Back to Archived Journals » Neurobehavioral HIV Medicine » Volume 7
Chronic distress and the vulnerable host: a new target for HIV treatment and prevention?
Authors Contoreggi C, Chrousos GP, Di Mascio M
Received 9 April 2015
Accepted for publication 5 August 2015
Published 28 December 2016 Volume 2016:7 Pages 53—75
DOI https://doi.org/10.2147/NBHIV.S86309
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Stephen Ferrando
Carlo Contoreggi,1 George P Chrousos,2 Michele Di Mascio3
1Intramural Research Program (IRP), National Institute on Drug Abuse (NIDA), National Institutes of Health (NIH), Baltimore, MD, USA; 2Department of Pediatrics, Aghia Sophia Children’s Hospital, National and Kapodistrian University of Athens Medical School, Athens, Greece; 3AIDS Imaging Research Section, Division of Clinical Research, National Institute of Allergy and Infectious Diseases (NIAID), National Institutes of Health (NIH), Bethesda, MD, USA
Abstract: Pathologic stress (distress) disturbs immune, cardiovascular, metabolic, and behavioral homeostasis. Individuals living with HIV and those at risk are vulnerable to stress disorders. Corticotropin-releasing hormone (CRH) is critical in neuroendocrine immune regulation. CRH, a neuropeptide, is distributed in the central and peripheral nervous systems and acts principally on CRH receptor type 1 (CRHR1). CRH in the brain modulates neuropsychiatric disorders. CRH and stress modulation of immunity is two-pronged; there is a direct action on hypothalamic–pituitary–adrenal secretion of glucocorticoids and through immune organ sympathetic innervation. CRH is a central and systemic proinflammatory cytokine. Glucocorticoids and their receptors have gene regulatory actions on viral replication and cause central and systemic immune suppression. CRH and stress activation contributes to central nervous system (CNS) viral entry important in HIV-associated neurocognitive disorders and HIV-associated dementia. CNS CRH overproduction short-circuits reward, executive, and emotional control, leading to addiction, cognitive impairment, and psychiatric comorbidity. CRHR1 is an important therapeutic target for medication development. CRHR1 antagonist clinical trials have focused on psychiatric disorders with little attention paid to neuroendocrine immune disorders. Studies of those with HIV and those at risk show that concurrent stress-related disorders contribute to higher morbidity and mortality; stress-related conditions, addiction, immune dysfunction, and comorbid psychiatric illness all increase HIV transmission. Neuropsychiatric disease, chronic inflammation, and substance abuse are endemic, and chronic distress is a pathologic factor. It is being understood that stress and CRH are fundamental to neuroendocrine immunity; therapeutic interventions with existing and novel agents hold promise for restoring homeostasis, reducing morbidity and mortality for those with HIV and possibly reducing future disease transmission.
Keywords: corticotropin-releasing hormone, stress, neuroendocrine immunology, hypothalamic–pituitary–adrenal, cytokine, glucocorticoid resistance
Introduction
Individuals who engage in high-risk behaviors remain the principal reservoirs of HIV, allowing continued viral dissemination into the community. Men who have sex with men (MSM) and drug abusers remain the principal pools of HIV, though increasing numbers of women and men contract the disease through heterosexual contacts. Currently, MSM and injection drug users (IDU) represent approximately 55% and 15%, respectively, of people living with HIV/AIDS. Newly infected populations, in 2010, comprised 63% MSM, 8% IDU, approximately 25% heterosexuals, and 3% MSM–IDU. Statistics vary, but injection and non-injection are approximately 30% of those with HIV/AIDS. Thus, MSM, heterosexuals, and IDU remain the pools with the highest risk of contracting and disseminating HIV.1,2 Substance abusers, especially IDU, remain a difficult group to attract to and maintain in effective HIV treatment.3
Although there have been many successes in prevention, education, and screening in some groups and communities, risk behaviors still account for the vast majority of new infections, and reaching these individuals and groups remains a priority to curb spread.4 The risks of sexual exposure and drug abuse are not confined to any specific group or behaviors. Finding more effective strategies to connect with and reduce viral transmission is critical at this stage of the epidemic. Neuroendocrine immunology and the study of stress is an evolving discipline seeking to find pathophysiologic commonalities in at-risk populations. How stress contributes to behavioral, neuroendocrine, and immune dysfunction may offer new therapeutic approaches for those with HIV and at high risk. Clinical and preclinical studies show an activated stress system has deleterious effects and increases susceptibility to neurocognitive and psychiatric disease, diminishes immune integrity, and increases metabolic and cardiovascular disease. These contribute to weakened host defenses.5–7 Cytokines and stress hormone markers of neuroendocrine immune dysfunction are often present prior to and once HIV infection has occurred.
The central nervous system (CNS) regulates stress through corticotropin-releasing hormone (CRH), as well as through other neurotransmitters and neuromodulators. Stress engages the hypothalamic–pituitary–adrenal (HPA) axis, the extended amygdala (EA), and other limbic structures, as well as neocortical areas responsible for cognition. This also increases vulnerability to infections and weakens immunity directly and indirectly by promoting high-risk behaviors, enhancing susceptibility to cognitive and affective disorders, generating systemic inflammation, and offering a permissive neuroendocrine environment with adverse effects on host defenses.
With HIV infection, the HPA axis is hyperactive, increasing adrenal glucocorticoid (GC)/cortisol secretion and activation of GC receptors (GCr). At a molecular level, this increases viral replication in lymphocytes and other viral reservoirs. This causes apoptosis of normal lymphocytes, decreases in circulating CD4+ cells, adverse shifts in cytokine patterns, and weakening of mucosal defenses, all increasing immune compromise and disease progression.8–12
This review examines the elements that confer noninfected high-risk groups greater susceptibility to HIV and how chronic distress hastens HIV progression, increasing morbidity and mortality. Can interventions limiting the effects of stress be potentially therapeutic in slowing the progression of HIV and can they improve prevention efforts? New molecular targets that modulate homeostasis may provide molecular probes into neuroimmunity and also eventually offer additional behavioral and pharmacologic therapeutic options for managing complications and improving survival.5,13,14
Vulnerable populations
Persons living with HIV and some who are at risk show chronic activation of stress hormone secretion.15–18 MSM who have frequent or infrequent sex without barrier protection have a high risk of contracting and/or spreading HIV. Concurrent drug use increases the frequency of casual sex and the likelihood of infection and transmission. Finlayson et al,19 reporting from the National Behavioral Surveillance System, estimated 54% of MSM have unprotected sex, with 25% in casual circumstances. Additionally, concurrent drug use was reported in 46% of their subjects. Others have found similar results in relevant populations.20,21
Despite several studies focusing on sociobehavioral characteristics and the prevalence of risk behaviors within the MSM population(s) impacting disease transmission and progression, little is known about pre-seroconversion immune functions in this vulnerable population. Longitudinal studies of non-HIV-infected seronegative MSM show long-term dysfunction in cellular immunity equivalent to the impairment seen in early HIV infection with CD4 counts >500.22 Palmer et al reported higher levels of immune activation in a group of HIV-negative self-identified “high-risk” MSM compared to age- and ethnicity-matched self-identified “healthy, low-risk” male control individuals.23 These observations need to be further validated in larger trials and substantiated in particular by studies focusing on the etiological factors of the immune compromise in this vulnerable population. HIV clinical studies have indeed shown that pre-seroconversion elevated levels of immune activation are associated with increased risk for development of AIDS after HIV-1 seroconversion.24 In active IDU, the pre-seroconversion state of immune dysfunction and reduced CD4+ counts negatively predict the pace of peripheral CD4+ decline once infected. The type of substance(s) used did not affect immune decline, though, as one might expect, the injecting frequency did correlate with immune deterioration.25 Low levels of CD4+ cellular activation, a favorable measure of immune integrity, is associated with diminished susceptibility to HIV seroconversion.26
Sexual promiscuity (sexual activity with multiple partners without barriers), continuous exposure to other antigens in community networks with high prevalence of STD infections, use of drugs, and chronic stress are all potential contributors to the establishment of an altered immune activation in this vulnerable population.
Long-term chronic antigenic stimulation, eg, from receptive rectal sex without barriers, is exacerbated by chronic drug use and increases vulnerability to infection. The levels of plasma lipopolysaccharide have been reported to be significantly higher in MSM than in controls and inversely associated with time since most recent sexual encounter without a condom. The levels of plasma lipopolysaccharide (and of other microbial products) correlate with systemic immune activation, which drives chronic HIV infection and disease progression.27–29
When MSM actively use and/or inject drugs, this combination of behaviors may lead to greater susceptibility to seroconversion as well as being more efficient vectors of the HIV into noninfected MSM, non-MSM partners, and non-drug users.30 Chronic antigenic and immune stimulation in this population is a lead risk factor; when these patients also have concurrent psychiatric disease, their immune response is further compromised. These changes are accompanied by a neuroendocrine milieu associated with systemic and local/regional immune compromise.6,7 Inflammation increases susceptibility of target tissues to viral infection and transmission. Immune activation also increases HIV-1 transmission efficiency.28 Zhang et al have shown that, at the site of simian immunodeficiency virus (SIV) infections, principally resting CD4+ T-cells are infected. They hypothesized that initial low-level replication in resting CD4+ T-cells sustains local infection; activated CD4+ T-cells generate greater viral replication, which enables spread of infection from the initial site through regional lymphatics and systemically. High levels of activated CD4+ T-cells at the site of HIV-1 exposure may increase HIV-1 transmission efficiency, while low levels of activated CD4+ T-cells may be associated with a persistent HIV-seronegative status despite high-risk sexual behavior.31
Injection drug use can result in direct transmission of HIV through contaminated syringes and paraphernalia. Both injection and non-injection drug use increases high-risk hetero- and homosexual activity; exchanging sex for drugs and drug abuse among sex workers facilitates HIV infection.32–34 Central dysfunction of the stress system through overproduction of CRH and adrenal cortisol is integral to induction to, maintenance of, and relapse to drug abuse.35–37
Populations living with HIV/AIDS and those at high risk with relevant comorbidities show central and peripheral impairment of the immune and stress systems, which has implications for disease susceptibility and progression. Social milieu, sexual practices, substance abuse, and associated behaviors form the interface of central stress and somatic immune dysfunction; this is a critical facet in a new integrated understanding of these processes.
CRH, a critical neuropeptide, orchestrates stress responses
The central stress system
The neuroendocrine immune system is a multifaceted network of neuropeptides, neurotransmitters, and endocrine hormones, which regulate and are regulated by a complex array of immune modulators and hormones. These are multiple feedback and feedforward circuits that regulate homeostasis that are differentially activated under acute and chronic conditions. CRH is of particular interest as it has been identified as a principal (but far from the only) stress activator with CRH through CRH receptor type 1 (CRHR1), a critical molecular target. As mentioned elsewhere, CRH and CRHR1 are found in the CNS, in the peripheral nervous system, and throughout the immune system.38–40
CRH is a member of a family of stress-related peptides with two main receptors with multiple active and inactive subtypes.41 Stress peptides and their receptors show wide species variation in both their actions and anatomic distribution. In the primate, CRH has high affinity and physiologic activity at the CRHR1. In the CNS, CRH and CRHR1 are highly concentrated in the hypothalamus, areas of the cortex related to cognition, and the EA. The hypothalamus regulates stress hormones from the pituitary and adrenals. Salience, behavioral arousal, and the sympathetic nervous system (SNS) are controlled by brainstem structures, principally the pontomedullary and raphe nuclei and the locus caeruleus. Hypothalamic CRH is released to the anterior pituitary, activates CRHR1, and, with vasopressin, stimulates adrenocorticotropin (ACTH) release. ACTH is a potent adrenal secretagogue releasing cortisol and adrenal androgens, including dehydroepiandrosterone (DHEA). The hypothalamus activates the systemic SNS and the adrenal medullary catecholamines epinephrine and norepinephrine (NE). Direct autonomic innervation and circulating epinephrine and NE stimulate visceral and lymphoid tissues and activate metabolic, cardiovascular, immune, and other physiologic responses to adverse stimuli.
In addition to the hypothalamus, pituitary, and SNS, CRH and CRHR1 are expressed in the neocortex, which is important for executive and cognitive processing and motivational behaviors. The EA is a complex structure composed of discrete but tightly linked nuclei. The bed nucleus of the stria terminalis; central, basolateral, and medial nuclei of the amygdala; insulae; and medial nucleus accumbens form the hub of the EA.42,43 The EA forms a functional interface between the higher cognitive functions of the cortex to the endocrine hypothalamus and to autonomic regulation from the brainstem to the peripheral nervous system. The EA is the seat of memory, emotion, and motivational behaviors, fundamental to impulsivity, drug abuse, and sexual drive. Functional disruption of the EA is seen in stress-related disease such as major depressive disorder (MDD), generalized anxiety disorder (GAD), posttraumatic stress disorder (PTSD), chronic pain, metabolic syndrome, chronic medical conditions, substance abuse, addiction, and other compulsive disorders.6,7
CRH as cytokine and stress immune modulator
Peripheral CRH actions
CRH is secreted peripherally as a proinflammatory cytokine. It is expressed in lymphocytes, eosinophils, mast cells, monocytes, microglia, enterochromaffin cells, lymphatic and stromal cells, parasympathetic ganglia, cellular elements regulating the blood–brain barrier (BBB), colonic epithelium, gut immune tissues, integument, and others.44–50 It acts in a paracrine manner and increases inflammatory responses to infection and autoimmune diseases. CRH is mobilized in infection and inflammation, causes disruption of mucosal membranes and the BBB, and is a locus of tissue injury in autoimmune diseases.51–53
CRH and CRHR1 are important in gut immune integrity, being highly concentrated in intestinal mucosa, lymphoid tissue, and parasympathetic ganglia. CRH is secreted locally and then systemically in response to regional inflammation and infection. CNS activation of the SNS and parasympathetic nervous system stimulates regional CRH secretion. Mucosal tissues show high CD4+ expression, and the gastrointestinal (GI) mucosa is an especially critical organ for HIV exposure and infection.44,54–57 CRH activates CD4+ lymphocytes, and CRH blockade may represent a target to diminish CD4+ activation to limit systemic HIV dissemination.39,46 It remains somewhat controversial as to the exact mechanism(s) by which a proinflammatory immune system affects disease progression, even though inflammation has undisputed deleterious mucosal effects. Inflammation increases GI mucosal permeability and increases microbial translocation, increasing systemic bacterial load and circulating lipopolysaccharide, a biomarker for systemic immune activation, bacterial invasion, and systemic HIV progression.29,58
CRH stimulates the HPA axis, and excess GC/cortisol modulates immune responsivity. Under chronic stress, integrated daily cortisol secretion is elevated; compared to normal individuals, GC/cortisol concentrations may approach pharmacologic levels.59 Diminished sensitivity of the HPA axis and the CRH-containing hypothalamic neurons to GC/cortisol negative feedback is at least partially responsible for GC/cortisol overproduction. In the acute setting, endogenous cortisol as well as synthetic GC acutely causes immunosuppression, diminishing the inflammatory response to infection and inflammatory and other immune insults. Acute activation of the HPA axis and GC release are very effective in suppressing inflammation. When the stress system and the HPA axis are chronically activated, immune responsivity can convert to a smoldering proinflammatory state; this can be seen in autoimmune disorders, malignancy, atherosclerosis, and metabolic conditions. Responses to acute inflammation and infectious insults may be blunted, resulting in a suboptimal host defense during chronic stress-related proinflammatory states.5,38,39,60 Immune suppression can stem from cortisol overproduction causing acquired dysfunction of the GCr. GC hyper-sensitivity or GC resistance (R) can be tissue specific and may be present in the individual simultaneously, inhibiting or stimulating an inflammatory host response.61 GC hyper-sensitivity or GCR has been associated with neuroimmune dyshomeostasis and is also a marker of host vulnerability and disease progression.62
Immune modulation of the CNS CRH stress system
Stress activation regulates cytokines at multiple sites in the neuroendocrine immune cascade. When a proinflammatory environment is present, there is both passive and active transport of cytokines/chemokines and other inflammatory mediators into the brain. In areas where the BBB is absent (circumventricular organs) or less restrictive (median eminence), there is direct cytokine penetration.63–65 In addition, carrier transport proteins have been identified for some interleukins (ILs) and for tumor necrosis factor (TNF)-alpha.62,66,67 Rodent models show that proinflammatory mediators IL-1-beta, IL-2, IL-6, and interferon (IFN)-alpha activate hypothalamic CRH that stimulate pituitary ACTH and adrenal cortisol; also, cytokines increase CRH production in the EA and other subcortical brain structures.68–71
CRH stimulates mast cells. Mast cells are highly concentrated in the stroma of the BBB and, when activated by CRH, they are degranulated and increase BBB permeability.72–74 CRHR1 knockout animals show decreased BBB penetrability and peripheral immune activation.75 This is important in HIV. Observations that neuro-AIDS is not correlated with peripheral circulating viral load suggest that active and passive viral transport due to increased BBB permeability may be a factor in early viral colonization and the development of CNS HIV infection.76,77
Figure 1 demonstrates some of the neurohormonal interactions with the immune system in the setting of immune activation during HIV-1 infection.
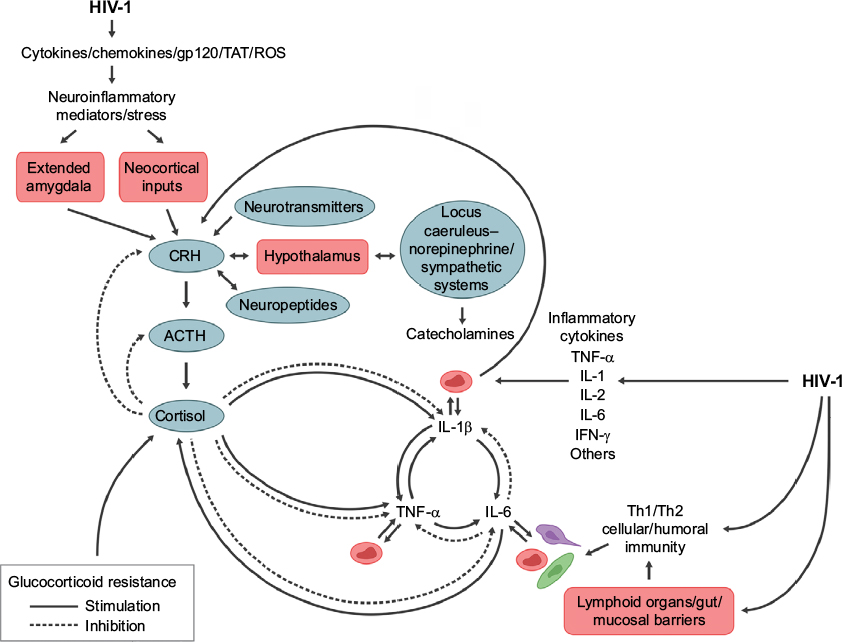 | Figure 1 The complex feedback and feedforward relations of neuroendocrine immune mediators in regulating the central nervous system and peripheral immune responses to HIV infection. Notes: Disruption of homeostatic mechanisms by infection is evident at multiple control points in stress and immune system regulation. HIV infection has direct and indirect effects on non-hypothalamic CRH and the hypothalamic–pituitary–adrenal axis to disrupt immune defenses. Abbreviations: ACTH, adrenocorticotropin; CRH, corticotropin-releasing hormone; IL, interleukin; IFN-γ, interferon gamma; ROS, reactive oxygen species; TAT, transactivator of transcription; TNF-α, tumor necrosis factor-alpha. |
Neuroinflammation and CNS HIV disease
Molecular actions within the infected brain
Delineation of the CNS as a potential target for anti-stress therapies stems from the high prevalence of neurologic complications with HIV infection. HIV-associated neurocognitive disorder and HIV-associated dementia prevalence has dropped in those living with HIV/AIDS with the adoption of highly active antiretroviral therapies (HAARTs). Prior to HAART, the prevalence of dementia in HIV+ individuals was estimated at >15% and is now estimated to be <5%. Less severe forms of “asymptomatic” neurocognitive disorders have increased to >30% despite HAART and controlled viremia.62,76–78
Microglial activation is seen in HIV-associated dementia, “asymptomatic” cognitive disruption, and IDU. Illicit drug-associated neuroinflammation impairs cognition, may contribute to ongoing risk behaviors, and may be additive after infection.79 The prevalence of addiction, depression, and other psychiatric disorders in HIV+ patients are independent causes of neuroinflammation, and all may contribute to the neuroimmune burden.80–82 The mechanism by which HIV-1 enters and infects cells in the CNS is complex. HIV-1 invades the CNS early after systemic infection and productively infects macrophages, microglial cells, and monocytes.
During HIV infection, CNS viral replication occurs principally in macrophages and microglia. These cells express CCR5 receptors necessary for viral entry.83 The two most potent effects of microglial and macrophage infection are the production of new virions and the induction of proinflammatory cytokine production. Microglia, macrophages, and monocytes release viral gp120 and transactivator of transcription (TAT) proteins, which are cytotoxic to neurons by generating free glutamate (GLU), an excitotoxic neurotransmitter, as well as by increasing free radical production.83 Along with extracellular calcium influx and cytokine/chemokine production, these insults can disable neuronal function and accelerate apoptosis. Neuronal death furthers neuroinflammation with the release of additional cytokines/chemokines, toxic cell membrane and synaptic molecules, free radicals, and more GLU.
Figure 2 shows the interface of HPA axis activation and bipartite actions of GCs/cortisol on inflammation in neuronal elements and neurons during HIV infection.
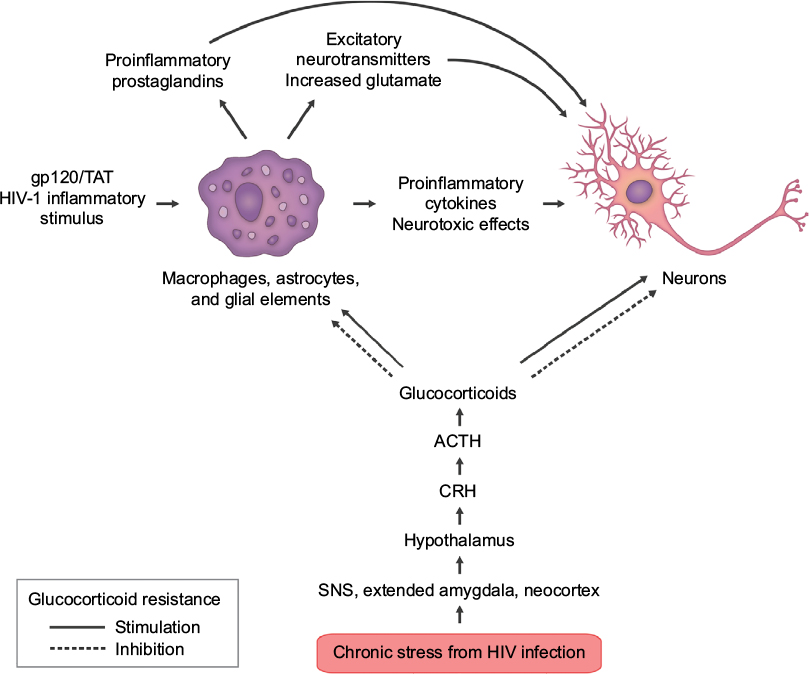 | Figure 2 Some of the complex interactions of HIV infection in the central nervous system. Notes: HIV infection triggers chronic stress hormone release; elevated levels of cortisol can trigger glucocorticoid resistance in susceptible immune cells and tissues. This engenders a proinflammatory environment, which generates a feedforward cascade of neuroinflammation. Inflammatory stimuli from virions, viral proteins gp120 and TAT, increased glutamate, cytokines/chemokines, free radicals, and membrane destabilization agents all contribute to neurodegeneration. This culminates in locoregional neuronal dysfunction on a microscale, which eventually translates to both structural and functional deficits leading to clinical neurologic and cognitive impairment. Abbreviations: ACTH, adrenocorticotropin; CRH, corticotropin-releasing hormone; SNS, sympathetic nervous system; TAT, transactivator of transcription. |
The inflammatory milieu and the presence of toxic viral proteins (VP) have demonstrable neurotoxic effects.62 Between 1% and 20% of astrocytes are reported to be HIV-1 DNA-positive in HIV-1-infected patients, eg, at a level similar to what is reported for lymphocytes in lymph nodes.83 There is no evidence of newly synthesized virions or VP generated from infected astrocytes in vivo. Microglial elements harbor HIV, and this reservoir can be activated and produce infectious virions, making it a potentially long-lived and significant reservoir even in successfully treated patients.84–87 Early entry of virus through the BBB suggests initial infection at a vulnerable time for colonization. CRH at the BBB may be a target for early intervention with CRHR1 antagonists to prevent or limit viral entry. Administering CRHR1 antagonists at or near the time of seroconversion might be optimal, though logistically difficult. The importance of CRH in BBB permeability is unknown; testing CRHR1 antagonists in early SIV infection may prove useful in understanding CRH and the potential efficacy of antagonists in maintaining barrier function.71–75
Clinical premorbid conditions impact HIV CNS progression
Depression studies show inflammation is activated through several pathways; markers of inflammation involve T-cell activation with a Th1 shift from Th2. This facilitates CNS release of cytokines, stimulating astrocytes and microglia. In depression and schizophrenia, as in HIV/AIDS cognitive impairment, microglia activation provokes a proinflammatory cascade increasing prostaglandin E (PGE) and quinolinic and kynurenic acid production, as well as cyclooxygenase-2 expression. Arachidonic acid (AA) and cell membrane changes related to inflammation are critical components of HIV neurodegeneration and other neuropsychiatric disorders.88–92
Sickness syndrome manifests with stress activation, which contributes to the symptomatology and morbidity in patients with HIV, as well as other chronic illnesses. Inflammatory PG and AA are implicated in sickness syndrome. Subtypes of PGE receptor are found throughout the hypothalamus, striatum, brainstem, and EA, where they are colocalized on CRH-, dopamine (DA)-, NE-, and serotonin (5HT)-containing neurons.93–96 Activation of PGE receptors stimulates neurons and neurotransmitter release in these structures and their projections. PGE and AA facilitate CRH-stimulated ACTH release and cytokine synthesis, release, and action, as well as febrile responses to inflammatory challenge, sickness syndrome symptoms, and stress-related behaviors. As important as PGE and AA are in maintaining physiologic homeostasis, it is not clear how chronic stress impacts AA and PGE generation and functionality.90–93
The mechanisms of neuroinflammation and stress may be supra-additive in HIV infection. Early inflammatory changes seen in CNS HIV disease add additional risks for psychiatric disease. Activation of membrane inflammatory pathways enhances degradation of 5HT and its precursor tryptophan, due to increased indoleamine 2,3-dioxygenase activity. GLU production is increased, with devastating effects on neuronal integrity and neurodegeneration (Supplementary materials). DA and N-methyl-D-aspartate (NMDA) signaling is also disrupted. Much of the current pharmacology for depression and affective disorders involves increasing 5HT, NE, DA, and other neurotransmitters from a purported depleted state by decreasing transmitter degradation, lengthening synaptic residence, increasing direct receptor stimulation, and/or stimulating transmitter synthesis. Inflammation increases the production of two neurotransmitters: kynurenic (a GLU agonist) and quinolinic acid, resulting in increased glutamatergic/excitatory neurotoxic neurotransmission through the NMDA receptor. These neurotransmitters are markers of inflammation and increase susceptibility to psychiatric disorders as they can deplete 5HT, DA, and NE. Other essential neurotransmitters such as acetylcholine at muscarinic and nicotinic cholinergic receptors are also impacted.93,94
GLU, over time, is important in the neurodegenerative processes, degrading neural plasticity and generating a hostile neurochemical milieu.95,96 The impact of these molecular events on CNS HIV progression is unknown, but may be cumulative with other known pathologic processes. These mechanisms would be expected to be additive to the progressive neurodegenerative processes in HIV. Demonstration of these effects will require both prospective observation studies and improved methods to track these effects in vivo.
Additive effects of substance abuse on neuroinflammation
Drugs of abuse, including psychostimulants, compromise BBB integrity and promote oxidative stress damage; methamphetamine (MA) is a commonly abused drug. SIV models of MA abuse show both higher CNS viral load and disruption of the BBB.97 MA use is independently associated with neurodegeneration through several mechanisms, including oxidative stress and DA neurotoxicity.97–101
Opiates have deleterious effects on neuroimmune function, and in vitro studies suggest that there is the potential for adverse clinical outcomes.102,103 Opiates can accelerate HIV replication, principally through mu-opiate receptor activation. Independent of HIV infection, opiates induce microglial, astrocyte, and neuronal inflammation. This causes glial cytokine and chemokine expression and accelerated macrophage apoptosis, which limit immune surveillance.104 In the setting of CNS HIV infection, opiate abusers show enhanced microglial activation.105,106 Opiate receptor subtypes can have opposing effects on CNS immune modulation; kappa receptor agonists (endogenous dynorphin peptides) can inhibit HIV replication and reduce microglial activation.107,108 Mu agonists (ie, endogenous endorphin, encephalin peptides; drugs: heroin, morphine, methadone, hydrocodone, codeine) increase viral production.102–105 Excitatory amino acid production, GLU, kynurenic acid, and NMDA are synergetic with mu-opioid agonist effects, which could enhance HIV replication and DA neuronal damage.109
Alcohol abuse and dependence are frequent in HIV+ infected individuals and is associated with high-risk sexual activity.110 Alcohol abuse increases the severity of neurocognitive or neuropsychological impairment in HIV infection, and alcohol independently has deleterious effects on cognition, immune function, and nutrition.111 Alcohol has neurotoxic effects by sensitizing the brain to excitatory neurotoxic amino acid neurotransmission, a common mechanism of glial and neuronal inflammation. NMDA antagonists, which block excitatory neurotransmission, can reduce neurotoxic effects from chronic alcohol exposure.112 CRH is a critical factor in alcohol abuse and dependence; under conditions of stress activation, CRHR1 knockout animals showed increased alcohol intake. This was linked to functional upregulation of the NMDA receptor, increasing sensitivity to ethanol.113–114 Blockade of CRHR1 blocks alcohol intake in rodents by modulating neurotransmitters associated with reward and reinforcement.115
Stress activation is critical for inducing and maintaining substance abuse and regulating neurochemical systems that modulate CNS neurotoxicity. CRH stimulation of NMDA and glutamatergic pathways sustains inflammation.116 This, with other stress dysfunction, increases behavioral susceptibility to drug use. Stress, both acute and chronic, heightens vulnerability to the acquisition and maintenance of substance abuse. The mechanisms whereby CRH impacts addictive behaviors and HIV are complex; the vulnerability to HIV and disease progression involve underlying neuropathologic and psychiatric processes, the pharmacology of abused substance(s), immune integrity, and other as-yet ill-defined factors.30,116–119 Chronic exposure to drugs generates a chronic activation of the HPA axis with resultant hypercortisolemia associated with diminished feedback responsivity, leading to increased overall GC actions, leading to immune and neurotransmitter dysfunction. Overproduction of CRH in EA and the neocortex distorts executive functioning, impulsivity, and motivational behaviors, which are all critical in maintaining addiction.30,118 The molecular actions of drugs of abuse in the setting of CNS HIV colonization likely constitute a supra-additive neuroinflammatory milieu with clinical relevance in those with addictive illness.
The epigenetics of stress, inflammation, and behavior
Stress and CRH have both central and peripheral immune-modulating effects. As noted in the CRH as cytokine and stress immune modulator section, this is relevant in active infection and those susceptible to HIV transmission. Tung et al examined epigenetic immune gene regulation in a rhesus macaque model of dominant and subordinate social hierarchy.120 Subordinate individuals in social groups with behavioral and HPA axis markers of stress, presumably regulated through CNS CRH overproduction, suffer chronic stress associated with behavioral, immune, reproductive, and metabolic morbidity.121,122 A proinflammatory state was seen in subordinate animals;120 proinflammatory cytokine/chemokine gene activation in peripheral blood mononuclear cells was associated with group social ranking. Enhanced IL signaling, T-cell activation, and proinflammatory cytokine and chemokine overexpression with GCR was prevalent in subordinate, behaviorally stressed macaques. Lower social rank also correlated with higher cortisol levels.
During social isolation, a standard measure of inescapable stress, subordinate animals showed maladaptive responses. This poses clinical relevance, as social isolation and conditions of inescapable stress provide established models of psychiatric illness in nonhuman primates. When animals reentered social groups in a dominant rank, proinflammatory genes and peripheral markers were suppressed with immune reconstitution. Epigenetic analysis showed a majority of immune-regulating histones were demethylated, causing gene reactivation and shift to a noninflammatory state in response to a new dominant social rank. Reducing social stress leads to dynamic epigenetic changes in the expression of immune defense genes. Maladaptive IL signaling, T-cell activation, proinflammatory cytokine/chemokine overexpression and GCR, found in subordinate macaques, reversed, with possible long-term survival benefits.120
Tung et al’s study of epigenetic immune regulation120 is a clinically relevant translational model of immune competence. This study links chronic stress and immune dysfunction to established markers of CRH overproduction, including elevated cortisol secretion and conditioned fear behaviors, suggesting centrally mediated chronic stress has a potent influence on immune physiology. These patterns of neuroendocrine disruption are frequently clinically demonstrated, and the behaviors tested here (conditioned helplessness) in similar stress models are responsive to intervention with a high-affinity CRHR1 antagonist, such as antalarmin.6,123 Preclinical experiments such as these are commonly used to model mental illness. The sociobehavioral milieu is critical to immune defense and supports clinical observations of increased morbidity and mortality in HIV-infected and vulnerable groups and how effective psychiatric interventions can restore immune and neuroendocrine homeostasis.5–7,59,120–124 These observations and molecular mechanisms will show increasing importance as the field of neuroendocrine immunology matures and, with that, their relevance to those living with HIV and individuals at risk.
Molecular actions of stress hormones
Chronic stress activation, CRH/HPA axis, and inflammation
Clinical studies show progressive increases in cortisol production are associated with impaired immunity and advancing disease progression.8,59,124–130 Chronic exposure to elevated levels of cortisol can adversely shift cytokine production and cellular immunity. In addition, in the setting of inflammation and infection, peripheral CRH together with other immune activators, such as histamine and catecholamines, induces production of IL-1, IL-6, IL-8, IL-18, TNF-alpha, and C-reactive protein, thereby driving inflammatory processes.131–133
When proinflammatory cytokines cross the BBB, they stimulate the HPA axis.39,69 Disease states characterized by chronic inflammation, such as granulomatous and autoimmune diseases, are frequently associated with behavioral and cognitive dysfunction and sickness syndrome.134,135 Depression is common with active inflammation, and clinical disease remission often results in symptom resolution.136 Sickness syndrome seen in chromic medical disorders can be partially attributed to inflammation and cytokine actions in the CNS.39,134–140 Proinflammatory biologics (ie, IFN and ILs) used in treatment of malignancy, multiple sclerosis, hepatitis C, and other disorders have well-recognized and frequent psychiatric and cognitive complications; concurrent treatment with antidepressants and other psychotropics is often recommended.136–142 With discontinuation of these therapies, CNS symptoms often resolve.
Chronic stress, the GCr, and GCR
GCR syndromes are important in HIV pathology. Excess cortisol can induce and exacerbate target organ disease such as visceral obesity, metabolic syndrome, GI disorders, immune suppression, opportunistic and other infections, atherosclerosis, and bone loss, conditions all seen in HIV infection.5,6,142–144 Tissue sensitivity to cortisol enhances risk to these and related disorders in susceptible and infected populations. Sensitivity to cortisol is a critical factor in multiple immune tissues. Lymphocytes, eosinophils, mast cells, monocytes, and dendritic cells all express GCr and have been demonstrated to manifest GCR in a wide variety of immune disorders.5,61 The interactions between GC and development of GCR in cells, tissues, and immune organs and how they induce and amplify stress-related disorders are not well understood.
Expression of GCr in the CNS and pituitary play a counterbalancing regulatory function enabling negative feedback integrity for controlling and muting physiologic stress responses. Receptors in hypothalamic nuclei and the anterior pituitary direct negative feedback control on ACTH and adrenal cortisol. Changes in GCr expression and sensitivity are seen in the hippocampus, EA, and cortical areas and, in the setting of chronic inflammation, in neuropsychiatric and neurodegenerative diseases leading to GCR. CRHR1 and GCr are frequently coexpressed on neurons where they are upstream modulators of neurotransmitters including GLU, 5HT, DA, and NE, as well as numerous neuropeptides and brain-derived neurotropic factors.5,145–150
GCR can be caused by competition for cortisol for the GCr binding by inducing expression of nonfunctional receptor isoforms, reducing ligand availability to functional native receptors. Reduced ligand binding affinity, decreased nuclear translocation of the activated GC–GCr complex, and decreased efficacy in modulating responsive transcription factors are key in the development of tissue GCR.150–152
GCR and HIV replication
GCs with their receptor(s) translocate to nuclei of HIV-infected T-cells. GCr activation has multiple cellular regulatory functions in HIV-infected cells and tissues, and GCR impacts HIV infection, replication, and progression. Cytokines and chemokines induce GCR and are, in turn, suppressed by GCR.153–155 IFN, TNF-alpha, IL-2, IL-6, IL-15, and IL-3 act to increase production of inactive competing GCr isoforms and decrease GCr HIV nuclear translocation, DNA transcription, and RNA synthesis and can reduce ligand receptor sensitivity up to fivefold in GCR HIV patients.5,8 The GC/GCr complex is synergistic with gp120 and can induce apoptosis in immunocompetent lymphocytes, hastening HIV progression.10,11 GCr exerts transcriptional control over viral replication.155,156 Early in the course of infection, VP with the GC/GCr complex enable proviral migration from cytoplasm into the nucleus, allowing RNA integration into the host cell.12 As infection progresses in CD4+ cells, GCr co-regulates critical aspects of viral replication through complexing with VP within cells. Ligand-bound GCr and VP complex can suppress cellular protective genes, ie, NF-κB, which can upregulate proinflammatory genes. An activated GCr-VP can also stimulate transcription of poly (ADP-ribose) polymerase 1 (PARP-1), a protein which can induce DNA repair, aiding HIV genome nuclear integration and propagation; activating PARP-1 increases cell depletion and immunosuppression.153 Activated GCr shows a synergistic effect on viral replication in the presence of proinflammatory cytokines.9,15 Activating the GCr complex with the GC dexamethasone potentiates TNF-alpha and IL-6 HIV transcription, viral RNA expression, and viral assembly. In vitro, the GCr antagonist RU 486 inhibited the GC stimulatory effects on replication.157 Despite the fact that GCr activation is critical in HIV replication, RU 486 failed to reduce HIV viral load in vivo.158 The reason for failure of reduction in viral load is unclear; RU 486 blockade of GCr in the cell may not be complete or sufficient to block intracellular trafficking of GC/GCr complex to inhibit viral replication. The molecular mechanism(s) of how RU 486 and GCr interact are far from being fully understood.
Modulation of the stress–immune system interaction: treatment implications
Stress, inflammation, and neuropsychiatric disorders in HIV
Affective disorders, ie, MDD, GAD, PTSD, and substance abuse, are overrepresented in at-risk populations.19–21,159,160 Behavioral and hormonal dysfunction characterizes these disorders with pathologic HPA axis activation from overproduction of CNS CRH in critical neurocognitive and limbic sites.6,161
The interface of CRH, neuropeptide, and neurotransmitter disruption in depression is well recognized; neurocognitive deficits in depression and affective disorders show commonalities with the neuropsychological effects of HIV neuroinfection. How CRH overproduction and central stress axis derangements impact neurocognitive decline with and without comorbid psychiatric disease needs better delineation.
The incidence of psychiatric comorbidity in substance abusers is high. A large study of IDU found high prevalence of MDD, GAD, alcohol dependence, personality disorders, and multiple substance use.162 Similar psychiatric disorders are found in MSM with negative societal bias; cultural milieu has a profound influence on psychological state and development of depression.163–164 Depressive affect is associated with higher mortality in infected men and was found to predict a more rapid decline in CD4 count.165,166
Changing demographics in 2013 found increasing numbers of women contract HIV through heterosexual exposures. The majority of those living with HIV remain white MSM, but African American (AfAm) MSM are overrepresented in HIV populations, as are Hispanic MSM. AfAm heterosexual women comprise the largest number of new infections, at 20%, and IDU make up 8% of new cases.167 HIV is a leading cause of death in AfAm and Hispanic women aged 25–44 years.168 In these women, nearly half reported chronic depressive symptoms and had nearly a fourfold increased prevalence of psychiatric disorders compared with matched noninfected women.134,169 Similar psychiatric disorders are observed in women as are seen in men. Chronically depressed women were found to be twice as likely to die as women without persistent symptoms; in addition, the trajectory of CD4 decline was significantly faster.134 Untreated depression delays women accessing care and instituting HAART and reduces their adherence with medical therapy. Comorbid substance abuse impairs effective treatment compliance further.170,171
The presence of a positive affect and resolution of depressive symptoms in longitudinal studies of HIV-infected men improves peripheral inflammation, including serological markers of active infection; increases CD4; lowers viral loads; and improves medication adherence. Importantly, improved mental health decreased disease mortality over a 3-year period.134,136,172
Somatic and psychological stressors have been linked to higher viral loads and more rapid disease progression despite HARRT.173–177 Leserman et al reported that cortisol elevations were independent of psychosocial factors as a risk for disease progression.136,176 Reductions in psychosocial stress and decreases in cortisol secretion are reported to precede immune reconstitution in the setting of behavioral therapy.130,177 These data suggest that neuroendocrine immunity and its behavioral manifestations may act autonomously in hastening the transition of HIV to AIDS, and stress activators may be credible molecular targets for controlling disease.
Depression, anxiety, and PTSD are common in HIV patients.178 Carrico and Antoni analyzed 31 randomized controlled trials of various psychological interventions in HIV patients. Fourteen studies included physiological measures.177 Though studies varied in treatment and study design, those involving psychological interventions showed improvement in patients’ well-being and neuroendocrine markers of stress and immunity. The authors concluded that psychosocial and behavioral therapies provide benefit, though larger and more standardized investigations are necessary.
Pharmacologic and behavioral psychiatric interventions
Membrane rescue agents
Preclinical studies support a role for mood stabilizers as membrane rescue agents. Mania, though far from fully understood, appears to have membrane inflammation as an important part of the disease constellation.89,93 Membrane stabilization by decreasing AA synthesis and turnover through decreased brain cyclooxygenase-2 activity and PGE are possible ancillary mechanisms for mood stabilizers (ie, lithium, valproic acid, lamotrigine, and carbamazepine); this may contribute to their anti-seizure effects.91,92,94,96 Reducing inflammation and stabilization of cell membranes may offer therapeutic options for treatment of cognitive disorders and related comorbid psychiatric conditions. HIV proteins compromise cell membrane integrity and have detrimental effects on neurotransmitters (ie, GLU, DA) and stress activation.60 Lithium, valproic acid, lamotrigine, and carbamazepine are primary and adjunct medications for major mental illness, so their use could be a future consideration for HIV pharmacotherapy.
Modulation of GCR and HPA axis homeostasis
Central CNS actions of GC and GCR have been incompletely explored, though normalization in cortisol and resultant reduction in nuclear GCr decrease cytokine expression and translation.179 Stress levels of GC have been shown in vitro to be toxic to cortical and hippocampal neurons.179–181 Delineation of epigenetic mechanisms within the CNS in vivo awaits substantial refinements in pharmacology, surrogate markers of gene expression, and imaging technologies. In vivo imaging of second messenger and related gene/receptor expression in oncology may allow future applications in neuroscience.182–184 Stress activation, HPA axis, and GCR markers would offer measurable refinements to correlate CNS immune activation with possible synergistic therapeutic future targets.185,186
Preclinical studies have shown that anti-GC medications such as RU 486 are neuroprotective.185,186 Administration of RU 486 in immune and behavioral models of neuroinflammation partially inhibits or completely blocks glial inflammation. Clinical studies with RU 486 show varying degrees of efficacy in treatment of major and psychotic depression, though the mechanisms for these effects are unknown.187–190
In GCR, an activated HPA axis with CRH overproduction can contribute to neuroinflammation; might GC antagonists such as RU 486 reduce CNS inflammation? In vivo imaging of glial and neuronal inflammation is progressing; current agents will benefit from increased sensitivity, specificity from these technologies, and comprehensive clinical validation. Imaging probes to study CNS disease progression await more refinement for assessment of treatment response and clinical efficacy of neuropharmacologic intervention.191–195 Further studies are needed to discern the actions of GC antagonists (and other therapeutics) in the progression of CNS HIV infection.
CRHR1 antagonist medications
Clinical depression is the best studied area for CRH and CRHR1 medications. No in vivo marker for CRHR1 is yet available, so surrogate measures and autopsy material are best validated and most frequently studied. Increased cerebral spinal fluid CRH and/or decreased CNS binding density of CRHR1 postmortem has been identified in patients with suicide, depression, addiction, PTSD, and other disorders.196–201 These findings may extrapolate to immune-compromised individuals with psychiatric and comorbid medical illness. As shown, CRH overproduction with resultant excess adrenal cortisol secretion is important in the genesis of mental illness, addiction, and inflammatory changes mediated by the GCr.15,36,37
Its many physiologic actions and associations with disease have made CRHR1 an important molecular target for drug development. There has been a concerted effort by the pharmaceutical industry to develop non-peptide CRHR1 antagonists as antidepressant and anti-stress medications.201 The first clinical trial by Zobel et al with the CRHR1 antagonist R121919 showed some efficacy in treatment of patients with MDD.202 This was an open-label trial, and its design is frequently criticized; however, when medication was withdrawn, some patients had symptom relapse. A number of other non-peptide antagonists were developed and brought to clinical trials. Indications to date have been depression, anxiety, and irritable bowel syndrome (IBS). The exact number of sponsored trials is unknown, though multiple Phase I and Phase II trials have been registered on ClinicalTrials.gov. The results of the clinical studies to date have been disappointing.201 No successful Phase III trials have been reported. CRHR1 antagonists have not been consistently successful as antidepressants, though newer-generation compounds are being developed with better pharmacologic characteristics.201,203,204 There are many speculations as to the reasons for the clinical trial failures; first, studies have been conducted on patients with chronic psychiatric disorders which may complicate the interpretation of the data. Second, these agents may have limited efficacy in reconstitution of homeostasis, as opposed to disease prevention and progression. Yet, despite challenges, interest remains in this molecular target for these and other indications relevant to HIV.
The CRHR1 antagonist antalarmin is effective in susceptible rodent strains in reducing gut inflammation and joint destruction in an adjuvant-induced model of rheumatoid arthritis.52,61
CRH acts as an inflammatory cytokine at the involved joints. Antalarmin blocks CRHR1 expressed on immune cells/tissues as well as ameliorating the chronic activation of the HPA axis; it regularizes circadian rhythms disrupted in chronic stress.205 Reducing GC production may also contribute to improvements in target tissue GCR seen in autoimmune disorders. Administration of the CRHR1 peptide antagonist alpha-CRH blocked distension and electrically induced abdominal pain in patients with IBS. IBS is characterized by peristaltic dysregulation, intestinal inflammation, and increased gut permeability with a lowered threshold to gut-induced pain. Anticipatory anxiety is a prominent feature of IBS.46,206,207 CRHR1 antagonist peptides do not cross the BBB and show peripheral actions through locoregional anti-inflammatory, SNS, and/or other non-CNS effects.29,46 How and if these observations are relevant to HIV proliferation in the gut are unknown; further assessment of CRHR1 antagonists as anti-inflammation agents in this and other conditions may guide future clinical investigations.
Overproduction of CNS CRH in cognitive processing, executive decision making, and emotional response areas of the brain causes CRHR1 downregulation in preclinical models of depression, anxiety, and addiction, as well as in patients with depression and stress-related diseases.197–200 Cerebral spinal fluid CRH, a surrogate marker, decreases with effective depression therapy, but this has not been demonstrated in HIV-related depression. Treatment of depression in HIV patients with medications and/or behavioral therapy improves viral load, CD4+ counts, inflammation, and immune integrity. The dual and potentially synergistic actions of reducing peripheral inflammation and CNS CRHR1 action may prove beneficial in the HIV-infected host. Improving emotional and cognitive symptoms and normalizing the HPA axis and cortisol, GCR, and proinflammatory states, especially in the gut and brain, hold promise for these agents; testing in SIV could be a productive next step.
Behavioral therapies
In randomized controlled trials of cognitive behavioral therapy, stress reduction, and other psychological interventions in HIV, patients showed benefits in neuroendocrine stress and immune markers.178 Positive affect significantly reduces the risk of dying from HIV.174,175,208 Pharmacotherapy is a common adjunct with psychotherapy for MDD, GAD, and PTSD.
Cognitive behavioral therapy with stress reduction administered to HIV-positive men improved neuroendocrine and immune markers of active infection in those who successfully completed 10 weeks of therapy. These included reductions of stress hormones and catecholamines and increased gonadal and adrenal androgens. Increased CD8, CD4, and antibody production were also seen along with reductions in herpes simplex type 2 antibodies.136,174
Symptomatology of chronic medical illness and psychiatric disease is often blurred. The usual tools and instruments for evaluation of psychiatric and neurocognitive impairment have limited validation in populations with chronic medical conditions and HIV (ie, Center for Epidemiologic Studies Depression Scale, the Hospital Anxiety and Depression Scale, the Beck Depression Inventory II, and the Patient Health Questionnaire).209–214 These populations may share common biological mechanisms of medical disease; psychiatric symptomatology can exacerbate the somatic manifestation(s).212–214 Immune activation is important in sickness syndrome and PGE, AA, CRH, and stress activation are factors in disease presentation and progression.81,89,139,215,216 Neocortical dysfunction impacts cognitive and executive functioning, adversely shaping patient decisions for seeking and adhering to treatment. Targeting these systems with anti-stress medications with combination therapies makes theoretical sense, though application and efficacy remain unknown.
Does substance abuse add greater risk?
Substance abuse, especially injection drug use, is associated with twice the mortality rate when compared with non-IDU HIV-positive individuals; also, there is less adherence to effective treatments in infected IDU, compromising therapeutic response.217–220 Pharmacologic interventions and substance abuse treatment have been generally less effective in HIV patients and add vulnerability to those at very high risk. Stabilization of patients with illicit or prescription opiate abuse using substitution therapy, ie, methadone or buprenorphine, results in reduced stress activation after months of treatment.221 Despite reductions in HPA axis hormones, treatment with chronic opiate therapy has a detrimental effect on neuronal integrity through increased glial and astrocyte stimulation and neuroimmune cell proapoptosis.222–226 This can accelerate progression to dementia in some individuals.227
There is no established effective pharmacology for stimulants such as MA and cocaine, though new molecular treatment targets continue to be explored.228 Associated comorbidity of drug use with other psychiatric disorders is associated with poorer outcomes, especially mood disorders such as concurrent MDD and PTSD. There is limited consensus on effective pharmacotherapies for the dual-diagnosed patient.228 Approaching patients with comorbid psychiatric disease and those with and at risk for HIV adds further diagnostic and therapeutic challenges. These individuals would be expected to have a higher incidence of central and peripheral stress activation based on infectious immune activation, addiction, and concurrent mental illness. Animal models of substance abuse show CRHR1 antagonists reduce self-administration, susceptibility to environmental stressors, and stress-induced drug relapse.36,37 Clinical studies of CRHR1 antagonists to prevent stress-induced relapse may prove to be effective.
Preclinical models of psychoimmune disorders have not been developed, so this area is unexplored. The drug abuse literature is replete with primate drug self-administration, neuro receptor imaging, and relapse models; there is potential to adapt these to SIV infection.216,229 Effectiveness of these agents in comorbid populations, let alone those with immunodeficiency, has yet to be established.
Antidepressants and immune recovery
In addition to behavioral effects, 5HT targeted and mixed monoamine medications have been shown to increase GCr expression, translocation, and gene transcription, resulting in diminished GCR.230,231 Effective antidepressant therapy also reverses proinflammatory cytokine production.232–237 The mechanisms of immune reconstitution from antidepressants and other medications are largely based on observational clinical and in vitro studies.229,238–243 Distinctions in immune effect between classes of medications have not been ascertained, principally because of the limited number of controlled studies and overriding clinical indications for patient therapy. Reductions in C-reactive protein are a commonly reported outcome measure in these observational clinical studies.240 Use of pharmacotherapy for treatment of psychiatric disease in HIV is associated with better survival and adherence to treatment.173,209 Mechanistic understanding of these agents in the HIV-infected host requires active investigation.
Peripheral inflammation: a therapeutic role for anti-stress medications?
CRHR1 antagonists have shown anti-inflammatory action in preclinical models of rheumatoid arthritis.52 Limited clinical studies with peripheral CRHR1 peptide blockers show these may have benefit in IBS.44,46,48,53–56 Many inflammatory disorders manifest both chronic inflammation and sickness syndrome.6,39,61 Target organs for HIV transmission represent critical sites for infection and progression. Bowel inflammation can increase susceptibility to HIV infection, and BBB integrity is critical to CNS transmission; CRHR1 antagonists could target both. HIV is characterized by a systemic proinflammatory state and by neuropsychiatric illness, so reduction in peripheral inflammation may be synergistic.
Anti-GC therapeutics
Administration of the GCr antagonist RU 486 inhibited the GC stimulatory effects on HIV replication in vitro.158 However, its in vivo administration to HIV infected patients did not show benefits in decreasing plasma viral load.159
This study did not assess behavioral effects or include patients with psychiatric illness. Activation of the stress axis with resultant GCR alters tissue GC sensitivity and can promote neuroinflammation. Some subgroups of patients with major and psychotic depression show response to these agents, with GCR as a possible factor.244 The degree to which GCR is responsible for HIV-related disease may predict clinical response. The actions of GC are complex with HIV, as they are with other medical and psychiatric disorders; further elucidation of the mechanism of action is needed.
Individuals at risk for infection show behavioral and immunologic vulnerability. Stress levels of GC have been shown in vitro to be toxic to cortical and hippocampal neurons.179,245,246 Substance abuse, depression, and immune stimulation with associated chronic stress activation may have as a common underpinning factor the development of GCR, as noted in sections on HPA and excess GC secretion.
Conclusion
Individuals living with HIV and those at high risk for infection carry a disease burden that involves neuropsychiatric, neuroendocrine, and immune dysfunction. The discovery of new molecular targets in the stress system, such as CRH, may be the intersection of seemingly disparate functions and disease manifestations. The stress system provides a commonality to better understand these disorders and for future therapeutic interventions. Central and peripheral stress mediators maintain homeostasis, and CRH has a central role in the choreography of neuroendocrine immunity.
Individuals at heightened risk for contracting and spreading HIV have frequent comorbidity with psychiatric disease, high-risk sexual behaviors, and substance abuse. These contribute to neuroendocrine immune dysfunction and may be supra-additive in determining risk for disease acquisition and progression.
The importance of CRH and its associated receptors is belied by their widespread distribution and varied physiologic functions. They are found in high concentration in the central and peripheral nervous system, immune cells and tissues, and organs integral to immune integrity. CRH is a psychosomatic molecular link critical in cognitive, emotive, immune, endocrine, and autonomic regulation. Central and peripheral feedback and feed forward pathways to and from the HPA axis, autonomic nervous system, immune system and CNS allow both acute and chronic modulation of homoeostasis to adjust to internal and external environmental insults.
Immune regulation by cytokines/chemokines provides a control circuitry to the CNS stress system critical in neuro-AIDS and neuroinflammation. The CNS remains vulnerable to infection in ways not yet clear; CRH and the proinflammatory milieu enhance BBB permeability to viral entry, and CRH may be an important preventive target. New approaches are needed to help explain the mechanism of neural infection and inflammation and why peripheral viral loads do not predict the development and progression of cognitive dysfunction despite HAART. Membrane inflammatory markers (PGE, AA, gp120) from microglial activation and neurotransmitter shifts (GLU neurotoxicity, monoamine depletion) are associated with CNS disease. CRH and central stress activation is seen, though whether these are primary or secondary events is uncertain. Membrane integrity contributes to both psychiatric and somatic manifestations of disease such as depression and “sickness syndrome” evident in other chronic inflammatory conditions (ie, other neurodegenerative conditions/autoimmune disorders); this is an important area for future studies.
Substance abuse with other psychiatric comorbidities as well as direct pharmacologic actions of drugs result in hypothalamic CRH, HPA axis disequilibrium, neuroinflammation which accelerate HIV progression and risk susceptibility. Addiction, with associated neural and peripheral inflammation and psychiatric and adverse metabolic consequences, increases HIV morbidity and mortality. CRHR1 antagonists may have the potential for substance abuse treatment; however, possible benefits to immune integrity are unknown.
Chronic stress causes detrimental epigenetic changes in behavior and peripheral immunity; reversal engenders recovery in preclinical models. How behavioral therapies and conventional and/or anti-stress drugs improve immune function remains to be demonstrated.
Molecular actions of cortisol are critical in viral replication. GCR from chronic overproduction of GC has detrimental effects on immunity and viral production. Our understanding of GCR is incomplete; simple blockade of the GCr with RU 486 holds no clinical benefits. The molecular complexity of GCR is considerable, and the interactions with neuroendocrine immune functions remain unclear.
Clinical observations and trials demonstrate improvements in psychiatric disease from behavioral and pharmaceutical therapies, with concomitant normalization of neuroendocrine immune integrity. Assuming similarities with MDD and GAD, CRH antagonists may provide a viable if only adjunctive target in well-selected populations.
The field of HIV and immunology awaits the development of new, more pharmacologically active CRHR1 antagonists as anti-stress medications and improved clinical trial methodology. When better drugs are found, trial study design should include assessment of immune parameters, the neuroendocrine stress state, presence of GCR, and neuroinflammation; improved standardized testing methodology for psychosocial disorders and neurocognition in these populations is critical. Preclinical investigations of CNS and regional immunity with CRHR1 blocker compounds can be performed in well-characterized models of SIV infection using basic immune methodologies. Studies of neuroinflammation, cellular immunity, and neuropharmacology will lead to improved translational models. The actions of CRH and the pharmacology of CRHR1 antagonists hold promise in improving health outcomes for those at risk and with disease, though much lies ahead.
Disclosure
The authors report no conflicts of interest in this work.
References
Centers for Disease Control and Prevention. HIV Among Gay and Bisexual Men. [webpage on the Internet]. Available from: http://www.cdc.gov/hiv/group/msm/index.html Accessed Nov 11, 2016. | ||
Available from: http://www.cdc.gov/nchhstp/newsroom/docs/2012/HIV-Infections-2007–2010.pdf. Accessed Nov 11 , 2016. | ||
Who is at risk for HIV Infection and Which Populations are Most Affected? [webpage on the Internet]. National Institute on Drug Abuse [updated July 2012]. Available from: http://www.drugabuse.gov/publications/hivaids/who-risk-hiv-infection-which-populations-are-most-affected. Accessed Nov 11, 2015. | ||
Persson A, Ellard J, Newman C, Holt M, de Wit J. Human rights and universal access for men who have sex with men and people who inject drugs: a qualitative analysis of the 2010 UNGASS narrative country progress reports. Soc Sci Med. 2011;73(3):467–474. | ||
Silverman MN, Sternberg EM. Glucocorticoid regulation of inflammation and its functional correlates: from HPA axis to glucocorticoid receptor dysfunction. Ann N Y Acad Sci. 2012;1261:55–63. | ||
Chrousos GP. Stress and disorders of the stress system. Nat Rev Endocrinol. 2009;5(7):374–381. | ||
Chrousos GP. Stressors, stress, and neuroendocrine integration of the adaptive response. The 1997 Hans Selye Memorial Lecture. Ann N Y Acad Sci. 1998;851:311–335. | ||
Norbiato G, Bevilacqua M, Vago T, Taddei A, Clerici. Glucocorticoids and the immune function in the human immunodeficiency virus infection: a study in hypercortisolemic and cortisol-resistant patients. J Clin Endocrinol Metab. 1997;82:3260–3263. | ||
Wang J, Harada A, Matsushita S, et al. IL-4 and a glucocorticoid up-regulate CXCR4 expression on human CD4+ T lymphocytes and enhance HIV-1 replication. J Leukoc Biol. 1998;64(5):642–649. | ||
Nair MPN, Mahajan S, Hou J, Sweet AM, Schwartz SA. The stress hormone, cortisol, synergizes with HIV-1 gp-120 to induce apoptosis of normal human peripheral blood mononuclear cells. Cell Mol Biol (Noisy-le-grand). 2000;46(7):1227–1238. | ||
Nair MP, Schwartz SA. Synergistic effect of cortisol and HIV-1 envelope peptide on the NK activities of normal lymphocytes. Brain Behav Immun. 1995;9(1):20–30 | ||
Wiegers K, Schwarck D, Reimer R, Bohn W. Activation of the glucocorticoid receptor releases unstimulated PBMCs from an early block in HIV-1 replication. Virology. 2008;375(1):73–84. | ||
“[No authors listed]. Glucocorticoids and mood: clinical manifestations, risk factors and molecular mechanisms. Proceedings of a meeting. June 20–21, 2008. La Jolla, California, USA. Ann N Y Acad Sci. 2009 Oct;1179:vii–viii, 1–233. | ||
Chrousos GP, Kino T. Glucocorticoid action networks and complex psychiatric and/or somatic disorders. Stress. 2007;10(2):213–219. | ||
Biglino A, Limone P, Forno B, et al. Altered adrenocorticotropin and cortisol response to corticotropin-releasing hormone in HIV-1 infection. Eur J Endocrinol. 1995;133(2):173–179. | ||
Kumar M, Kumar AM, Waldrop D, Antoni MH, Schneiderman N, Eisdorfer C. The HPA axis in HIV-1 infection. J Acquir Immune Defic Syndr. 2002;31(Suppl 2):S89–S93. | ||
Chittiprol S, Kumar AM, Satishchandra P, et al. Progressive dysregulation of autonomic and HPA axis functions in HIV-1 clade C infection in South India. Psychoneuroendocrinology. 2008;33(1):30–40. | ||
Zapanti E, Terzidis K, Chrousos G. Dysfunction of the hypothalamic-pituitary-adrenal axis in HIV infection and disease. Hormones (Athens). 2008;7(3):205–216. | ||
Finlayson TJ, Le B, Smith A, et al; Centers for Disease Control and Prevention (CDC). HIV risk, prevention, and testing behaviors among men who have sex with men – National HIV Behavioral Surveillance System, 21 U.S. cities, United States, 2008. MMWR Surveill Summ. 2011;60(14):1–34. | ||
Pappas MK, Halkitis PN. Sexual risk taking and club drug use across three age cohorts of HIV-positive gay and bisexual men in New York City. AIDS Care. 2011;23(11):1410–1416. | ||
Ghanem A, Little SJ, Drumright L, Liu L, Morris S, Garfein RS. High-risk behaviors associated with injection drug use among recently HIV-infected men who have sex with men in San Diego, CA. AIDS Behav. 2011;15(7):1561–1569. | ||
Barabás E, González R, Nagy K, Várkonyi V, Horváth A. No change in impaired cellular immune response of HIV-negative homosexuals after 15 years of HIV epidemic in Eastern/Central European region. J Investig Allergol Clin Immunol. 2001;11(3):172–175. | ||
Palmer CD, Tomassilli J, Sirignano M, et al. Enhanced immune activation linked to endotoxemia in HIV-1 seronegative MSM. AIDS. 2014;28(14):2162–2166. | ||
Hazenberg MD, Otto SA, van Benthem BH, et al. Persistent immune activation in HIV-1 infection is associated with progression to AIDS. AIDS. 2003;17(13):1881–1888. | ||
van Asten L, Danisman F, Otto SA, et al. Pre-seroconversion immune status predicts the rate of CD4 T cell decline following HIV infection. AIDS. 2004;18:1885–1893. | ||
Koning FA, Otto SA, Hazenberg MD, et al. Low-level CD4+ T cell activation is associated with low susceptibility to HIV-1 infection. J Immunol. 2005;175:6117–6122. | ||
Sandler NG, Douek DC. Microbial translocation in HIV infection: causes, consequences and treatment opportunities. Nat Rev Microbiol. 2012;10(9):655–666. | ||
Jiang W, Lederman MM, Hunt P, et al. Plasma levels of bacterial DNA correlate with immune activation and the magnitude of immune restoration in persons with antiretroviral-treated HIV infection. J Infect Dis. 2009;199(8):1177–1185. | ||
Douek D. HIV disease progression: immune activation, microbes, and a leaky gut. Top HIV Med. 2007;15(4):114–117. | ||
Sutton AJ, House T, Hope VD, Ncube F, Wiessing L, Kretzschmar M. Modelling HIV in the injecting drug user population and the male homosexual population in a developed country context. Epidemics. 2012;4:48–56. | ||
Zhang ZQ, Wietgrefe SW, Li Q, et al. Roles of substrate availability and infection of resting and activated CD4+ T cells in transmission and acute simian immunodeficiency virus infection. Proc Natl Acad Sci U S A. 2004;101(15):5640–5645. | ||
Muñoz FA, Pollini RA, Zúñiga ML, et al. Condom access: associations with consistent condom use among female sex workers in two northern border cities of Mexico. AIDS Educ Prev. 2010;22(5):455–465. | ||
Strathdee SA, Lozada R, Martinez G, et al. Social and structural factors associated with HIV infection among female sex workers who inject drugs in the Mexico-US border region. PLoS One. 2011;6(4):e19048. | ||
Surratt HL, Inciardi JA. An effective HIV risk-reduction protocol for drug-using female sex workers. J Prev Interv Community. 2010;38(2):118–131. | ||
Koob GF. The role of CRF and CRF-related peptides in the dark side of addiction. Brain Res. 2010;1314:3–14. | ||
Shalev U, Erb S, Shaham Y. Role of CRF and other neuropeptides in stress-induced reinstatement of drug seeking. Brain Res. 2010;1314:15–28. | ||
Shaham Y, Shalev U, Lu L, De Wit H, Stewart J. The reinstatement model of drug relapse: history, methodology and major findings. Psychopharmacology (Berl). 2003;168(1–2):3–20. | ||
Elenkov IJ. Neurohormonal-cytokine interactions: implications for inflammation, common human diseases and well-being. Neurochem Int. 2008;52(1–2):40–51. | ||
Calcagni E, Elenkov I. Stress system activity, innate and T helper cytokines, and susceptibility to immune-related diseases. Ann N Y Acad Sci. 2006;1069:62–76. | ||
Elenkov IJ, Wilder RL, Chrousos GP, Vizi ES. The sympathetic nerve--an integrative interface between two supersystems: the brain and the immune system. Pharmacol Rev. 2000;52(4):595–638. | ||
Grammatopoulos DK, Chrousos GP. Functional characteristics of CRH receptors and potential clinical applications of CRH-receptor antagonists. Trends Endocrinol Metab. 2002;13(10):436–444. | ||
Alheid GF, Heimer L. New perspectives in basal forebrain organization of special relevance for neuropsychiatric disorders: the striatopallidal, amygdaloid, and corticopetal components of substantia innominata. Neuroscience. 1988;27(1):1–39. | ||
Heimer L, Van Hoesen GW. The limbic lobe and its output channels: implications for emotional functions and adaptive behavior. Neurosci Biobehav Rev. 2006;30(2):126–147. | ||
Zheng PY, Feng BS, Oluwole C, et al. Psychological stress induces eosinophils to produce corticotrophin releasing hormone in the intestine. Gut. 2009;58(11):1473–1479. | ||
Kohno M, Kawahito Y, Tsubouchi Y, et al. Urocortin inflammatory activity. J Clin Endocrinol Metab. 2001;86(9):4344–4352. | ||
Taché Y, Martinez V, Wang L, Million M. CRF1 receptor signaling pathways are involved in stress-related alterations of colonic function and viscerosensitivity: implications for irritable bowel syndrome. Br J Pharmacol. 2004;141(8):1321–1330. | ||
Wlk M, Wang CC, Venihaki, M et al. Corticotropin-releasing hormone antagonists possess anti-inflammatory effects in the mouse ileum. Gastroenterology. 2002;123(2):505–515. | ||
Kokkotou E, Torres D, Moss AC, et al. Corticotropin-releasing hormone receptor 2- deficient mice have reduced intestinal inflammatory responses. J Immunol. 2006;177(5):3355–3361. | ||
O’Kane M, Murphy EP, Kirby B. The role of corticotropin-releasing hormone in immune- mediated cutaneous inflammatory disease. Exp Dermatol. 2006;15(3):143–153. | ||
Agelaki S, Tsatsanis C, Gravanis A, Margioris AN. Corticotropin-releasing hormone augments proinflammatory cytokine production from macrophages in vitro and in lipopolysaccharide-induced endotoxin shock in mice. Infect Immun. 2002;70(11):6068–6074. | ||
Kim BJ, Kayembe K, Simecka JW, Pulse M, Jones HP. Corticotropin-releasing hormone receptor-1 and 2 activity produces divergent resistance against stress-induced pulmonary Streptococcus pneumoniae infection. J Neuroimmunol. 2011;237(1–2):57–65. | ||
Webster EL, Barrientos RM, Contoreggi C, et al. Corticotropin releasing hormone (CRH) antagonist attenuates adjuvant induced arthritis: role of CRH in peripheral inflammation. J Rheumatol. 2002;29(6):1252–1261. | ||
Goebel M, Stengel A, Wang L, Reeve J Jr, Taché Y. Lipopolysaccharide increases plasma levels of corticotropin-releasing hormone in rats. Neuroendocrinology. 2011;93(3):165–173. | ||
Buckinx R, Adriaensen D, Nassauw LV, Timmermans JP. Corticotrophin-releasing factor, related peptides, and receptors in the normal and inflamed gastrointestinal tract. Front Neurosci. 2011;5:54. | ||
Wallon C, Söderholm JD. Corticotropin-releasing hormone and mast cells in the regulation of mucosal barrier function in the human colon. Ann N Y Acad Sci. 2009;1165:206–210. | ||
Fukudo S. Role of corticotropin-releasing hormone in irritable bowel syndrome and intestinal inflammation. J Gastroenterol. 2007;42(Suppl 17):48–51. | ||
Marchetti G, Bellistrì GM, Borghi E, et al. Microbial translocation is associated with sustained failure in CD4+ T-cell reconstitution in HIV-infected patients on long-term highly active antiretroviral therapy. AIDS. 2008;22(15):2035–2038. | ||
Vassallo M, Mercié P, Cottalorda J, Ticchioni M, Dellamonica P. The role of lipopolysaccharide as a marker of immune activation in HIV-1 infected patients: a systematic literature review. Virol J. 2012;9:174. | ||
Elias AN, Meshkinpour H, Valenta LJ, Grossman MK. Pseudo-Cushing’s syndrome: the role of alcohol. J Clin Gastroenterol. 1982;4(2):137–139. | ||
Pace TW, Hu F, Miller AH. Cytokine-effects on glucocorticoid receptor function: relevance to glucocorticoid resistance and the pathophysiology and treatment of major depression. Brain Behav Immun. 2007;21(1):9–19. | ||
Sternberg EM. Neural regulation of innate immunity: a coordinated nonspecific host response to pathogens. Nat Rev Immunol. 2006;6(4):318–328. | ||
Wang T, Rumbaugh JA, Nath A. Viruses and the brain: from inflammation to dementia. Clin Sci (Lond). 2006;110(4):393–407. | ||
Churchill M, Nath A. Where does HIV hide? A focus on the central nervous system. Curr Opin HIV AIDS. 2013;8(3):165–169. | ||
Watkins LR, Maier SF, Goehler LE. Cytokine-to-brain communication: a review & analysis of alternative mechanisms. Life Sci. 1995;57:1011–1026. | ||
Dantzer R, Aubert A, Bluthé RM, et al. Mechanisms of the behavioural effects of cytokines. In: Dantzer R, Wollman EE, Yirmiya R, editors. Cytokines, Stress, and Depression. New York: Kluwer Academic/Plenum Publishers; 1999:83–105. | ||
Banks WA, Farr SA, Morley JE. Entry of blood-borne cytokines into the central nervous system: effects on cognitive processes. Neuroimmunomodulation. 2002–2003;10(6):319–327. | ||
Banks WA, Kastin AJ, Broadwell RD. Passage of cytokines across the blood–brain barrier. Neuroimmunomodulation. 1995;2(4):241–248. | ||
Tsigos C, Chrousos GP. Hypothalamic-pituitary adrenal axis, neuroendocrine factors and stress. J Psychosom Res. 2002;53:865–871. | ||
Raber J, Sorg O, Horn TF, et al. Inflammatory cytokines: putative regulators of neuronal and neuro-endocrine function. Brain Res Brain Res Rev. 1998;26(2–3):320–326. | ||
Kronfol Z, Remick DG. Cytokines and the brain: implications for clinical psychiatry. Am J Psychiatry. 2000;157(5):683–694. | ||
Marques AH, Cizza G, Sternberg E. Interações imunocerebrais e implicações nos transtornos psiquiátricos. [Brain-immune interactions and implications in psychiatric disorders]. Rev Bras Psiquiatr. 2007;29(Suppl 1):S27–S32. Portuguese. | ||
Theoharides TC, Rozniecki JJ, Sahagian G, et al. Impact of stress and mast cells on brain metastases. J Neuroimmunol. 2008;205(1–2):1–7. | ||
Theoharides TC, Konstantinidou AD. Corticotropin-releasing hormone and the blood- brain-barrier. Front Biosci. 2007;12:1615–1628. | ||
Esposito P, Gheorghe D, Kandere K, et al. Acute stress increases permeability of the blood-brain-barrier through activation of brain mast cells. Brain Res. 2001;888(1):117–127. | ||
Benou C, Wang Y, Imitola J, et al. Corticotropin-releasing hormone contributes to the peripheral inflammatory response in experimental autoimmune encephalomyelitis. J Immunol. 2005;174(9):5407–5413. | ||
Heaton RK, Franklin DR, Ellis RJ, et al; CHARTER Group; HNRC Group. HIV-associated neurocognitive disorders before and during the era of combination antiretroviral therapy: differences in rates, nature, and predictors. J Neurovirol. 2011;17(1):3–16. | ||
Sacktor N, Lyles RH, Skolasky R, et al; Multicenter AIDS Cohort Study. HIV-associated neurologic disease incidence changes: Multicenter AIDS Cohort Study, 1990–1998. Neurology. 2001;56(2):257–260. | ||
Simioni S, Cavassini M, Annoni JM, et al. Cognitive dysfunction in HIV patients despite long-standing suppression of viremia. AIDS. 2010;24(9):1243–1250. | ||
Anand P, Springer SA, Copenhaver MM, Altice FL. Neurocognitive impairment and HIV risk factors: a reciprocal relationship. AIDS Behav. 2010;14(6):1213–1226. | ||
Bonnet F, Amieva H, Marquant F, et al; S CO3 Aquitaine Cohort. Cognitive disorders in HIV-infected patients: are they HIV-related? AIDS. 2013;27(3):391–400. | ||
Anderson E, Zink W, Xiong H, Gendelman HE. HIV-1-associated dementia: a metabolic encephalopathy perpetrated by virus-infected and immune-competent mononuclear phagocytes. J Acquir Immune Defic Syndr. 2002;31(Suppl 2):S43–S54. | ||
Nath A, Hauser KF, Wojna V, et al. Molecular basis for interactions of HIV and drugs of abuse. J Acquir Immune Defic Syndr. 2002;31(Suppl 2):S62–S69. | ||
Churchill MJ, Wesselingh SL, Cowley D, et al. Extensive astrocyte infection is prominent in human immunodeficiency virus-associated dementia. Ann Neurol. 2009;66:253–258. | ||
Hu NW, Ondrejcak T, Rowan MJ. Glutamate receptors in preclinical research on Alzheimer’s disease: update on recent advances. Pharmacol Biochem Behav. 2012;100(4):855–862. | ||
Brack-Werner R, Kleinschmidt A, Ludvigsen A, et al. Infection of human brain cells by HIV-1: restricted virus production in chronically infected human glial cell lines. AIDS. 1992;6(3):273–285. | ||
Tornatore C, Nath A, Amemiya K, Major EO. Persistent human immunodeficiency virus type 1 infection in human fetal glial cells reactivated by T-cell factor(s) or by the cytokines tumor necrosis factor alpha and interleukin-1 beta. J Virol. 1991;65(11):6094–6100. | ||
Valcour V, Chalermchai T, Sailasuta N, et al; RV254/SEARCH 010 Study Group. Central nervous system viral invasion and inflammation during acute HIV infection. J Infect Dis. 2012;206:275–282. | ||
Rao JS, Lee HJ, Rapoport SI, Bazinet RP. Mode of action of mood stabilizers: is the arachidonic acid cascade a common target? Mol Psychiatry. 2008;13(6):585–596. | ||
Farooqui AA, Ong WY, Horrocks LA. Inhibitors of brain phospholipase A2 activity: their neuropharmacological effects and therapeutic importance for the treatment of neurologic disorders. Pharmacol Rev. 2006;58(3):591–620. | ||
Müller N, Myint AM, Schwarz MJ. Inflammatory biomarkers and depression. Neurotox Res. 2011;19(2):308–318. | ||
Bazinet RP. Is the brain arachidonic acid cascade a common target of drugs used to manage bipolar disorder? Biochem Soc Trans. 2009;37(Pt 5):1104–1109. | ||
Leonard B, Maes M. Mechanistic explanations how cell-mediated immune activation, inflammation and oxidative and nitrosative stress pathways and their sequels and concomitants play a role in the pathophysiology of unipolar depression. Neurosci Biobehav Rev. 2012;36(2):764–785. | ||
Kim K, Lee SG, Kegelman TP, et al. Role of excitatory amino acid transporter-2 (EAAT2) and glutamate in neurodegeneration: opportunities for developing novel therapeutics. J Cell Physiol. 2011;226(10):2484–2493. | ||
Müller N, Schwarz MJ. The immune-mediated alteration of serotonin and glutamate: towards an integrated view of depression. Mol Psychiatry. 2007;12(11):988–1000. | ||
de Pablos RM, Herrera AJ, Espinosa-Oliva AM, et al. Chronic stress enhances microglia activation and exacerbates death of nigral dopaminergic neurons under conditions of inflammation. J Neuroinflammation. 2014;11:34. | ||
Vázquez-Santiago FJ, Noel RJ Jr, Porter JT, Rivera-Amill V. Glutamate metabolism and HIV-associated neurocognitive disorders. J Neurovirol. 2014;20(4):315–331. | ||
Kousik SM, Napier TC, Carvey PM. The effects of psychostimulant drugs on blood brain barrier function and neuroinflammation. Front Pharmacol. 2012;3:121. | ||
Sharma HS, Sjöquist PO, Ali SF. Drugs of abuse-induced hyperthermia, blood- brain barrier dysfunction and neurotoxicity: neuroprotective effects of a new antioxidant compound H-290/51. Curr Pharm Des. 2007;13(18):1903–1923. | ||
Sharma HS, Muresanu D, Sharma A, Patnaik R. Cocaine-induced breakdown of the blood-brain barrier and neurotoxicity. Int Rev Neurobiol. 2009;88:297–334. | ||
Silverstein PS, Shah A, Gupte R, et al. Methamphetamine toxicity and its implications during HIV-1 infection. J Neurovirol. 2011;17(5):401–415. | ||
Flora G, Lee YW, Nath A, Maragos W, Hennig B, Toborek M. Methamphetamine-induced TNF-alpha gene expression and activation of AP-1 in discrete regions of mouse brain: potential role of reactive oxygen intermediates and lipid peroxidation. Neuromolecular Med. 2002;2(1):71–85. | ||
Noel RJ Jr, Rivera-Amill V, Buch S, Kumar A. Opiates, immune system, acquired immunodeficiency syndrome, and nonhuman primate model. J Neurovirol. 2008;14(4):279–285. | ||
Anthony IC, Ramage SN, Carnie FW, Simmonds P, Bell JE. Does drug abuse alter microglial phenotype and cell turnover in the context of advancing HIV infection? Neuropathol Appl Neurobiol. 2005;31(3):325–338. | ||
Singhal PC, Sharma P, Kapasi AA, Reddy K, Franki N, Gibbons N. Morphine enhances macrophage apoptosis. J Immunol. 1998;160(4):1886–1893. | ||
Wang X, Zhang T, Ho WZ. Opioids and HIV/HCV infection. J Neuroimmune Pharmacol. 2011;6(4):477–489. | ||
El-Hage N, Wu G, Wang J, et al. HIV-1 Tat and opiate-induced changes in astrocytes promote chemotaxis of microglia through the expression of MCP-1 and alternative chemokines. Glia. 2006;53(2):132–146. | ||
El-Hage N, Wu G, Ambati J, Bruce-Keller AJ, Knapp PE, Hauser KF. CCR2 mediates increases in glial activation caused by exposure to HIV-1 Tat and opiates. J Neuroimmunol. 2006;178(1–2):9–16. | ||
Khurdayan VK, Buch S, El-Hage N, et al. Preferential vulnerability of astroglia and glial precursors to combined opioid and HIV-1 Tat exposure in vitro. Eur J Neurosci. 2004;19(12):3171–3182. | ||
Chao CC, Gekker G, Hu S, Sheng WS, Portoghese PS, Peterson PK. Upregulation of HIV-1 expression in cocultures of chronically infected promonocytes and human brain cells by dynorphin. Biochem Pharmacol. 1995;50(5):715–722. | ||
Chao CC, Gekker G, Hu S, et al. Kappa opioid receptors in human microglia downregulate human immunodeficiency virus 1 expression. Proc Natl Acad Sci U S A. 1996;93(15):8051–8056. | ||
Chao CC, Hu S, Gekker G, Lokensgard JR, Heyes MP, Peterson PK. U50, 488 protection against HIV-1-related neurotoxicity: involvement of quinolinic acid suppression. Neuropharmacology. 2000;39(1):150–160. | ||
Baldwin JA, Maxwell CJ, Fenaughty AM, Trotter RT, Stevens SJ. Alcohol as a risk factor for HIV transmission among American Indian and Alaska Native drug users. Am Indian Alsk Native Ment Health Res. 2000;9(1):1–16. | ||
Tyor WR, Middaugh LD Do alcohol and cocaine abuse alter the course of HIV- associated dementia complex? J Leukoc Biol. 1999;65(4):475–481. | ||
Rudolph JG, Lemasters JJ, Crews FT. Effects of chronic ethanol exposure on oxidation and NMDA-stimulated neuronal death in primary neuronal cultures. Alcohol Clin Exp Rex 1998;22(9):2080–2085. | ||
Chandler LJ, Newsom H, Sumners C, Crews F. Chronic ethanol exposure potentiates NMDA excitotoxicity in cerebral cortical neurons. J Neurochem. 1993;60(4):1578–1581. | ||
Sillaber I, Rammes G, Zimmermann S, et al. Enhanced and delayed stress-induced alcohol drinking in mice lacking functional CRH1 receptors. Science. 2002;296(5569):931–933. | ||
Kaur S, Li J, Stenzel-Poore MP, Ryabinin AE. Corticotropin-releasing factor acting on corticotropin-releasing factor receptor type 1 is critical for binge alcohol drinking in mice. Alcohol Clin Exp Res. 2012;36(2):369–376. | ||
Lowery EG, Spanos M, Navarro M, Lyons AM, Hodge CW, Thiele TE. CRF-1 antagonist and CRF-2 agonist decrease binge-like ethanol drinking in C57BL/6J mice independent of the HPA axis. Neuropsychopharmacology. 2010;35:1241–1252. | ||
Sparta DR, Sparrow AM, Lowery EG, Fee JR, Knapp DJ, Thiele TE. Blockade of the corticotropin releasing factor type 1 receptor attenuates elevated ethanol drinking associated with drinking in the dark procedures. Alcohol Clin Exp Res. 2008;32:259–265. | ||
Gehlert DR, Cippitelli A, Thorsell A, et al. 3-(4-Chloro-2-morpholin-4-yl-thiazol-5-yl)-8-(1-ethylpropyl)-2,6-dimethyl-imidazo[1,2-b]pyridazine: a novel brain-penetrant, orally available corticotropin-releasing factor receptor 1 antagonist with efficacy in animal models of alcoholism. J Neurosci. 2007;27:2718–2726. | ||
Arango JC, Simmonds P, Brettle RP, Bell JE. Does drug abuse influence the microglial response in AIDS and HIV encephalitis? AIDS. 2004;18(Suppl 1):S69–S74. | ||
Kreek MJ, LaForge KS Butelman E. Pharmacotherapy of addictions. Nat Rev Drug Discov. 2002;1:710–726. | ||
Logrip ML, Koob GF, Zorrilla EP. Role of corticotropin-releasing factor in drug addiction: potential for pharmacological intervention. CNS Drugs. 2011;25(4):271–287. | ||
Refojo D, Schweizer M, Kuehne C, et al. Glutamatergic and dopaminergic neurons mediate anxiogenic and anxiolytic effects of CRHR1. Science. 2011;333(6051):1903–1907. | ||
Tung J, Barreiro LB, Johnson ZP, et al. Social environment is associated with gene regulatory variation in the rhesus macaque immune system. Proc Natl Acad Sci U S A. 2012;109(17):6490–6495. | ||
Sapolsky RM. The influence of social hierarchy on primate health. Science. 2005;308:648–652. | ||
Sapolsky RM. Social status and health in humans and other animals. Annu Rev Anthropol. 2004;33:393–418. | ||
Habib KE, Weld KP, Rice KC, et al. Oral administration of a corticotropin-releasing hormone receptor antagonist significantly attenuates behavioral, neuroendocrine, and autonomic responses to stress in primates. Proc Natl Acad Sci U S A. 2000;97(11):6079–6084. | ||
Antoni MH, Cruess S, Cruess DG, et al. Cognitive-behavioral stress management reduces distress and 24-hour urinary free cortisol output among symptomatic HIV- infected gay men. Ann Behav Med. 2000;22(1):29–37. | ||
Ickovics JR, Hamburger ME, Vlahov D, et al; HIV Epidemiology Research Study Group. Mortality, CD4 cell count decline, and depressive symptoms among HIV-seropositive women: longitudinal analysis from the HIV Epidemiology Research Study. JAMA. 2001;285(11):1466–1474. | ||
Ickovics JR, Milan S, Boland R, Schoenbaum E, Schuman P, Vlahov D; HIV Epidemiology Research Study (HERS) Group. Psychological resources protect health: 5-year survival and immune function among HIV-infected women from four US cities. AIDS. 2006;20(14):1851–1860. | ||
Cruess DG, Kalichman SC, Amaral C, Swetzes C, Cherry C, Kalichman MO. Benefits of adherence to psychotropic medications on depressive symptoms and antiretroviral medication adherence among men and women living with HIV/AIDS. Ann Behav Med. 2012;43(2):189–197. | ||
Cruess DG, Antoni MH, Kumar M, Schneiderman N. Reductions in salivary cortisol are associated with mood improvement during relaxation training among HIV- seropositive men. J Behav Med. 2000;23(2):107–122. | ||
Antoni MH. Stress management effects on psychological, endocrinological, and immune functioning in men with HIV infection: empirical support for a psychoneuroimmunological model. Stress. 2003;6(3):173–188. | ||
Leserman J, Petitto JM, Golden RN, et al. Impact of stressful life events, depression, social support, coping, and cortisol on progression to AIDS. Am J Psychiatry. 2000;157(8):1221–1228. | ||
Clerici M, Galli M, Bosis S, Gervasoni C, Moroni M, Norbiato G. Immunoendocrinologic abnormalities in human immunodeficiency virus infection. Ann N Y Acad Sci. 2000;917:956–961. | ||
Schiepers OJ, Wichers MC, Maes M. Cytokines and major depression. Prog Neuropsychopharmacol Biol Psychiatry. 2005;29(2):201–217. | ||
Müller N, Ackenheil M. Psychoneuroimmunology and the cytokine action in the CNS: implications for psychiatric disorders. Prog Neuropsychopharmacol Biol Psychiatry. 1998;22(1):1–33. | ||
Bruce TO. Comorbid depression in rheumatoid arthritis: pathophysiology and clinical implications. Curr Psychiatry Rep. 2008;10(3):258–264. | ||
Siegert RJ, Abernethy DA. Depression in multiple sclerosis: a review. J Neurol Neurosurg Psychiatry. 2005;76(4):469–475. | ||
Valentine AD, Meyers CA, Kling MA, Richelson E, Hauser P. Mood and cognitive side effects of interferon-alpha therapy. Semin Oncol. 1998;25(1 Suppl 1):39–47. | ||
Bonaccorso S, Puzella A, Marino V, et al. Immunotherapy with interferon-alpha in patients affected by chronic hepatitis C induces an intercorrelated stimulation of the cytokine network and an increase in depressive and anxiety symptoms. Psychiatry Res. 2001;105(1–2):45–55. | ||
Meyers CA. Mood and cognitive disorders in cancer patients receiving cytokine therapy. In: Dantzer R, Wollman EE, Yirmiya R, editors. Cytokines, Stress, and Depression. New York: Kluwer Academic/Plenum Publishers; 1999:75–81. | ||
Miller AH, Maletic V, Raison CL. Inflammation and its discontents: the role of cytokines in the pathophysiology of major depression. Biol Psychiatry. 2009;65(9):732–741. | ||
Walker Harris V, Brown TT. Bone loss in the HIV-infected patient: evidence, clinical implications, and treatment strategies. J Infect Dis. 2012;205(Suppl 3):S391–S398. | ||
Giannarelli C, Klein RS, Badimon JJ. Cardiovascular implications of HIV- induced dyslipidemia. Atherosclerosis. 2011;219(2):384–389. | ||
McComsey GA, Tebas P, Shane E, et al. Bone disease in HIV infection: a practical review and recommendations for HIV care providers. Clin Infect Dis. 2010;51(8):937–946. | ||
Chiba S, Numakawa T, Ninomiya M, Richards MC, Wakabayashi C, Kunugi H. Chronic restraint stress causes anxiety- and depression-like behaviors, downregulates glucocorticoid receptor expression, and attenuates glutamate release induced by brain- derived neurotrophic factor in the prefrontal cortex. Prog Neuropsychopharmacol Biol Psychiatry. 2012;39(1):112–119. | ||
Wise RA, Morales M. A ventral tegmental CRF-glutamate-dopamine interaction in addiction. Brain Res. 2010;1314:38–43. | ||
Borgland SL, Ungless MA, Bonci A. Convergent actions of orexin/hypocretin and CRF on dopamine neurons: emerging players in addiction. Brain Res. 2010;1314:139–144. | ||
Lowry CA, Moore FL. Regulation of behavioral responses by corticotropin- releasing factor. Gen Comp Endocrinol. 2006;146(1):19–27. | ||
Pace TW, Miller AH. Cytokines and glucocorticoid receptor signaling. Relevance to major depression. Ann N Y Acad Sci. 2009;1179:86–105. | ||
Butts CL, Jones YL, Lim JK, Salter CE, Belyavskaya E, Sternberg EM. Tissue expression of steroid hormone receptors is associated with differential immune responsiveness. Brain Behav Immun. 2011;25(5):1000–1007. | ||
Muthumani K, Choo AY, Zong WX, et al. The HIV-1 Vpr and glucocorticoid receptor complex is a gain-of-function interaction that prevents the nuclear localization of PARP-1. Nat Cell Biol. 2006;8(2):170–179. | ||
Kino T, Gragerov A, Kopp JB, Stauber RH, Pavlakis GN, Chrousos GP. The HIV-1 virion-associated protein vpr is a coactivator of the human glucocorticoid receptor. J Exp Med. 1999;189(1):51–62. | ||
Kinter AL, Biswas P, Alfano M, et al. Interleukin-6 and glucocorticoids synergistically induce human immunodeficiency virus type-1 expression in chronically infected U1 cells by a long terminal repeat independent post-transcriptional mechanism. Mol Med. 2001;7(10):668–678. | ||
Corley PA. Interleukin-1 receptor antagonist as a treatment of HIV infection. Med Hypotheses. 2000;54(4):513–518. | ||
Llorens-Martín M, Trejo JL. Mifepristone prevents stress-induced apoptosis in newborn neurons and increases AMPA receptor expression in the dentate gyrus of C57/BL6 mice. PLoS One. 2011;6(11):e28376. | ||
Donia M, Anzaldi M, Di Marco R, et al. Phase II study of the antiretroviral activity and safety of the glucocorticoid receptor antagonist mifepristone in HIV-1- infected patients. Int J Mol Med. 2011;28(3):437–442. | ||
Campos LN, Guimarães MD, Remien RH. Anxiety and depression symptoms as risk factors for non-adherence to antiretroviral therapy in Brazil. AIDS Behav. 2010;14(2):289–299. | ||
DeLorenze GN, Satre DD, Quesenberry CP, Tsai AL, Weisner CM. Mortality after diagnosis of psychiatric disorders and co-occurring substance use disorders among HIV-infected patients. AIDS Patient Care STDS. 2010;24(11):705–712. | ||
Handwerger K. Differential patterns of HPA activity and reactivity in adult posttraumatic stress disorder and major depressive disorder. Harv Rev Psychiatry. 2009;17(3):184–205. | ||
Mackesy-Amiti ME, Donenberg GR, Ouellet LJ. Prevalence of psychiatric disorders among young injection drug users. Drug Alcohol Depend. 2012;124(1–2):70–78. | ||
Logie CH, Newman PA, Chakrapani V, Shunmugam M. Adapting the minority stress model: associations between gender non-conformity stigma, HIV-related stigma and depression among men who have sex with men in South India. Soc Sci Med. 2012;74(8):1261–1268. | ||
Dyer TP, Shoptaw S, Guadamuz TE, et al. Application of syndemic theory to black men who have sex with men in the Multicenter AIDS Cohort Study. J Urban Health. 2012;89(4):697–708. | ||
Mayne TJ, Vittinghoff E, Chesney MA, Barrett DC, Coates TJ. Depressive affect and survival among gay and bisexual men infected with HIV. Arch Intern Med. 1996;156(19):2233–2238. | ||
Burack JH, Barrett DC, Stall RD, Chesney MA, Ekstrand ML, Coates TJ. Depressive symptoms and CD4 lymphocyte decline among HIV-infected men. JAMA. 1993:270(21):2568–2573. | ||
Anderson RN, Smith BL. Deaths: leading causes for 2001. Natl Vital Stat Rep. 2003;52:1–85. | ||
Risk by Gender [webpage on the Internet]. Atlanta: Centers for Disease Control and Prevention (CDC) [updated June 29, 2015]. Available from: http://www.cdc.gov/hiv/group/gender/index.html. Accessed July 20, 2015. | ||
Orlando M, Burnam MA, Beckman R, et al. Re-estimating the prevalence of psychiatric disorders in a nationally representative sample of persons receiving care for HIV: results from the HIV Cost and Services Utilization Study. Int J Methods Psychiatr Res. 2002;11(2):75–82. | ||
Cook JA, Grey D, Burke-Miller J, et al. Effects of treated and untreated depressive symptoms on highly active antiretroviral therapy use in a US multi-site cohort of HIV-positive women. AIDS Care. 2006;18(2):93–100. | ||
Cook JA, Grey DD, Burke-Miller JK, et al. Illicit drug use, depression and their association with highly active antiretroviral therapy in HIV-positive women. Drug Alcohol Depend. 2007;89(1):74–81. | ||
Kumar V, Encinosa W. Effects of antidepressant treatment on antiretroviral regimen adherence among depressed HIV-infected patients. Psychiatr Q. 2009;80(3):131–141. | ||
Cole SW. Psychosocial influences on HIV-1 disease progression: neural, endocrine, and virologic mechanisms. Psychosom Med. 2008;70(5):562–568. | ||
Creswell JD, Myers HF, Cole SW, Irwin MR. Mindfulness meditation training effects on CD4+ T lymphocytes in HIV-1 infected adults: a small randomized controlled trial. Brain Behav Immun. 2009;23(2):184–188. | ||
Ironson G, O’Cleirigh C, Fletcher MA, et al. Psychosocial factors predict CD4 and viral load change in men and women with human immunodeficiency virus in the era of highly active antiretroviral treatment. Psychosom Med. 2005;67(6):1013–1021. | ||
Leserman J, Petitto JM, Gu H, et al. Progression to AIDS, a clinical AIDS condition and mortality: psychosocial and physiological predictors. Psychol Med. 2002;32(6):1059–1073. | ||
Carrico AW, Antoni MH. Effects of psychological interventions on neuroendocrine hormone regulation and immune status in HIV-positive persons: a review of randomized controlled trials. Psychosom Med. 2008;70(5):575–584. | ||
Merali Z, Du L, Hrdina P, et al. Dysregulation in the suicide brain: mRNA expression of corticotropin-releasing hormone receptors and GABA(A) receptor subunits in frontal cortical brain region. J Neurosci. 2004;24(6):1478–1485. | ||
Iyer AM, Brooke SM, Sapolsky RM. Glucocorticoids interact with gp120 in causing neurotoxicity in striatal cultures. Brain Res. 1998;808(2):305–309. | ||
Lee AL, Ogle WO, Sapolsky RM. Stress and depression: possible links to neuron death in the hippocampus. Bipolar Disord. 2002;4(2):117–128. | ||
Brogan J, Li F, Li W, He Z, Huang Q, Li CY. Imaging molecular pathways: reporter genes. Radiat Res. 2012;177(4):508–513. | ||
Smith TA. Towards detecting the HER-2 receptor and metabolic changes induced by HER-2-targeted therapies using medical imaging. Br J Radiol. 2010;83(992):638–644. | ||
Zhang Y, Hong H, Cai W. PET tracers based on Zirconium-89. Curr Radiopharm. 2011;4(2):131–139. | ||
de Pablos RM, Villarán RF, Argüelles S, et al. Stress increases vulnerability to inflammation in the rat prefrontal cortex. J Neurosci. 2006;26(21):5709–5719. | ||
Frank MG, Thompson BM, Watkins LR, Maier SF. Glucocorticoids mediate stress-induced priming of microglial pro-inflammatory responses. Brain Behav Immun. 2012;26(2):337–345. | ||
Watson S, Gallagher P, Porter RJ, Smith MS, Herron LJ, Bulmer S; North-East Mood Disorders Clinical Research Group, Young AH, Ferrier IN. A randomized trial to examine the effect of mifepristone on neuropsychological performance and mood in patients with bipolar depression. Biol Psychiatry. 2012;72(11):943–949. | ||
DeBattista C, Belanoff J, Glass S, et al. Mifepristone versus placebo in the treatment of psychosis in patients with psychotic major depression. Biol Psychiatry. 2006;60(12):1343–1349. | ||
Gallagher P, Young AH. Mifepristone (RU-486) treatment for depression and psychosis: a review of the therapeutic implications. Neuropsychiatr Dis Treat. 2006;2(1):33–42. | ||
Belanoff JK, Rothschild AJ, Cassidy F, et al. An open label trial of C-1073 (mifepristone) for psychotic major depression. Biol Psychiatry. 2002;52(5):386–392. | ||
Kiferle L, Politis M, Muraro PA, Piccini P. Positron emission tomography imaging in multiple sclerosis-current status and future applications. Eur J Neurol. 2011;18(2):226–231. | ||
Ratchford JN, Endres CJ, Hammoud DA, et al. Decreased microglial activation in MS patients treated with glatiramer acetate. J Neurol. 2012;259(6):1199–1205. | ||
Venneti S, Lopresti BJ, Wang G, et al. PET imaging of brain macrophages using the peripheral benzodiazepine receptor in a macaque model of neuroAIDS. J Clin Invest. 2004;113(7):981–989. | ||
Wiley CA, Lopresti BJ, Becker JT, et al. Positron emission tomography imaging of peripheral benzodiazepine receptor binding in human immunodeficiency virus-infected subjects with and without cognitive impairment. J Neurovirol. 2006;12(4):262–271. | ||
Hammoud DA, Endres CJ, Chander AR, et al. Imaging glial cell activation with [11C]-R-PK11195 in patients with AIDS. J Neurovirol. 2005;11(4):346–355. | ||
Adinoff B, Anton R, Linnoila M, Guidotti A, Nemeroff CB, Bissette G. Cerebrospinal fluid concentrations of corticotropin-releasing hormone (CRH) and diazepam-binding inhibitor (DBI) during alcohol withdrawal and abstinence. Neuropsychopharmacology. 1996;15(3):288–295. | ||
Arató M, Bánki CM, Bissette G, Nemeroff CB. Elevated CSF CRF in suicide victims. Biol Psychiatry. 1989;25(3):355–359. | ||
Nemeroff CB, Owens MJ, Bissette G, Andorn AC, Stanley M. Reduced corticotropin releasing factor binding sites in the frontal cortex of suicide victims. Arch Gen Psychiatry. 1988;45(6):577–579. | ||
Banki CM, Bissette G, Arato M, O’Connor L, Nemeroff CB. CSF corticotropin- releasing factor-like immunoreactivity in depression and schizophrenia. Am J Psychiatry. 1987;144(7):873–877. | ||
Koob GF, Zorrilla EP. Update on corticotropin-releasing factor pharmacotherapy for psychiatric disorders: a revisionist view. Neuropsychopharmacology. 2012;37(1):308–309. | ||
Zorrilla EP, Koob GF. Progress in corticotropin-releasing factor-1 antagonist development. Drug Discov Today. 2010;15(9–10):371–383. | ||
Zobel AW, Nickel T, Künzel HE, et al. Effects of the high-affinity corticotropin-releasing hormone receptor 1 antagonist R121919 in major depression: the first 20 patients treated. J Psychiatr Res. 2000;34(3):171–181. | ||
Fleck BA, Hoare SR, Pick RR, Bradbury MJ, Grigoriadis DE. Binding kinetics redefine the antagonist pharmacology of the corticotropin-releasing factor type 1 receptor. J Pharmacol Exp Ther. 2012;341(2):518–531. | ||
Ramsey SJ, Attkins NJ, Fish R, van der Graaf PH. Quantitative pharmacological analysis of antagonist binding kinetics at CRF1 receptors in vitro and in vivo. Br J Pharmacol. 2011;164(3):992–1007. | ||
Bornstein SR, Webster EL, Torpy DJ, et al. Chronic effects of a nonpeptide corticotropin-releasing hormone type I receptor antagonist on pituitary-adrenal function, body weight, and metabolic regulation. Endocrinology. 1998;139(4):1546–1555. | ||
Martinez V, Taché Y. CRF1 receptors as a therapeutic target for irritable bowel syndrome. Curr Pharm Des. 2006;12(31):4071–4088. | ||
Hubbard CS, Labus JS, Bueller J, et al. Corticotropin-releasing factor receptor 1 antagonist alters regional activation and effective connectivity in an emotional-arousal circuit during expectation of abdominal pain. J Neurosci. 2011;31(35):12491–12500. | ||
Moskowitz JT. Positive affect predicts lower risk of AIDS mortality. Psychosom Med. 2003;65(4):620–626. | ||
Benton TD. Depression and HIV/AIDS. Curr Psychiatry Rep. 2008;10:280–285. | ||
Woods SP, Rippeth JD, Frol AB, et al. Interrater reliability of clinical ratings and neurocognitive diagnoses in HIV. J Clin Exp Neuropsychol. 2004;26(6):759–778. | ||
Heaton RK, Kirson D, Velin RA, Grant I, The HNRC Group. The utility of clinical ratings for detecting cognitive change in HIV infection. In: Grant I, Martin A, editors. Neuropsychology of HIV Infection. New York: Oxford University Press; 1994:188–205. | ||
Heaton RK, Taylor MJ, Manly JJ. Demographic effects and use of demographically corrected norms with the WAIS-III and WMS-III. In: Tulsky DS, Saklofske DH, Chelune GJ, et al, editors. Clinical Interpretation of the WAIS-III and WMS-III. San Diego: Academic Press; 2003:183–209. | ||
Heaton RK, Miller SW, Taylor MJ, Grant I. Revised comprehensive norms for an expanded halstead-reitan battery: demographically adjusted neuropsychological norms for african american and caucasian adults, professional manual. Lutz: Psychological Assessment Resources; 2004. Available from: http://www4.parinc.com/Products/Product.aspx?ProductID=RCNAAC/ Accessed November 24, 2016. | ||
Evans DL, Charney DS, Lewis L, et al. Mood disorders in the medically ill: scientific review and recommendations. Biol Psychiatry. 2005;58(3):175–189. | ||
Kissane DW, Clarke DM, Street AF. Demoralization syndrome – a relevant psychiatric diagnosis for palliative care. J Palliat Care. 2001;17:12–21. | ||
Dunn AJ, Swiergiel AH, de Beaurepaire R. Cytokines as mediators of depression: what can we learn from animal studies? Neurosci Biobehav Rev. 2005;29(4–5):891–909. | ||
O’Brien SM, Scott LV, Dinan TG. Cytokines: abnormalities in major depression and implications for pharmacological treatment. Hum Psychopharmacol. 2004;19(6):397–403. | ||
Matsuoka Y, Furuyashiki T, Yamada K, et al. Prostaglandin E receptor EP1 controls impulsive behavior under stress. Proc Natl Acad Sci U S A. 2005;102(44):16066–16071. | ||
Lacosta S, Merali Z, Anisman H. Central monoamine activity following acute and repeated systemic interleukin-2 administration. Neuroimmunomodulation. 2000;8(2):83–90. | ||
Murray M, Hogg RS, Lima VD, et al; Antiretroviral Therapy Cohort Collaboration (ART-CC). The effect of injecting drug use history on disease progression and death among HIV-positive individuals initiating combination antiretroviral therapy: collaborative cohort analysis. HIV Med. 2012;13(2):89–97. | ||
Cohn SE, Jiang H, McCutchan JA, et al. Association of ongoing drug and alcohol use with non-adherence to antiretroviral therapy and higher risk of AIDS and death: results from ACTG 362. AIDS Care. 2011;23(6):775–785. | ||
Obel N, Omland LH, Kronborg G, et al. Impact of non-HIV and HIV risk factors on survival in HIV-infected patients on HAART: a population-based nationwide cohort study. PLoS One. 2011;6(7):e22698. | ||
Rodríguez-Arenas MA, Jarrín I, del Amo J, et al; CoRIS-MD. Delay in the initiation of HAART, poorer virological response, and higher mortality among HIV-infected injecting drug users in Spain. AIDS Res Hum Retroviruses. 2006;22(8):715–723. | ||
Nava F, Caldiroli E, Premi S, Lucchini A. Relationship between plasma cortisol levels, withdrawal symptoms and craving in abstinent and treated heroin addicts. J Addict Dis. 2006;25(2):9–16. | ||
Dave RS, Khalili K. Morphine treatment of human monocyte-derived macrophages induces differential miRNA and protein expression: impact on inflammation and oxidative stress in the central nervous system. J Cell Biochem. 2010;110(4):834–845. | ||
Hauser KF, El-Hage N, Stiene-Martin A, et al. HIV-1 neuropathogenesis: glial mechanisms revealed through substance abuse. J Neurochem. 2007;100(3):567–586. | ||
Hauser KF, El-Hage N, Buch S, et al. Impact of opiate-HIV-1 interactions on neurotoxic signaling. J Neuroimmune Pharmacol. 2006;1(1):98–105. | ||
Hauser KF, El-Hage N, Buch S, et al. Molecular targets of opiate drug abuse in neuroAIDS. Neurotox Res. 2005;8(1–2):63–80. | ||
Hauser KF, Fitting S, Dever SM, Podhaizer EM, Knapp PE. Opiate drug use and the pathophysiology of neuroAIDS. Curr HIV Res. 2012;10(5):435–452. | ||
Najt P, Fusar-Poli P, Brambilla P. Co-occurring mental and substance abuse disorders: a review on the potential predictors and clinical outcomes. Psychiatry Res. 2011;186(2–3):159–164. | ||
Kelly TM, Daley DC, Douaihy AB. Treatment of substance abusing patients with comorbid psychiatric disorders. Addict Behav. 2012;37(1):11–24. | ||
Gould RW, Porrino LJ, Nader MA. Nonhuman Primate Models of Addiction and PET Imaging: Dopamine System Dysregulation. In: Carter CS, Dalley JW, editors. Brain Imaging in Behavioral Neuroscience. Springer-Verlag Berlin Heidelberg. 2011;25–44. | ||
Yau JL, Noble J, Thomas S, et al. The antidepressant desipramine requires the ABCB1 (Mdr1)-type p-glycoprotein to upregulate the glucocorticoid receptor in mice. Neuropsychopharmacology. 2007;32(12):2520–2529. | ||
Pariante CM, Makoff A, Lovestone S, et al. Antidepressants enhance glucocorticoid receptor function in vitro by modulating the membrane steroid transporters. Br J Pharmacol. 2001;134(6):1335–1343. | ||
Pariante CM, Kim RB, Makoff A, Kerwin RW. Antidepressant fluoxetine enhances glucocorticoid receptor function in vitro by modulating membrane steroid transporters. Br J Pharmacol. 2003;139(6):1111–1118. | ||
Kenis G, Maes M. Effects of antidepressants on the production of cytokines. Int J Neuropsychopharmacol. 2002;5(4):401–412. | ||
Eller T, Vasar V, Shlik J, Maron E. Pro-inflammatory cytokines and treatment response to escitalopram in major depressive disorder. Prog Neuropsychopharmacol Biol Psychiatry. 2008;32(2):445–450. | ||
Lanquillon S, Krieg JC, Bening-Abu-Shach U, Vedder H. Cytokine production and treatment response in major depressive disorder. Neuropsychopharmacology. 2000;22(4):370–379. | ||
Lanquillon S, Krieg JC, Bening-Abu-Shach U, Vedder H. Cytokine production and treatment response in major depressive disorder. Neuropsychopharmacology. 2000;22(4):370–379. | ||
Castanon N, Leonard BE, Neveu PJ, Yirmiya R. Effects of antidepressants on cytokine production and actions. Brain Behav Immun. 2002;16(5):569–574. | ||
Leonard BE. The immune system, depression and the action of antidepressants. Prog Neuropsychopharmacol Biol Psychiatry. 2001;25(4):767–780. | ||
Goebel M, Stengel A, Wang L, Reeve J Jr, Taché Y. Lipopolysaccharide increases plasma levels of corticotropin-releasing hormone in rats. Neuroendocrinology. 2011;93(3):165–173. | ||
Buckinx R, Adriaensen D, Nassauw LV, Timmermans JP Corticotrophin-releasing factor, related peptides, and receptors in the normal and inflamed gastrointestinal tract. Front Neurosci. 2011;5:54. | ||
Wallon C, Söderholm JD. Corticotropin-releasing hormone and mast cells in the regulation of mucosal barrier function in the human colon. Ann N Y Acad Sci. 2009;1165:206–210. | ||
Fukudo S. Role of corticotropin-releasing hormone in irritable bowel syndrome and intestinal inflammation. J Gastroenterol. 2007;42(Suppl 17):48–51. | ||
Iyer AM, Brooke SM, Sapolsky RM. Glucocorticoids interact with gp120 in causing neurotoxicity in striatal cultures. Brain Res. 1998;808(2):305–309. | ||
Kling MA, Coleman VH, Schulkin J. Glucocorticoid inhibition in the treatment of depression: can we think outside the endocrine hypothalamus? Depress Anxiety. 2009;26(7):641–649. | ||
McEwen BS. Plasticity of the hippocampus: adaptation to chronic stress and allostatic load. Ann N Y Acad Sci. 2001;933:265–277. | ||
Bremner JD. Stress and brain atrophy. CNS Neurol Disord Drug Targets. 2006;5(5):503–512. |
Supplementary materials
HIV-induced neurodegeneration
Neurotoxicity from inflammation in the HIV-infected host comprises a highly complex series of molecular events; its severity and trajectory can be altered by pathologic conditions in a vulnerable host prior to infection. Despite highly active antiretroviral therapies (HAARTs), earlier intervention, and more effective disease control, neurocognitive impairment and dementia remain major causes of disability, increased morbidity, and mortality.1,2 Underlying chronic distress and neuroinflammation add to disease burden and susceptibility in those at risk. Psychiatric disorders, addiction (especially with stimulants and opiates), high-risk sexual behaviors, sickness syndrome, and other chronic medical conditions characterize patient populations infected with HIV or those at high risk of HIV infection. Progressive neuronal injury leads to clinical sequelae of neural degeneration and cognitive impairment.3
Loss of blood–brain barrier (BBB) integrity
Those with early HIV infection and those at risk suffering from medical and behavioral conditions, ie, chemical/pharmacologic (drug) insults, psychiatric disorders, and medical illness, show blood–brain barrier (BBB) compromise. Stress-associated mast cell activation with corticotropin-releasing hormone (CRH) release, “leaky” basement membranes, permeability of perivascular endothelial cells, and active transport of high concentrations of circulating cytokines/chemokines contribute to BBB failure.4 Infected macrophages and T-cells invade the central nervous system (CNS) extracellular space and brain parenchyma. The loss of BBB integrity is complex and incompletely understood. This cascade of inflammatory events results in early HIV colonization of the brain.
Multifaceted neurotoxicity
The trajectory of HIV neurotoxicity involves HIV transit into the CNS by T-cells and perivascular and mobile macrophages which infect resident monocytes, macrophages, and microglia; astrocytes, though not shown to harbor HIV, are target cells that promote neuronal injury. Infected monocytes and macrophages release cytokines/chemokines, intact virions, and component viral proteins, mainly gp120 and transactivator of transcription (TAT), which perpetuate infection and neurotoxicity. Noninfected monocytes/macrophages respond to immune insults releasing cytokines/chemokines and other agents, which disrupt astrocyte glutamate (GLU) regulation. GLU dysregulation with excess production in astrocytes is a critical component of neuronal degeneration.5,6
Astrocytes are critical in maintaining GLU homeostasis. GLU is a toxic excitatory neurotransmitter which becomes disrupted at multiple steps in its metabolic pathways. Increased production and reduced clearance and metabolism of GLU elicit toxic neuronal activation. Excitatory amino acid transporters (types 1 and 2) are downregulated, decreasing glial and astrocyte GLU uptake. The enzyme glutaminase is upregulated, which generates glutamine, a GLU precursor to GLU, increasing intracellular/extracellular and intrasynaptic GLU. GLU increases from downregulation of GLU degradation enzymes (GCP I and GCP II). GLU astrocyte exocytosis increases extracellular GLU. GLU is excitotoxic through N-methyl-D-aspartate (NMDA) receptors which, when activated, increase intracellular Ca+2 release. Excess Ca+2 activates second messenger pathways responsible for gene transcription and signaling, ie, nuclear factor kappa B (NF-κB), JNK/STAT-1, and ERK 1/2. These trigger genomic events that accelerate production of proinflammatory agents, apoptosis, and oxidative stress, as well as further increasing GLU production and release. Cumulatively, these events decrease synaptic integrity and plasticity and perpetuate neuron apoptosis.
Excess GLU also enhances glial inflammation (gliosis) in concert with neural dysfunction; mounting glial activation generates a cascading effect that perpetuates neuroinflammation, neuronal dysfunction, network integrity, and cognitive decline.
CNS HIV infection of neuronal support cells and tissues generates a proinflammatory microenvironment. Neuronal damage is multifactorial, with toxic insults from increased intracellular, extracellular, and intrasynaptic GLU; cytokines. Oxidative stress from reactive oxygen radicals/nitric oxides; and membrane destabilization from prostaglandins/arachidonic acid promotes neuronal damage. This is magnified by depletion of intracellular antioxidant detoxification molecules, ie, glutathione. Active viral replication maintains and progressively compromises CNS immunity with deteriorating neuron viability and neuronal networks. Degeneration of neocortical and limbic structures and their connectivity are principal sites responsible for cognitive decline and increased susceptibility to neuropsychiatric conditions.
Figure S1 represents some of the multiple molecular mediators generated by HIV infection within the CNS and the multiple neurotoxic and inflammatory effects on both neuronal support cells/tissues and neurons.
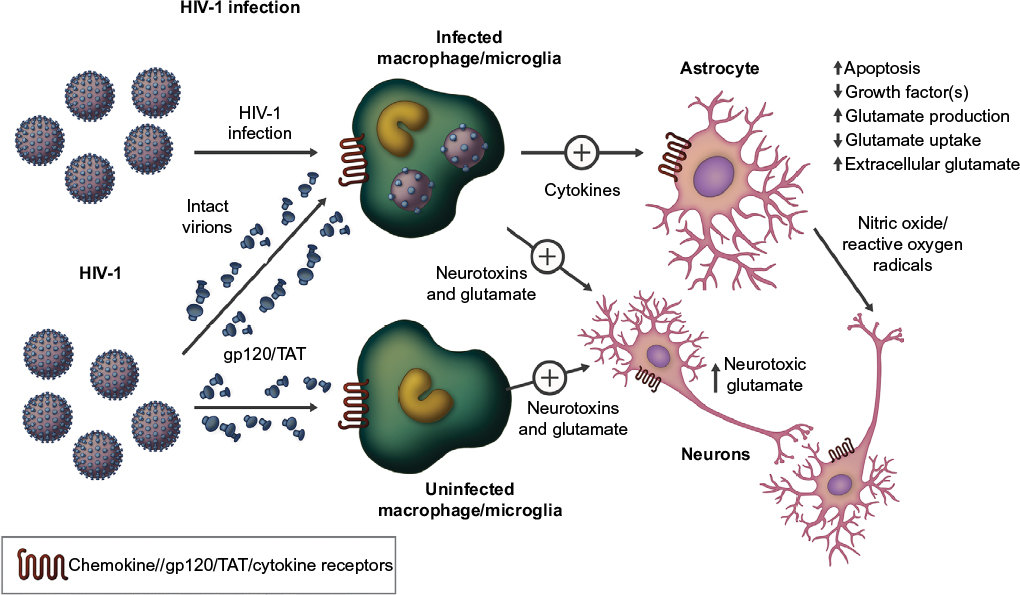 | Figure S1 HIV infection in the central nervous system involves both HIV-infected and noninfected neuronal support cells. Notes: Macrophages, monocytes, and glial elements modulate actions of neurons through astrocytes. Virions and viral protein components (gp120, TAT), cytokines, membrane destabilization molecules, and glutamate (with other agents) stimulate remaining glial and other immune cells/tissues, generating a proinflammatory neurotoxic state. These adversely affect astrocytes, diminishing their support of neurons. This promotes neuronal dysfunction with loss of synaptic integrity and plasticity, morphologic and functional impairment, and apoptosis. Continued HIV invasion and replication perpetuate a cascading cycle of increasing inflammation and neurodegeneration. Macroscopic structural deficits develop with loss of cortical and subcortical functions and neuronal connectivity leading to clinical sequelae. Abbreviation: TAT, transactivator of transcription. |
Effects of chronic stress
During acute stress, glucocorticoids (GC) provide neuroprotection, blunting glial and other CNS inflammatory responses, indicative of their immunosuppressive effects. Induction of chronic stress in models of Parkinson’s disease results in long-term pathologically elevated concentrations of GC. Chronic exposure to GC during conditions of prolonged stress induces GC resistance (GCR) in immune/lymphoid tissues and organs. This progressively increases inflammation from HIV in the CNS. There is a limited understanding of GCR; excess GC has complex interactions with intracytoplasmic GC receptors. Activated GC receptors upregulate genes for proinflammatory cytokine production, viral replication in T-cells as well as in monocytes/macrophages, and glial and other support elements;7–10 GCR also enhances astrocyte GLU neurotransmission. In Parkinson’s disease, this contributes to dopamine neuronal toxicity and cell death; this pattern is seen in other neuroinflammatory conditions.
Chronic neurodegeneration
Once infection is established, viral reservoirs remain within the brain despite low/absent cerebral spinal fluid viral load. Glial elements, monocytes, and perivascular/circulating macrophages are long-lived in the CNS, with glial cells lasting years to lifetime. With the advent of HAART, the prevalence and severity of neurocognitive disorders have decreased, with dementia dropping from >15% to less than 5%. “Asymptomatic” neurocognitive impairment, however, has increased to >30% over time. How pre- and post-infection comorbid conditions from disrupted stress homeostasis influence CNS HIV infection is largely unknown.
For progressive cognitive impairment, current recommendations include utilization of antiretrovirals with improved BBB penetration to decrease viral replication, free virion passage, and viral component (gp120/TAT) production. When effective, HAART blocks GLU production/action and the proinflammatory milieu.11,12 Reduced viral replication and medications purported to improve general cognition in practice have limited clinical utility.
Future directions
HIV neurotoxicity involves critical action points that determine the rate and severity of neurocognitive deterioration. When HIV enters the CNS, infection of monocytes, microglia, and macrophages; generation of intact virions and viral components (gp120/TAT); and cytokines/chemokines all contribute to activate astrocytes. GLU toxicity leads to neuronal damage, apoptosis, and overall loss of cognitive function. Once initiated, these processes feed forward, increasing involvement of more and more neuronal networks, with greater disability and functional impairment. To date, targeted therapies have been disappointing. Most antiretrovirals lack physicochemical properties to effectively cross the BBB and suppress the virus. GLU/NMDA receptor antagonists and synthesis inhibitors have not demonstrated therapeutic effects. Anticholinergics (used in Alzheimer’s disease), serotonin, and noradrenergic agonists have also failed to show significant benefits. These findings are not unexpected, given the inability to modify the course of other neurodegenerative disorders. Targeting chronic stress, GCR and CRH may offer limited potential as adjunctive therapies, but the overall complexities of these systems, the chronicity of ongoing infection, and the relative lack of sensitive surrogate markers for inflammation will limit treatment options in the near and mid-term future. Advances in basic neuroscience will be necessary to determine better therapeutic interventions for HIV-related and other neurodegenerative disorders.
References
Brabers NA, Nottet HS. Role of the pro-inflammatory cytokines TNF-alpha and IL-1beta in HIV-associated dementia. Eur J Clin Invest. 2006; 36(7):447–458. | ||
Cysique LA, Vaida F, Letendre S, et al. Dynamics of cognitive change in impaired HIV-positive patients initiating antiretroviral therapy. Neurology. 2009;73(5):342–348. | ||
Croteau D. Psychology and AIDS Exchange Newsletter. January 2013. | ||
de Pablos RM, Herrera AJ, Espinosa-Oliva AM, et al. Chronic stress enhances microglia activation and exacerbates death of nigral dopaminergic neurons under conditions of inflammation. J Neuroinflammation. 2014;11:34. | ||
Erdmann N, Tian C, Huang Y>, et al. In vitro glutaminase regulation and mechanisms of glutamate generation in HIV-1-infected macrophage. J Neurochem. 2009;109(2):551–561. | ||
Erdmann NB, Whitney NP, Zheng J. Potentiation of excitotoxicity in HIV-1 associated dementia and the significance of glutaminase. Clin Neurosci Res. 2006;6(5):315–328. | ||
Frank MG, Hershman SA, Weber MD, Watkins LR, Maier SF. Chronic exposure to exogenous glucocorticoids primes microglia to pro-inflammatory stimuli and induces NLRP3 mRNA in the hippocampus. Psychoneuroendocrinology. 2014;40:191–200. | ||
Frank MG>, Thompson BM, Watkins LR, Maier SF. Glucocorticoids mediate stress-induced priming of microglial pro-inflammatory responses. Brain Behav Immun. 2012;26(2):337–345. | ||
Gold SM, Sasidhar MV, Lagishetty V, et al. Dynamic development of glucocorticoid resistance during autoimmune neuroinflammation. J Clin Endocrinol Metab. 2012;97(8):E1402–E1410. | ||
Sugama S, Takenouchi T, Fujita M, Kitani H, Conti B, Hashimoto M. Corticosteroids limit microglial activation occurring during acute stress. Neuroscience. 2013;232:13–20. | ||
Tian C, Erdmann N, Zhao J, Cao Z, Peng H, Zheng J. HIV-infected macrophages mediate neuronal apoptosis through mitochondrial glutaminase. J Neurochem. 2008;105(3):994–1005. | ||
Vázquez-Santiago FJ, Noel RJ Jr, Porter JT, Rivera-Amill V. Glutamate metabolism and HIV-associated neurocognitive disorders. J Neurovirol. 2014;20(4):315–331. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.