Back to Journals » International Medical Case Reports Journal » Volume 9
Chorea-acanthocytosis: a case report
Authors Lekhjung Thapa L ![]() , Bhattarai S, Shrestha MP, Panth R, Gongal D, Devkota U
, Bhattarai S, Shrestha MP, Panth R, Gongal D, Devkota U
Received 6 September 2015
Accepted for publication 9 December 2015
Published 23 February 2016 Volume 2016:9 Pages 39—42
DOI https://doi.org/10.2147/IMCRJ.S95882
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Professor Ronald Prineas
Video abstract presented by Lekhjung Thapa
Views: 7295
Lekhjung Thapa,1 Suman Bhattarai,1 Milan P Shrestha,1 Rajesh Panth,2 Dinesh Nath Gongal,3 Upendra Prasad Devkota,3
1Department of Neurology, 2Department of Pathology, 3Department of Neurosurgery, National Institute of Neurological and Allied Sciences, Kathmandu, Nepal
Abstract: Neuroacanthocytosis is a group of rare disorders. We report a 36-year-old right-handed female who presented with gradually progressive abnormal facial movements, generalized weakness, and lower-lip biting starting 4 years ago. On examination, she had lower-lip ulcer, orofacial dyskinesias, and peripheral neuropathy. Her peripheral blood smears showed acanthocytosis and magnetic resonance imaging revealed atrophied head of caudate nuclei and putaminal hyperintensities on T2-weighted and fluid attenuated inversion recovery images. Work-up for autoimmune and metabolic causes was negative. She was diagnosed with chorea-acanthocytosis, an entity under neuroacanthocytosis syndrome and the patient was offered symptomatic treatment.
Keywords: acanthocytes, lip-biting, neuroacanthocytosis, orofacial dyskinesia, movement disorder
Introduction
Neuroacanthocytosis (NA) is a group of very rare disorders estimated to have affected ~1,000 people worldwide. NA are complex, progressive, and incurable disorders and their treatment is symptomatic. The complex presentations that include a hyperkinetic movement disorder, a polyneuropathy, cognitive decline, and elevated creatine kinase (CK) raise the suspicion of a NA syndrome. To our knowledge, this is the first case report of NA from Nepal.
Case report
A 36-year-old right-handed female had presented with gradually worsening abnormal facial movements associated with intermittent rapid brief forceful eyes closure, perioral movements, and lower-lip biting. Her husband reported a history of difficulty in speaking, swallowing, and weakness of all four limbs, and she was wheelchair-bound. She was born to a nonconsanguineous parent and lacked a family history of neurological disease. She was not exposed to long-term medications known to cause extrapyramidal dysfunction.
Examination revealed orofaciolingual dyskinesias mimicking suckling and grimacing, and lower-lip ulcer because of repeated biting (Figure 1). Mini Mental Status Examination (MMSE) could not be performed because of unintelligible speech. She had wasting of all four limbs, diminished power (4/5) and reflexes. Her fundus and sensory examinations were normal. Kayser–Fleicher (KF) ring, cerebellar signs, and autonomic dysfunction were absent.
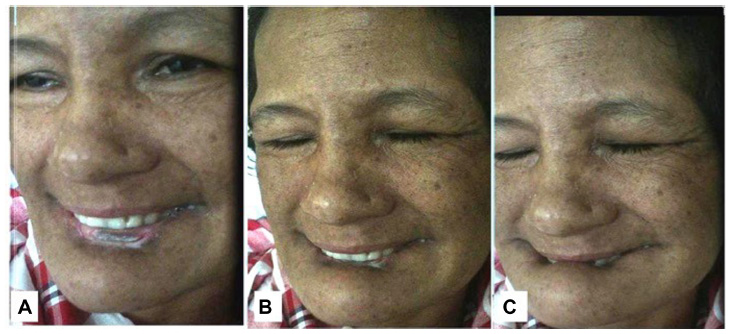 | Figure 1 (A–C) Sequential snapshots from video demonstrating involuntary movements with closure of both eyes and lower-lip biting. |
Peripheral smear examination performed following standard protocol1 showed acanthocytes (Figure 2) and magnetic resonance imaging showed degenerative changes in basal ganglia (Figure 3). Her CK was raised (528 U/L [Normal: up to 165 U/L]). Nerve conduction study showed peripheral mixed axonal neuropathy. Complete blood count, liver function test, random blood sugar, renal function test, antinuclear antibody, double stranded DNA, serum copper, serum ceruloplasmin, HIV, hepatitis B antigen, hepatitis C, cerebrospinal fluid analysis, USG abdomen, ECG, and echocardiography were normal.
 | Figure 2 Peripheral blood smear showing acanthocytes (red arrows). |
 | Figure 3 MRI showing caudate atrophy and striatal hyperintensities in both T2WI and FLAIR sequences. |
She was treated with haloperidol and clonazepam. On follow-up at 3 months, her condition was static.
Ethics
Written informed consent was obtained from the patient to publish case details and pictures. Ethical clearance was also obtained from the National Institute of Neurological and Allied Sciences Ethical Review Board.
Discussion
NA syndrome disorders are all exceedingly rare, but also very likely to be underdiagnosed.2 It is estimated that 500–1,000 people worldwide have chorea-acanthocytosis (ChAc), an entity under NA syndrome.3 ChAc is more prevalent in Japan.4 As literature search did not reveal any documentation of ChAc from Nepal, we believe that this is the first reported case of ChAc from Nepal.
ChAc is a progressive neurodegenerative autosomal recessive disorder. Its neurological symptoms usually manifest in the twenties.5 NA was first described in 1960 as “Levine–Critchley syndrome”6 and as mentioned earlier, ChAc is one of the entities under NA. Other entities include: McLeod syndrome (MLS), Huntington’s disease like 2 (HDL2), and pantothenate kinase–associated neurodegeneration.
Our diagnosis of ChAc was based on clinical ground and laboratory findings. Clinically, our patient had adult-onset illness, hence, our diagnosis was restricted to either ChAc or MLS because HDL2 and pantothenate kinase–associated neurodegeneration have a childhood or juvenile onset, and HDL2 is usually found in patients with African ancestry. MLS is an X-linked disorder that has predominant cardiovascular manifestations (our patient’s cardiovascular status was normal). Also, lip biting is exceedingly rare in MLS. This feature with female sex in our case rules out MLS.
ChAc consists of choreoathetoid movement disorder with orofacial dyskinesia and progressive cognitive decline. The movement problems worsen with age. In few cases, seizures may precede movement disorders by a decade.7 The characteristic phenotype includes chorea, a very peculiar “feeding dystonia” with tongue protrusion,8 orofacial dyskinesias, involuntary vocalizations, dysarthria, and involuntary tongue- and lip-biting. All these mentioned features were prominent manifestations of our case. The gait of ChAc patients may have a “rubber man” appearance with truncal instability and sudden, violent trunk spasms.9 We could not examine gait because of weakness. Behavioral changes are a common feature and may be the first presenting feature.
Clinical neuromuscular manifestations include areflexia, sensorimotor neuropathy, and variable weakness and atrophy.4 Most ChAc patients have elevated levels of CK.4 Our patient had all of these findings known to be typical manifestation of ChAc.
ChAc is caused by various mutations of a 73 exon gene on chromosome 9, VPS13A, coding for chorein.6,10 Confirmatory DNA analysis of the large VPS13A gene is difficult, due to the large gene size and heterogeneity of mutation sites.11–13 However, absence of chorein in erythrocytes can be demonstrated on Western blot.14 Chorein is implicated in intracellular protein sorting but its physiological functions are not yet known.4 We did not test for chorein because of financial constraints although protein testing is available free of charge.15
We diagnosed the case as ChAc based on history and also because acanthocytes were readily documented in peripheral smear with elevated serum CK level. The elevated CK level suggested presence of myopathy, an important diagnostic clue to NA syndrome, however, her relative declined electromyography test. Acanthocytosis may be absent in peripheral blood smears and a negative screen does not reliably exclude an NA syndrome.16
Neurodegeneration affects predominantly the caudate nucleus, putamen, and globus pallidus. In ChAc, thalamus and substantia nigra are also involved.4 In magnetic resonance imaging, we observed changes in all these mentioned regions. Computed tomography scan did not show calcification in the aforementioned regions.
So far no curative or disease-modifying treatments are available. Recognition of treatable complications such as seizures, and swallowing problems are essential. Neuropsychiatric issues are more amenable to pharmacotherapy. Dopamine antagonists or depleters such as clozapine or tetrabenazine may ameliorate the movement disorders. Results of deep brain stimulation (DBS) in ChAc are mixed.17–19 NA disorders have a relentlessly progressive course and are eventually fatal. Sudden death may be attributable to seizure or autonomic dysfunction. Early identification of the case may avoid the need for invasive and nondiagnostic tests such as muscle, bone marrow, or liver biopsy and help guide the proper management and counseling.
Conclusion
This rare disorder should be considered in the differential diagnosis of atypical cases of epilepsy, peripheral neuropathy, and behavioral disorder associated with movement disorder.
Acknowledgment
The authors would like to thank Prof Madhu Dixit Devkota, Dr Samarth Singh, and all the Magister Chirurgiae Registrars of National Institute of Neurological and Allied Sciences.
Disclosure
The authors report no conflicts of interest in this work.
References
Storch A, Kornhass M, Schwarz J. Testing for acanthocytosis A prospective reader-blinded study in movement disorder patients. J Neurol. 2005;252:84–90. | |
Jung HH, Danek A, Walker RH. Neuroacanthocytosis syndromes. Orphanet J Rare Dis. 2011;6:68. | |
Chorea-acanthocytosis. Genetic home reference. Available from: http://ghr.nlm.nih.gov/condition/chorea-acanthocytosis. Accessed May 30, 2015. | |
Ueno S, Maruki Y, Nakamura M, et al. The gene encoding a newly discovered protein, chorein, is mutated in chorea-acanthocytosis. Nat Genet. 2001;28(2):121–122. | |
Walker RH, Jung HH, Dobson-Stone C, et al. Neurologic phenotypes associated with acanthocytosis. Neurology. 2007;68:92–98. | |
Danek A, editor. Neuroacanthocytosis Syndromes. Dordrecht, Springer Verlag; 2005:5–7. | |
Al-Asmi A, Jansen AC, Badhwar A, et al. Familial temporal lobe epilepsy as a presenting feature of choreoacanthocytosis. Epilepsia. 2005;46(8):1256–1263. | |
Bader B, Walker RH, Vogel M, et al. Tongue protrusion and feeding dystonia: a hallmark of chorea-acanthocytosis. Mov Disord. 2010;25(1):127–129. | |
Schneider SA, Lang AE, Moro E, Bader B, Danek A, Bhatia KP. Characteristic head drops and axial extension in advanced chorea-acanthocytosis. Mov Disord. 2010;25(10):1487–1491. | |
Rampoldi L, Dobson-Stone C, Rubio JP, et al. A conserved sorting-associated protein is mutant in chorea-acanthocytosis. Nat Genet. 2001;28(2):119–120. | |
Allen FH, Krabbe SMR, Corcoran PA. A new phenotype (McLeod) in the Kell blood-group system. Vox Sang. 1961;6:555–560. | |
Marsh WL, Taswell HF, Øyen R, Nichols ME, Vergera MS, Pineda AA. Kx antigen of the Kell system and its relationship to chronic granulomatous disease. Evidence the Kx gene is X-linked. Transfusion. 1975;15:527. | |
Marsh WL, Marsh NJ, Moore A, et al. Elevated serum creatine phosphokinase in subjects with McLeod syndrome. Vox Sang. 1981;40(6):403–411. | |
Dobson-Stone C, Velayos-Baeza A, Filippone LA, et al. Chorein detection for the diagnosis of chorea-acanthocytosis. Ann Neurol. 2004;56(2):299–302. | |
EHDN-Neuroacanthocytosis submodule. Available from: https://www.euro-hd.net/edit/na/network/docs/na-blood-sampling-instructions.pdf. Accessed August 23, 2015. | |
Sorrentino G, De Renzo A, Miniello S, Nori O, Bonavita V. Late appearance of acanthocytes during the course of chorea-acanthocytosis. J Neurol Sci. 1999;163(2):175–178. | |
Wihl G, Volkmann J, Allert N, Lehrke RE, Sturm V, Freund HJ. Deep brain stimulation of the internal pallidum did not improve chorea in a patient with neuro-acanthocytosis. Mov Disord. 2001;16(3):572–575. | |
Burbaud P, Rougier A, Ferrer X, et al. Improvement of severe trunk spasms by bilateral high-frequency stimulation of the motor thalamus in a patient with chorea-acanthocytosis. Mov Disord. 2002;17(1):204–207. | |
Burbaud P, Vital A, Rougier A, et al. Minimal tissue damage after stimulation of the motor thalamus in a case of chorea-acanthocytosis. Neurology. 2002;59(12):1982–1984. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.