABL001 and K0706 (in many countries), TALL-104 (ClinicalTrials.gov identifier: NCT00415909), BAY43-9006 (sorafenib), AP24534, and Zileuton (in USA, ClinicalTrials.gov identifier: NCT02047149). There are many clinical studies that check the effectiveness of TKI in combination, eg, with pegIFNα, venetoclax, ruxolitinib (RUX), pioglitazone, and DNA methyltransferase inhibitor such as decitabine (Table 3).
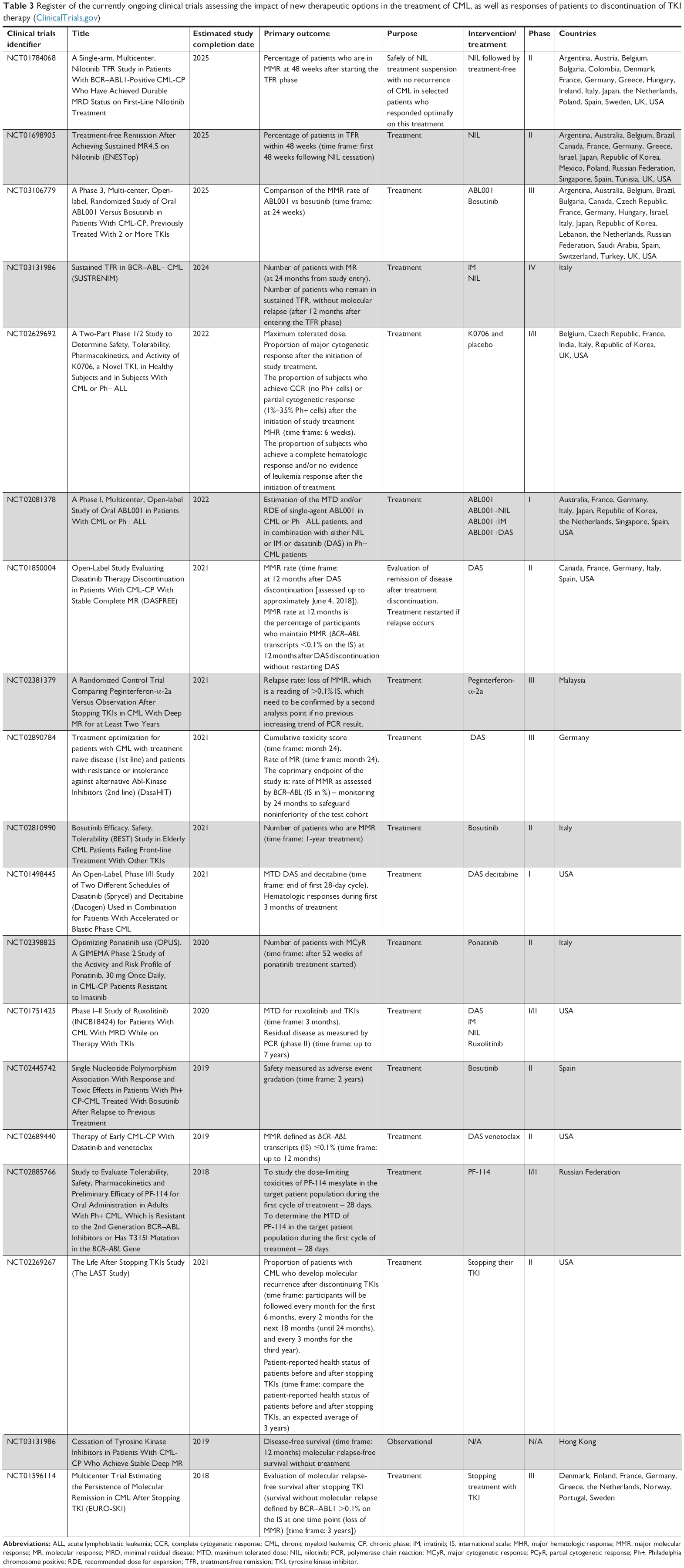 | Table 3 Register of the currently ongoing clinical trials assessing the impact of new therapeutic options in the treatment of CML, as well as responses of patients to discontinuation of TKI therapy (ClinicalTrials.gov) |
Role of BCR–ABL kinase in deregulation of crucial cell signaling pathways
The major cellular events initiated by BCR–ABL kinase important in the malignant transformation include altered adhesion to stroma cells and the extracellular matrix, constitutively active mitogenic signaling, reduced apoptosis, and altered DNA repair mechanisms. However, some new factors such as elevated levels of reactive oxygen species (ROS) and autophagy seem to play important roles in CML development and progression.
Neoplastic transformation, which occurs through the BCR–ABL, is associated with constitutive activation of the Ras/MAPK signaling pathway. The Ras protein activates the serine–threonine Raf kinase, which initiates a signaling cascade through serine–threonine kinases Mek1/Mek2 and Erk1/Erk2, belonging to MAPKs. Kinases migrate to the cell nucleus where phosphorylation and activation of transcription factors such as c-Jun, c-Myc, and c-Fos occur, resulting in expression of the genes responsible for proliferation processes.3 On the contrary, to sustain the proliferative properties of BCR–ABL-positive cells, an active PI3K is required. PI3K is activated by forming multimeric complexes with BCR–ABL, Cbl, and adapter molecules such as Crk and Crkl. This leads to activation of another substrate of the cell signaling cascade such as serine–threonine kinase Akt.32
An active Akt kinase promotes cell survival by phosphorylation of proapoptotic Bad protein at serine 136. Phosphorylated Bad is trapped and maintained in the complex with 14-3-3 protein; therefore, it cannot interact with antiapoptotic proteins such as Bcl-2 and BCl-xL. In this situation, free BCl-xL binds to the proapoptotic Bax protein preventing the formation of proapoptotic Bax homodimers in the mitochondrial outer membrane. There is no promotion of mitochondrial outer membrane permeabilization or the release of proapoptotic factors such as cytochrome c and SMAC/DIABLO from the mitochondria into the cytosol to activate the caspase cascade. Moreover, Akt kinases may directly phosphorylate caspase-9 on serine 196 and thereby prevent Apaf-1/cytochrome c activation of caspase-9. This activity of Akt is tightly linked with BCR–ABL kinase activity. Additionally, BCR–ABL increases the expression of antiapoptotic Bcl-2 family members such as Bcl-2 and Bcl-xL, probably through direct activation, without the involvement of Janus kinases (JAK), of the transcription factor STAT5.33–35 Pleiotropic action of BCR–ABL kinase on the PI3K/AKT, MAPK, and other signaling pathways results in changes in ROS regulation in HSC. Under physiological conditions, these pathways support the residence of HSC in bone marrow/osteoblastic niches in hypoxia and low ROS levels. An upregulation of signaling pathways by the constitutive activity of BCR–ABL leads to elevation of ROS levels. This results in nonspecific oxidative damage to biomolecules such as oxidative DNA damage, single- and double-stranded DNA breaks (DSB), as well as DNA repair proteins leading to increased genomic instability.36 Genomic instability acts as a mutator, ie, this has a potential to create other mutations. This is closely connected with the incorrect action of mechanisms of DNA repair such as nonhomologous end joining (NHEJ) and homologous recombination, which are responsible for DSB repair. Unfortunately, both mechanisms work with an increased frequency of error leading to mutations. However, some lines of evidence show that in CML cells the key players of NHEJ, Artemis, and DNA ligase IV are downregulated. In this situation, an alternative pathway of NHEJ (a-NHEJ) is activated, involving DNA ligase IIIα (LIG3) and the protein deleted in Werner’s syndrome, a protein that is upregulated in CML cells. This suggests that as long as the a-NHEJ pathway contributes to ROS-induced DSB repair the genomic instability increases, leading to the progression of the disease from the chronic to the BP.37,38 Additionally, it was shown by Muvarak et al that c-MYC transcription factor is a key downstream target of BCR–ABL kinase. It was demonstrated that c-MYC binds to the promoter regions of both LIG3 and poly(ADP-ribose) polymerase 1 (PARP1), thereby increasing transcriptional activity, leading to an increased a-NHEJ in BCR–ABL-positive cells. Thus, the authors showed other interactions of BCR–ABL kinase, which led to genomic instability through increased expression of proteins essential for an error-prone alternative NHEJ.39
The pivotal deregulated control mechanisms of cells in cancer development are apoptosis and autophagy processes. The former has been briefly described in this paper, while it is described in more detail in the scientific literature. The latter process in recent years has received more attention from researchers.
Autophagy (“self-eating”) is a fundamental process necessary for cell survival and maintenance of homeostasis, and it is activated as a result of various intrinsic and extrinsic cellular stress conditions, such as nutrient deprivation, oxidative stress (ROS accumulation), DNA damage, and hypoxia. Activation of autophagy serves to clear the damaged protein aggregates and impaired organelles as well as intracellular pathogens. So far, the role of autophagy in tumor suppression has also been unquestioned. However, some lines of evidence suggest the dualistic mechanism of action of autophagy, ie, that autophagy can prevent tumor initiation as well as play a protective role in enhancing the survival of drug-treated cells.40
Generally, the main key players of autophagy are Beclin-1, hVps34, and/or Ulk1 complexes. The first of these is activated in response to cellular stress (eg, ROS), while the last is regulated by the metabolic status (eg, starvation) of the cells regulated by proteins such as mammalian targets of rapamycin (mTOR) complex 1 (mTORC1) and AMP-activated protein kinase.
In CML cells, autophagy is downregulated by constitutively activated BCR–ABL kinase. The mechanism responsible for this inhibition correlates with upregulation of the mTOR pathway. mTOR is an atypical serine/threonine protein kinase belonging to the PI3K-related kinase family. This molecule interacts with several proteins to form two distinct complexes, referred to as mTORC1 and mTOR complex 2. The major negative regulator of autophagy seems to be mTORC1, which interacts with and phosphorylates Ulk1, the mammalian uncoordinated-51-like protein kinase (Figure 2).41 Sheng et al showed that BCR–ABL, through the PI3K/Akt pathway, stimulates mTORC1 kinase that is required for autophagy inhibition. Moreover, the authors revealed that PI3K/Akt signaling also stimulates mTOR activity through a complex protein phosphorylation pathway that involves several other components including tuberous sclerosis complex proteins TSC1 and TSC2. This means that the regulation of mTORC1 kinase activity by BCR–ABL occurs on both the transcriptional and the protein level.44
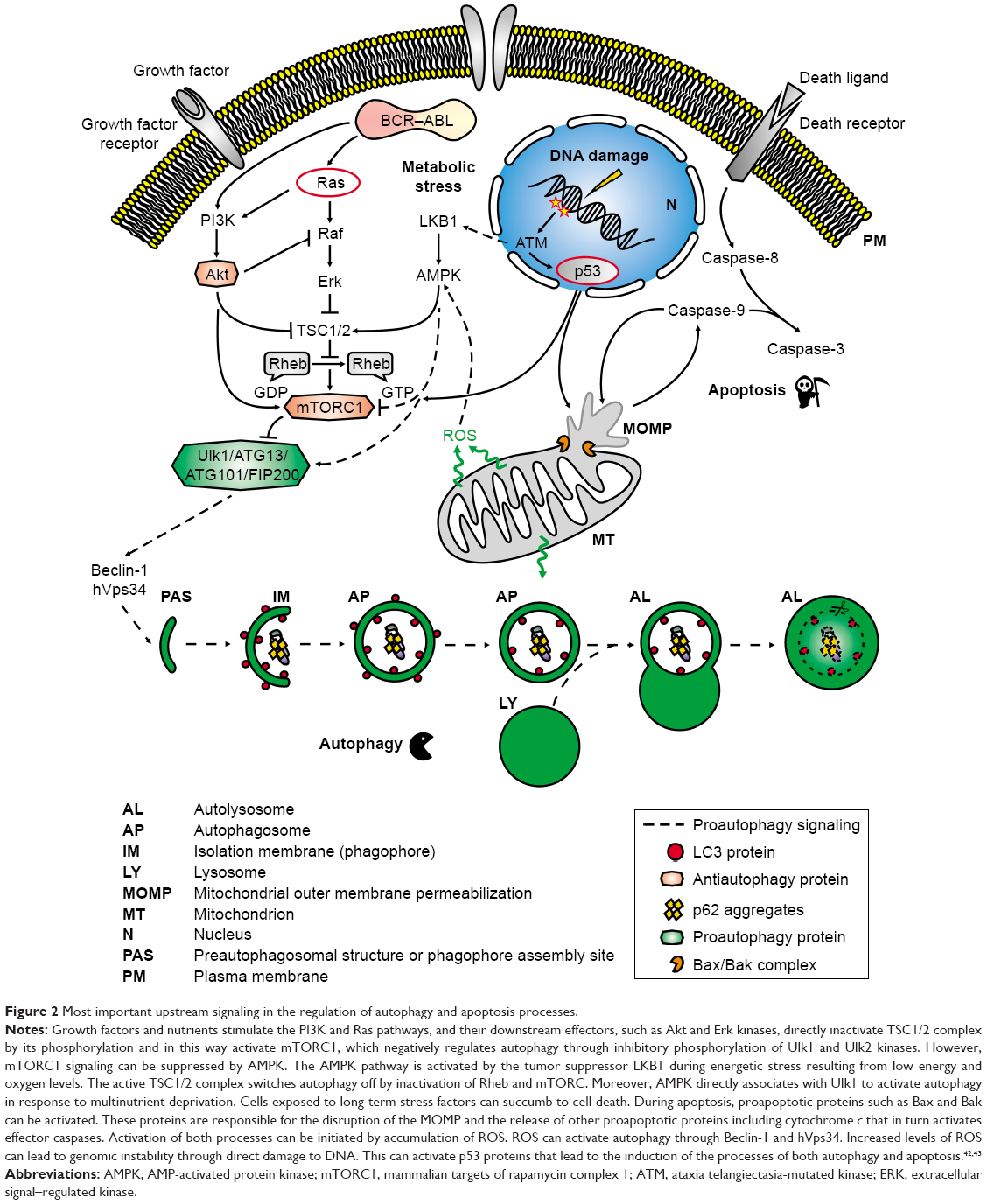 | Figure 2 Most important upstream signaling in the regulation of autophagy and apoptosis processes. |
All described changes/deregulation in the signaling pathways or cellular processes are important for proper cell function and development or progression of diseases. Decoding of the characteristic aberrations of particular disease units is the key to their treatment. A breakthrough in the treatment of CML patients was the introduction of BCR–ABL TKIs, which significantly improved and prolonged the lives of patients. Unfortunately, TKIs, contrary to expectations, do not eliminate the disease. The complicated relationship/dependence of changes in cellular signaling, resulting from the emergence of a mutation in the course of the disease, has produced the need to search for new therapeutic options.
Stem cells – a key therapeutic goal
The theory of the cancer stem cell (CSC) was postulated in the 1970s on the example of CML, which proved that initiation of the disease starts at the level of HSCs by activity of BCR–ABL kinase. CSCs are a subpopulation of cancer cells that are capable of self-renewal, proliferation, and differentiation. However, these cells are also responsible for cancer progression and clinical outcome in cancer patients.45 As mentioned in the previous section above, CML is an LSC-derived disease. LSCs (CD34+38−) are able to generate large numbers of LPCs (CD34+38+), which do not possess the ability to self-renew and differentiate into mature cells. These cells are mainly eliminated in CML-CP patients during TKI therapy, whereas LSCs can persist and may be responsible for treatment failure and relapse. Patients with CML-CP can initially have ~5×107 leukemia cells demonstrating primary resistance to IM, and even PCR-negative patients may harbor up to 106 malignant cells.46 LSCs seem to be the main target of CML therapy, but to develop a proper strategy the mechanism underlying disease persistence has to be understood. Much of the information concerning the modulation of various signaling pathways by BCR–ABL kinase activity has been described. LSCs have a survival advantage and are able to escape cell death in response to drug treatment. Therefore, research teams are working on novel drugs against targets in key signaling pathways being under control of BCR–ABL activity. In the future, these drugs could be administered alone or in combination with TKIs. In this part of the review, we would like to further highlight few key signaling pathways, deregulation of which is important for maintaining the CML stem cell survival.
As was mentioned above, activated BCR–ABL drives a variety of downstream prosurvival, growth, and antiapoptosis signaling pathways. Development of new molecules or compounds can generate new possibilities in CML treatment. Consideration of molecules downstream of BCR–ABL as additional therapeutic targets would seem to be a sound approach. Therefore, direct inhibition of proteins or their synthesis has been the target of extensive research.
Xin et al showed that NVP-BEZ235, the dual inhibitor of PI3K and mTOR, inhibits the growth of CML cells by inducing cell cycle arrest in the G0/G1 phase, inducing apoptosis and reactivating autophagy in K562 cells. NVP-BEZ235 in combination with IM inhibits the growth of K562 cells as well as the primary T315I mutant cells KBM7R. NVP-BEZ235 also suppresses colony formation by murine cells derived from mice that have undergone reconstitution with HSC transduced with BCR–ABL.47 Similar effects have been observed for other compounds affecting the PI3K/AKT/mTOR pathway such as GDC0941, a pan PI3K inhibitor, and KU-0063794, a dual mTORC1/2 inhibitor.48
An interesting approach in CML treatment seems to be the possibility of using tigecycline, a glycylcycline class of antibiotic, which induces autophagy in CML cells by downregulating the PI3K/AKT/mTOR signaling pathway. On the contrary, combination of tigecycline with chloroquine, an autophagy inhibitor, enhances the cytotoxic effect of the former. Moreover, knockdown of ATG5 gene by siRNA to ablate autophagy at the genetic level significantly increases the sensitivity of CML cells with T315I point mutation to the inhibitory effect of tigecycline.49
Due to the “double-face” of autophagy, it is difficult to unequivocally state which therapeutic approach, enhancement, or inhibition of this process will be the best in CML treatment. Autophagy is used by various types of cancer cells as a defense mechanism to evade apoptosis. For CML, treatment with TKI induces autophagy in LSC as a survival mechanism.50 Inhibition of autophagy by RNA interference-mediated knockdown of either ATG5 or ATG7 genes, encoding proteins belonging to the ubiquitin-like conjugation system, which is required for preautophagosome expansion and completion,51 results in sensitization of K562 cells to IM-induced apoptosis as well as enhancing the colony formation inhibitory effect of IM to CML CD34+ cells.52
Therefore, blocking of this process seems to be an attractive therapeutic target for eradicating LSC, especially in combination with TKI treatment.
JAK2 kinase is also a popular pharmacological target in CML. Okabe et al demonstrated that inhibition of JAK2 by either JAK2 siRNA transfection or the JAK2 inhibitor TG101348 downregulated STAT5 activity and enhanced IM efficacy even when leukemia cells were protected by hematopoietic stromal cell-mediating cytokine/chemokine production.53 Gallipoli et al provided preclinical evidence for effective targeting of JAK2 in CML CD34+ cells using the JAK2 inhibitor RUX in combination with the TK inhibitor NIL. Treatment with a combination of RUX and NIL led to inhibition of JAK2/STAT5 activity, as shown by the correlative changes in both p-JAK2 and p-STAT5 levels, associated with correlative changes in STAT5 target genes. The authors also showed that a combination of RUX and NIL led to a reduction in the viability, colony output, and proliferation of CML CD34+ cells in vitro and reduced engraftment of CML CD34+ cells in vivo.54
Interesting results were also obtained by Yeh et al. These authors showed that a novel microtubule inhibitor, MPT0B002, causes growth inhibition and apoptosis induction in wild-type BCR–ABL and T315I-mutated BCR–ABL. The authors showed that MPT0B002 significantly inhibits the phosphorylation of BCR–ABL, Crkl, and STAT5 proteins in both cell types. This suggests that MPT0B002 may reduce downstream signaling of BCR–ABL. However, the important conclusion of this paper is that MPT0B002 overcomes IM resistance caused by T315I mutation.55
Numerous studies have demonstrated essential role of the Wnt/β-catenin pathway in the HSC self-renewal as well as in the pathogenesis of CML especially in the transition from chronic to blastic phase, what may be connected with higher level of β-catenin expression. There are a number of preclinical data showing that BCR–ABL directly stabilizes β-catenin through its kinase activity. Reduction of β-catenin level by inhibition of prostaglandin E2 level as a result of inhibition of cyclooxygenase-2 activity decreases CML cell resistance to TKIs. Such observation suggests requirements of β-catenin for the maintenance of TKI-resistant LSC.56 These results imply that inhibition of β-catenin by small molecules or genetic ablation could be of some interest in clinical approach. Currently, PRI-724 agent is under intense investigations as the potent Wnt/β-catenin inhibitor. The in vitro and in vivo studies provide evidence that PRI-724 by inhibition of β-catenin restores sensitivity to TKI in TKI-resistant BCR–ABL gene-mutated CML cells.57 Currently, PRI-724 is being studied to treat subjects with advanced myeloid malignancies (ClinicalTrials.gov identifier: NCT01606579) in combination with dasatinib.58 From the point of view of new therapeutic possibilities, it is also worth to mention about ectopic viral integration site 1 (EVI1) protein. Experimental and clinical studies have provided evidence that EVI1 activation enhanced self-renewal capacity to HSC, leading to leukemia development. In the case of CML, the enhanced expression of EVI1 gene resulting from its translocation seems to be responsible for the disease progression to blastic phase.59 Sato et al using single-cell analysis of primitive or differentiated subsets from primary CML-CP samples showed that EVI1 is highly expressed in stem cell-enriched CD34+ CD38– cells. On the contrary, the authors showed that EVI1-high CML progenitor cells have a superior proliferative potential in vitro, a superior leukemia-initiating capacity in vivo, and NIL resistance. This latter finding possesses important clinical meaning that high EVI1 is related to TKI resistance; therefore, EVI1 may be considered as a valuable therapeutic target for CML stem cells.60
Another major pathway influencing the development of CML is the hedgehog (Hh) signaling pathway, which is involved in a number of cellular mechanisms such as stem cell renewal, proliferation, and survival. A fundamental component of this pathway is the smoothened (SMO) transmembrane protein. It was also demonstrated that Hh signaling is increased in BCR–ABL-positive progenitor cells and that it is further upregulated with disease progression.61 Irvine et al have shown that targeting the Hh pathway using the SMO inhibitor LDE225 in combination with NIL is an effective strategy for targeting CP-CML LSC. The authors highlighted that the heterogeneity of Hh pathway gene expression and response to the SMO inhibitor LDE225 indicate that further studies are required, especially for identifying a proper subpopulation of patients who would be more likely to benefit from combination therapy of SMO inhibitor and TKI and perhaps with lower toxicity.62
Instead of genetic alteration in leukemogenesis, the epigenetic modifications such as DNA methylation, histone modifications, and miRNA play a pivotal role in changes of gene expression. In CML, these epigenetic processes are known to be critical for stem cell maintenance and development. Abnormal hypermethylation of ABL promoter appears to correlate closely with clonal evolution and is thought to reflect the genetic instability of the highly proliferative CML progenitors. Many results indicate that hypermethylation of ABL promoter may be a marker of disease progression and a kind of factor initiating changes in the expression of other genes.63 Also tumor suppressor gene MTSS1 is downregulated by higher DNA methylation level at its promoter in LSC and progenitor cells. Repression of MTSS1 that probably promotes CML LSC and progenitor cell proliferation and motility can be reversed by the 5-azacytidine demethylating agent as shown in in vitro studies conducted on leukemic cell lines. Therefore, the use of compounds that inhibit DNA methyltransferase activity is considered as another therapeutic target in CML. The situation is similar with the use of inhibitors for acetyl- or methyl-transferases (HATs and HMTs) as well as histone deacetylases or demethylases (HDACs and HDMs), enzymes responsible not only for biochemical post-translational histone modifications of genes responsible for survival and proliferation signaling pathways but also for the modification of proteins important for basic cellular processes. In CML, a number of these histone tagging systems at specific amino acid residues are dysregulated. It was demonstrated that Sirtuin 1 deacetylase (SIRT1), which regulates the acetylation of several transcription factors (ie, p53 and FoxOs), is able to promote the acquisition of genetic mutations implicated in drug resistance and survival of CML LSCs.64,65 It was shown that altering the balance between HAT and HDAC activity in CML cells may increase acetylation of p53 protein at residue, blocking its translocation to the cytoplasm upon DNA damage and in this way prevent mitochondrial-dependent induction of apoptosis. Table 4 summarizes the well-established potential therapeutic targets in CML stem cells. Part of them is investigated in the clinical trials (Table 4). Searching for the new combination of non-ABL-directed inhibitors with TKIs seems to be a very important issue. The main reason for such approach is a high effectiveness of TKIs, which are well tolerated by most patients, against the bulk of leukemic cells but not for LSCs, which are independent from BCR–ABL activity. The selected results presented above carry significant meaning in understanding the mechanisms of the development and progression of CML disease as well as the possibilities for its treatment. That is why combining TKIs with compounds that work on other molecular targets turns out to be so essential. This type of therapy may give a better chance of successful treatment, consisting of eliminating not only differentiating progenitor cells but first of all LSCs responsible for the failure of therapy and relapse of the disease.
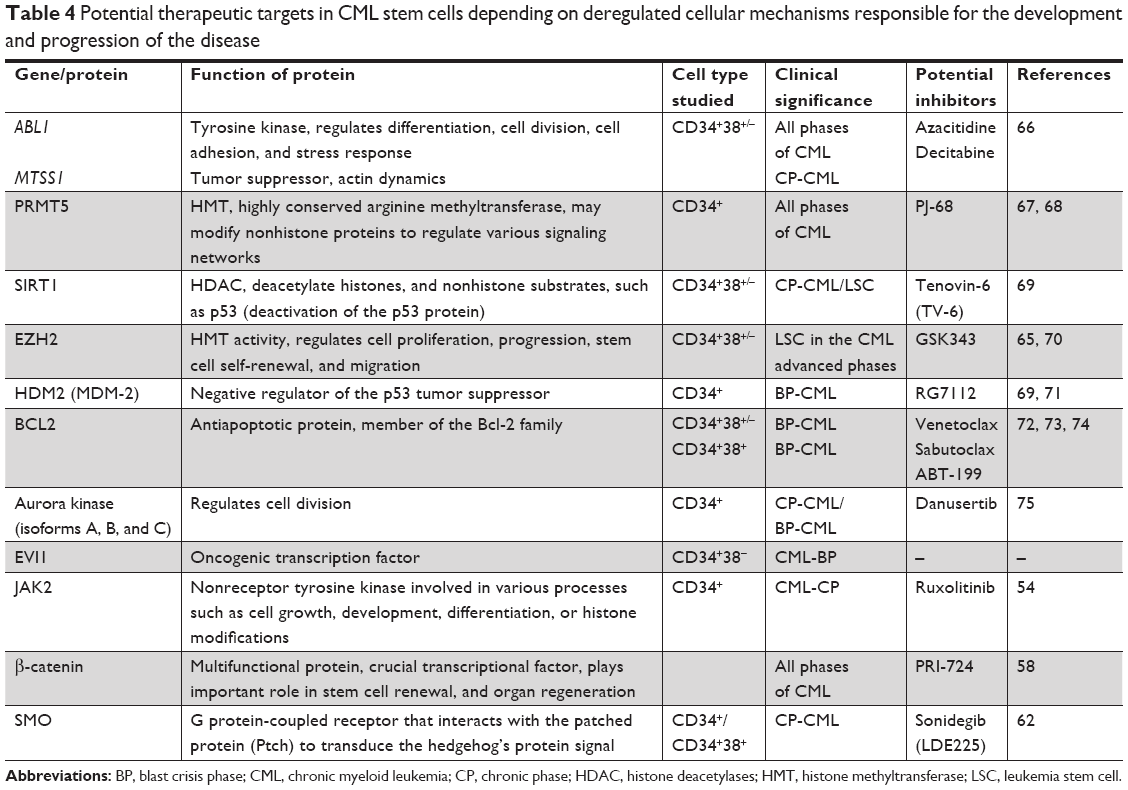 | Table 4 Potential therapeutic targets in CML stem cells depending on deregulated cellular mechanisms responsible for the development and progression of the disease |
The results selected from many of the papers published in recent years seem to present promising compounds against CML and carry significant meaning in understanding the mechanisms of CML development and progression as well as the possibilities for its treatment. Despite the success of TKIs, the search for new therapeutic options in this disease entity is still important due to the emergence of primary or secondary resistance to treatment in patients and the difficulty of eradicating CML stem cells as the main “culprit” of the disease. Therefore, CML is still an important area of research aimed at improving the effectiveness of patient treatment.
Acknowledgment
This work was supported by the National Science Centre of Poland (grant no 2013/11/B/NZ7/02248; http://www.ncn.gov.pl/).
Disclosure
The authors report no conflicts of interest in this work.
References
Soverini S, De Benedittis C, Mancini M, Martinelli G. Best practices in chronic myeloid leukemia monitoring and management. Oncologist. 2016;21(5):626–633. | ||
Goldman JM. Chronic myeloid leukemia: a historical perspective. Semin Hematol. 2010;47(4):302–311. | ||
Deininger MW, Goldman JM, Melo JV. The molecular biology of chronic myeloid leukemia. Blood. 2000;96(10):3343–3356. | ||
Bruns I, Czibere A, Fischer JC, et al. The hematopoietic stem cell in chronic phase CML is characterized by a transcriptional profile resembling normal myeloid progenitor cells and reflecting loss of quiescence. Leukemia. 2009;23(5):892–899. | ||
Shet AS, Jahagirdar BN, Verfaillie CM. Chronic myelogenous leukemia: mechanisms underlying disease progression. Leukemia. 2002;16(8):1402–1411. | ||
Żołnierowicz J, Kawiak J, Hoser G. Pathogenesis of chronic myeloid leukemia – from gene to targeted therapy. Hematologia. 2010;1:195–218. | ||
Marley SB, Gordon MY. Chronic myeloid leukaemia: stem cell derived but progenitor cell driven. Clin Sci (Lond). 2005;109(1):13–25. | ||
Perrotti D, Jamieson C, Goldman J, Skorski T. Chronic myeloid leukemia: mechanisms of blastic transformation. J Clin Invest. 2010;120(7):2254–2264. | ||
Arber DA, Orazi A, Hasserjian R, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. 2016;127(20):2391–2405. | ||
Baccarani M, Deininger MW, Rosti G, et al. European LeukemiaNet recommendations for the management of chronic myeloid leukemia: 2013. Blood. 2013;122(6):872–884. | ||
Hehlmann R. How I treat CML blast crisis. Blood. 2012;120(4):737–747. | ||
Hasford J, Baccarani M, Hoffmann V, et al. Predicting complete cytogenetic response and subsequent progression-free survival in 2060 patients with CML on imatinib treatment: the EUTOS score. Blood. 2011;118(3):686–692. | ||
Sacha T, Lewandowski K, Hellmann A, et al. Polish adult leukemia Group recommendations for diagnosis and treatment of chronic myeloid leukemia in 2013. Acta Haematol Pol. 2013;44:345–362. | ||
Műller M. The role of second-generation TKIs as first-line treatments in CML. In: EXcel Preceptorship. Interactive Forum on CML; September 16–17, 2013; Mannheim, Germany: University Medical Center; 2013. | ||
Gong Z, Medeiros LJ, Cortes JE, et al. Cytogenetics-based risk prediction of blastic transformation of chronic myeloid leukemia in the era of TKI therapy. Blood Adv. 2017;1(26):2541–2552. | ||
Wang W, Cortes JE, Tang G, et al. Risk stratification of chromosomal abnormalities in chronic myelogenous leukemia in the era of tyrosine kinase inhibitor therapy. Blood. 2016;127(22):2742–2750. | ||
Jabbour E, Cortes J, Nazha A, et al. EUTOS score is not predictive for survival and outcome in patients with early chronic phase chronic myeloid leukemia treated with tyrosine kinase inhibitors: a single institution experience. Blood. 2012;119(19):4524–4526. | ||
Larson RA, Hochhaus A, Hughes TP, et al. Nilotinib vs imatinib in patients with newly diagnosed Philadelphia chromosome-positive chronic myeloid leukemia in chronic phase: ENESTnd 3-year follow-up. Leukemia. 2012;26(10):2197–2203. | ||
Kantarjian HM, Shah NP, Cortes JE, et al. Dasatinib or imatinib in newly diagnosed chronic-phase chronic myeloid leukemia: 2-year follow-up from a randomized phase 3 trial (DASISION). Blood. 2012;119(5):1123–1129. | ||
Sacha T, Szmit S, Zozulińska-Ziółkiewicz D, Prejzner W, Góra-Tybor J. Recommendations for assessment of co-morbidities and tyrosine kinase inhibitor choice in patients suffering from chronic myeloid leukemia. Acta Haemat Pol. 2016;47:184–196. | ||
Sureda A, Bader P, Cesaro S, et al. Indications for allo- and auto-SCT for haematological diseases, solid tumours and immune disorders: current practice in Europe, 2015. Bone Marrow Transplant. 2015;50(8):1037–1056. | ||
National Comprehensive Cancer Network [homepage on the Internet]. NCCN clinical practice guidelines in oncology. CML. Version 2. 2017. Available from: www.nccn.org. Accessed February, 2018. | ||
Khoury HJ, Cortes J, Baccarani M, et al. Omacetaxine mepesuccinate in patients with advanced chronic myeloid leukemia with resistance or intolerance to tyrosine kinase inhibitors. Leuk Lymphoma. 2015;56(1):120–127. | ||
Pavlů J, Apperley JF. Allogeneic stem cell transplantation for chronic myeloid leukemia. Curr Hematol Malig Rep. 2013;8(1):43–51. | ||
Saglio G, Hochhaus A, Goh YT, et al. Dasatinib in imatinib-resistant or imatinib-intolerant chronic myeloid leukemia in blast phase after 2 years of follow-up in a phase 3 study: efficacy and tolerability of 140 milligrams once daily and 70 milligrams twice daily. Cancer. 2010;116(16):3852–3861. | ||
Hughes T, Deininger M, Hochhaus A, et al. Monitoring CML patients responding to treatment with tyrosine kinase inhibitors: review and recommendations for harmonizing current methodology for detecting BCR-ABL transcripts and kinase domain mutations and for expressing results. Blood. 2006;108(1):28–37. | ||
Marin D, Hedgley C, Clark RE, et al. Predictive value of early molecular response in patients with chronic myeloid leukemia treated with first-line dasatinib. Blood. 2012;120(2):291–294. | ||
Chen Z, Medeiros LJ, Kantajian HM, et al. Differential depth of treatment response required for optimal outcome in patients with blast phase versus chronic phase of chronic myeloid leukemia. Blood Cancer J. 2017;7(2):e521. | ||
Mahon FX, Richter J, Guilhot J, et al. Interim analysis of a pan European stop tyrosine kinase inhibitor trial in chronic myeloid leukemia: the EURO-SKI study. Blood. 2014;124:151. | ||
Rea D, Nicolini FE, Tulliez M, et al. Discontinuation of dasatinib or nilotinib in chronic myeloid leukemia: interim analysis of the STOP 2G-TKI study. Blood. 2017;129(7):846–854. | ||
DESTINY (De-Escalation and Stopping Treatment of Imatinib, Nilotinib or sprYcel) study. ASH. 2016; Abstract 938. | ||
Chávez-González A, Avilés-Vázquez S, Moreno-Lorenzana D, Mayani H. Hematopoietic stem cells in chronic myeloid leukemia. Chapter 8. In: Stem Cell Biology in Normal Life and Diseases Stem. UK: Kamran Alimoghaddam, IntechOpen; 2013. | ||
Cardone MH, Roy N, Stennicke HR, et al. Regulation of cell death protease caspase-9 by phosphorylation. Science. 1998;282(5392):1318–1321. | ||
Steelman LS, Pohnert SC, Shelton JG, Franklin RA, Bertrand FE, McCubrey JA. JAK/STAT, Raf/MEK/ERK, PI3K/Akt and BCR-ABL in cell cycle progression and leukemogenesis. Leukemia. 2004;18(2):189–218. | ||
Deming PB, Schafer ZT, Tashker JS, Potts MB, Deshmukh M, Kornbluth S. BCR-ABL-mediated protection from apoptosis downstream of mitochondrial cytochrome c release. Mol Cell Biol. 2004;24(23):10289–10299. | ||
Antoszewska-Smith J, Pawlowska E, Blasiak J. Reactive oxygen species in BCR-ABL1-expressing cells – relevance to chronic myeloid leukemia. Acta Biochim Pol. 2017;64(1):1–10. | ||
Sallmyr A, Fan J, Rassool FV. Genomic instability in myeloid malignancies: increased reactive oxygen species (ROS), DNA double strand breaks (DSBs) and error-prone repair. Cancer Lett. 2008;270(1):1–9. | ||
Dueva R, Iliakis G. Alternative pathways of non-homologous end joining (NHEJ) in genomic instability and cancer. Transl Cancer Res. 2013;2:163–177. | ||
Muvarak N, Kelley S, Robert C, et al. C-myc generates repair errors via increased transcription of alternative-NHEJ factors, LIG3 and PARP1, in tyrosine kinase-activated leukemias. Mol Cancer Res. 2015;13(4):699–712. | ||
White E, DiPaola RS. The double-edged sword of autophagy modulation in cancer. Clin Cancer Res. 2009;15(17):5308–5316. | ||
Hu Y, Liu J, Wu YF, et al. mTOR and autophagy in regulation of acute lung injury: a review and perspective. Microbes Infect. 2014;16(9):727–734. | ||
Altman BJ, Rathmell JC. Metabolic stress in autophagy and cell death pathways. Cold Spring Harb Perspect Biol. 2012;4(9):a008763. | ||
Macintosh RL, Ryan KM. Autophagy in tumour cell death. Semin Cancer Biol. 2013;23(5):344–351. | ||
Sheng Z, Ma L, Sun JE, Zhu LJ, Green MR. BCR-ABL suppresses autophagy through ATF5-mediated regulation of mTOR transcription. Blood. 2011;118(10):2840–2848. | ||
Ramos EK, Hoffmann AD, Gerson SL, Liu H. New opportunities and challenges to defeat cancer stem cells. Trends Cancer. 2017;3(11):780–796. | ||
Skorski T. Chronic myeloid leukemia cells refractory/resistant to tyrosine kinase inhibitors are genetically unstable and may cause relapse and malignant progression to the terminal disease state. Leuk Lymphoma. 2011;52(Suppl 1):23–29. | ||
Xin P, Li C, Zheng Y, et al. Efficacy of the dual PI3K and mTOR inhibitor NVP-BEZ235 in combination with imatinib mesylate against chronic myelogenous leukemia cell lines. Drug Des Devel Ther. 2017;11:1115–1126. | ||
Schuster K, Zheng J, Arbini AA, Zhang CC, Scaglioni PP. Selective targeting of the mTORC1/2 protein kinase complexes leads to antileukemic effects in vitro and in vivo. Blood Cancer J. 2011;14(9):e34. | ||
Lu Z, Xu N, He B, et al. Inhibition of autophagy enhances the selective anti-cancer activity of tigecycline to overcome drug resistance in the treatment of chronic myeloid leukemia. J Exp Clin Cancer Res. 2017;36(1):43. | ||
Sinclair A, Latif AL, Holyoake TL. Targeting survival pathways in chronic myeloid leukaemia stem cells. Br J Pharmacol. 2013;169(8):1693–1707. | ||
Arakawa S, Honda S, Yamaguchi H, Shimizu S. Molecular mechanisms and physiological roles of Atg5/Atg7-independent alternative autophagy. Proc Jpn Acad Ser B Phys Biol Sci. 2017;93(6):378–385. | ||
Bellodi C, Lidonnici MR, Hamilton A, et al. Targeting autophagy potentiates tyrosine kinase inhibitor-induced cell death in Philadelphia chromosome-positive cells, including primary CML stem cells. J Clin Invest. 2009;119(5):1109–1123. | ||
Okabe S, Tauchi T, Katagiri S, Tanaka Y, Ohyashiki K. Combination of the ABL kinase inhibitor imatinib with the Janus kinase 2 inhibitor TG101348 for targeting residual BCR-ABL-positive cells. J Hematol Oncol. 2014;7:37. | ||
Gallipoli P, Cook A, Rhodes S, et al. JAK2/STAT5 inhibition by nilotinib with ruxolitinib contributes to the elimination of CML CD34+ cells in vitro and in vivo. Blood. 2014;124(9):1492–1501. | ||
Yeh YY, Liou JP, Lee YL, Lin JY, Huang HM. MPT0B002, a novel microtubule inhibitor, downregulates T315I mutant BCR-ABLl and induces apoptosis of imatinib-resistant chronic myeloid leukemia cells. Invest New Drugs. 2017;35(4):427–435. | ||
Heidel FH, Bullinger L, Feng Z, et al. Genetic and pharmacologic inhibition of β-catenin targets imatinib-resistant leukemia stem cells in CML. Cell Stem Cell. 2012;10(4):412–424. | ||
Zhou H, Mak PY, Mu H, et al. Combined inhibition of β-catenin and Bcr-Abl synergistically targets tyrosine kinase inhibitor-resistant blast crisis chronic myeloid leukemia blasts and progenitors in vitro and in vivo. Leukemia. 2017;31(10):2065–2074. | ||
Cayssials E, Guilhot F. Beyond tyrosine kinase inhibitors: combinations and other agents. Best Pract Res Clin Haematol. 2016;29(3):271–283. | ||
Kataoka K, Kurokawa M. Ecotropic viral integration site 1, stem cell self-renewal and leukemogenesis. Cancer Sci. 2012;103(8):1371–1377. | ||
Sato T, Goyama S, Kataoka K, et al. EVI1 defines leukemia-initiating capacity and tyrosine kinase inhibitor resistance in chronic myeloid leukemia. Oncogene. 2014;33(42):5028–5038. | ||
Zhao C, Chen A, Jamieson CH, et al. Hedgehog signalling is essential for maintenance of cancer stem cells in myeloid leukaemia. Nature. 2009;458(7239):776–779. | ||
Irvine DA, Zhang B, Kinstrie R, et al. Deregulated hedgehog pathway signaling is inhibited by the smoothened antagonist LDE225 (Sonidegib) in chronic phase chronic myeloid leukaemia. Sci Rep. 2016;6:25476. | ||
Asimakopoulos FA, Shteper PJ, Krichevsky S, et al. ABL1 methylation is a distinct molecular event associated with clonal evolution of chronic myeloid leukemia. Blood. 1999;94(7):2452–2460. | ||
Wang Z, Yuan H, Roth M, et al. SIRT1 deacetylase promotes acquisition of genetic mutations for drug resistance in CML cells. Oncogene. 2013;32(5):589–598. | ||
Arrigoni E, Del Re M, Galimberti S, et al. Concise review: chronic myeloid leukemia: stem cell niche and response to pharmacologic treatment. Stem Cells Transl Med. 2018;7(3):305–314. | ||
Kantarjian HM, O’Brien S, Cortes J, et al. Results of decitabine (5-aza-2′deoxycytidine) therapy in 130 patients with chronic myelogenous leukemia. Cancer. 2003;98(3):522–528. | ||
Jin Y, Zhou J, Xu F, et al. Targeting methyltransferase PRMT5 eliminates leukemia stem cells in chronic myelogenous leukemia. J Clin Invest. 2016;126(10):3961–3980. | ||
Koschmieder S, Vetrie D. Epigenetic dysregulation in chronic myeloid leukaemia: a myriad of mechanisms and therapeutic options. Semin Cancer Biol. 2018;51:180–197. | ||
Massimino M, Stella S, Tirrò E, et al. Non ABL-directed inhibitors as alternative treatment strategies for chronic myeloid leukemia. Mol Cancer. 2018;17(1):56. | ||
Scott MT, Korfi K, Saffrey P, et al. Epigenetic reprogramming sensitizes CML stem cells to combined EZH2 and tyrosine kinase inhibition. Cancer Discov. 2016;6(11):1248–1257. | ||
Peterson LF, Mitrikeska E, Giannola D, et al. p53 stabilization induces apoptosis in chronic myeloid leukemia blast crisis cells. Leukemia. 2011;25(5):761–769. | ||
Carter BZ, Mak PY, Mu H, et al. Combined targeting of Bcl-2 and Bcr-Abl tyrosine kinase eradicates chronic myeloid leukemia stem cells. Sci Transl Med. 2016;8(355):355ra117. | ||
Goff DJ, Court Recart A, Sadarangani A, et al. A Pan-BCL2 inhibitor renders bone-marrow-resident human leukemia stem cells sensitive to tyrosine kinase inhibition. Cell Stem Cell. 2013;12(3):316–328. | ||
Ko TK, Chuah CT, Huang JW, Ng KP, Ong ST. The BCL2 inhibitor ABT-199 significantly enhances imatinib-induced cell death in chronic myeloid leukemia progenitors. Oncotarget. 2014;5(19):9033–9038. | ||
Gontarewicz A, Balabanov S, Keller G, et al. Simultaneous targeting of Aurora kinases and Bcr-Abl kinase by the small molecule inhibitor PHA-739358 is effective against imatinib-resistant Bcr-Abl mutations including T315I. Blood. 2008;111(8):4355–4364. |
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.