Back to Journals » International Journal of Nanomedicine » Volume 14
Chitosan gold nanoparticles induce different ROS-dependent cell death modalities in leukemic cells
Authors Martínez-Torres AC ![]() , Lorenzo-Anota HY
, Lorenzo-Anota HY ![]() , García-Juárez MG, Zarate-Triviño DG
, García-Juárez MG, Zarate-Triviño DG ![]() , Rodríguez-Padilla C
, Rodríguez-Padilla C ![]()
Received 29 June 2019
Accepted for publication 3 August 2019
Published 4 September 2019 Volume 2019:14 Pages 7173—7190
DOI https://doi.org/10.2147/IJN.S221021
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Thomas Webster
Ana Carolina Martínez-Torres*, Helen Yarimet Lorenzo-Anota*, Martín Gerardo García-Juárez*, Diana G Zarate-Triviño, Cristina Rodríguez-Padilla
Universidad Autónoma De Nuevo León, Facultad De Ciencias Biológicas, Laboratorio De Inmunología Y Virología, Monterrey, Nuevo Leon, Mexico
*These authors contributed equally to this work
Correspondence: Ana Carolina Martínez-Torres
Universidad Autónoma de Nuevo León, Facultad de Ciencias Biológicas, Laboratorio de Inmunología y Virología, Monterrey 66455, México
Tel +52 8 121 4115
Fax +52 818 352 4212
Email [email protected]
Background: Nanotechnology proposes the use of gold nanoparticles (AuNPs) for drug delivery, diagnosis, and treatment of cancer. Leukemia is a type of hematopoietic cancer that results from the malignant transformation of white blood cells. Chitosan-coated AuNPs (CH-AuNPs) are cell death inductors in HeLa and MCF-7 cancer cells without affecting peripheral blood mononuclear cells (PBMC). Considering the selectivity and versatile cytotoxicity of CH-AuNPs, we evaluated whether their selectivity is due to the cell lineage or the characteristics of the cancer cells, by assessing its cytotoxicity in leukemic cells. Moreover, we further examined the cell death mechanism and assessed the implication of nuclear damage, autophagosome formation, and the cell death mechanism induced in leukemic cells.
Materials and methods: We synthesized CH-AuNPs by chemical methods and analyzed their cell death capacity in a T-acute lymphocytic leukemia cell line (CEM), in a chronic myeloid leukemia cell line (K562), and in healthy cells from the same lineage (PBMC and bone marrow, BM, cells). Then, we assessed ROS generation and mitochondrial and nuclear damage. Finally, we evaluated whether cell death occurred by autophagy, apoptosis, or necroptosis, and the role of ROS in this mechanism.
Results: We found that CH-AuNPs did not affect PBMC and BM cells, whereas they are cytotoxic in a dose-dependent manner in leukemic cells. ROS production leads to mitochondrial and nuclear damage, and cell death. We found that CH-AuNPs induce apoptosis in CEM and necroptosis in K562, both undergoing autophagy as a pro-survival mechanism.
Conclusion: CH-AuNPs are selective cell death inductors in hematologic cancer cells, without affecting their healthy counterparts. Cell death induced by CH-AuNPs is independent of the cancer cell type; however, its mechanism is different depending on the type of leukemic cells.
Keywords: AuNPs, leukemia, nuclear alterations, apoptosis, necroptosis, autophagy
Introduction
Nanotechnology is a multidisciplinary field that involves different areas such as biomedicine. Within the nanostructures, gold nanoparticles (AuNPs) have shown therapeutic and diagnostic potential in different diseases, including several types of cancer.1 Recently, AuNPs have been used as drug delivery agents because of their surface properties, which can be functionalized with molecules such as DNA, peptides, antibodies, and anticancer drugs.2 It is known that these properties depend on the size, shape, medium, cell type, charge, and the reducing agent used for the nanoparticle synthesis.3–6 However, to be aware of the best combinatory possibilities or its potential in theragnostics, it is important to evaluate the effect of AuNPs alone before they are combined with other agents. Chitosan (CH), a polysaccharide derived from deacetylation of chitin, has been used as a cationic reducing agent used to synthesize AuNPs. This application is possible, thanks to its many biological properties, which include cancer cytotoxicity, biocompatibility, and its capacity for diagnosis and as a carrier for the delivery of cancer therapies.7
Leukemia is a type of cancer that affects white blood cells and, according to the American Cancer Society, it can be classified into several types according to the affected cell lineage and its progression. Acute lymphoblastic leukemia (ALL) is the most common cancer in children younger than 5 years, whereas chronic myeloid leukemia (CML) affects adults older than 64 years. It is known that mutations or deletions in proteins related to regulated cell death (RCD) induce resistance to conventional treatments.8 The first-line therapies for leukemia consist of chemotherapies and corticosteroids, kinase inhibitors, and stem cell transplants; however, these treatments are aggressive and have many side effects in the immune system.8
We know that the nature and therapies for blood cancers are different than the ones used for solid cancers due to their unspecific localization, tumor microenvironment, and immunosuppression.9 In a previous report, we characterized chitosan-coated AuNPs (CH-AuNPs) and found that they induced selectively RCD in cervical (HeLa) and breast (MCF-7) cancer cells through ROS production. We also used peripheral blood mononuclear cells (PBMC) as control of healthy cells and observed selectivity to cancer cells.10 However, the cancerous and healthy donor-derived cells were not from the same cell type; here, we analyzed if the selectivity of CH-AuNPs is due to the cell lineage or the characteristics of the cancer cells. For this reason, we analyzed the cytotoxic effect of CH-AuNPs in healthy immune system cells (PBMC and bone marrow, BM) and their cancerous counterpart, leukemic cells (T-acute lymphoid leukemia cell line, CEM, and CML cell line, K562), which are derived from the same lineage. Furthermore, we delved in the mechanism of cell death induced by CH-AuNPs. We assessed ROS generation (DCFDA), mitochondrial (tetramethylrhodamine ethyl ester, TMRE) and nuclear (ϒ-H2Ax, cell cycle, and DNA degradation) alterations, caspase activation, autophagosome formation, and the role of ROS in the mechanism of cell death.
Materials and methods
Nanoparticles
CH-AuNPs and sodium citrate (SC)-AuNPs were synthesized by the Turkevich method as previously described.10 First, an acid solution of CH (2% w/w in acetic acid 0.4 M) was obtained by dissolving CH (medium molecular weight, 300,000 g/mol, with 75–85% of deacetylation) in 2 mM hydrochloroauric acid solution (HAuCl4), and it was homogenized at room temperature for 15 mins on a magnetic plate at 80–90 rpm. Then, the solution was heated (100°C ±5°C in glycerol bath for 15 mins on a magnetic plate at 80–90 rpm) until it changed color to a wine red and then it was diluted in RPMI 1640 medium (GIBCO® by Life Technologies). SC-AuNPs were obtained by heating hydrochloroauric acid solution (2 mM) to 140°C (±5ºC) in glycerol bath for 5 mins on a magnetic plate at 80–90 rpm, and then 1% sodium citrate solution (Milliporesigma, St. Louis, MO, USA) was added by slow drip until it changed to a wine red color.
The physicochemical properties of nanoparticles (such as size, charge, and dose) are important to consider its safeness,5 and thus we characterized CH-AuNPs by analyzing surface plasmon resonance by ultra visible spectroscopy using a Nanodrop spectrophotometer 2000c (Thermo Fisher Scientific, Bartlesville, OK, USA). Mean particle diameter and Z potential were measured by dynamic light scattering using Nanosizer NS90 (Siemens, Malvern, PA, USA). The sample was dispersed in distilled water (1:1,000). Table 1 shows the physicochemical properties of CH-AuNPs and SC-AuNPs, which are similar to our previous report.10 They exhibit the typical surface plasmon resonance of AuNPs, at 520 nm.11 The Z potential is +28 mV upon synthesis and the size range is 3–10 nm (Table 1). The polydispersity of CH-AuNPs was 0.3 (Table 1). In Figure S1, we show size (Figure S1A) and surface plasmon resonance of CH-AuNPs (Figure S1B).
 |
Table 1 Overview of AuNPs |
Cells and cell culture
Chronic myelogenous leukemia cells, K562 (ATCC® CCL-243™), and T-ALL cells, CEM (ATCC® CCL-119™), were obtained from the American Type Culture Collection (ATCC, Manassas, VA, USA) and maintained under suggested conditions: cultured in sterile plastic flasks (Corning Inc. Costar®, USA) at 37ºC in 5% CO2 atmosphere, using RPMI 1640 medium (GIBCO® by Life Technologies) supplemented with 1 µg/mL amphotericin B, 1 µg/mL penicillin and 2.5×10−3 µg/mL streptomycin (GIBCO® by Life Technologies), and 10% of FBS (GIBCO® by Life Technologies).
PBMC were obtained from healthy donors after obtaining written informed consent. This study was approved by the Institutional Ethics Committee at the Universidad Autónoma de Nuevo León, College of Biological Sciences. PBMC were isolated by density gradient centrifugation with Ficoll-Hypaque1119 (Milliporesigma, Eugene, OR, USA) and maintained at 4×106 cells/mL in cell culture plates at 37ºC in 5% CO2 atmosphere, using RPMI 1640 medium (GIBCO® by Life Technologies) supplemented with 1 µg/mL amphotericin B, 1 µg/mL penicillin and 2.5×10−3 µg/mL streptomycin (GIBCO® by Life Technologies), and 10% of FBS (GIBCO® by Life Technologies).
The animal study was approved by the Animal Ethical Committee (CEIBA), Number: 01/2015. All experiments were conducted according to Mexican regulation NOM-062-ZOO-1999. Bone morrow cultures were obtained from only one femur and tibia per healthy mouse (male, 6–8 weeks) and maintained at 4×106 cells/mL in cell culture plates at 37ºC in 5% CO2 atmosphere, using RPMI 1640 medium (GIBCO® by Life Technologies) supplemented with 1 µg/mL amphotericin B, 1 µg/mL penicillin and 2.5×10−3 µg/mL streptomycin (GIBCO® by Life Technologies), and 10% of FBS (GIBCO® by Life Technologies).
Cell death analysis
Cell death was determined after 24 hrs of treatment with CH-AuNPs, CH, HACl4, or SC-AuNPs. Cells were seeded at 5×104 cells per well in 96-well plates (Corning Inc. Costar®, USA) and exposed at the indicated concentrations of CH-AuNPs, CH, HACl4, or SC-AuNPs. The concentrations of SC-AuNPs or CH-AuNPs (µM) were determined based on the µM concentration of the precursor salt (HACl4) used for the synthesis of AuNPs. The median cytotoxic concentration of CH-AuNPs required to reduce cell viability by 50% (CC50) was used in subsequent assays. After 24 hrs of treatment, the cells were washed with PBS and then resuspended in 200 µL of binding buffer (10 mM HEPES/NaOH pH 7.4, 140 mM NaCl, 2.5 mM CaCl2) and stained with Annexin V-allophycocyanin (AnnV; BD Biosciences Pharmingen, San Jose, CA, USA) following manufacturer’s instructions and propidium iodide (PI; 0.5 μg/mL; Milliporesigma, Eugene, OR, USA). Cells were then assessed by flow cytometry (Fluorescence-Activated Cell Sorting [FACS]; BDAccury6; Becton Dickinson, San Jose, CA, USA) and analyzed using FlowJo Software (Tree Star Inc., Ashland, OR, USA).
ROS analysis
To measure ROS production levels, we used dichlorodihy drofluorescein diacetate (DCFDA; Invitrogen, St Louis, MO, USA) staining by flow cytometry.12 Cells were seeded at 5×104 cells per well in 96-well plates (Corning Inc. Costar®, USA) and treated with CC50 of CH-AuNPs for 24 hrs. Then, the cells were washed with PBS and stained with DCFDA. Finally, we measured ROS levels by flow cytometry and then analyzed using FlowJo Software (Tree Star Inc., Ashland, OR, USA).
Mitochondrial membrane potential analysis
The loss of mitochondrial membrane potential was detected by (TMRE; Sigma, Aldrich, Darmstadt, Germany). The cells were seeded at 1×105 cells per well in 96-well plates (Corning Inc. Costar®, USA) and treated with CH-AuNPs (CC50). After 24 hrs of treatment, cells were rinsed with PBS and stained with TMRE (50 nM). Finally, cells were measured by flow cytometry and analyzed using FlowJo Software (Tree Star Inc., Ashland, OR, USA).
DNA damage analysis
The DNA damage was assessed through γ-H2AX quantification.13 About 5×105 cells/mL were seeded in 6-well plates (Corning Inc. Costar®, USA) and treated with the CC50 of CH-AuNPs for each cell line, for 24 hrs. Then, the cells were washed with PBS, fixed by bubbling with methanol (100%) and incubated at −20ºC overnight. Then, the cells were blocked for 30 mins at 4ºC and washed (FACS buffer). Then, cells were marked with a primary antibody specific to the phosphorylated form of H2AX (γ-H2AX; ABCAM, Cambridge, UK) in a 1:100 dilution, for 1 hr at room temperature with constant agitation. Then, the secondary antibody (anti-mouse/FITC; ABCAM, Cambridge, UK) was added in a 1:100 dilution and incubated for 30 mins at room temperature and then overnight at 4ºC. Negative controls were incubated only with the secondary antibody. Analysis was done by flow cytometry and analyzed using FlowJo Software (Tree Star Inc., Ashland, OR, USA).
Cell cycle analysis
Cell cycle was evaluated by quantifying intracellular DNA using PI staining through flow cytometry.12 About 5×105 cells were seeded in 6-well dishes and then treated with CC50 of CH-AuNPs for 24 hrs. Later, we washed the cells with PBS and fixed them in 70% ethanol. Additionally, cells were washed again with PBS and incubated with RNase (Sigma-Aldrich, USA) for 30 mins and then with PI (10 μg/mL; Milliporesigma) for 30 mins. Cell DNA contents for cell cycle and DNA degradation were measured by flow cytometry and analyzed in FlowJo Software (Tree Star Inc., Ashland, OR, USA). For DNA degradation, we quantified the SubG1 population.
LC3-II detection
To evaluate autophagosome formation, we measured LC3-II levels through flow cytometry using a commercial specific autophagy detection kit (ABCAM; Cambridge, UK).14 First, cells were seeded at 5×104 cells per well in 96-well plates (Corning Inc. Costar®, USA); then, we exposed them at CC50 of CH-AuNPs for 24 hrs. Cells were then recovered and stained following the manufacturer’s instructions. Analysis was done by flow cytometry and analyzed using FlowJo Software (Tree Star Inc., Ashland, OR, USA).
Caspase-3 analysis
Cleaved caspase-3 was determined by flow cytometry using specific detection kit, FITC-DEV-FMK (ABCAM; Cambridge, UK).12 The cells were then seeded at 1×105 cells per well in 96-well plates (Corning Inc. Costar®, USA) and treated with CC50 of CH-AuNPs for 24 hrs. Cells were then recuperated and stained following the manufacturer’s instructions. Cells were then assessed by flow cytometry and analyzed using FlowJo Software (Tree Star Inc., Ashland, OR, USA).
Cell death inhibitors
To determine cell death mechanism, we used different inhibitors: N-acetyl-cysteine (NAC, 5mM; Sigma-Aldrich, St Louis, MO, USA) as ROS inhibitor,12 QVD-OPh (QVD; 10 μM; BioVision, Milpitas, CA, USA) as pan-caspase inhibitor,15 Spautin-1 (0.75 μM; Sigma-Aldrich, St Louis, Missouri, USA) as an autophagy inhibitor,16 and Necrostatin-1 (5 μM; Sigma-Aldrich, St Louis, Missouri, USA) as necroptosis inhibitor.17 The inhibitors were added 30 mins before treatment with CH-AuNPs. All stock solutions were wrapped in foil and stored at −20°C.
Statistical analysis
All data were analyzed using GraphPad Prism (GraphPad Software, San Diego, CA, USA). The results given in this study represent the mean of at least three independent experiments done in triplicate (mean±SD). Statistical analysis was done using a paired Student’s t-test. The statistical significance was defined as p<0.05.
Results
CH-AuNPs are selective cell death inductors in leukemic cells
We previously demonstrated that CH-AuNPs, but not SC-AuNPs, induce selective ROS-dependent cell death in cervical and breast cancer cell lines, without affecting immune system cells (PBMC).10 Here, we assessed whether the selective cytotoxicity of CH-AuNPs could be due to cell lineage or due to the characteristics of cancer cells. For this, we analyzed cell death in immature immune system cells (BM) and PBMC, using K562 and CEM cell lines as hematological cancer cells. To evaluate cell death, we assessed phosphatidylserine exposure (AnnV) and membrane permeabilization (PI) after 24 hrs of treatment with different concentrations of CH-AuNPs. As shown in Figure 1, CH-AuNPs induce an increase in cell death, with double-positive population for AnnV and PI staining (Figure 1A) at 25 µM which increased in a concentration-dependent way; the mean cytotoxic concentration, CC50, was 25 μM in K562 and CEM cells after 24 hrs of treatment (Figure 1B). However, in PBMC and BM cells, we did not observe cell death induction (Figure 1). Neither CH, HAuCl4, nor SC-AuNPs induced cell death in leukemic cells (Figure S2A) or PBMC (Figure S2B), highlighting the selective cytotoxic effect of CH-AuNPs and not its separate components. These results demonstrated that CH-AuNPs are selective cell death inductors in leukemic cell lines, affecting them at the same concentration, but without affecting healthy immune system cells, thus confirming the selectively to cancer cells.
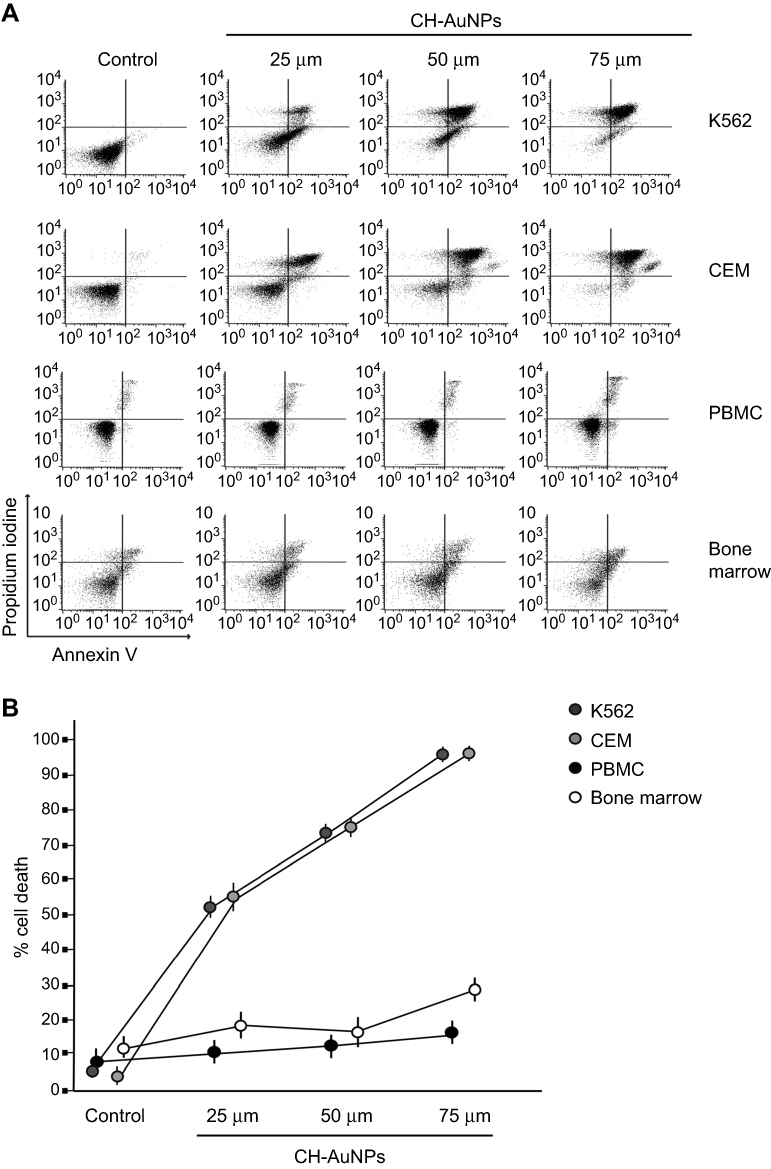 |
Figure 1 Phosphatidylserine exposure and membrane permeability of K562, CEM, PBMC, and bone marrow cells after CH-AuNPs treatment. (A) Cell death analysis by flow cytometry using Annexin-V and propidium iodide (PI) staining in K562, CEM, and non-cancer cells, PBMC, and bone marrow treated with different concentrations of CH-AuNPs for 24 hrs. (B) Quantification of cell death assessed as in (A). |
CH-AuNPs induce ROS production
As CH-AuNPs induce selective cell death in HeLa and MCF-7 cells through ROS production10; therefore, we tested ROS production in leukemic cells treated with CH-AuNPs, using DCFDA staining by flow cytometry. In Figure 2, results exhibit that CH-AuNPs increase ROS production from 3.5% to 40% in K562 cells (Figure 2A) and from 3.5% to 35% in CEM cells (Figure 2B). This indicates that CH-AuNPs induce the same ROS production in both types of leukemia cell lines. Furthermore, we demonstrated that CH-AuNPs do not induce ROS production in PBMC (Figure S3).
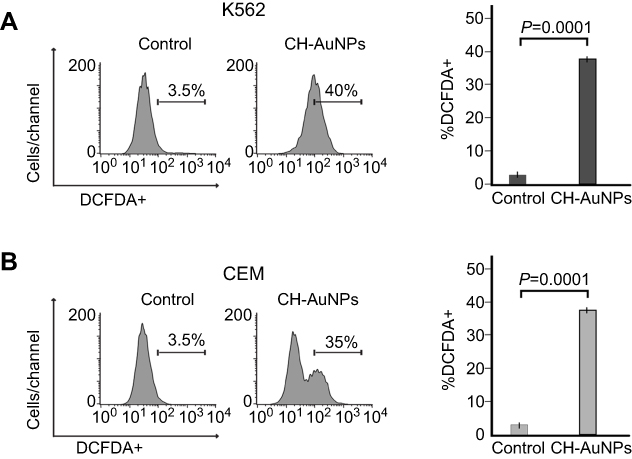 |
Figure 2 CH-AuNPs induce ROS production. Analysis (left) and quantification (right) of ROS production by flow cytometry using DCFDA staining in K562 and CEM cells treated with CH-AuNPs. |
CH-AuNPs induce mitochondrial and nuclear alterations
Mitochondrial and nuclear dysfunction and alterations play a central role in RCD.18,19 Thus, to determine the mechanism of cytotoxicity induced by CH-AuNPs in leukemia cells, we tested mitochondrial and nuclear damage. We assessed the loss of mitochondrial membrane potential (LMMP) with TMRE staining and found that CH-AuNPs induce LMMP in K562 from 5% to 60% and in CEM from 3% to 99% (Figure 3A), indicating that CH-AuNPs induce mitochondrial impairment.
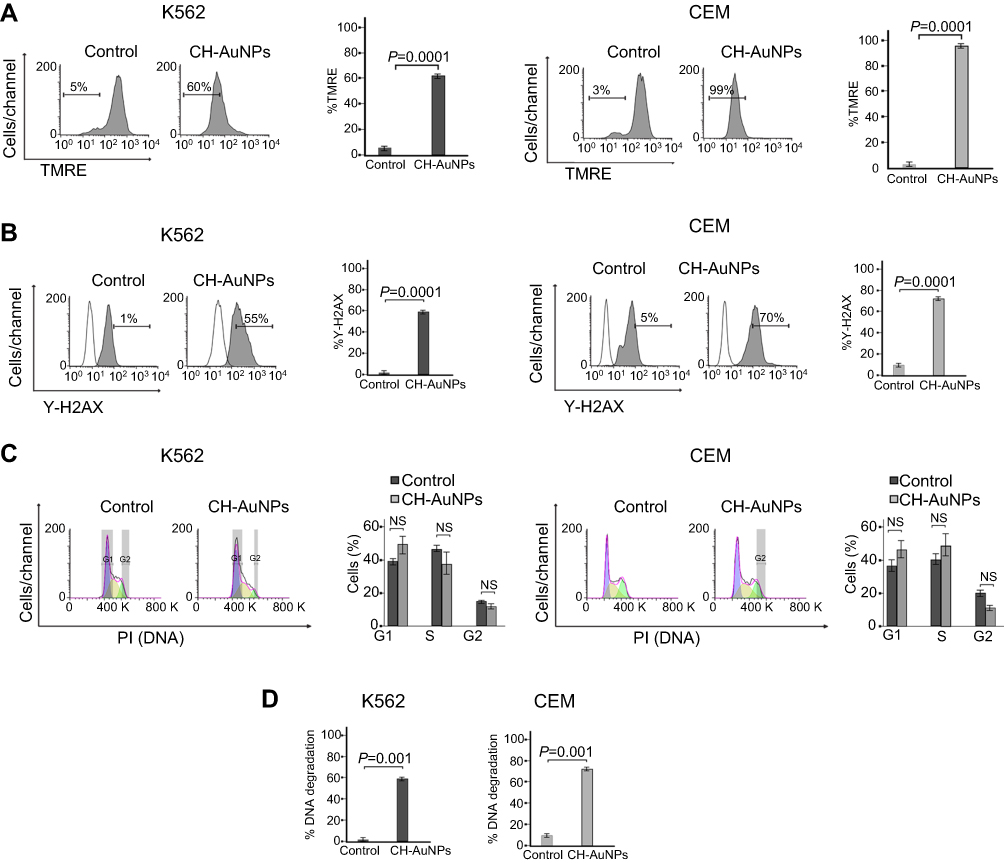 |
Figure 3 Mitochondrial and nuclear alterations in leukemic cells upon treatment with CH-AuNPs. (A) Mitochondrial membrane potential loss analysis (left) and quantification (right), using TMRE by flow cytometry in K562 and CEM cells after 24 hrs of treatment with CH-AuNPs. (B) Nuclear damage analysis (left) and quantification (right) measured through γ-H2AX by Flow cytometry in K562 and CEM cells. (C) Cell cycle analysis (left) and quantification (right), using propidium iodide (PI) staining in K562 and CEM cells, by flow cytometry after 24 hrs of treatment with CH-AuNPs. (D) DNA degradation analysis, using SubG1 population obtained as in A, by flow cytometry after 24 hrs of treatment with CH-AuNPs. |
Nuclear alterations can induce the production of γ-H2AX, the first step in recruiting and localizing DNA repair proteins, which can lead to cell cycle arrest, followed by DNA degradation and cell death.20 To evaluate these processes, we first assessed phosphorylation of H2AX (γ-H2Ax), and we found that treatment with CH-AuNPs increases the percentage of γ-H2AX-positive cells from 1% to 55% in K562 (left) and from 5% to 77% in CEM (right) cells (Figure 3B), indicating that CH-AuNPs induce DNA damage. To determine whether CH-AuNPs induce cell cycle alterations, we quantified DNA in leukemic cells using PI staining. Our analysis shows that CH-AuNPs did not induce cell cycle modifications in K562 and CEM cells, with respect to control cells (Figure 3C). Finally, endonucleases can induce DNA fragmentation in different cell death modalities.21 DNA degradation was analyzed by quantification of a sub-G1 population in leukemic cells treated with CH-AuNPs. Results exhibit 57% of DNA degradation in K562 and 65% in CEM cells treated with CH-AuNPs for 24 hrs (Figure 3D). These results demonstrate that CH-AuNPs induce γH2AX and DNA degradation, without cell cycle arrest in leukemic cells.
CH-AuNPs exhibit biochemical characteristics of autophagy and apoptosis
Due to the characteristics previously observed, we further tried to determine whether cell death was occurring through two of the principal cell death modalities: apoptosis and autophagy.22 The formation of autophagosomes (double-membrane vesicles) is a widely accepted characteristic of autophagy,23 whereas caspase activation is the principal characteristic of apoptosis.24 Thus, we quantified levels of LC3-II, a molecule recruited for the formation of autophagosome membranes during autophagy, and cleaved caspase-3, the principal effector caspase in apoptosis. Results showed that cells treated with CH-AuNPs increase expression of LC3-II from 5% to 75% in K562 and from 2% to 30% in CEM (Figure 4A). In Figure 4B, we show the analysis of cleaved caspase-3 induced by CH-AuNPs. We can observe a slight increase in caspase-3 activation, from 1% to 30% in K562, and from 1% to 40% in CEM cells. This indicates that CH-AuNPs induce autophagosome formation and caspase activation in leukemic cells.
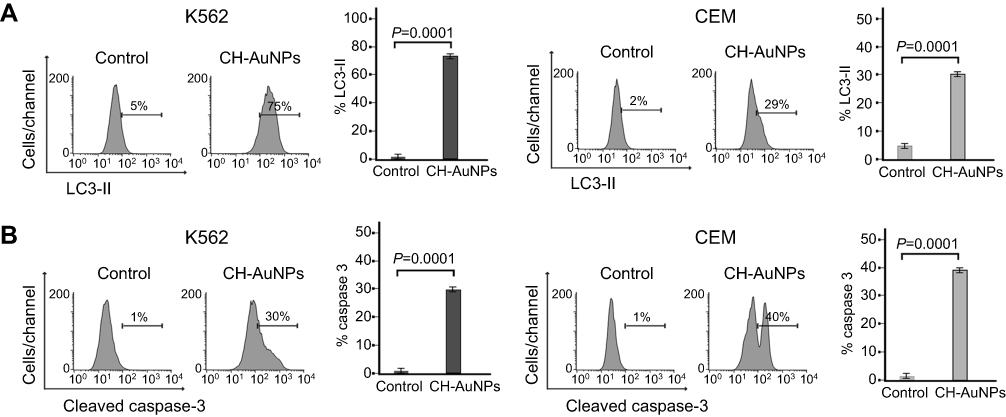 |
Figure 4 CH-AuNPs induce autophagosomes and caspase-3 activation in leukemic cells. (A) Autophagy assessment through LC3-II analysis (left) and quantification (right), by flow cytometry in K562 and CEM cells treated for 24 hrs with CH-AuNPs. (B) Cleaved caspase-3 analysis (left) and quantification (right), by flow cytometry using FITC-DEVD-FMK staining in K562 and CEM cells at 24 hrs of treatment with CH-AuNPs. |
CH-AuNPs induce pro-survival autophagy in cancer cells
To understand if autophagosomes were required for cell death or were activated as a pro-survival mechanism, we used a specific autophagy inhibitor (Spautin-1).22 First, we analyzed autophagosome formation, and we show in Figure 5A that autophagosomes induced by CH-AuNPs decrease in the presence of Spautin-1 in K562 and CEM cells. Then, cell death analysis demonstrated that during autophagy inhibition with Spautin-1 there is an increase in cell death of K562 and CEM cells (Figure 5B) passing from 53% to 79% and from 49% to 75%, respectively. These results indicate that leukemic cell lines produce autophagosomes as a pro-survival mechanism during CH-AuNPs treatment.
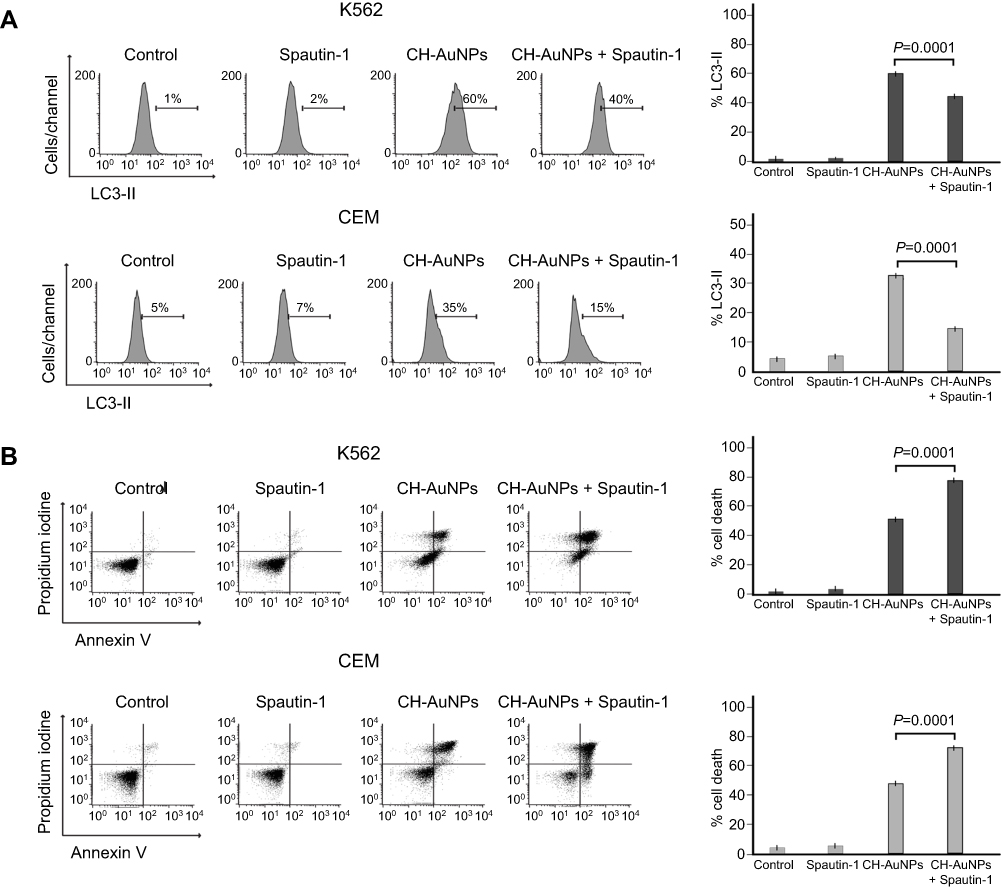 |
Figure 5 CH-AuNPs treatment induces pro-survival autophagy in K562 and CEM cells. (A) Autophagy assessment through LC3-II analysis (left) and quantification (right), by flow cytometry using Spautin-1 as a specific inhibitor of autophagosomes in K562 and CEM cells treated for 24 hrs with CH-AuNPs. (B) Cell death analysis (left) and quantification (right) by flow cytometry using Annexin-V and propidium iodide (PI) staining in K562 and CEM cells treated with CH-AuNPs and Spautin-1. |
CH-AuNPs trigger apoptosis in CEM cells
We then assessed whether cell death induced by CH-AuNPs was caspase-dependent apoptosis. First, we evaluated if a pan-caspase inhibitor (QVD) inhibited activation of caspase-3, the principal biochemical marker of apoptosis.25 The result shows that QVD inhibits cleaved caspase-3 in K562 cells and CEM cells, decreasing from 25% to 15%, and from 45% to 10%, respectively (Figure 6A). Then, we evaluated caspase dependency in cell death using QVD, and analyzed cell death. In Figure 6B, we can observe that cell death induced by CH-AuNPs in K562 cells is not significantly inhibited in the presence of QVD; this result suggests that the mechanism of death is independent of the activation of caspases. In CEM cells, we show that when using QVD, the percentage of cell death diminishes from 50% to 34%, indicating that the mechanism of cell death induced by CH-AuNPs is caspase-dependent (Figure 6B).
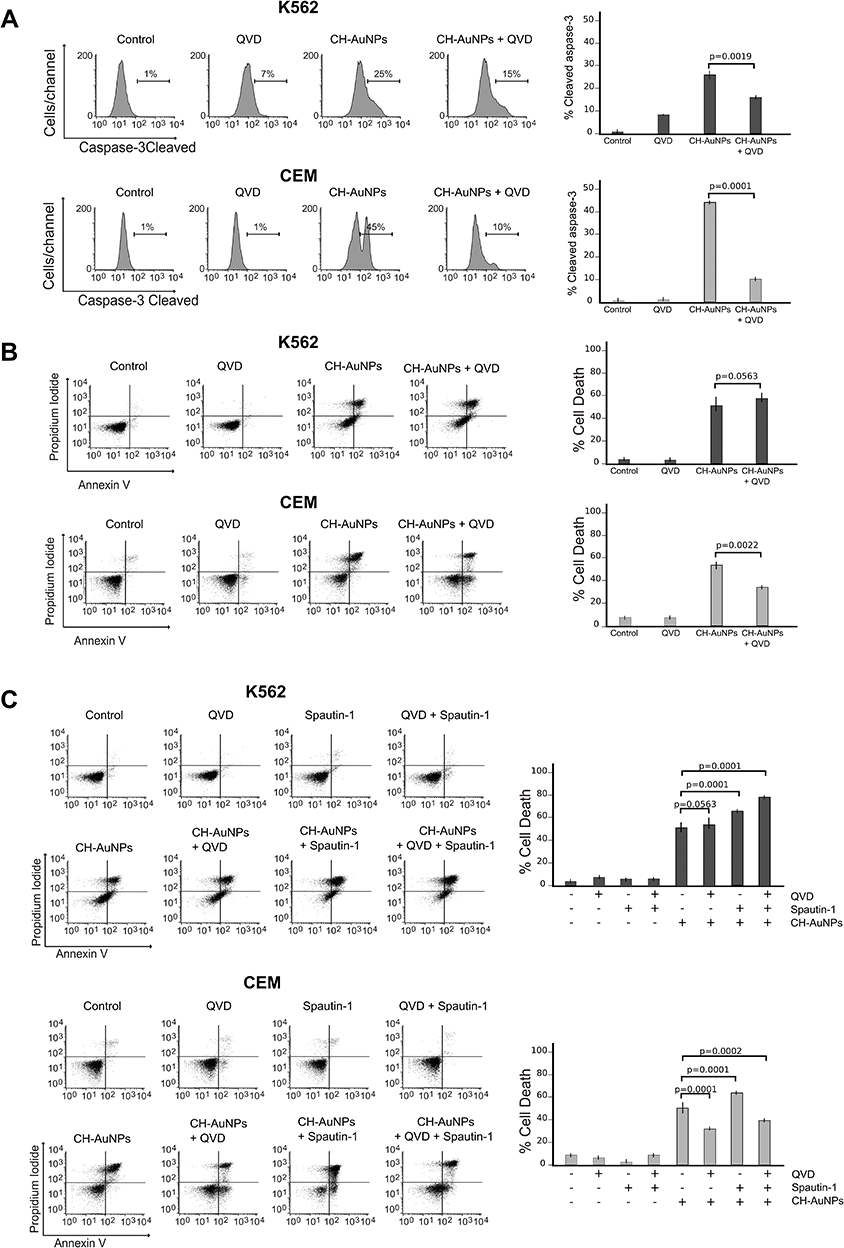 |
Figure 6 Role of apoptosis and autophagy in K562 and CEM cells after CH-AuNPs treatment. (A) Cleaved caspase-3 analysis (left) and quantification (right), using QVD as a pan-caspase inhibitor, in K562 and CEM cells at 24 hrs of treatment with CH-AuNPs. (B) Cell death analysis (left) and quantification (right) using QVD in K562 and CEM cells after CH-AuNPs for 24 hrs. (C) Cell death analysis (left) and quantification (right) during inhibition with Spautin-1 (autophagy inhibitor) and QVD (pan-caspase inhibitor) in K562 and CEM cells treated with CH-AuNPs. |
Then, we wanted to know if during autophagy inhibition, CH-AuNPs were inducing caspase-dependent apoptosis. Thus, we evaluated cell death using Spautin-1 and QVD. Interestingly, the results show that only in CEM cells the percentages of cell death induced by Spautin-1 were reduced in the presence of QVD, from 63% to 38%, whereas in K562 we observed a slight increase in cell death (Figure 6C). These results suggest that autophagy inhibition promotes apoptosis in CEM cells, which was not observed in K562 cells.
CH-AuNPs induce necroptosis in K562 cells
It has been shown that AuNPs can induce different cell death mechanisms.5,26 Necroptosis is a cell death mechanism that plays an important role in mammalian embryonic development and the immune system maturation. This mechanism of cell death involves the recruitment of receptor-interacting proteins (RIP) that are crucial mediators by the formation of the necrosome. We found that CH-AuNPs induce apoptosis in CEM cells even during autophagy inhibition, but not in K562 cells, we decided to analyze whether this cell death mechanism could be induced by CH-AuNPs in K562. For this, we used Necrostatin-1 (Nec-1), a RIPK1 inhibitor.17 Figure 7A shows that the percentage of cell death in K562 diminishes in the presence of Nec-1, even during co-inhibition with QVD; however, in CEM cells, the percentage of cell death increased in both cases. In Figure 7B, we confirmed cell death inhibition with Nec-1 in K562 cells, alone and during treatment with Spautin-1 and CH-AuNPs, decreasing from 63% to 38% (when also using Nec-1), whereas in CEM we saw a slight increase in cell death. These results suggest that CH-AuNPs induce necroptosis in K562 cells and that autophagy inhibition promotes necroptosis in K562 cells, but not in CEM. These results underline that CH-AuNPs induce different mechanisms of cell death in cancer cells.
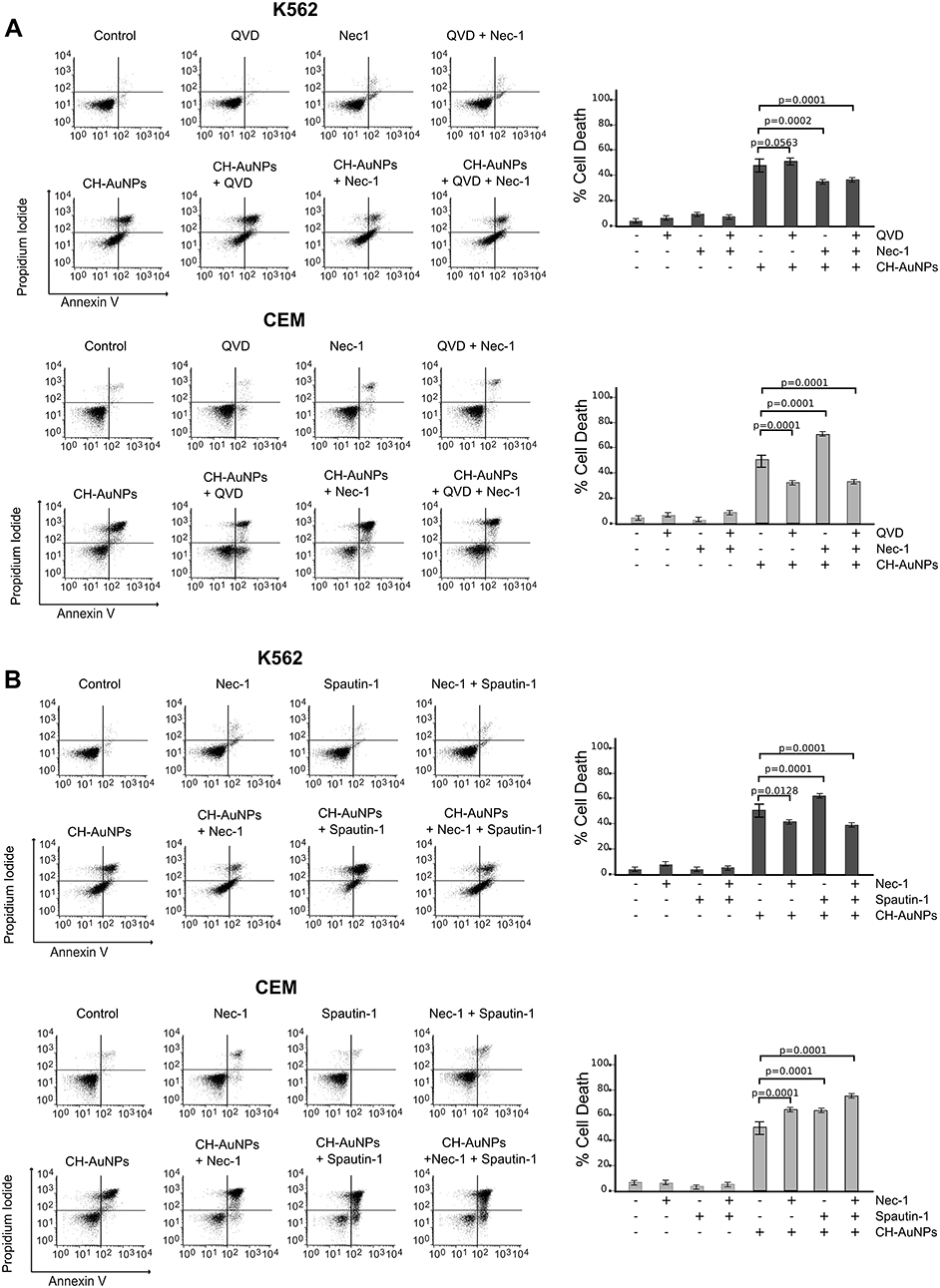 |
Figure 7 Role of necroptosis and autophagy in leukemic cells treated with CH-AuNPs. (A) Cell analysis (left) and quantification (right) using Necrostatin-1 (Nec-1) as a necrosome inhibitor and QVD (pan-caspase inhibitor) in K562 and CEM cells treated with CH-AuNPs. (B) Cell death analysis (left) and quantification (right) during inhibition with Spautin-1 (autophagy inhibitor) and Nec-1 (necrosome inhibitor) in K562 and CEM cells treated with CH-AuNPs. |
CH-AuNPs induce ROS production, triggering mitochondrial and nuclear damage, caspase-3 activation, and cell death in leukemic cells
ROS have an important role in cell death, as they can orchestrate different mechanisms of cell death. To analyze the implication of ROS in the biochemical characteristics of cell death induced by CH-AuNPs, we used NAC, an antioxidant that increases intracellular glutathione levels and possesses thiol-disulfide exchange activity,10 and then we assessed ROS, mitochondrial and DNA damage, and caspase activity. The results demonstrate that NAC was able to inhibit ROS production during CH-AuNPs treatment with CC50 (25 μM) in K562 and CEM cells (Figure 8A). Mitochondrial membrane potential analyses show that ROS inhibition with NAC blocks mitochondrial damage after treatment with CH-AuNPs in both cell lines (Figure 8B). Then, we show that the DNA damage (γ-H2AX) diminished in K562 and CEM cells during ROS inhibition (Figure 8C). Finally, we analyzed whether ROS production activates caspase-3 in CH-AuNPs-treated cells. Figure 8D shows that in the presence of NAC inhibits caspase-3 activation by CH-AuNPs in both cell lines.
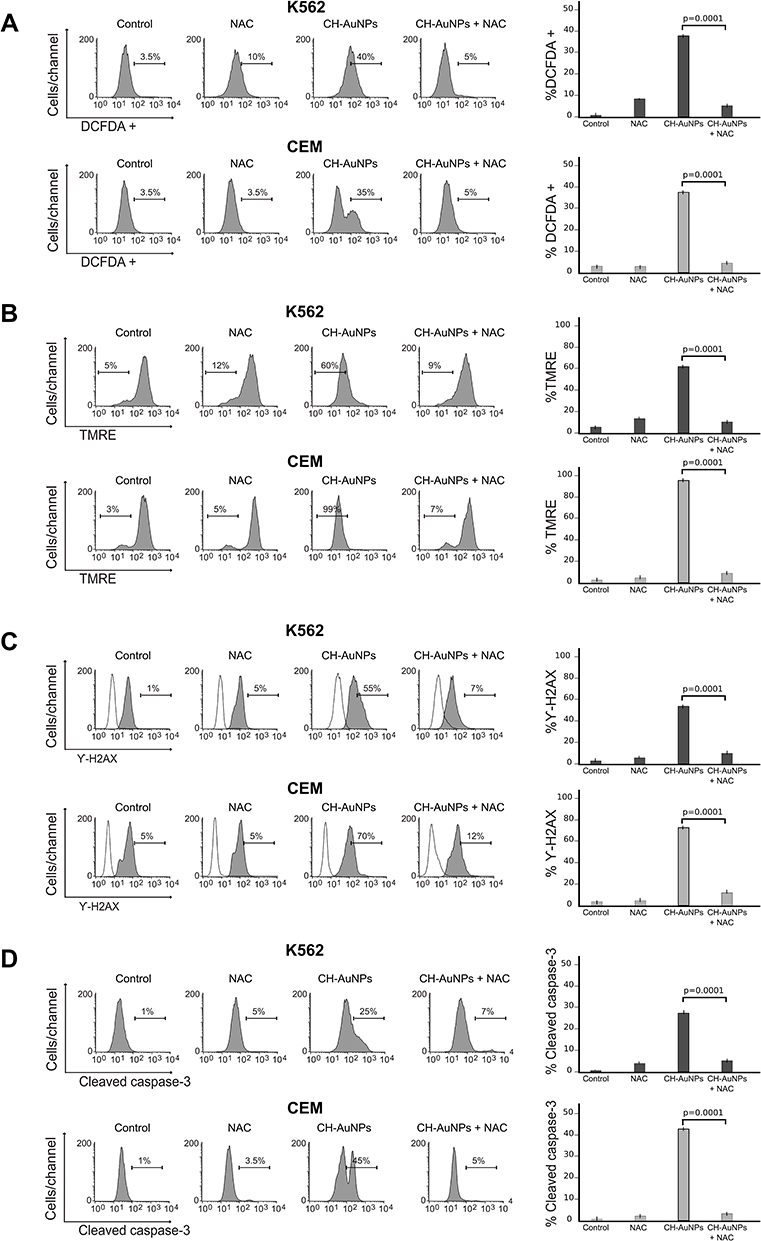 |
Figure 8 Effect of ROS production in the biochemical characteristics of cell death induced by CH-AuNPs in K562 and CEM cells. (A) Analysis and quantification ROS by flow cytometry using DCFDA staining and N-acetyl-cysteine (NAC) as a ROS inhibitor in K562 and CEM cells treated with CH-AuNPs. (B) Mitochondrial membrane potential loss analysis and quantification by Flow cytometry using TMRE staining and NAC (ROS inhibitor) in K562 and CEM cells after 24 hrs of treatment with CH-AuNPs. (C) Nuclear damage analysis and quantification measured through γ-H2AX using NAC in K562 and CEM cells. (D) Cleaved caspase-3 analysis (left) and quantification (right) during ROS inhibition (NAC) by flow cytometry in K562 and CEM cells after 24 hrs of treatment with CH-AuNPs. |
We demonstrated that CH-AuNPs induce ROS production, which plays an important role in mitochondrial and nuclear damage, and ROS can be related to AuNPs' cytotoxicity.27,28 We analyzed if ROS production was playing a role in the cell death of leukemic cells. In Figure 9, we show that cell death induced by CH-AuNPs is blocked during ROS inhibition in both cell lines, K562 and CEM cells. This reveals that CH-AuNPs induce ROS-dependent cell death in leukemic cells, demonstrating that ROS production is an important and conserved characteristic of cell death induced by CH-AuNPs in cancer cells, independently of the cell death mechanism activated.
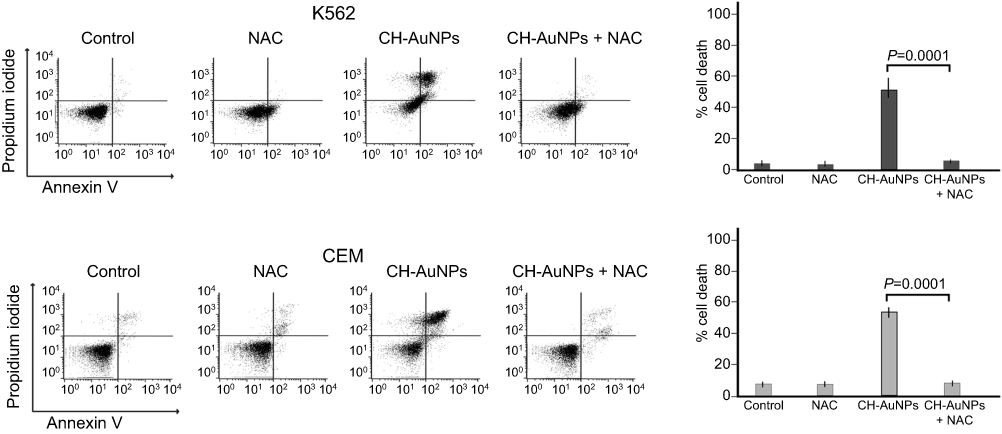 |
Figure 9 Role of ROS in the cell death induced by CH-AuNPs in leukemic cells. Cell death analysis and quantification by flow cytometry using Annexin-V and propidium iodide (PI) staining in K562 and CEM cells treated with different concentrations of CH-AuNPs for 24 hrs during ROS inhibition with NAC. |
Discussion
We evaluated the cytotoxic mechanism of CH-AuNPs in two different leukemic cell lines: CEM, from T-ALL, and K562, from CML. We obtained CH-AuNPs of 3–10 nm with a surface plasmon resonance at 520 nm and assessed the effect of CH-AuNPs in leukemic and healthy cells. We observed that CH-AuNPs induce PS exposure, loss of membrane permeability, and cytotoxicity in a time- and concentration-dependent manner in leukemic cells. Additionally, leukemic cells were more sensitive to CH-AuNPs compared to HeLa and MCF-7 cell lines, as the CC50 in leukemic cell lines is 25 µM, while in HeLa and MCF-7 cells it was of 75 µM.10 Cell death analysis showed that CH-AuNPs have a selective cytotoxic effect in leukemic cells but not in PBMC nor in murine BM cells, at the same concentration and times, indicating that CH-AuNPs’ cytotoxicity is due to the characteristics of cancer cells, and not the cell type.10 This selectivity has been observed also with nanoparticles of other metals.29–31 It has been recently shown that silver nanoparticles green-synthetized using the desert plant Cyperus conglomeratus31 or walnut green husk30 also present selectivity against cancer cells, without affecting non-cancerous cells.
Pan et al evaluated the toxicity of CH-AuNPs and observed that the cellular response is dependent on their concentration in A549, NCI-H460, and A431 cell lines.32 On the other hand, AuNPs (18 nm) obtained using cetyl trimethylammonium bromide showed low cytotoxicity in K562 cells. These differences can be due to the reducing agent, as it has been reported that medium-molecular-weight CH is cytotoxic per se in U939, K562, HL60, and THP-1 cell lines.33 Liu et al used seleno-short-chain CH (SSCC) in K562 and observed that it significantly suppressed the growth of K562 cells in a dose-dependent manner, by inducing caspase-dependent apoptosis.34
Another important observation we had was that CH-AuNPs did induce changes in the cell cycle of leukemic cells, as we determined previously in HeLa and MCF-710 and as shown in a study done in A549 lung cancer cells treated with CH-AuNPs.35 However, although CH-AuNPs do not induce cell cycle arrest in different cell types, SSCC induced cell cycle arrest in G2 phase in K562 cells34 and in MCF-7 and BT-20 cells.36 These differences could be due to the purity or structure of CH molecule itself, which is different from the AuNPs.
We also showed that CH-AuNPs induce the loss of MMP and ROS production in both CEM and K562, cell lines, which correspond with other studies where AuNPs induce mitochondrial damage and oxidative stress.4,5,28,37 Furthermore, we observed that cell death was dependent on ROS production. This effect has been observed to be produced by CH in fibrosarcoma cells,38 and by AuNPs on human leukemia (HL-60) and hepatoma (HepG2) cell lines,39 and in MCF-7 and HeLa cells.10
DNA damage has been barely assessed after nanoparticle treatment, and here we assessed γH2AX and observed that CH-AuNPs increased γH2AX positive cells, indicating DNA damage. Ross I Berbeco et al also observed that AuNPs enhanced DNA damage (through γH2AX) after irradiation in Hela cells.40 In another study, AuNPs coated with grafted galactose and polyethylene glycol induced radiosensitivity, confirmed by elevated levels of DNA damage compared with naked AuNPs and the control group.41
It has been reported that AuNPs can induce apoptosis through mitochondrial and DNA damage.32,42–44 As caspases are effectors of apoptosis, we analyzed caspase-3 activity in both cell lines and observed higher levels of cleaved caspase-3. Caspase inhibition showed that CH-AuNPs induce caspase-independent cell death in K562 and caspase-dependent cell death in CEM. Xia et al reported two different mechanisms of cell death for polystyrene AuNPs. They observed LAMP-1-mediated endocytosis, calcium release, proapoptotic protein expression, mitochondrial damage, and caspase activation in Raw 264.7. They also observed Caveolin-1 mediated endocytosis and did not observe caspase activation in bronchial epithelium (BEAS-2B) and concluded that the uptake and cell death mechanism depend on the endocytosis pathway.45
Autophagosome formation has been barely studied in cells treated with AuNPs. Here we found that autophagy was induced after treatment with CH-AuNPs as a prosurvival mechanism, as we could enhance cell death by inhibiting autophagy. This feature has been also observed in lung fibroblasts (MRC5 cells) treated with AuNPs,46 and in several cell lines treated with chemotherapies.47,48
Our results also show that NAC inhibits cell death in leukemic cells treated with CH-AuNPs. It has been reported that ROS inhibition reduces cell death induced by AuNPs in several models, including HepG2 cells,49 HL-60 cells,39 and HeLa and MCF-7 cells,10 yet this does not occur in normal human dermal fibroblast cells, despite the observed increase in ROS production after 24 hrs of treatment.50 These results reinforce our results that indicate that CH-AuNPs activate a different mechanism in cancerous and normal cells.
Conclusion
Our results indicate that CH-AuNPs induce selective cytotoxicity in leukemia cells compared with their healthy counterpart and reveal that CH-AuNPs induce necroptosis in K562 cells (Figure 10A) and caspase-dependent cell death in CEM cells (Figure 10B), including pro-survival autophagy activation in both cases (Figure 10). Although cell death relies on different effector molecules, we observed that in both cases the mechanism is dependent on ROS production. This study improves the understanding of the cytotoxicity of CH-AuNPs and their selectivity to cancer cells, irrespective of the cell lineage, extending their effects to hematopoietic cancers.
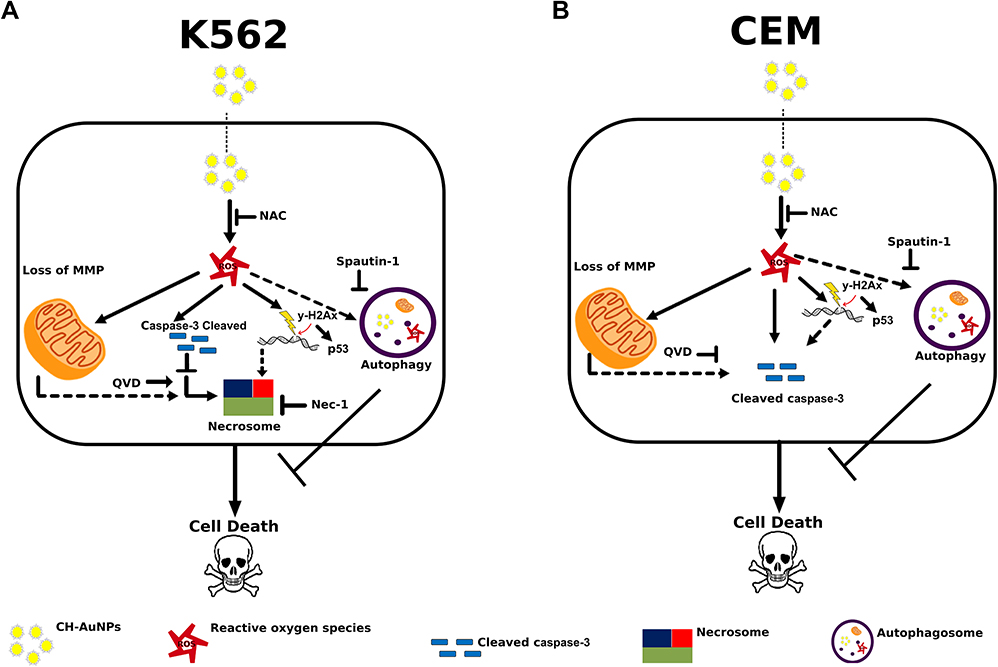 |
Figure 10 Cell death mechanism induced by CH-AuNPs in leukemic cells. (A) In K562 cells, CH-AuNPs induce ROS production that are inhibited using NAC. ROS production leads to loss of mitochondrial membrane potential, caspase-3 activation, and nuclear alterations. Nuclear alterations include DNA damage (γ-H2Ax) and degradation. During, apoptosis inhibition with QVD, cell death is maintained while it is inhibited with necrostatin-1. (B) In CEM cells, CH-AuNPs generate ROS production (inhibited with NAC) leading to caspase-dependent cell death. ROS production induces loss of mitochondrial membrane potential, DNA damage (γ-H2Ax) and degradation, and cleaved of caspase-3 to trigger cell death. Finally, QVD inhibits the cell death induced by CH-AuNPs. In A and B, pro-survival autophagy is activated. |
Ethics approval and informed consent
This study was approved by the Institutional Ethics Committee at the Universidad Autónoma de Nuevo León, College of Biological Sciences. The animal study was approved by the Animal Ethical Committee (CEIBA), Number: 01/2015.
Data sharing statement
All datasets generated during the current study are available from the corresponding author on reasonable request.
Acknowledgments
We thank Andrea Avila-Avila for overall help, Sergio Galindo (Departamento de Química de la Facultad de Ciencias Biológicas, UANL) for technical help and Alejandra Arreola-Triana for article revision. HYLA and MGGJ thank CONACYT for scholarship. We thank CONACYT for funding and the Laboratorio de Inmunología y Virología for the funding and the facilities provided to perform this work. This work was supported by a research grant from the Consejo Nacional de Ciencia y Tecnología, Ciencia Básica (CB 252017) to C Rodriguez-Padilla, and by the Laboratorio de Inmunología y Virología.
Author contributions
All authors contributed to data analysis, drafting or revising the article, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Disclosure
Dr Ana Carolina Martinez-Torres reports grants from CONACYT, during the conduct of the study. Dr Diana G. Zarate-Triviño reports grants from CONACYT, outside the submitted work. The authors report no other conflicts of interest in this work.
References
1. Jain S, Hirst DG, O’Sullivan JM. Gold nanoparticles as novel agents for cancer therapy. Br J Radiol. 2012;85(1010):101–113. doi:10.1259/bjr/59448833
2. Rana S, Bajaj A, Mout R, Rotello VM. Monolayer coated gold nanoparticles for delivery applications ☆. Adv Drug Deliv Rev. 2012;64(2):200–216. doi:10.1016/j.addr.2011.08.006
3. Chithrani BD, Chan WCW. Elucidating the mechanism of cellular uptake and removal of protein-coated gold nanoparticles of different sizes and shapes. Nano Lett. 2007;7(6):1542–1550. doi:10.1021/nl070363y
4. Boyles MSP, Kristl T, Andosch A, et al. Chitosan functionalisation of gold nanoparticles encourages particle uptake and induces cytotoxicity and pro-inflammatory conditions in phagocytic cells, as well as enhancing particle interactions with serum components. J Nanobiotechnol. 2015;13(1):84. doi:10.1186/s12951-015-0146-9
5. Pan Y, Leifert A, Ruau D, et al. Gold nanoparticles of diameter 1. 4 nm trigger necrosis by oxidative stress and mitochondrial damage. Small. 2009:2067–2076. doi:10.1002/smll.200900466
6. Maiorano G, Sabella S, Sorce B, et al. Effects of cell culture media on the dynamic formation of protein-nanoparticle complexes and influence on the cellular response. ACS Nano. 2010;4(12):7481–7491. doi:10.1021/nn101557e
7. Zhu J, Li Z, Luo J, Wang J. Chitosan–gold nanoparticles as peroxidase mimic and their application in glucose detection in serum. RSC adv. 2017:44463–44469. doi:10.1039/c7ra08967h
8. Martinez-Torres AC, Quiney C, Attout T, et al. CD47 agonist peptides induce programmed cell death in refractory chronic lymphocytic leukemia B cells via PLCγ1 activation: evidence from mice and humans. PLoS Med. 2015;12(3):1–37. doi:10.1371/journal.pmed.1001796
9. Journal NE, Medicine I, Med AI. Difference between hematological malignancy and Solid tumor research articles published in four major medical journals. Leukemia. 2006:1655–1657. doi:10.1038/sj.leu.2404369
10. Martínez-Torres AC, Zarate-Triviño DG, Lorenzo-Anota HY, Ávila-Ávila A, Rodríguez-Abrego C, Rodríguez-Padilla C. Chitosan gold nanoparticles induce cell death in hela and MCF-7 cells through reactive oxygen species production. Int J Nanomedicine. 2018;13:3235–3250. doi:10.2147/IJN.S165289
11. Hüttmann G, Rudnitzki F, Endl E, et al. Bleaching of plasmon-resonance absorption of gold nanorods decreases efficiency of cell destruction. J Biomed Opt. 2012;17(5):058003. doi:10.1117/1.jbo.17.5.058003
12. Martínez-Torres AC, Reyes-Ruiz A, Benítez-Londoño M, Franco-Molina MA, Rodríguez-Padilla C. Immunepotent CRP induces cell cycle arrest and caspase-independent regulated cell death in HeLa cells through reactive oxygen species production. BMC Cancer. 2018;18(1):13. doi:10.1186/s12885-017-3954-5
13. Kim EH, Kim M-S, Song HS, et al. Gold nanoparticles as a potent radiosensitizer in neutron therapy. Oncotarget. 2017;8(68):112390–112400. doi:10.18632/oncotarget.19837
14. Tusskorn O, Khunluck T, Prawan A, Senggunprai L, Kukongviriyapan V. Mitochondrial division inhibitor-1 potentiates cisplatin-induced apoptosis via the mitochondrial death pathway in cholangiocarcinoma cells. Biomed Pharmacother. 2019;111:109–118. doi:10.1016/j.biopha.2018.12.051
15. Martínez-Torres AC, Calvillo-Rodríguez KM, Uscanga-Palomeque AC, et al. PKHB1 tumor cell lysate induces antitumor immune system stimulation and tumor regression in syngeneic mice with tumoral T lymphoblasts. J Oncol. 2019;2019. doi:10.1155/2019/9852361
16. Shao S, Li S, Qin Y, et al. Spautin-1, a novel autophagy inhibitor, enhances imatinib-induced apoptosis in chronic myeloid leukemia. Int J Oncol. 2014;44(5):1661–1668. doi:10.3892/ijo.2014.2313
17. Cui H, Zhu Y, Yang Q, et al. Necrostatin-1 treatment inhibits osteocyte necroptosis and trabecular deterioration in ovariectomized rats. Sci Rep. 2016;6:1–13. doi:10.1038/srep33803
18. Bortul R, Zweyer M, Billi AM, et al. Nuclear changes in necrotic HL-60 cells. J Cell Biochem. 2001;81:19–31. doi:10.1002/jcb.1073
19. Rogalinska M. Alterations in cell nuclei during apoptosis. Cell Mol Biol Lett. 2002;7:995–1018.
20. Kuo LJ, Yang LX. Gamma-H2AX- A novel biomaker for DNA double-strand breaks. Vivo (Brooklyn). 2008;22(3):305–310. doi:0258-851X/2008
21. He F, Wei R, Wang J, et al. RIP3 induces ischemic neuronal DNA degradation and programmed necrosis in rat via AIF. Sci Rep. 2016;6(1):1–11. doi:10.1038/srep29362
22. Glick D, Macleo SB, Macleod KF. Autophagy: cellular and molecular mechanisms Danielle. Circ Res. 2006;3–12. doi:10.1161/01.RES.0000243584.45145.3f
23. Olsson S, Yamamoto A, Nishino I, et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy. 2016;12(1):1–222. doi:10.1080/15548627.2015.1100356
24. Kroemer G, Galluzzi L, Vandenabeele P, et al. Classification of cell death 2009. Cell Death Differ. 2009;16(1):3–11. doi:10.1038/cdd.2008.150.Classification
25. Galluzzi L, Vitale I, Aaronson SA, et al. Molecular mechanisms of cell death: recommendations of the nomenclature committee on cell death 2018. Cell Death Differ. 2018;25(3):486–541. doi:10.1038/s41418-017-0012-4
26. Fadeel B, Torres F. Programmed cell death : molecular mechanisms and implications for safety of nanomaterials. Acc Chem Res. 2013;46(3):733–742. doi:10.1021/ar300020b
27. Zhao Y, Gu X, Ma H, He X, Liu M, Ding Y. Association of glutathione level and cytotoxicity of gold nanoparticles in lung cancer cells. J Phys Chem C. 2011;115(26):12797–12802. doi:10.1021/jp2025413
28. Piryazev AP, Azizova OA, Aseichev AV, Dudnik LB, Sergienko VI. Effect of gold nanoparticles on production of reactive oxygen species by human peripheral blood leukocytes stimulated with opsonized zymosan. Bull Exp Biol Med. 2013;156(1):101–103. doi:10.1007/s10517-013-2288-9
29. Mioc M, Pavel IZ, Ghiulai R, et al. The cytotoxic effects of betulin-conjugated gold nanoparticles as stable formulations in normal and melanoma cells. Front Pharmacol. 2018;9:1–16. doi:10.3389/fphar.2018.00429
30. Khorrami S, Zarrabi A, Khaleghi M, Danaei M, Mozafari MR. Selective cytotoxicity of green synthesized silver nanoparticles against the MCF-7 tumor cell line and their enhanced antioxidant and antimicrobial properties. Int J Nanomed. 2018;13:8013–8024. doi:10.2147/IJN.S189295
31. Al-nuairi AG, Mosa KA, Mohammad MG, El-keblawy A, Soliman S. Biosynthesis, characterization, and evaluation of the cytotoxic effects of biologically synthesized silver nanoparticles from cyperus conglomeratus root extracts on breast cancer cell line MCF-7. Biol Trace Elem Res. 2019. doi:10.1007/s12011-019-01791-7
32. Choi SY, Jang SH, Park J, et al. Cellular uptake and cytotoxicity of positively charged chitosan gold nanoparticles in human lung adenocarcinoma cells. J Nanoparticle Res. 2012;14:12. doi:10.1007/s11051-012-1234-5
33. Kim MO, Moon DO, Kang CH, Choi YH, Lee JD, Kim GY. Water-soluble chitosan sensitizes apoptosis in human leukemia cells via the downregulation of bcl-2 and dephosphorylation of akt. J Food Biochem. 2013;37(3):270–277. doi:10.1111/j.1745-4514.2011.00628.x
34. Liu A, Song W, Cao D, Liu X, Jia Y. Growth inhibition and apoptosis of human leukemia K562 cells induced by Seleno-short-chain chitosan. Methods Find Exp Clin Pharmacol. 2008;30(3):181. doi:10.1358/mf.2008.30.3.1213209
35. Ramalingam V, Revathidevi S, Shanmuganayagam T, Muthulakshmi L. Advances Biogenic gold nanoparticles induce cell cycle arrest through oxidative stress and sensitize mitochondrial membranes in A549 lung cancer cells. RSC Adv. 2016;6:20598–20608. doi:10.1039/C5RA26781A
36. Wu D, Zhao Y, Fu S, Zhang J, Wang W, Yan Z. Seleno-Short-Chain Chitosan induces apoptosis in human breast cancer cells through mitochondrial apoptosis pathway in vitro. Cell Cycle. 2018:4101. doi:10.1080/15384101.2018.1464845
37. Liu M, Gu X, Zhang K, et al. Gold nanoparticles trigger apoptosis and necrosis in lung cancer cells with low intracellular glutathione. J Nanoparticle Res. 2013;15(8). doi:10.1007/s11051-013-1745-8
38. Kong CS, Kim JA, Ahn B, Byun HG, Kim SK. Carboxymethylations of chitosan and chitin inhibit MMP expression and ROS scavenging in human fibrosarcoma cells. Process Biochem. 2010;45(2):179–186. doi:10.1016/j.procbio.2009.09.004
39. Haza AI, Mateo D, Morales P, Alicia A. Oxidative stress contributes to gold nanoparticle-induced cytotoxicity in human tumor cells. Toxicol Mech Methods. 2014;6516(3):161–172. doi:10.3109/15376516.2013.869783
40. Berbecoa RI, Koridecka H, Ngwaa W, et al. DNA damage enhancement from gold nanoparticles for clinical. Radiant Res. 2013;178(6):604–608. doi:10.1667/RR3001.1.DNA
41. Zheng Q, Zhu C, Xu H-F, et al. Synthesis of novel galactose functionalized gold nanoparticles and its radiosensitizing mechanism. J Nanobiotechnol. 2015;13(1):1–11. doi:10.1186/s12951-015-0129-x
42. Mohan JC, Praveen G, Chennazhi KP, Jayakumar R, Nair SV. Functionalised gold nanoparticles for selective induction of in vitro apoptosis among human cancer cell lines. J Exp Nanosci. 2013;8(1):32–45. doi:10.1080/17458080.2011.557841
43. Selim ME, Hendi A. Gold nanoparticles induce apoptosis in MCF-7 human breast cancer cells. Asian Pac J Cancer Prev. 2012;13(4):1617–1620. doi:10.7314/APJCP.2012.13.4.1617
44. Mukherjee P, Bhattacharya R, Bone N, et al. Potential therapeutic application of gold nanoparticles in B-chronic lymphocytic leukemia (BCLL): enhancing apoptosis. J Nanobiotechnol. 2007;5(Cll):4. doi:10.1186/1477-3155-5-4
45. Xia T, Kovochich M, Liong M, Zink JI, Nel AE. Cationic polystyrene nanosphere toxicity depends on cell-specific endocytic and mitochondrial injury pathways. ACS Nano. 2008;2(1):85–96. doi:10.1021/nn700256c
46. Li JJ, Hartono D, Ong CN, Bay BH, Yung LYL. Autophagy and oxidative stress associated with gold nanoparticles. Biomaterials. 2010;31(23):5996–6003. doi:10.1016/j.biomaterials.2010.04.014
47. Sui X, Chen R, Wang Z, et al. Autophagy and chemotherapy resistance: a promising therapeutic target for cancer treatment. Cell Death Dis. 2013;4(10):1–12. doi:10.1038/cddis.2013.350
48. Fulda S. Autophagy in Cancer Therapy. Front Oncol. 2017;7:1–4. doi:10.3389/fonc.2017.00128
49. Hamed S, Emara M, Shawky RM, El-domany RA, Youssef T. N-Acetylcysteine Potentiates the Antimicrobial Activity of Gold Nanoparticles: A Paradigm Shift in Treatment of Multidrug Resistant Pathogens. EC Microbiology. 2017;3:110–121.
50. Mateo D, Morales P, Ávalos A, Haza AI. Comparative cytotoxicity evaluation of different size gold nanoparticles in human dermal fibroblasts. J Exp Nanosci. 2015;10:1401–1417. doi:10.1080/17458080.2015.1014934
Supplementary materials
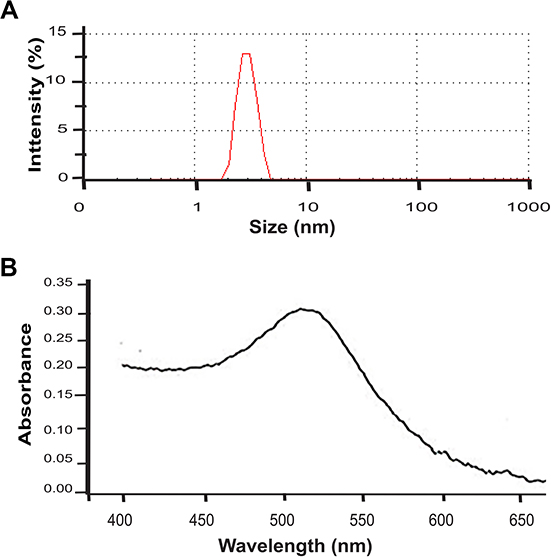 |
Figure S1 Size and plasmon resonance.Note: Representative size distribution obtained from DLS analysis. (B) UV-Vis absorption spectrum. |
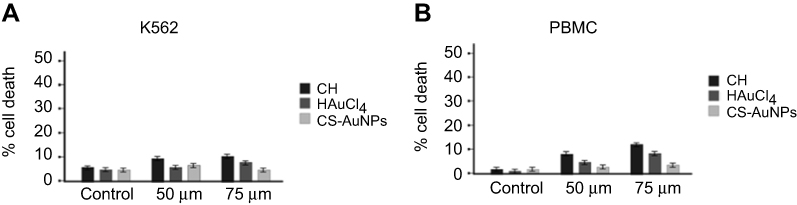 |
Figure S2 Effect of Chitosan, AuHCl4, and SC-AuNPs on K562 and PBMC cell viability.Notes: K562 (A), and PBMC (B) were treated with Chitosan (CH), HAuCl4, and Sodium-citrate nanoparticles (SC-AuNPs) for 24 hours. Cell death was measured by Flow cytometry, and the means of triplicates were graphed. |
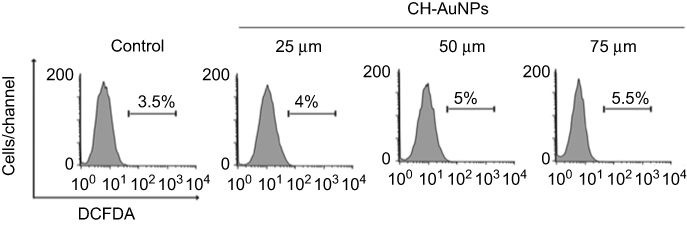 |
Figure S3 ROS analysis in PBMC cells treated with CH-AuNPs.Note: Analysis of ROS by Flow cytometry using DCFDA staining in PBMC treated with CH-AuNPs. |
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.