Back to Journals » Journal of Asthma and Allergy » Volume 9
Chronic hypersensitivity pneumonitis
Authors Pereira C, Gimenez A, Kuranishi L, Storrer K
Received 26 September 2015
Accepted for publication 19 January 2016
Published 21 September 2016 Volume 2016:9 Pages 171—181
DOI https://doi.org/10.2147/JAA.S81540
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Amrita Dosanjh
Carlos AC Pereira,1 Andréa Gimenez,2 Lilian Kuranishi,2 Karin Storrer 2
1Interstitial Lung Diseases Program, 2Pulmonology Postgraduate, Federal University of São Paulo, São Paulo, Brazil
Abstract: Hypersensitivity pneumonitis (HSP) is a common interstitial lung disease resulting from inhalation of a large variety of antigens by susceptible individuals. The disease is best classified as acute and chronic. Chronic HSP can be fibrosing or not. Fibrotic HSP has a large differential diagnosis and has a worse prognosis. The most common etiologies for HSP are reviewed. Diagnostic criteria are proposed for both chronic forms based on exposure, lung auscultation, lung function tests, HRCT findings, bronchoalveolar lavage, and biopsies. Treatment options are limited, but lung transplantation results in greater survival in comparison to idiopathic pulmonary fibrosis. Randomized trials with new antifibrotic agents are necessary.
Keywords: interstitial lung diseases, extrinsic allergic alveolitis, diffuse lung disease, lung immune response, HRCT, farmers lung
Introduction
In a National Heart Lung and Blood Institute/Organization for Rare Diseases workshop, hypersensitivity pneumonitis (HSP) was defined as a complex syndrome of varying intensity, clinical presentation, and natural history.1 HSP is the result of an immunologically induced inflammation of the lung parenchyma (specifically, the disease involves the alveoli, terminal bronchioli, and interstitium) that occurs in susceptible individuals in response to a variety of antigens.2
The prevalence varies considerably around the world, depending on disease definition, diagnostic methods, type and intensity of exposure, geographical conditions, agricultural and industrial practices, and host risk factors.3 In Brazil, HSP is a common interstitial lung disease (ILD). In our database, which includes 3,168 cases of ILDs, HSP was the second most common disease (15%), after connective tissue diseases (17%), followed by idiopathic pulmonary fibrosis (IPF) and sarcoidosis (14% each, data not published). In a study of 431 incident cases in central Denmark, HSP was the third most common ILD (7%), after IPF (28%) and connective tissue diseases (14%).4
Typically, HSP has been classified as acute, subacute, and chronic. However, the imaging findings do not necessarily correlate with the duration of symptoms. In general, both subacute and chronic HSP develop after a low but long exposure to antigens, due to molds or birds at home. In general, there is an insidious onset of dyspnea, weight loss, and cough that develops over several weeks or months, or even years.
Textbooks of radiology commonly describe subacute HSP based on a variable combination of ground-glass opacities, poorly defined centrilobular nodules, and mosaic pattern and chronic HSP based on findings indicative of the presence of fibrosis. A cluster analysis of a large cohort showed that most of the cases examined fit best into a two-cluster model.5 The study also showed that subacute HSP is particularly difficult to define because the features in this subset overlap with both the “acute” and “chronic” components. Patients in cluster 1 had features of acute HSP, whereas those in cluster 2 showed features of chronic HSP, including fibrosis on a high-resolution computed tomography (HRCT) scan. The presence of fibrosis in specimens of lung biopsy or on HRCT portends a worse prognosis, as shown in several studies. Based on these findings, we propose to classify HSP into acute and chronic forms. Chronic HSP can be further classified into fibrosing and nonfibrosing.
Pathogenic mechanisms
HSP is an immunopathological disorder occurring in susceptible individuals, where both humoral and cellular mechanisms participate in the development of lung lesions. However, the genetic basis of the disease is poorly understood.
Familial cases can be found (17.5% in a series from Japan).6 Both environmental and genetic factors can be involved in these cases.
If exposed to agents capable of inducing HSP, most individuals develop immune tolerance, and inhalation of the antigen may result in a mild increase in local lymphocytes, without clinical significance.2 Low levels of interleukin-17 were detected in sera and bronchoalveolar lavage (BAL) from both normal and asymptomatic individuals, whereas measurable levels were found in patients. Regulatory T-cells may be involved in antigen tolerance in asymptomatic subjects. Defective regulatory T-cell function, potentially caused by increased interleukin-17 production, could account for the exacerbated immune response characteristic of HSP.7 However, according to a “two-hit” hypothesis, the coexistence of inducing factors (eg, antigens) and promoting factors (eg, genetic abnormalities or additional environmental exposures) may lead to the development of an exaggerated immune reaction that results in marked lung inflammation.3
Following antigen exposure, the BAL fluid shows increased numbers of neutrophils, which peaks after 48 hours. This is then followed by an increase in lymphocytes. Both cluster of differentiation 4 (CD4) and cluster of differentiation 8 (CD8) are involved in the pathogenesis of HSP. T-cell responses are controlled by the molecular interaction between the clonotypically expressed αβ T-cell receptor and cognate peptide–major histocompatibility complex (MHC) antigen. Typically, CD8 T-cells recognize peptides bound to MHC class I molecules and mediate direct target cell lysis, whereas CD4 T-cells recognize peptide–MHC class II-restricted ligands.
Natural killer T (NKT) cells are a heterogeneous population of cells with elements of both innate and adaptive immune systems, capable of rapid responses to antigens with cytotoxic NK cell activity and production of T-helper 1 and T-helper 2 cytokines. NKT cells are elevated in BAL from patients with HSP.8
In HSP, the immune processes that lead to persistent disease and progression to fibrosis are less clear. However, features associated with chronic HSP include an increase in CD4+ T-cells and in the CD4+/CD8+ ratio, a skewing toward Th2 T-cell differentiation and cytokine profile, and an exhaustion of CD8+ T-cells.9 Increased T-helper 17 cells following chronic inhalation of aerosolized antigens may also contribute to the development of lung fibrosis (by promoting collagen deposition).2 Despite this progress, the reason for some patients showing resolution of disease and others progressing to fibrosis even without further antigen exposure is still unknown.
Histologic findings
Histologic findings of HSP have been recently revised.10 The diagnosis of chronic nonfibrosing HSP is best made on a wedge biopsy, but the diagnosis can occasionally be suggested on a transbronchial biopsy with appropriate clinical correlation, when peribronchiolar chronic inflammation and small granulomas or giant cells are found.
The histopathological features comprise chronic bronchiolocentric inflammation and poorly formed nonnecrotizing granulomas. Bronchiolitis is common and can be cellular, follicular, or obliterans. Peribronchiolar metaplasia is a prominent finding, especially in airway-centered interstitial fibrosis (ACIF), and should point to diagnosis.11
The granulomas characteristic of HSP are small, loose, and epithelioid and may be difficult to find. In fact, up to 30% of biopsies obtained from patients with clinically documented HSP lack granulomas. The granulomas and giant cells are typically located in the bronchiolar wall or interstitium, but can also be found in alveolar spaces.12 In HSP, due to nontuberculous mycobacteria, granulomas can be large and well-formed.
Foci of organizing pneumonia are common. Sometimes the disease presents as a pattern of organizing pneumonia.13 Foamy macrophage accumulation may also be present within alveolar spaces due to airway obstruction. In some instances, the chronic interstitial inflammation may be relatively diffuse and mimic cellular nonspecific interstitial pneumonia (NSIP).14 In these cases, a careful search for giant cells and granulomas must be made, but they can be absent.15,16 In some cases, an isolated granulomatous bronchiolitis is seen.
Chronic fibrosing HSP can be diagnostically challenging as the findings overlap with other chronic lung diseases, such as usual interstitial pneumonia (UIP) and NSIP, and it can present with an ACIF, all with or without granulomas or giant cells.
Centrilobular fibrosis, characteristic bridging fibrosis (fibrosis between the respiratory bronchioles and the interlobular septa, with adjacent respiratory bronchioles or with subpleural areas), and organizing pneumonia are characteristic features of chronic HSP with a UIP-like pattern.17 Features more typical of subacute HSP may be present in some cases, particularly if more than one lobe is biopsied including areas devoid of fibrosis in CT.
ACIF is a common pathologic pattern observed in chronic HSP. The main diagnostic criteria include a fibrosis, predominantly bronchiolocentric, associated with bronchiolar or peribronchiolar inflammation and peribronchiolar metaplasia. In a study conducted by our group, 68 cases with ACIF were evaluated.11 Hypersensitivity pneumonitis and GERD, isolated or combined, were the most common etiologies. Granulomas were absent in all cases; giant cells were observed in a small percentage of cases, but were not specific.
In a study from Spain, 20 (43%) out of the 46 patients with a diagnosis of IPF according to 2011 guidelines had a subsequent diagnosis of chronic HSP.18 In 16 out of the 20 patients, there were histopathological features on surgical lung biopsy that were consistent with the diagnosis of HSP.
Multidisciplinary review is critical as HRCT scans may show findings suggesting HSP, even if granulomas are not identified on biopsy.
Differential features from UIP in surgical biopsies should be carefully looked for.17,19
A case of fibrotic HSP submitted to surgical lung biopsy, with fragments retrieved from three lobes at right, is shown in Figure 1. The diversity of pathologic findings is illustrated in this case.
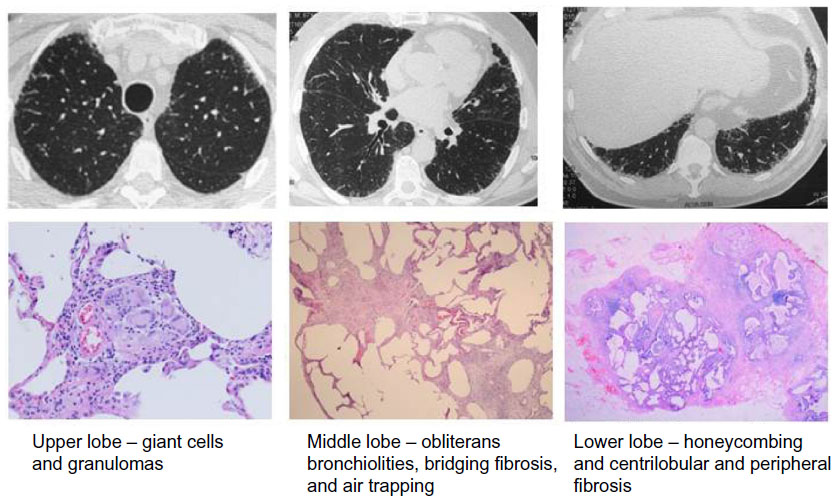 | Figure 1 Chronic HSP caused by feather pillows. |
BAL
The BAL cell profile in HSP is characterized by a significant increase in the total cell count, especially in the percentage of lymphocytes. The presence of plasma cells, mast cells, and macrophages with a foamy cytoplasm (expressing bronchiolitis) reinforces the diagnosis.
In the appropriate clinical context, lymphocytic alveolitis is a major criterion for the diagnosis of HSP in the absence of pathological confirmation. BAL lymphocytosis can be characterized up to ≥30% in non-and ex-smokers and ≥20% in current smokers.20
It was suggested that in the absence of lymphocytic alveolitis, the diagnosis of HSP would be excluded.21,22 However, in the chronic fibrosing disease, an increased percentage of lymphocytes is absent in a significant proportion of cases. In a large study from Japan, increased numbers of lymphocytes in BAL were absent in many cases of chronic HSP due to bird-related HSP and home-related HSP.23 In two small series involving patients with fibrotic HSP, increased numbers of lymphocytes in BAL were found in seven out of 16 (43%) cases.24,25 When the pathologic pattern is “usual interstitial pneumonia-like”, increased numbers of lymphocytes are usually absent in BAL.15 This correlates with the absence of granulomas, a finding which is also common in ACIF.11
Typically, BAL shows an accumulation of activated CD8+ suppressor/cytotoxic lymphocytes, with a CD4/CD8 ratio lower than 1.00. Nevertheless, as shown in an extensive review not all HSP patients display CD8 alveolitis.20 Several reasons may account for this variability, such as the type and dose of inhaled antigen, time elapsed since last exposure, tobacco smoking, and forms of HSP. In fibrosing chronic HSP, an elevated CD4/CD8 ratio is more common.26
The ATS guideline recommends that lymphocyte subset analysis should not be a routine component of BAL cellular analysis.22
Exposures
More than 200 antigens have been identified as causal agents for HSP.27 The vast majorities of causative antigens of HSP are derived from fungal, bacterial, protozoal, and animal proteins, or are small-molecular-weight chemical compounds. HSP has been classified in the past as an occupational disease, because most reports were based on the workplace and antigens particular to that environment (farmer’s lung, suberosis, sauna taker’s lung, fish meal worker’s lung, and others). Over many years, the home slowly emerged as the primary causative environment from exposures to ordinary antigens under common living conditions in individual cases rather than reports of large outbreaks.28–30 New sources of airborne organic particles are recognized every year.31
Bird fanciers’ lung (BFL) is a common form of HSP. It has been reported that the prevalence of BFL among pigeon breeders is between 0.5% and 22%.32 Antigen exposure from many different types of birds may cause BFL. BFL is most commonly reported after exposure to parrots, pigeons, lovebirds, and budgerigars. Some cases of BFL result from exposure to poultry, most commonly canaries. Excreted intestinal mucin and immunoglobulins A and G from bird droppings and bloom (a waxy keratinous powder that coats the feathers of pigeons and serves as waterproofing) are highly antigenic and are the likely major sources for inhalant bird antigen.33 Both bird droppings and pigeon bloom are major reservoirs for pigeon intestinal mucin. The latter has been previously isolated from mucus and pigeon intestines and is implicated as an important antigen in pigeon-breeder’s lung. The intensity of exposure to avian bioaerosol is also important, for example, cleaning of bird cages and walking through enclosed pigeon cages are associated with more intense exposure and hence a possibly increased risk for BFL in the susceptible individual.33 Avian antigens gradually decline after extensive environmental control measures and can be detected for several months.34
Feather duvets and pillows can induce acute and chronic BFL. In these cases, specific antibodies against avian antigens are positive in acute BFL patients, but can be negative in chronic BFL cases. The diagnosis can be made by antigen-induced lymphocyte proliferation in peripheral blood or BAL and an environmental or inhalation provocation test.35
An important study highlights the importance of occult exposure as a cause of disease and the ability of HSP to mimic IPF.18 In that study, 60 consecutive patients diagnosed with diagnosis of IPF were prospectively followed up for 6 years. Almost half of the patients were subsequently diagnosed with chronic HSP, and most of these cases were attributed to exposure to occult avian antigens from commonly used feather bedding.18
Large diversity of molds may occur within and between countries, depending on climates, seasonal changes, lifestyles, and operation of home or workplace environments. Traditionally, HSP has been associated with occupational environments, but more recently, exposures in domestic environment are more recognized.
In the search for antigen at home, particular attention should be paid to possible microbial products: visible contamination with molds or mildew on walls, floors, furniture, air conditioning vents or filters, presence of musty odors in the home or workplace, liquid sources that could allow growth – humidifiers, vaporizers, hot tubs, and pools, and potential sources of water contamination – known water damage from floods, leaks, and broken pipes in the home or workplace.19
Hot-tub lung represents HSP due to inhalational exposure to Mycobacterium avium complex. These bacteria have been recovered from hot and cold water in showers and from showerheads.36–38
Another cause of occupational HSP has been described in the last decade, due to metal working fluid exposure contaminated with Mycobacterium chelonae or Mycobacterium immunogenum. This exposure became the most commonly reported cause of occupational HSP in the UK.39 Based on the evidence base collected during a large UK outbreak, a case definition was proposed.40
Farmer’s lung is a classic form of HSP that results from repeated exposure to inhaled antigens from moldy hay or straw. The species more commonly implicated are thermophilic actinomycetes species, including Saccharopolyspora rectivirgula (formerly Micropolyspora faeni), Thermoactinomyces vulgaris, Thermoactinomyces viridis, and Thermoactinomyces sacchari, among others. These organisms flourish in areas of high humidity and prefer temperatures of 40°C–60°C. An incidence of 8–540 cases per 100,000 persons per year has been reported in farmers. Many cases are still seen in the USA, UK, France, and Sweden. In Brazil, farmer’s lung is uncommon.
Synthetic low-molecular-weight compounds, such as isocyanates, insecticides, and epoxy resins, can form bonds with human protein molecules and incite an immunologic response leading to HSP. Common exposures include toluene diisocyanate and diphenylmethane diisocyanate, specifically toluene typically found in paints, foams, and sealants.41
Cases of HSP due to wind instruments contaminated by bacteria and molds have been published.42,43 A well-documented case of HSP due to sensitization to fungi- and mite-contaminated flours in a baker was recently published.44 Other rare causes of HSP have been discovered as chacinero’s lung.45
Serum precipitins
The presence of specific immunoglobulin G antibodies reflects an immune response to a specific exposure and in the appropriate clinical setting supports the diagnosis of HSP. Conversely, the absence of serum precipitins does not rule out HSP.2
Precipitating antibodies against potential antigens may be present in serum and BAL specimen in patients with HSP and in asymptomatic subjects exposed to antigens responsible for HSP. A study that evaluated 43 exposed asymptomatic dairy farmers demonstrated that asymptomatic subjects who had specific serum antibodies to farmer’s lung antigens or a lymphocytic alveolitis did not develop abnormal respiratory outcomes after a 20-year follow up.46
The diversity of antigens and immunological techniques used and the interval since last exposure to the causative antigen are responsible for low sensitivity and specificity.3 In addition, sensitivity declines when the underlying pathologic pattern is UIP.15
Recently, a study evaluated the presence of antibody to antigen collected in the environment of individuals with HSP and controls.47 Nineteen individuals with HSP participated in the study with 15 of them classified as having fibrotic disease. Of the seven individuals who were tested positive to one or more environmental samples, three had a positive response to more than one antigen from the environmental sample (range: 1–9). Several antigens from bacteria fungi and yeasts resulted in positive responses. A significant association existed between the results of interviews/site evaluations and the ability to collect antigens eliciting a positive response (P<0.001).
Clinical manifestations and diagnostic criteria
HSP is often unrecognized or misdiagnosed. Exposure to a relevant antigen is a key component of clinical evaluation. In some series, an exposure is not identified in many cases.48,49 In these patients, the diagnosis is suspected based on histopathology, BAL findings, and HRCT characteristics. We hypothesize that exposure to molds at home was not recognized in many of these cases. The inability to identify an inhaled antigen is independently associated with shortened survival.48,49
Exposure conditions vary from country to country. The most common causes of HSP in Brazil are exposure to bird’s feathers and droppings (including feather pillows), molds at home, or both, and exposure to isocyanates.50 In other countries, farmer’s lung and pigeon breeder’s lung are more common, whereas summer-type HSP is limited to Japan.
Cigarette smoking has a protective role against the development of HSP, but this does not apply to ex-smokers. Nicotine is thought to inhibit macrophage activation and lymphocyte proliferation and function.51 On the other hand, when smokers develop HSP, fibrosing disease with a worse prognosis is more common.52
HSP is more common in females (due to greater exposure at home). Patients are younger in comparison to IPF patients.50 Dyspnea and cough are the most common symptoms, and both may improve during periods away from the inciting antigen. Wheezing, a less common symptom in other ILDs (except sarcoidosis), was referred by 33% in a large series, in comparison to 11% of controls (P<0.001)53 and in physical examination in 13% in another series.54 In sarcoidosis and in HSP, bronchial hyperresponsiveness seems to be related to an extensive epithelial damage in airways.55 Some of the agents that cause occupational asthma, such as diisocyanates and fungal spores, can also cause HSP.56 Although uncommon, HSP can be associated with wheezing, airway hyperresponsiveness, and a normal chest radiograph. Patients with chronic HSP can refer recurrent episodes of symptoms that are misdiagnosed as recurrent pneumonia. Similar to IPF, in chronic HSP, the clinical examination can reveal velcro crackles at lung bases as well digital clubbing. On lung auscultation, a suggestive clinical finding in chronic HSP is the presence of inspiratory squeaks, which are caused by coexisting bronchiolitis.21 However, in fibrosis resulting from microaspiration and in rheumatoid arthritis, conditions where the coexistence of bronchiolitis and ILD is common, inspiratory squeaks can also be heard. Weight loss is common in chronic HSP.
Like other ILDs, the most frequent lung function abnormalities are a restrictive ventilatory impairment and impaired gas exchange (decreased diffusing capacity or increasing hypoxemia during exercise).
Only a few patients show obstruction of the peripheral airways in lung function tests. The correlation between pulmonary function abnormalities and extension of disease in HRCT scans is poor.
In HSP, the chest radiograph and even HRCT scans may be normal.57 HSP can present as isolated bronchiolitis with HRCT scan showing air trapping, which is present only in expiration. In these cases, residual volume should be measured by plethysmography.
CT features play a central role in the diagnosis of HSP. Histologically, ground-glass opacification is thought to represent either active interstitial inflammation or fine fibrosis, which can be reversible or not.58
Ground-glass opacification is frequently found in association with other CT abnormalities, such as centrilobular nodules or air trapping, findings indicative of peribronchiolar infiltration and bronchiolitis. In nonsmokers, the combination of ground-glass with poorly defined centrilobular nodules or focal areas of decreased attenuation on inspiratory HRCT and/or air trapping on expiratory HRCT should suggest immediately the diagnosis of HSP. Lobular areas of decreased attenuation and vascularity should be bilateral and present in three or more lobes.59 Their extension correlates with severity of air trapping, as indicated by an increased residual volume.60 When air-trapping areas, ground-glass opacities, and normal lung are seen at the same HRCT section, the pattern is called “headcheese sign”. The CT imaging pattern is reminiscent of the variegated appearance of headcheese cold cut meat.
Irregular linear opacities, architectural distortion traction bronchiectasis and bronchiolectasis, lobar volume loss, and honeycombing usually indicate fibrosis. Suggestive findings of fibrosing HSP in HRCT are upper lobe predominance, peribronchovascular distribution, and relative subpleural sparing in the lung immediately adjacent to the pleura in the dorsal regions of the lower lobes or relative sparing of the lung below the level of the dome of the diaphragm.61,62 Although upper lobe predominance of fibrosis is suggestive of HSP, this is seen in <25% of cases of fibrosing HSP. The remaining cases had a similar percentage of distribution, diffuse or predominant in lower lobes. In some cases, the findings are similar to those seen in IPF-lower lobe predominance, with subpleural honeycombing.63
ACIF has, in many cases, a peribronchovascular distribution on HRCT.11 HSP and fibrosis due to microaspiration should be entertained in these cases.
In patients with a reticular pattern on CT, without honeycombing (“possible UIP”), some studies have suggested that in the appropriate clinical context, the diagnosis of IPF can be accepted.64–66 In our center, these cases are submitted to surgical lung biopsy, and in several patients, the final diagnosis was chronic HSP.67 Increased numbers of lymphocytes in BAL can be absent.
There is no gold standard for diagnosis of HSP. In a classical study, exposure to a known offending antigen, positive precipitating antibodies, recurrent episodes of symptoms, inspiratory crackles, symptoms 4–8 hours after exposure, and weight loss were found to be indicative findings of HSP.53 By logistic regression, exposure to a known offending antigen had the greatest odds ratio (38.8). In the absence of a unique gold standard defining the presence or absence of HSP, the final diagnosis relied on findings of BAL, HRCT, and, if needed, other diagnostic procedures. BAL lymphocytosis and bilateral ground-glass or poorly defined centrilobular nodular opacities on HRCT were required for a diagnosis of HSP to be accepted without resorting to additional diagnostic procedures.
In some centers, a specific inhalation challenge is available. A positive result is specific for diagnosis.
Current criteria for the diagnosis of HSP apply only to the classic acute presentation and are of limited value in the chronic forms. The presence and extension of fibrosis seen in lung biopsy and HRCT is a major determinant of survival in HSP (see the section “Natural history and prognosis”). In Table 1, we suggest criteria for chronic nonfibrosing HSP (“subacute disease”) and chronic fibrosing HSP. In HRCT, ground-glass opacification can express interstitial inflammation or fine fibrosis,58 and hence the implication of this finding in the absence of surgical lung biopsy must be dictated by evolution.
 | Table 1 Comparative findings between chronic nonfibrosing and fibrosing HSP (relevant exposure and two or more criteria from other separated categories are suggested as diagnostic.) |
Exposure to relevant antigens and two or more findings are suggested as sufficient for the diagnosis in each category. These criteria should be validated in a prospective trial.
Findings on HRCT and lung biopsy in nonfibrosing and fibrosing chronic HP are shown in Figures 2 and 3, respectively.
 | Figure 2 Chronic nonfibrotic HSP. |
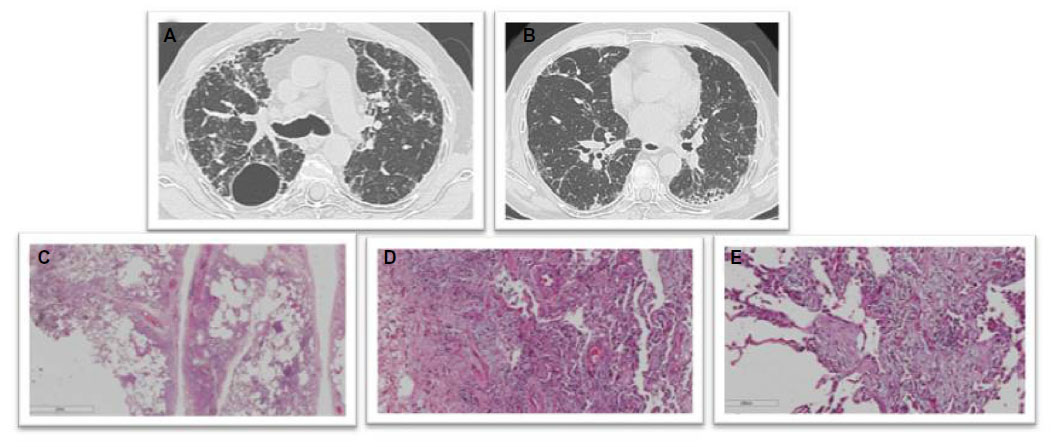 | Figure 3 Chronic fibrosing HSP. |
Pulmonary hypertension (PH)
ILDs are common causes of precapillary pulmonary hypertension (PH) and are classified into group III of the international etiological classification of PH. The prevalence of PH varies widely among the different forms of ILD. Data on PH in chronic HSP are scarce. A study from our center was the first to determine the prevalence of PH in fibrotic HSP patients with hemodynamic data.68 Precapillary PH was found in 44% of 50 cases. Patients with precapillary PH had lower forced vital capacity, diffusing capacity for carbon monoxide, arterial oxygen tension, and saturation after the 6-minute walk test. The predictive value of PH on survival was not determined.
Treatment
Detecting relevant exposures and removing offending antigens have prognostic and therapeutic implications.69
The most important intervention in managing HSP is to avoid the inciting antigen, but exposure removal is not always possible, because frequently, the causal antigen cannot be identified or complete avoidance may mean major changes in occupation, hobbies, and in the domestic environment.48,49 Continuous exposure carries the risk of progressive pulmonary impairment.70 The levels of exposure to avian antigens were related to disease progression and prognosis in chronic bird-related HSP.71
Currently, courses of corticosteroids are frequently prescribed for HSP patients, when there are symptoms and physiologic abnormalities, but randomized controlled trials of prednisone and immunossupressants in fibrotic HSP patients have not been performed.72,73 The best available evidence comes from a randomized placebo-controlled trial in farmer’s lung that showed a faster improvement in lung function with corticosteroids, in comparison to avoidance only, but there were no differences in the long-term outcomes between the two groups.74
A subset of patients develop progressive fibrosis, even after removing the causative antigen.75 In these cases, prolonged courses of oral corticosteroids are often required.76 Therefore, each patient’s treatment needs to be individualized.
The conventional dose for oral corticosteroids is 0.5–1 mg/kg/d of prednisone for a month, followed by a gradual reduction until a maintenance dose of 10–15 mg/d is reached.2,76 In patients with symptoms of cough, wheezing, or airflow obstruction, the use of inhaled bronchodilators and inhaled steroids is appropriate.73,76
In patients with hot-tub lung, antimycobacterial therapy does not appear to be necessary. Although corticosteroids may be helpful in the treatment of severely affected patients, others can be managed by avoidance of additional exposure alone.77
In chronic progressive HSP, immunosuppressor drugs may be added to corticosteroids, but no controlled trials are available to support this strategy.78,79
More recently, rituximab was used as alternative treatment in a case of progressive HSP, refractory to conventional treatment. Compared with the prerituximab, diffusing lung capacity for carbon monoxide (DLCO) and forced vital capacity (FVC) values increased significantly.80 In a study of six patients with progressive HSP and severe physiological impairment, treatment with rituximab resulted in stabilization in three cases.81 More studies are necessary to confirm the utility of this expensive drug in HSP.
There is no evidence to support the prescription of antifibrotic agents (nintedanib and pirfenidone) in the treatment of chronic HSP. Randomized studies in this common ILD are urgently necessary.
In chronic progressive HSP cases that do not respond to corticosteroid and/or immunosuppressant therapy, lung transplantation should be considered.
A study compared the outcomes in 31 HSP patients submitted to lung transplantation to those in 91 subjects with IPF.82 Survival rates at 1, 3, and 5 years after lung transplantation in HSP patients compared with IPF patients were 96%, 89%, and 89% vs 86%, 67%, and 49%, respectively. Subjects with HSP manifested a reduced adjusted risk for death, when compared with subjects with IPF (hazard ratio, 0.25; 95% confidence interval (CI), 0.08–0.74; P=0.013).
Further studies should be conducted aiming to determine response to immunosuppressive agents in the individual patient and the best time to refer to lung transplantation in HSP patients.
Natural history and prognosis
The prognosis of HSP varies greatly and depends on the type and duration of antigen exposure, the dose of the inhaled antigen, and the clinical form of disease. Some patients may experience progression, despite avoiding exposure and undergoing treatment.19 In a long-term follow-up study of patients with pigeon breeders’ lung, those who were asymptomatic had stable spirometric data, in contrast to those who were symptomatic.83
The presence and extension of fibrosis seen on lung biopsy and HRCT is a major determinant of survival in HSP.16,48,50,70,84,85 In lung biopsies, cases with NSIP pattern have a better prognosis and patients with UIP have the worst.50,85 Patients with ACIF and typical features on surgical lung biopsies have an intermediate prognosis.50,84,85
In a study comprising 103 patients from our center, by univariate analysis, several findings were predictors of mortality: older age, velcro crackles, higher FEV1/FVC ratio, lower oxygen saturation during exercise, absence of mosaic pattern/air trapping, and presence of fibrosis on HRCT.50
In a larger and more recent study involving 177 cases of HSP, clinical predictors of mortality included a history of cigarette smoking, crackles on lung examination, lower baseline FVC, and an elevated FEV1/FVC ratio.85
Another study investigated HRCT scans and pulmonary function test results for determining prognosis in patients with chronic fibrotic HSP in 92 patients.86 There were 42 deaths during the study period. Increasing severity of traction bronchiectasis was the strongest predictor of mortality (hazard ratio 1.10, P<0.001, 95% CI 1.04–1.16). Increasing global interstitial disease extent and honeycombing were also independent predictors of mortality. In contrast, no individual PFT variable was predictive of mortality, once HRCT patterns were accounted for.86
Disclosure
The authors report no conflicts of interest in this work.
References
Fink JN, Ortega HG, Reynolds HY, et al. Needs and opportunities for research in hypersensitivity pneumonitis. Am J Respir Crit Care Med. 2005;171(7):792–798. | |
Spagnolo P, Rossi G, Cavazza A, et al. Hypersensitivity pneumonitis: a comprehensive review. J Investig Allergol Clin Immunol. 2015;25(4):237–250. | |
Selman M, Pardo A, King TE Jr. Hypersensitivity pneumonitis: insights in diagnosis and pathobiology. Am J Respir Crit Care Med. 2012;186(4):314–324. | |
Hyldgaard C, Hilberg O, Muller A, Bendstrup E. A cohort study of interstitial lung diseases in central Denmark. Respir Med. 2014;108(5):793–799. | |
Lacasse Y, Selman M, Costabel U, et al; HP Study Group. Classification of hypersensitivity pneumonitis: a hypothesis. Int Arch Allergy Immunol. 2009;149(2):161–166. | |
Okamoto T, Miyazaki Y, Tomita M, Tamaoka M, Inase N. A familial history of pulmonary fibrosis in patients with chronic hypersensitivity pneumonitis. Respiration. 2013;85(5):384–390. | |
Girard M, Israël-Assayag E, Cormier Y. Impaired function of regulatory T-cells in hypersensitivity pneumonitis. Eur Respir J. 2011;37(3):632–639. | |
Rijavec M, Volarevic S, Osolnik K, Kosnik M, Korosec P. Natural killer T cells in pulmonary disorders. Respir Med. 2011;105(suppl 1):S20–S25. | |
Barrera L, Mendoza F, Zuñiga J, et al. Functional diversity of T-cell subpopulations in subacute and chronic hypersensitivity pneumonitis. Am J Respir Crit Care Med. 2008;177(1):44–55. | |
Grunes D, Beasley MB. Hypersensitivity pneumonitis: a review and update of histologic findings. J Clin Pathol. 2013;66(10):888–895. | |
Kuranishi LT, Leslie KO, Ferreira RG, et al. Airway-centered interstitial fibrosis: etiology, clinical findings and prognosis. Respir Res. 2015;16:55. | |
Castonguay MC, Ryu JH, Yi ES, Tazelaar HD. Granulomas and giant cells in hypersensitivity pneumonitis. Hum Pathol. 2015;46(4):607–613. | |
Ito T, Sugino K, Satoh D, et al. Birdfancier’s lung which developed in a pigeon breeder presenting organizing pneumonia. Intern Med. 2010;49(23):2605–2608. | |
Vourlekis JS, Schwarz MI, Cool CD, et al. Nonspecific interstitial pneumonitis as the sole histologic expression of hypersensitivity pneumonitis. Am J Med. 2002;112(6):490–493. | |
Ohtani Y, Saiki S, Kitaichi M, et al. Chronic bird fancier’s lung: histopathological and clinical correlation. An application of the 2002 ATS/ERS consensus classification of the idiopathic interstitial pneumonias. Thorax. 2005;60(8):665–671. | |
Churg A, Sin DD, Everett D, Brown K, Cool C. Pathologic patterns and survival in chronic hypersensitivity pneumonitis. Am J Surg Pathol. 2009;33(12):1765–1770. | |
Takemura T, Akashi T, Kamiya H, et al. Pathological differentiation of chronic hypersensitivity pneumonitis from idiopathic pulmonary fibrosis/usual interstitial pneumonia. Histopathology. 2012;61(6):1026–1035. | |
Morell F, Villar A, Montero MÁ, et al. Chronic hypersensitivity pneumonitis in patients diagnosed with idiopathic pulmonary fibrosis: a prospective case-cohort study. Lancet Respir Med. 2013;1(9):685–694. | |
Glazer CS. Chronic hypersensitivity pneumonitis: important considerations in the work-up of this fibrotic lung disease. Curr Opin Pulm Med. 2015;21(2):171–177. | |
Bronchoalveolar lavage constituents in healthy individuals, idiopathic pulmonary fibrosis, and selected comparison groups. The BAL Cooperative Group Steering Committee. Am Rev Respir Dis. 1990;141(5 pt 2):S169–S202. | |
Costabel U, Bonella F, Guzman J. Chronic hypersensitivity pneumonitis. Clin Chest Med. 2012;33(1):151–163. | |
Meyer KC, Raghu G, Baughman RP, et al; American Thoracic Society Committee on BAL in Interstitial Lung Disease. An official American Thoracic Society clinical practice guideline: the clinical utility of bronchoalveolar lavage cellular analysis in interstitial lung disease. Am J Respir Crit Care Med. 2012;185(9):1004–1014. | |
Okamoto T, Miyazaki Y, Ogura T, et al. Nationwide epidemiological survey of chronic hypersensitivity pneumonitis in Japan. Respir Investig. 2013;51(3):191–199. | |
Mooney JJ, Koth LL. Surgical lung biopsy over bronchoalveolar lavage in chronic hypersensitivity pneumonitis. Am J Respir Crit Care Med. 2014;189(3):371–372. | |
Espoladore LM, Gregório BB, Lima MS, de Pereira CA, Soares MR, Coletta EN. Cytological analysis of bronchoalveolar lavage in patients with interstitial lung diseases and the relation of cytological analysis to fibrosis in high-resolution computed tomography. Anal Quant Cytopathol Histpathol. 2014;36(4):206–212. | |
Murayama J1, Yoshizawa Y, Ohtsuka M, Hasegawa S.Lung fibrosis in hypersensitivity pneumonitis. Association with CD4+ but not CD8+ cell dominant alveolitis and insidious onset. Chest. 1993;104(1):38–43. | |
Mohr LC. Hypersensitivity pneumonitis. Curr Opin Pulm Med. 2004;10:401–411. | |
Jacobs RL, Andrews CP, Coalson J. Organic antigen-induced interstitial lung disease: diagnosis and management. Ann Allergy Asthma Immunol. 2002;88(2):30–41. | |
Jacobs RL, Andrews CP, Coalson JJ. Hypersensitivity pneumonitis: beyond class occupational disease– changing concepts of diagnosis and management. Ann Allergy Asthma Immunol. 2005;95(2):114–127. | |
Selman M. Hypersensitivity pneumonitis: a multifaceted deceiving disorder. Clin Chest Med. 2004;25(3):531–547. | |
Fishwick D. New occupational and environmental causes of asthma and extrinsic allergic alveolitis. Clin Chest Med. 2012;33(4):605–616. | |
Cooper CJ, Teleb M, Elhanafi S, Ajmal S, Hernandez GT. Bird fanciers’ lung induced by exposure to duck and goose feathers. Am J Case Rep. 2014;15:155–158. | |
Chan AL, Juarez MM, Leslie KO, Ismail HA, Albertson TE. Bird fancier’s lung: a state-of-the-art review. Clin Rev Allergy Immunol. 2012;43(1–2):69–83. | |
Craig TJ, Hershey J, Engler RJ, Davis W, Carpenter GB, Salata K. Bird antigen persistence in the home environment after removal of the bird. Ann Allergy. 1992;69(6):510–512. | |
Inase N, Ohtani Y, Sumi Y, et al. A clinical study of hypersensitivity pneumonitis presumably caused by feather duvets. Ann Allergy Asthma Immunol. 2006;96(1):98–104. | |
Falkinham JO. The changing pattern of nontuberculous mycobacterial disease. Can J Infect Dis. 2003;14(5):281–286. | |
Marras TK, Wallace RJ Jr, Koth LL, Stulbarg MS, Cowl CT, Daley CL. Hypersensitivity pneumonitis reaction to Mycobacterium avium in household water. Chest. 2005;127(2):664–671. | |
Hankwitz PE, Cervia JS, Thomas CF, Fink JN, Marras T, Tomic R. Nontuberculous mycobacterial hypersensitivity pneumonitis related to a home shower: treatment and secondary prevention. BMJ Case Rep. 2011;2011; doi:10.1136/bcr.06.2011.4360. | |
Cullinan P, D’Souza E, Tennant R, Barber C. Lesson of the month: extrinsic allergic (bronchiolo) alveolitis and metal working fluids. Thorax. 2014;69(11):1059–1060. | |
Barber CM, Burton CM, Hendrick DJ, et al. Hypersensitivity pneumonitis in workers exposed to metalworking fluids. Am J Ind Med. 2014;57(8):872–880. | |
Dhar S, Daroowalla F. Hypersensitivity pneumonitis. Clin Pulm Med. 2011;18(4):169–174. | |
Metersky ML, Bean SB, Meyer JD, et al. Trombone player’s lung: a probable new cause of hypersensitivity pneumonitis. Chest. 2010;138(3):754–756. | |
Metzger F, Haccuria A, Reboux G, Nolard N, Dalphin JC, De Vuyst P. Hypersensitivity pneumonitis due to molds in a saxophone player. Chest. 2010;138(3):724–726. | |
Gerfaud-Valentin M, Reboux G, Traclet J, Thivolet-Béjui F, Cordier JF, Cottin V: Occupational hypersensitivity peumonitis in a baker. Chest. 2014;145(4):856–858. | |
Morell F, Cruz MJ, Gómez FP, Rodriguez-Jerez F, Xaubet A, Muñoz X. Chacinero’s lung hypersensitivity pneumonitis due to dry sausage dust. Scand J Work Environ Health. 2011;37(4):349–356. | |
Cormier Y, Létourneau L, Racine G. Significance of precipitins and asymptomatic lymphocytic alveolitis: a 20-yr follow-up. Eur Respir J. 2004;23(4):523–525. | |
Millerick-May ML, Mulks MH, Gerlach KR, et al. Hypersensitivity pneumonitis and antigen identification – an alternate approach. Respir Med. 2016;112:97–105. | |
Hanak V, Golbin JM, Hartman TE, Ryu JH. High-resolution CT findings of parenchymal fibrosis correlate with prognosis in hypersensitivity pneumonitis. Chest. 2008;134(1):133–138. | |
Fernández Pérez ER, Swigris JJ, Forssén AV, et al. Identifying an inciting antigen is associated with improved survival in patients with chronic hypersensitivity pneumonitis. Chest. 2013;144(5):1644–1651. | |
Lima MS, Coletta EN, Ferreira RG, et al. Subacute and chronic hypersensitivity pneumonitis: histopathological patterns and survival. Respir Med. 2009;103(4):508–515. | |
Blanchet MR, Israël-Assayag E, Cormier Y. Inhibitory effect of nicotine on experimental hypersensitivity pneumonitis in vivo and in vitro. Am J Respir Crit Care Med. 2004;169(8):903–909. | |
Ohtsuka Y, Munakata M, Tanimura K, et al. Smoking promotes insidious and chronic farmer’s lung disease, and deteriorates the clinical outcome. Intern Med. 1995;34(10):966–971. | |
Lacasse Y, Selman M, Costabel U, et al. Clinical diagnosis of hypersensitivity pneumonitis. Am J Respir Crit Care Med. 2003; 168(8):952–958. | |
Hanak V, Golbin JM, Ryu JH. Causes and presenting features in 85 consecutive patients with hypersensitivity pneumonitis. Mayo Clin Proc. 2007;82(7):812–816. | |
Laitinen LA, Haahtela T, Kava T, Laitinen A. Non-specific bronchial reactivity and ultrastructure of the airway epithelium in patients with sarcoidosis andallergic alveolitis. Eur J Respir Dis Suppl. 1983; 131:267–284. | |
Cartier A, Sastre J. Clinical assessment of occupational asthma and its differential diagnosis. Immunol Allergy Clin North Am. 2011;31(4):717–728. | |
Lynch DA, Rose CS, Way D, King TE Jr. Hypersensitivity pneumonitis: sensitivity of high-resolution CT in a population-based study. AJR Am J Roentgenol. 1992;159(3):469–472. | |
Glazer CS, Rose CS, Lynch DA. Clinical and radiologic manifestations of hypersensitivity pneumonitis. J Thorac Imaging. 2002; 17(4):261–272. | |
Raghu G, Collard HR, Egan JJ, et al; ATS/ERS/JRS/ALAT Committee on Idiopathic Pulmonary Fibrosis. ATS/ERS/JRS/ALAT Committee on Idiopathic Pulmonary Fibrosis. An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management. Am J Respir Crit Care Med. 2011;183(6):788–824. | |
Hansell DM, Wells AU, Padley SP, Müller NL. Hypersensitivity pneumonitis: correlation of individual CT patterns with functional abnormalities. Radiology. 1996;199(1):123–128. | |
Lynch DA, Newell JD, Logan PM, King TE Jr, Müller NL. Can CT distinguish hypersensitivity pneumonitis from idiopathic pulmonary fibrosis? AJR Am J Roentgenol. 1995;165(4):807–811. | |
Silva CI, Müller NL, Lynch DA, et al. Chronic hypersensitivity pneumonitis: differentiation from idiopathic pulmonary fibrosis and nonspecific interstitial pneumonia by using thin-section CT. Radiology. 2008;246(1):288–297. | |
Adler BD, Padley SP, Müller NL, Remy-Jardin M, Remy J. Chronic hypersensitivity pneumonitis: high-resolution CT and radiographic features in 16 patients. Radiology. 1992;185(1):91–95. | |
Chung JH, Chawla A, Peljto AL, et al. CT scan findings of probable usual interstitial pneumonitis have a high predictive value for histologic usual interstitial pneumonitis. Chest. 2015;147(2):450–459. | |
Raghu G, Lynch D, Godwin JD, et al. Diagnosis of idiopathic pulmonary fibrosis with high-resolution CT in patients with little or no radiological evidence of honeycombing: secondary analysis of a randomised, controlled trial. Lancet Respir Med. 2014;2(4):277–284. | |
Lee JW, Shehu E, Gjonbrataj J, et al. Clinical findings and outcomes in patients with possible usual interstitial pneumonia. Respir Med. 2015;109(4):510–516. | |
Storrer K. Prognostic findings in fibrosing interstitial lung diseases submitted to surgical lung biopsy. [doctoral thesis]. Federal University of São Paulo; 2014:76p. | |
Oliveira RK, Pereira CA, Ramos RP, et al. A haemodynamic study of pulmonary hypertension in chronic hypersensitivity pneumonitis. Eur Respir J. 2014;44(2):415–424. | |
Wuyts W, Sterclovab M, Vasakova M. Pitfalls in diagnosis and management of hypersensitivity pneumonitis. Curr Opin Pulm Med. 2015;21(5):490–498. | |
Vourlekis JS, Schwarz MI, Cherniack RM, et al. The effect of pulmonary fibrosis on survival in patients with hypersensitivity pneumonitis. Am J Med. 2004;116(10):662–668. | |
Tsutsui T, Miyazaki Y, Kuramochi J, Uchida K, Eishi Y, Inase N: The amount of avian antigen in household dust predicts the prognosis of chronic bird-related hypersensitivity pneumonitis. Ann Am Thorac Soc. 2015;12(7):1013–1021. | |
Ismail T, Mcsharry C, Boyd G. Extrinsic allergic alveolitis. Respirology. 2006;11(3):262–268. | |
Lacasse Y, Cormier Y. Hypersensitivity pneumonitis. Orphanet J Rare Dis. 2006;1:25. | |
Kokkarinen JI, Tukiainen HO, Terho EO. Effect of corticosteroid treatment on the recovery of pulmonar function in farmer’s lung. Am Rev Respir Dis. 1992;145(1):3–5. | |
Yoshizawa Y, Miyake S, Sumi Y, Hisauchi K, Sato T, Kurup VP. A follow-up study of pulmonary function tests, bronchoalveolar lavage cells, and humoral and cellular immunity in bird fancier’s lung. J Allergy Clin Immunol. 1995;96(1):122–129. | |
Navarro C, Mejia M, Gaxiola M, et al. Hypersensitivity pneumonitis. Treat Respir Med. 2006;5(3):167–179. | |
Hanak V, Kalra S, Aksamit TR, Hartman TE, Tazelaar HD, Ryu JH. Hot tub lung: presenting features and clinical course of 21 patients. Respir Med. 2006;100(4):610–615. | |
Oshimo S, Bonella F, Guzman J, Costabel U. Hypersensitivity pneumonitis. Immunol Allergy Clin North Am. 2012;32(4):537–556. | |
Agache LO, Rogozea L. Management of hypersensivity pneumonitis. Clin Transl Allergy. 2013;3(1):5. | |
Lota HK, Keir GJ, Hansell DM, et al. Novel use of rituximab in hypersensitivity pneumonitis refractory to conventional treatment. Thorax. 2013;68(8):780–781. | |
Keir GJ, Maher TM, Ming D, et al. Rituximab in severe, treatment-refractory interstitial lung disease. Respirology. 2013;19(3):353–359. | |
Kern RM, Singer JP, Koth L, et al. Lung transplantation for hypersensitivity pneumonitis. Chest. 2015;147(6):1558–1565. | |
Schmidt CD, Jensen RL, Christensen LT, Crapo RO, Davis JJ. Longitudinal pulmonary function changes in pigeon breeders. Chest. 1988;93(2):359–363. | |
Gaxiola M, Buendía-Roldán I, Mejía M, et al. Morphologic diversity of chronic pigeon breeder’s disease: clinical features and survival. Respir Med. 2011;105(4):608–614. | |
Mooney JJ, Elicker BM, Urbania TH, et al. Radiographic fibrosis score predicts survival in hypersensitivity pneumonitis. Chest. 2013;144(2):586–592. | |
Walsh SL, Sverzellati N, Devaraj A, Wells AU, Hansell DM. Chronic hypersensitivity pneumonitis: high resolution computed tomography patterns and pulmonary function indices as prognostic determinants. Eur Radiol. 2012;22(8):1672–1679. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.