Back to Journals » Clinical, Cosmetic and Investigational Dermatology » Volume 16
Chronic Mucocutaneous Candidiasis: A Case Report
Received 16 November 2022
Accepted for publication 12 January 2023
Published 25 January 2023 Volume 2023:16 Pages 231—236
DOI https://doi.org/10.2147/CCID.S396802
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Jeffrey Weinberg
Zhensheng Wang,1 Yongfeng Zhang,1 Weiyuan Ma2
1Department of Pediatrics, Affiliated Hospital of Weifang Medical University, Weifang, People’s Republic of China; 2Department of Dermatology, Affiliated Hospital of Weifang Medical University, Weifang, People’s Republic of China
Correspondence: Weiyuan Ma, Department of Dermatology, Affiliated Hospital of Weifang Medical University, Weifang, People’s Republic of China, Tel +86-536-3081272, Email [email protected] Yongfeng Zhang, Department of Pediatrics, Affiliated Hospital of Weifang Medical University, Weifang, People’s Republic of China, Tel +86-536-3081502, Email [email protected]
Abstract: Chronic mucocutaneous candidiasis (CMC) is a rare infectious skin disease. This study reported a case of CMC in a child with clinical manifestations of oral mucosal leukoplakia and erythema and crust-like thick scabs on the skin of the face and upper limbs. Microscopic fungal examination revealed a large amount of pseudohyphae, and the fungal culture indicated Candida albicans. A drug sensitivity test indicated that it was sensitive to itraconazole and nystatin. Laboratory tests did not show significant immunodeficiency or endocrine abnormalities, and gene sequencing did not identify DNA gene mutations in the coiled-coil domain (CCD) or the DNA-binding domain (DBD) of signal transducer and activator of transcription 1 (STAT1). The skin lesions subsided after oral administration of itraconazole but relapsed 6 months later, and hypoparathyroidism occurred 1 year later. Patients with repeated superficial fungal infection should be alert to the possibility of CMC. CMC has numerous complications and a poor prognosis that requires the attention of clinicians. In this case, STAT1 mutation was not found, and parathyroid dysfunction was rare, providing reference for clinical diagnosis and treatment of CMC.
Keywords: chronic mucocutaneous candidiasis, children, Candida albicans, itraconazole, hypoparathyroidism
Introduction
Chronic mucosal candidiasis (CMC) is a rare primary immunodeficiency disease characterized by persistent or recurrent C. albicans infections of the skin, nails, and mucous membranes.1 This disease mostly occurs in children, and mutations in the signal transducer and activator of transcription 1 (STAT1) gene (gain-of-function mutations) are the most common genetic cause.2 In addition to invasive fungal diseases and viral and mycobacterial infections, patients with this disease have a higher incidence of autoimmune diseases and endocrine diseases.3 In this study, we report a case of CMC in a child, which mainly manifested as oral leukoplakia and erythema on the skin of the face and upper limbs along with thick crust-like scabs. C. albicans infection was confirmed by mycological examination, and the drug susceptibility test indicated that it was sensitive to itraconazole and nystatin. Gene sequencing did not reveal gene mutations in the coiled-coil structure domain (CCD) or the DNA-binding domain (DBD) of STAT1. It is hoped that this case can draw the attention of pediatricians to CMC. The possibility of CMC should be considered in children with recurrent skin and mucosal candidiasis. The diagnosis should be made as soon as possible, and the cause of the disease should be actively investigated. Other infections, autoimmune diseases, and endocrine diseases, which may be combined with CMC, should be investigated. The quality of life of children can be improved through active treatment.
Case Presentation
A 4.5-year-old male patient was diagnosed with a rash on his face and upper limbs and white pseudomembranes appearing on the oral mucosa for 2 months. Two months before presentation, with no obvious cause, white pseudomembranes appeared in the buccal mucosa, and no treatment was provided. Six weeks before presentation, perioral erythema, papules, and thick brown scabs appeared. The pediatric department of the local hospital diagnosed the patient with “impetigo”, and cefaclor was given orally for 7 days, but the condition was not alleviated. One month before presentation, the rash increased and affected the face and both upper limbs. The patient had a full-term normal delivery, with no airway restriction at birth, breastfeeding after birth, and a balanced diet. The parents were not consanguineous, and there was no similar patient in the family.
The child’s nutritional status was moderate, no developmental abnormalities were observed, and no obvious abnormalities were found in the physical examination. A dermatological examination revealed a scattered distribution of erythema and papules on the face, covered with yellow scabs, and the skin lesions around the mouth, nose, and right cheek showed granulomatous hyperplastic plaques, covered with crust-like thick brown scars, which could not be easily removed (Figure 1). Red bean-sized papules and coin-sized red plaques were scattered on both upper limbs; large white pseudomembranes were visible on the oral buccal mucosa, tongue, and palate. The fingers, toenails, and hair were normal. A large amount of pseudohyphae and blastospores were observed by direct microscopic examination of the pseudomembranes in the oral cavity and the skin lesions under the thick scabs of the face. A chromogenic CHROMagar Candida culture showed a green colony (Figure 2A). The sensitivity of C. albicans to antifungal agents was detected by K-B disk diffusion method. The antifungal sensitivity paper was produced by ROSCO Company in Sweden, and the experimental steps and results interpretation standards were carried out according to the instructions of the kit. The antifungal susceptibility test indicated sensitivity to itraconazole and nystatin and moderate sensitivity to terbinafine (Figure 2B). Fungal genomic DNA was extracted, and the internal transcribed spacer 1 (ITS1) region was amplified by PCR. Followed by Sanger sequencing, BLAST comparison showed the identity with C. albicans sequence was 100%. Meanwhile, the peripheral blood of patient was extracted and mononuclear cells were separated which genomic DNA was extracted. Sanger sequencing and BLAST comparison revealed no point mutations in the CCD and DBD of STAT1. Tables 1 and 2 shows the other laboratory tests. The patient was eventually diagnosed with CMC.
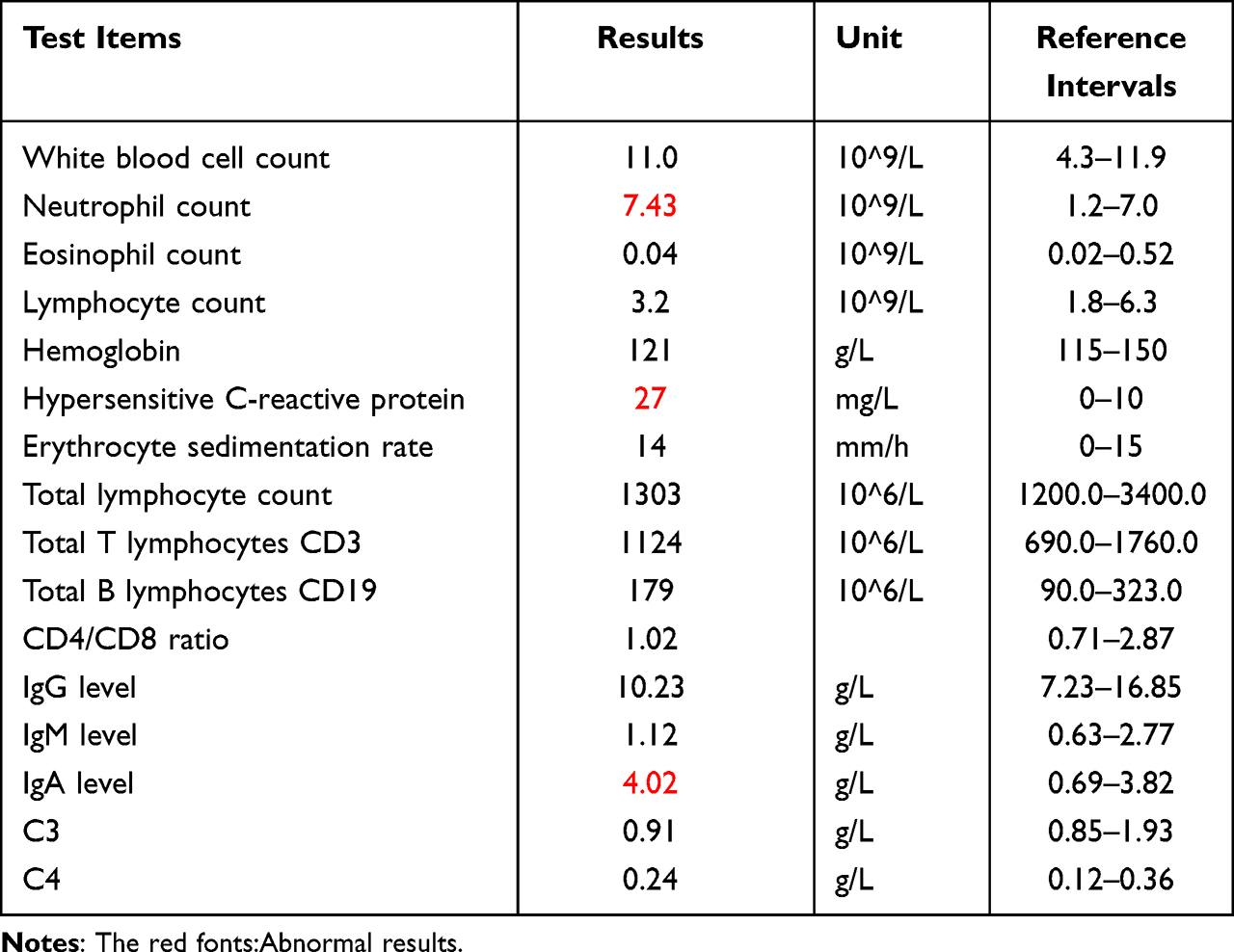 |
Table 1 Blood Laboratory Examinations of the Patient for Diagnosis |
 |
Table 2 Blood Laboratory Examinations of the Patient for Diagnosis |
 |
Figure 1 Clinical manifestations of CMC. (A) Facial lesions before treatment, with visible red plaques covered with thick brown scabs. (B) After 1 month of itraconazole treatment, the thick scabs had disappeared, and the plaque area was reduced and flattened. |
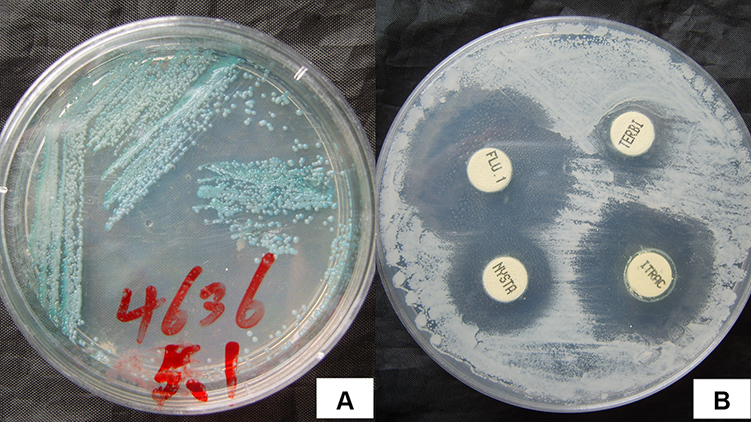 |
Figure 2 Microbiological examination of CMC. (A) CHROMagar chromogenic medium showed green colonies, which were uplifted, moist, smooth, and pearly. (B) The drug sensitivity test identified sensitivity to itraconazole, nystatin, and 5-fluorocytosine and moderate sensitivity to terbinafine. |
According to the drug sensitivity test, the patient was given oral itraconazole, 40 mg twice a day, and topical bifonazole cream twice a day. After 1 month of treatment, the white pseudomembranes in the oral cavity completely disappeared, and the red plaques on the face and upper limbs subsided but yellow crusts were still present. The microscopic examination of the skin lesions was still positive. Oral itraconazole solution and bifonazole cream were continued for 2 months; the skin lesions completely subsided; the microscopic examination and fungal culture were negative, and the treatment was discontinued. After 6 months of follow-up, a white pseudomembrane in the oral cavity relapsed. After 1 year of follow-up, the patient had skin lesions again with manifestations of hypoparathyroidism (hand and foot convulsions, hypocalcemia, and reduced parathyroid hormone (PTH)).
Discussion
CMC is a rare disease with congenital immunological or endocrine abnormalities and persistent or recurrent mucosal and skin Candida infections. Disease onset mostly occurs during childhood. Generally, the oral cavity is first affected, and then the symptoms spread to the head and face, trunk, hands, feet, and nail plate,4 which is manifested as red macules, plaques, thick yellow crust-like scabs on the surface, and even granulomatous hyperplasia. In addition to typical clinical skin lesions, the diagnosis of this disease requires a combination of microbiological examinations, including fungal microscopy, fungal culture, and drug sensitivity testing. Molecular biology techniques such as ITS and next generation sequencing (NGS) to identify pathogenic fungi can be conducted to confirm the diagnosis. In this case, the patient had typical skin lesions that were confirmed to be C. albicans infection by fungal culture, which was consistent with the diagnosis of CMC. CMC is often accompanied by a variety of autoimmune and endocrine diseases, such as systemic lupus erythematosus, inflammatory bowel disease, vitiligo, type 1 diabetes, hypothyroidism,3 and hypofunction of the adrenal cortex.5 The occurrence of CMC can also be accompanied by abnormal iron metabolism and iron deficiency anemia.6,7 These other diseases associated with CMC may gradually manifest with age. One year after the onset of CMC, the patient in this case had clinical manifestations of hypoparathyroidism. Therefore, long-term follow-up of CMC should be conducted to avoid missing comorbidities.
Under normal circumstances, the body mainly relies on T-cell-mediated cellular immunity to defend against Candida infection. CMC patients have T lymphocyte dysfunction.8 Although the number of T lymphocytes in most CMC patients is normal, they do not respond to Candida antigens. The Janus kinase/signal transducer and activator of transcription (JAK/STAT) signaling pathway is closely related to the function of T lymphocytes. In 2011, Liu et al first reported the presence of a STAT1 mutation in a patient with congenital hypothyroidism combined with chronic mucocutaneous candidiasis.9 Many subsequent studies10–12 have demonstrated that CMC is associated with STAT1 gain-of-function (GOF) autosomal dominant mutations. These mutations are mostly located in the CCD and DBD of STAT1.13 STAT1 GOF mutations block the function of STAT1, resulting in T helper cell 17 (Th17) differentiation disorder and reduced secretion of cytokines such as interleukin-17 (IL-17).14,15 Cytokines such as IL-17 can induce skin and mucosal cells to produce procytokines and chemokines, recruit neutrophils to the infection site, and eliminate Candida infection. Approximately half of CMC patients carry STAT1 GOF mutations,16 suggesting that the reduction in IL-17 secretion caused by this mutation may be the main molecular mechanism of CMC pathogenesis. In addition, biallelic TRAF3IP2 mutations16,17 and β-glucan receptor (dectin-1) mutations18 may also be involved in the development of CMC. The sequencing results of the CMC patient in our report revealed no gene mutations in the CCD and DBD regions of the STAT1 gene. It is speculated that the mutation may be located in other regions of the STAT1 gene or other genes and deserves further investigation.
CMC is the most difficult type of candidiasis to treat, and there is still a lack of effective treatment methods. Amphotericin B,19 fluconazole, itraconazole, voriconazole,20 topical nystatin, and other drug treatments can be used based on the results of a antifungal susceptibility test. After oral antifungal treatment, mucosal damage usually disappears within 5 to 7 days, and the skin lesions began to be relieved after 2 weeks. Because antifungal treatment does not change the immune function of the host, CMC relapses soon after drug withdrawal. Therefore, children often need long-term medication. To prevent pathogen resistance, antifungal treatment should have an appropriate withdrawal period or several alternative antifungal drugs should be used, and regular in vitro drug sensitivity tests should be performed to monitor the emergence of drug-resistant strains.21 Studies have confirmed that the JAK inhibitor ruxolitinib is effective in some cases, which is possibly related to stimulation of the increase in IL-17A and IL-17F secretion.22 However, long-term medication is required, and the disease may relapse after drug withdrawal.23 In addition, there are also reports of successful treatment with baricitinib.24 For cases that do not respond to conventional treatment, hematopoietic stem cell transplantation is another possible option.25
Conclusion
As a group of rare immunodeficiency diseases, CMC mainly manifests as recurrent or persistent oral, skin, and C. albicans infections. This disease is mostly associated with GOF mutations in the STAT1 gene and is often accompanied by various autoimmune diseases and endocrine diseases, with a poor prognosis. In this case, GOF mutations in the STAT1 gene were not found, and rare hypoparathyroidism was associated with it. In clinical practice, the possibility of CMC should be considered in patients with recurrent or persistent Candida infection, and careful physical examination and comprehensive laboratory tests should be performed. Once diagnosed, long-term follow-up is required to avoid missed diagnoses of complications. Antifungal drugs and some JAK enzyme inhibitors are effective for the treatment of CMC, and hematopoietic stem cell transplantation can be attempted for severely ill patients. In this case, the skin lesions of the patient were completely subsided after treatment with itraconazole, but it was easy to relapse after drug withdrawal. This suggests that the disease needs long-term follow-up and treatment.
Consent
The patient’s legal guardian had given written informed consent for the publication of the patient’s clinical details and accompanying images. Institutional approval is not required for this case study.
Funding
Research reported in this publication was supported by Traditional Chinese Medicine Science and Technology Project of Shandong Province (No. 2021Q093) and Doctoral Startup Fund of Affiliated Hospital of Weifang Medical University (No. 2021BKQ02).
Disclosure
The authors have no conflicts of interest to declare for this work.
References
1. Campois Z, Almeida Araujo D, de Almeida Araujo EJ, et al. Immunological and histopathological characterization of cutaneous candidiasis. J Med Microbiol. 2015;64(8):810–817. doi:10.1099/jmm.0.000095
2. Carey L, Lambourne J, Porter S, et al. Chronic mucocutaneous candidiasis due to gain-of-function mutation in STAT 1. Oral Dis. 2019;25(3):684–692. doi:10.1111/odi.12881
3. Toubiana O, Okada S, Hiller J, et al. Heterozygous STAT1 gain-of-function mutations underlie an unexpectedly broad clinical phenotype. Blood. 2016;127(25):3154–3164. doi:10.1182/blood-2015-11-679902
4. Khosravi M, Mansouri P, Saffarian Z, et al. Chronic mucocutaneous candidiasis, a case study and literature review. J Mycol Med. 2018;28(1):206–210. doi:10.1016/j.mycmed.2018.02.004
5. Perniola F, Fierabracci A, Falorni A. Autoimmune addison’s disease as part of the autoimmune polyglandular syndrome type 1: historical overview and current evidence. Front Immunol. 2021;12(606860). doi:10.3389/fimmu.2021.606860
6. Davidson H, Hayes JP, Hussein S. Recurrent genital candidiasis and iron metabolism. Br J Vener Dis. 1977;53(2):123–125. doi:10.1136/sti.53.2.123
7. Lu S-Y. Perception of iron deficiency from oral mucosa alterations that show a high prevalence of Candida infection. J Formos Med Assoc. 2016;115(8):619–627. doi:10.1016/j.jfma.2016.03.011
8. Kirkpatrick CH. Chronic mucocutaneous candidiasis. Pediatr Infect Dis J. 2001;20(2):197–206. doi:10.1097/00006454-200102000-00017
9. Liu O, Okada A, Kong S, et al. Gain-of-function human STAT1 mutations impair IL-17 immunity and underlie chronic mucocutaneous candidiasis. J Exp Med. 2011;208(8):1635–1648. doi:10.1084/jem.20110958
10. Bloomfield Z, Zentsova I, Milota I, et al. Immunoprofiling of monocytes in STAT1 gain-of-function chronic mucocutaneous candidiasis. Front Immunol. 2022;13(983977). doi:10.3389/fimmu.2022.983977
11. Martinot K, Korganow S, Wald AS, et al. Case report: a new gain-of-function mutation of STAT1 identified in a patient with chronic mucocutaneous candidiasis and rosacea-like demodicosis: an emerging association. Front Immunol. 2021;12(760019). doi:10.3389/fimmu.2021.760019
12. Zhang L, Jiang W. Single-cell RNA sequencing combined with whole exome sequencing reveals the landscape of the immune pathogenic response to chronic mucocutaneous candidiasis with STAT1 GOF mutation. Front Immunol. 2022;13:988766.
13. Soltesz T, Tóth B, Shabashova B, et al. New and recurrent gain-of-function STAT1 mutations in patients with chronic mucocutaneous candidiasis from Eastern and Central Europe. J Med Genet. 2013;50(9):567–578. doi:10.1136/jmedgenet-2013-101570
14. Nielsen K-O, Kofod-Olsen E, Spaun E, et al. A STAT1-gain-of-function mutation causing Th17 deficiency with chronic mucocutaneous candidiasis, psoriasiform hyperkeratosis and dermatophytosis. BMJ Case Rep;2015. bcr2015211372. doi:10.1136/bcr-2015-211372
15. Dhalla F, Fox H, Davenport H, et al. Chronic mucocutaneous candidiasis: characterization of a family with STAT-1 gain-of-function and development of an ex-vivo assay for Th17 deficiency of diagnostic utility. Clin Exp Immunol. 2016;184(2):216–227. doi:10.1111/cei.12746
16. Lobo B, Lei P, Pelham SJ, et al. Biallelic TRAF3IP2 variants causing chronic mucocutaneous candidiasis in a child harboring a STAT1 variant. Pediatr Allergy Immunol. 2021;32(8):1804–1812. doi:10.1111/pai.13603
17. Bhattad D, Dinakar N, Pinnamaraju C, et al. Chronic mucocutaneous candidiasis in an adolescent boy due to a novel mutation in TRAF3IP2. J Clin Immunol. 2019;39(6):596–599. doi:10.1007/s10875-019-00664-x
18. Ferwerda F, Ferwerda G, Plantinga G, et al. Human dectin-1 deficiency and mucocutaneous fungal infections. N Engl J Med. 2009;361(18):1760–1767. doi:10.1056/NEJMoa0901053
19. Desai U, Urban A, Swaim A, et al. Efficacy of cochleated amphotericin B in mouse and human mucocutaneous Candidiasis. Antimicrob Agents Chemother. 2022;66(7):e0030822. doi:10.1128/aac.00308-22
20. Rautemaa R, Richardson M, Pfaller M, et al. Activity of amphotericin B, anidulafungin, caspofungin, micafungin, posaconazole, and voriconazole against Candida albicans with decreased susceptibility to fluconazole from APECED patients on long-term azole treatment of chronic mucocutaneous candidiasis. Diagn Microbiol Infect Dis. 2008;62(2):182–185. doi:10.1016/j.diagmicrobio.2008.05.007
21. Zarrinfar H, Kord Z, Fata A. High incidence of azole resistance among Candida albicans and C. glabrata isolates in Northeastern Iran. Current Med Mycol. 2021;7(3):18–21. doi:10.18502/cmm.7.3.7801
22. Mossner D, Diering N, Bader N, et al. Ruxolitinib induces interleukin 17 and ameliorates chronic mucocutaneous Candidiasis caused by STAT1 gain-of-function mutation. Clin Infect Dis. 2016;62(7):951–953. doi:10.1093/cid/ciw020
23. Bloomfield K, Kanderová V, Parackova V, et al. Utility of ruxolitinib in a child with chronic mucocutaneous candidiasis caused by a novel STAT1 gain-of-function mutation. J Clin Immunol. 2018;38(5):589–601. doi:10.1007/s10875-018-0519-6
24. Borgstrom E, Edvinsson SA, Perez M, et al. Three adult cases of STAT1 gain-of-function with chronic mucocutaneous candidiasis treated with JAK inhibitors. J Clin Immunol. 2023;43(1):136–150. doi:10.1007/s10875-022-01351-0
25. Kiykim C, Charbonnier A, Akcay LM, et al. Hematopoietic stem cell transplantation in patients with heterozygous STAT1 gain-of-function mutation. J Clin Immunol. 2019;39(1):37–44. doi:10.1007/s10875-018-0575-y
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.