Back to Journals » Therapeutics and Clinical Risk Management » Volume 14
Childhood-onset growth hormone deficiency and the transition to adulthood: current perspective
Authors Ahmid M ![]() , Ahmed SF
, Ahmed SF ![]() , Shaikh MG
, Shaikh MG
Received 28 March 2018
Accepted for publication 19 August 2018
Published 23 November 2018 Volume 2018:14 Pages 2283—2291
DOI https://doi.org/10.2147/TCRM.S136576
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 5
Editor who approved publication: Professor Garry Walsh
M Ahmid, SF Ahmed, MG Shaikh
Development Endocrinology Research Group, School of Medicine, Dentistry and Nursing, University of Glasgow, Glasgow, UK
Abstract: Childhood-onset growth hormone deficiency (CO-GHD) is an endocrine condition associated with a broad range of health issues from childhood through to adulthood, which requires particular attention during the transition period from adolescence to young adulthood. There is uncertainty in the clinical practice of the management of CO-GHD during transition regarding the clinical assessment and management of individual patients during and after transition to obtain optimal follow-up and improved health outcomes. Despite the availability of clinical guidelines providing the framework for transition of young adults with CO-GHD, there remains substantial variation in approaching transitional care among pediatric and adult services. A well-structured and coordinated transitional plan with clear communication and direct collaboration between pediatric and adult health care to ensure optimal management of adolescents with CO-GHD during transition is needed.
Keywords: growth hormone deficiency, childhood-onset, transition, adolescents
Introduction
Transition is a planned process that aims to address the wider set of medical, psychological and educational requirements of young adults with chronic conditions as they move from pediatric to adult health services.1 Increased survival of children with complex health conditions has led to growing attention on the importance of the quality of services provided for adolescents during transition. Childhood-onset growth hormone deficiency (CO-GHD) is one of a number of complex endocrine conditions that may have an impact throughout life, starting in childhood and continuing into adulthood. Transition of patients with CO-GHD that have been treated with recombinant human growth hormone (rhGH) during childhood is a challenging phase for health care providers. This phase occurs in CO-GHD from mid-late teens until approximately 6 or 7 years after final height has been reached.2 This is a critical period for maximizing bone mass and muscle strength, which are important determinants for the risk of fractures related to osteoporosis occurring later in life.3 The presented data largely support the positive role of rhGH therapy during the transition period of adolescence and beyond.4,5 The effects of CO-GHD and the benefits of rhGH therapy during transition have been previously described in several studies.6–10 Although there are numerous publications that have discussed transition in general with some specifically discussing CO-GHD,2,6,11 a number of unanswered issues still remain.
Current practice during transition
Several clinical practice guidelines have described the theoretical framework for the transition of adolescents with CO-GHD.2,12–15 However, in the clinical practice setting, both adult and pediatric endocrinologists lack consistency when managing the adolescent patients with CO-GHD during transition. The clinical experience in Scotland highlighted various areas of uncertainty in clinical practice:16 the best method of evaluating the hypothalamic–pituitary axis, identification of patients to test, determining which test to use for diagnosing persistent growth hormone deficiency (GHD), identifying which patients would most benefit from continued rhGH treatment and methods of ascertaining if patients should continue treatment. In addition, it is unclear with whom the responsibility for the biochemical reevaluation of GHD lies, the pediatric endocrinologist or adult endocrinologist who will decide whether to continue rhGH therapy into adulthood and the involvement of patients and family in making the decision and management plan. Practice varies across the UK, particularly in cases when no organic cause for GHD is identified.16–18
The current guidelines classify CO-GHD according to the likelihood of persistent GHD.2,12,13 A previous history of organic disease, deficits in two or more additional pituitary hormones,19,20 hypothalamic–pituitary structural abnormalities as well as tumour-related organic GHD are strong predictors of persistent GHD.21–24 Patients who are considered having a high probability of persistent GHD may not require their growth hormone (GH) status to be reevaluated and should continue rhGH without interruption. Those who are less likely to have persistent GHD should undergo reevaluation of the GH axis before resuming rhGH replacement therapy.2 Although there are clearly stated criteria under both categories, other factors such as clinical observations and development of symptoms could influence the decision of how the patients are categorized and managed. In addition, there is no clear reevaluation pathway for those who exhibit a discordant GH and IGF-1 pattern.
The timing of retesting is another issue. According to guidelines, the decision when to reassess depends largely on attainment of adult height as defined (height velocity <2 cm/year). The recommended washout interval between stopping rhGH and retesting is from 1 to 3 months,2 although there are no existing data on the optimal shortest interval without rhGH for retesting reliability and health parameters. There is some evidence, however, that the longer interval and delay in reinstituting rhGH may have an impact on optimizing skeletal outcomes in these subjects,25 as well as other parameters, such as cardiovascular and lipid profiles.26 Moreover, a longer period off rhGH during reevaluation may increase the risk of losing patients to follow-up.27
GH stimulation tests are used to reevaluate GH levels taking into account the appropriate cutoff limits with various assay measurements.28,29 The insulin tolerance test (ITT) is considered the gold standard to reevaluate secretion of GH in these patients. Assuming adequate hypoglycemia is achieved, this test can distinguish GH deficiency from the sufficient GH secretion that is linked with obesity and normal aging. This test, however, is contraindicated in patients with epilepsy or heart disease. Given this precaution, the only alternative reliable stimulation tests that can be used are glucagon/growth hormone-releasing hormone (GHRH)+arginine, taking into account appropriate cutoff limits.2
Cutoff levels for peak GH responses to stimulation tests during the transition period are still arbitrary; studies use a peak GH cutoff <5.1 μg/L,23 <6.1 μg/L30 or <5.6 μg/L.31 The European Society for Pediatric Endocrinology recommended a cutoff <5 μg/L, using the ITT as an appropriate criterion for GH replacement during transition.2 In CO-GHD where persistent GHD is highly likely, confirmation of persistent GHD can be made by a low IGF-I level alone (<−2 SD for age and gender) measured after at least 1 month off rhGH therapy, without the need for any further stimulation testing.5 Figure 1 illustrates the current proposal workup of diagnosing and reevaluating CO-GHD patients during transition,2 highlighting the areas of uncertainty.
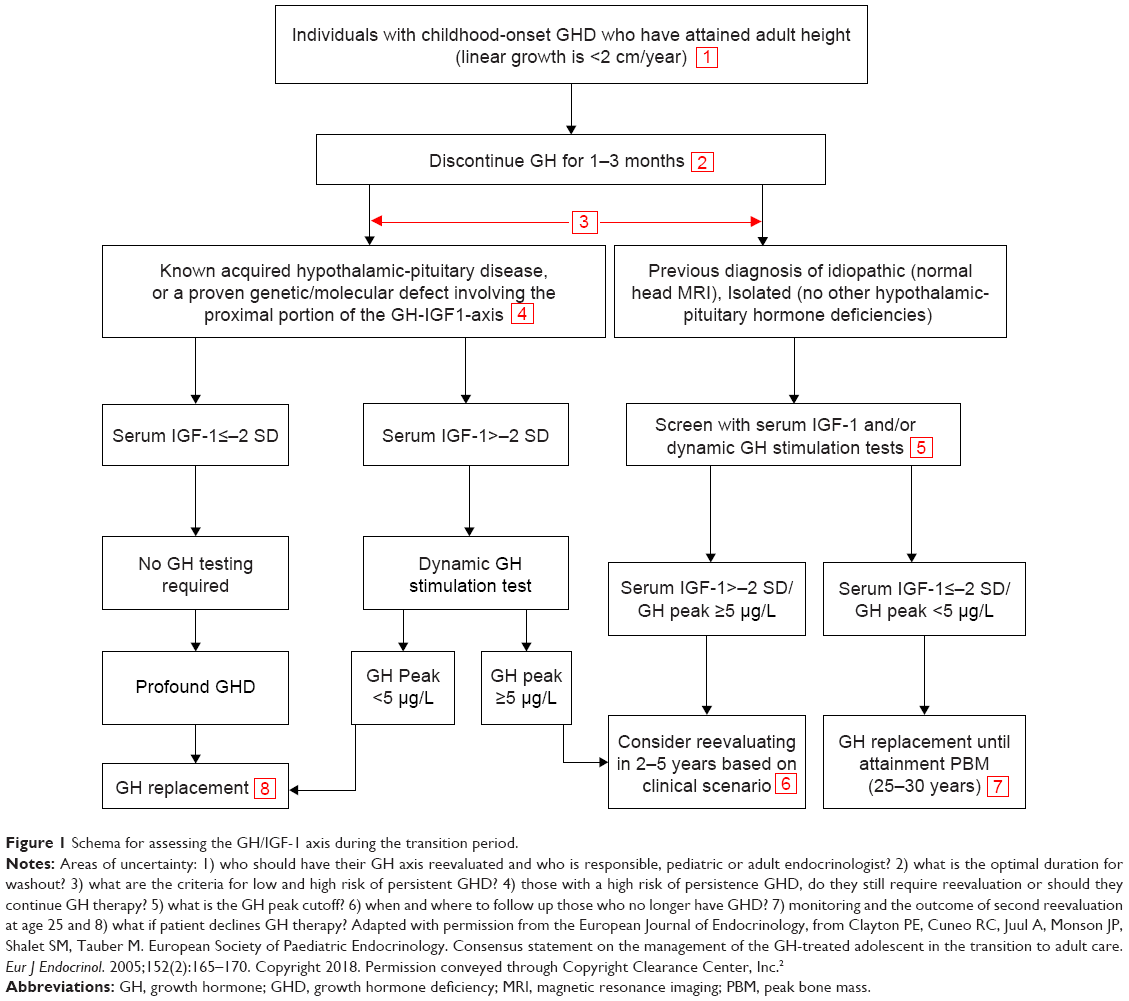 | Figure 1 Schema for assessing the GH/IGF-1 axis during the transition period. |
It is currently recommended to restart rhGH at a low dose (0.2–0.3 mg/day) and then titrate to attain IGF-1 levels that fall into the upper half of the normal range. The dose of rhGH is then adjusted during the transition to adult treatment, with young females receiving oral estrogen requiring higher rhGH doses. IGF-1 should be measured during dose adjustment at 1–2-month intervals and also a minimum of once a year during therapy in order for it to be kept at an age- and sex-appropriate level.2
Benefits of rhGH during transition
The broader benefits of rhGH for optimizing other health aspects during childhood and the period of the transition to adulthood are becoming more widely recognized (Figure 2).32,33
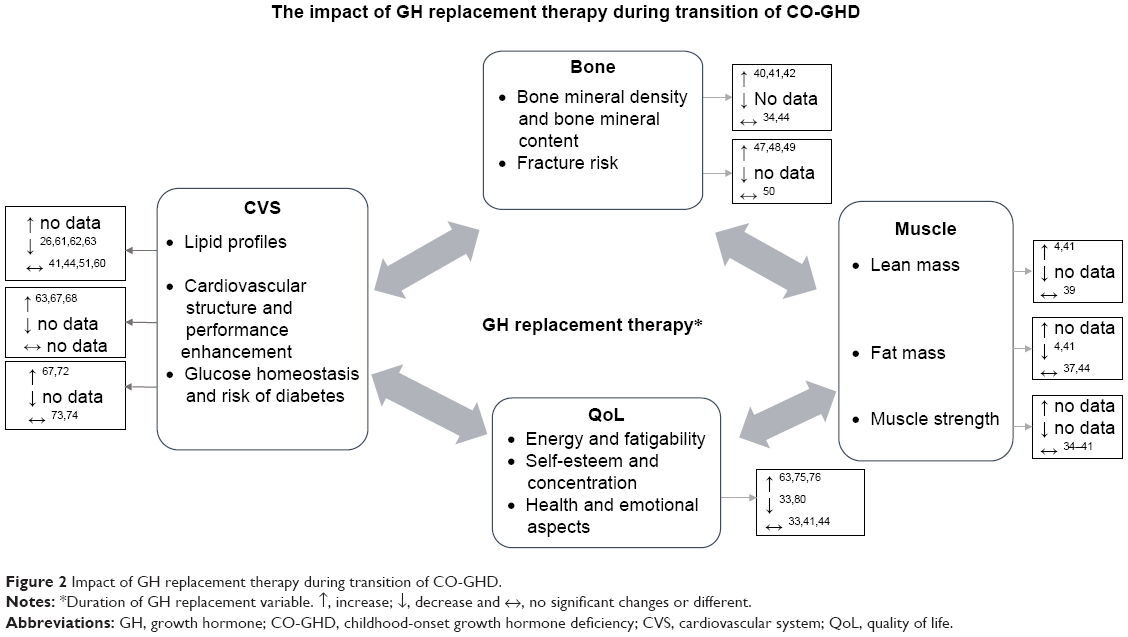 | Figure 2 Impact of GH replacement therapy during transition of CO-GHD. |
Bone and fracture risk
There is currently conflicting evidence on whether CO-GHD contributes to low bone mass and an increased risk of fracture in adulthood,34–36 although mechanisms and the pathophysiology of bone mass reduction are still far from fully understood. Indeed, many factors such as gender, height, age at onset, body composition, gonadal status, GHD severity and assessment methods may all contribute to inconsistent data on bone mass in patients with CO-GHD. Studies suggest that CO-GHD results in developmental bone mass deficits both at the time of diagnosis37,38 and at retesting when final height has been reached.39 Reinstitution of rhGH replacement beyond the attainment of final height in young adults who have persistent CO-GHD has been reported to have a positive effect on bone density with a net benefit change in bone mineral density (BMD) of lumber spine ranging from 3% to 6% after 1 year35 or 2 years.4,40–42 On the other hand, some studies did not find any change in BMD up to 2 years after either discontinuation of rhGH43 or after reinstituting rhGH replacement therapy after final height has been reached.34,44 In addition, there are scarce data on the link between bone density and fracture risk in adolescents and young adults with CO-GHD that were observed in adults with GHD and hypopituitarism.45,46 Previous studies have reported increased fracture risk in adults with CO-GHD47,48 and only in women with CO-GHD in another study,49 while another study did not find any difference in the risk of fracture in adolescents with CO-GHD compared with that in the normal population.50 Therefore, there is no clear evidence that the CO-GHD results in low bone mass or increases the risk of fracture.
Body composition and muscle strength
During transition after withdrawal of childhood rhGH replacement therapy, several but not all studies have showed a significant decrease in lean mass (LM; −8%) parallel by increased fat mass (FM; 10%–17%) in patients with CO-GHD who were diagnosed with persistent GHD and had stopped rhGH for at least 2 years.4,34,51,52 Restarting rhGH therapy resulted in a notable improvement in body composition, with LM increasing by 14% and FM being reduced by 7% over 2 years of therapy.4,41 In terms of muscle strength, previous studies have reported a lower maximum isotonic strength in young adults with CO-GHD, as measured by hand grip force,53,54 but the effects on muscle strength in adolescents with CO-GHD during transition still remain to be completely elucidated.55 There are some suggestions that reduced muscle mass was the cause of diminished muscle strength in GHD, as opposed to a reduction in contractile function.56 Furthermore, it has been proposed that CO-GHD may initially result in a reduction in muscle mass and force, which would ultimately have an impact on bone density and geometry.57 However, little research has been carried out into the relationship between muscle and bone strength in adult GHD patients,58 and there has been no existing study targeting CO-GHD in children and adolescents.
Cardiovascular risks and glucose homeostasis
It has been reported in some studies that adolescents with CO-GHD have adverse lipid profiles after they discontinue rhGH treatment upon reaching final height,26,51,52,59 but not all studies.41,44,51,60 These parameters have been found to improve after restarting rhGH replacement therapy.26,61–63 In addition, it has been noted that the longer the rhGH therapy is discontinued, the more abnormal the lipid profiles.64 It is possible that the alterations in lipid profiles observed when rhGH is discontinued may be partly explained by the short-term effects of rhGH therapy and therefore could not be analyzed independently of treatment duration during childhood.65 Studies of adults with GHD provided evidence for this assumption, revealing that only long periods of rhGH therapy (in the range of 5–10 years) were associated with improved lipid profiles in adult GHD patients.66,67 Further research is required to confirm this hypothesis in CO-GHD. In addition to aberrations in lipid profiles, direct impacts of CO-GHD on cardiac structure and function were reported in some echocardiographic studies. A study observed a decrease in all cardiac dimensions of adolescents with CO-GHD after withdrawal of rhGH during the transition period, when reinstituting rhGH results in a significant increase in left ventricular (LV) mass index and exercises capacity after 12–24 months63,68 with improvement in endothelial function within the first 6 months of restarting rhGH.69
Regarding glucose homeostasis parameters, children with CO-GHD have been reported to be more insulin sensitive at the time of initial diagnosis70 and after rhGH is withdrawn when they reach final height.51,71 However, adults with GHD have been found to have an increased likelihood of insulin resistance,67,72 but this is not the case for adolescents.63 It is not clear whether changes in glucose homeostasis in these subjects can be attributed to GHD itself, body composition and adiposity or both, with no clear evidence for an increased risk of developing diabetes.73,74
Quality of life (QoL)
GH/IGF-1 axis may play a role in normal brain structure, cognitive function, psychology and thereby QoL.75,76 A study of children with isolated-GHD revealed a significant reduction in white matter, corpus callosum and neural volumes.77 Given these findings, several clinical studies have been carried out to evaluate the effects of discontinuing and subsequently resuming rhGH treatment with regard to QoL elements in young adults with CO-GHD during transition from childhood to adulthood.
Some studies reported no changes in QoL in patients with CO-GHD, when measured at the time they discontinued rhGH after reaching final height and after 2 years remeasured while either off or resuming rhGH therapy.33,41,44 Another study further reported no impairment in QoL in short-stature adolescents and children with GHD and without GHD,78 which indicates that other unknown confounders could influence QoL other than height and GH levels.79 On the other hand, other studies have stated that adolescent CO-GHD patients who had been treated with rhGH and then discontinued therapy after attaining height suffered from some cognitive and psychological impairment, particularly in domains of attention, memory, energy drive, emotional reactions and social isolation.80,81 These measures significantly improved when rhGH treatment was restarted. An inverse relationship has been reported between QoL and rhGH therapy duration; a longer period without rhGH was associated with a poorer QoL, whereas restarting rhGH treatment had a significantly positive effect on health-related QoL aspects.64 There is also some evidence that discontinuing rhGH treatment resulted in decreased QoL within 6 months, which was improved in 3–6 months after recommencing rhGH therapy.81,82 Although individuals with GHD treated with rhGH reported having a higher self-esteem compared with short-stature and normal-stature children,83,84 the replacement of rhGH in adult CO-GHD patients resulted in less QoL improvement than in those with adult-onset GHD,33 even after undergoing rhGH therapy for long-term periods of 4–10 years.85
Challenges for future
The need for an appropriate transition service is increasingly recognized within health care systems. Generally, the purpose of transition is to offer adolescent patients continuity of care and promote mental and physical health through the exchange of information among health care workers in order to optimize their health and QoL aspects.11 Although the existing guidelines have theoretically described how to reevaluate CO-GHD during transition, there is no clear pathway about how to organize and deliver transitional care from pediatric to adult services. Much of the transition literature has focused on the risk of untreated GHD during transition in terms of health and QoL outcomes; however, none of the studies have identified a clear transition plan, before and after reevaluation follow-up. Several studies have highlighted the risks of adolescents who drop out specialist endocrine care during the transition to adulthood care. The Scottish audit demonstrated 21% of patients stopped rhGH replacement without comprehensive reevaluation and 18% of patients did not attend follow-up while on treatment and never returned to endocrine clinics. This study has also highlighted issues about the follow-up of patients who no longer have GHD and particularly those patients with persistent GHD who have chosen not to resume adult rhGH treatment.16 In another endocrine setting, transitional care was evaluated in patients with congenital adrenal hyperplasia reporting that 50% of the whole cohort were lost to follow-up following transfer to adult care,86 with only 10% of the expected numbers still attending the adult endocrine service in another study.87
The current transition of endocrine conditions including GHD has been reported to be of suboptimal quality in young adults with endocrine conditions.88,89 Such studies provide an insight into actual practice barriers and potential solutions for successful transition. The readiness of the patients and their caregivers, appropriate environmental setting and local resources were described as factors, which interfere with the precise format of the service and successful transition. There are various expert-based recommendations on how to improve transition and provide quality services for other chronic endocrine diseases, for example, type 1 diabetes mellitus,90 turner syndrome,91 and hypogonadism92. Other reports suggest some strategies for successful transition including standardized referral systems and establishment of transition centers to ensure that the needs of young adults with CO-GHD are being met.93,94 In addition, for optimizing transitional care, it was recommended that pediatric and adult endocrinologists collaborate and share information while understanding that some overlap and a joint transition service would optimize the follow-up and transfer of adolescents,94 although there are lack of data about patient attendance and follow-up after transition. The existing theoretical generic models have demonstrated structured processes of transitioning adolescents from pediatric to adult care.95,96 However, there appears to be clear difficulties in the feasibility of implementation of the all processes at every center and/or for every individual. Using the model designed by Gleeson et al, application of this model faces several challenges for effective transition of adolescents with CO-GHD as outlined in Table 1. In addition to the above highlighted areas of uncertainty in the schema of reevaluation and management of adolescents with CO-GHD, the model does not address issues concerning the readiness of patients and their caregiver to transition, transition clinic visits and frequency of appointments. The arrangement and duration to get the first adult care appointment, together with aspects to ensure the continuity of care, are also important factors. Therefore, there is a growing need to further review the current practices and develop more specific models for better planning and promoting the continuity of care during and after transition.
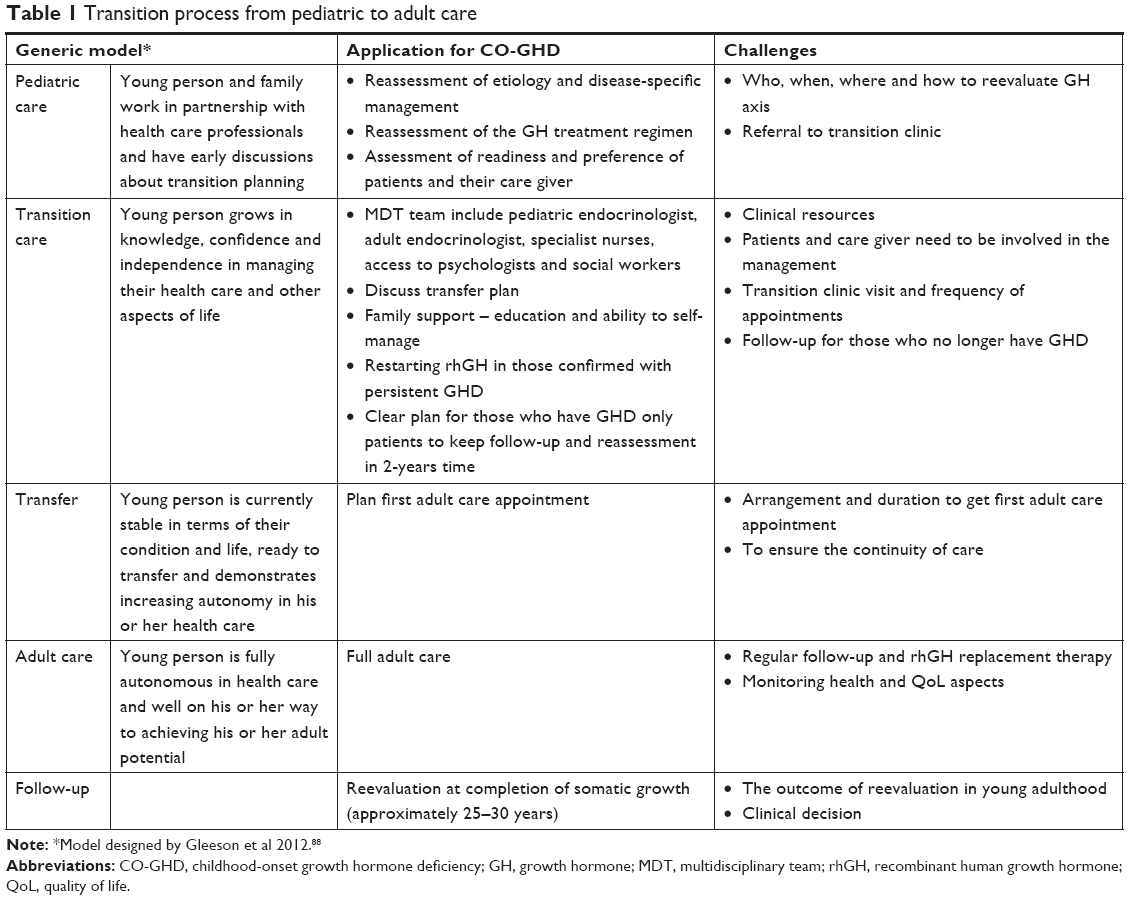 | Table 1 Transition process from pediatric to adult care |
Conclusion
Transition is a concept of a multidisciplinary approach that ensures continuity of health care while adolescents with complex health conditions are transferred from pediatric care to adult care. There is some evidence that recommencing rhGH treatment in CO-GHD during transition results in not only improved growth and bone health but also a better prognosis for metabolic and cardiovascular risks in the long term. Further studies are required on the current practice of assessing and managing CO-GHD, as well as research into the long-term follow-up of patients who have been confirmed to have persistent CO-GHD and continuous monitoring for optimizing somatic and psychological outcomes in patients. The major challenge during this stage, however, is appropriately identifying candidates for adult GH therapy. The current guidelines need to be reviewed, addressing areas of uncertainly as evidenced by variation in clinical practice. Hence, clearly a structured transition protocol is a vital key to establish the best practice of transitioning adolescents with CO-GHD.
Disclosure
MA was supported by the Government of Libya. SFA has received speaker fees from Novo Nordisk and Sandoz and consultancy fees from Pfizer. MGS has received speaker fees from Pfizer and educational grants from Novo Nordisk, Pfizer and Sandoz. The authors report no other conflicts of interest in this work.
References
Willis ER, Mcdonagh JE. Transition from children’s to adults’ services for young people using health or social care services (NICE Guideline NG43). Arch Dis Child Educ Pract Ed. Epub 2017, Dec 21. | ||
Clayton PE, Cuneo RC, Juul A, Monson JP, Shalet SM, Tauber M. European Society of Paediatric Endocrinology. Consensus statement on the management of the GH-treated adolescent in the transition to adult care. Eur J Endocrinol. 2005;152(2):165–170. | ||
Rizzoli R, Bianchi ML, Garabédian M, Mckay HA, Moreno LA. Maximizing bone mineral mass gain during growth for the prevention of fractures in the adolescents and the elderly. Bone. 2010;46(2):294–305. | ||
Attanasio AF, Shavrikova E, Blum WF, et al. Continued growth hormone (GH) treatment after final height is necessary to complete somatic development in childhood-onset GH-deficient patients. J Clin Endocrinol Metab. 2004;89(10):4857–4862. | ||
Molitch ME. Growth hormone treatment in adults with growth hormone deficiency: the transition. J Endocrinol Invest. 2011;34(2):150–154. | ||
Chamberlain Shea H, Levy RA. Transition care of patients with growth hormone deficiency from pediatric endocrinologists to adult endocrinologists. Endocr Pract. 2012;18(2):256–268. | ||
Nguyen VT, Misra M. Transitioning of children with GH deficiency to adult dosing: changes in body composition. Pituitary. 2009;12(2):125–135. | ||
Gleeson H. Transition from Puberty to Adulthood. In: Ho K, editor. Growth Hormone Related Diseases and Therapy. Contemporary Endocrinology. New York: Humana Press; 2011:187–210. | ||
Attanasio AF, Shalet SM. Growth hormone and the transition from puberty into adulthood. Endocrinol Metab Clin North Am. 2007;36(1):187–201. | ||
Ahmid M, Perry CG, Ahmed SF, Shaikh MG. Growth hormone deficiency during young adulthood and the benefits of growth hormone replacement. Endocr Connect. 2016;5(3):R1–R11. | ||
Gleeson H, Turner G. Transition to adult services. Arch Dis Child Educ Pract Ed. 2012;97(3):86–92. | ||
Radovick S, Divall S. Approach to the growth hormone-deficient child during transition to adulthood. J Clin Endocrinol Metab. 2007;92(4):1195–1200. | ||
Ho KK. Consensus guidelines for the diagnosis and treatment of adults with GH deficiency II: a statement of the GH Research Society in association with the European Society for Pediatric Endocrinology, Lawson Wilkins Society, European Society of Endocrinology, Japan Endocrine Society, and Endocrine Society of Australia. Eur J Endocrinol. 2007;157(6):695–700. | ||
Cook DM, Rose SR. A review of guidelines for use of growth hormone in pediatric and transition patients. Pituitary. 2012;15(3):301–310. | ||
Grimberg A, Allen DB. Growth hormone treatment for growth hormone deficiency and idiopathic short stature: new guidelines shaped by the presence and absence of evidence. Curr Opin Pediatr. 2017;29(4):466–471. | ||
Ahmid M, Fisher V, Graveling AJ, et al. An audit of the management of childhood-onset growth hormone deficiency during young adulthood in Scotland. Int J Pediatr Endocrinol. 2016;2016:6. | ||
Murray PG, Dattani MT, Clayton PE. Controversies in the diagnosis and management of growth hormone deficiency in childhood and adolescence. Arch Dis Child. 2016;101(1):96–100. | ||
Chinoy A, Murray PG. Diagnosis of growth hormone deficiency in the paediatric and transitional age. Best Pract Res Clin Endocrinol Metab. 2016;30(6):737–747. | ||
Juul A, Holm K, Kastrup KW, et al. Free insulin-like growth factor I serum levels in 1430 healthy children and adults, and its diagnostic value in patients suspected of growth hormone deficiency. J Clin Endocrinol Metab. 1997;82(8):2497–2502. | ||
Hartman ML, Crowe BJ, Biller BM, Ho KK, Clemmons DR, Chipman JJ; HyposCCS Advisory Board; U.S. HypoCCS Study Group. Which patients do not require a GH stimulation test for the diagnosis of adult GH deficiency? J Clin Endocrinol Metab. 2002;87(2):477–485. | ||
Tillmann V, Tang VW, Price DA, Hughes DG, Wright NB, Clayton PE. Magnetic resonance imaging of the hypothalamic-pituitary axis in the diagnosis of growth hormone deficiency. J Pediatr Endocrinol Metab. 2000;13(9):1577–1583. | ||
Kalina MA, Kalina-Faska B, Gruszczyńska K, Baron J, Małecka-Tendera E. Usefulness of magnetic resonance findings of the hypothalamic-pituitary region in the management of short children with growth hormone deficiency: evidence from a longitudinal study. Childs Nerv Syst. 2012;28(1):121–127. | ||
Bonfig W, Bechtold S, Bachmann S, et al. Reassessment of the optimal growth hormone cut-off level in insulin tolerance testing for growth hormone secretion in patients with childhood-onset growth hormone deficiency during transition to adulthood. J Pediatr Endocrinol Metab. 2008;21(11):1049–1056. | ||
Quigley CA, Zagar AJ, Liu CC, et al. United States multicenter study of factors predicting the persistence of GH deficiency during the transition period between childhood and adulthood. Int J Pediatr Endocrinol. 2013;2013(1):6. | ||
Tritos NA, Hamrahian AH, King D, et al. A longer interval without GH replacement and female gender are associated with lower bone mineral density in adults with childhood-onset GH deficiency: a KIMS database analysis. Eur J Endocrinol. 2012;167(3):343–351. | ||
Lanes R, Paoli M, Carrillo E, Villaroel O, Palacios A. Cardiovascular risk of young growth-hormone-deficient adolescents. Differences in growth-hormone-treated and untreated patients. Horm Res. 2003;60(6):291–296. | ||
Courtillot C, Baudoin R, Du Souich T, et al. Monocentric study of 112 consecutive patients with childhood onset GH deficiency around and after transition. Eur J Endocrinol. 2013;169(5):587–596. | ||
de Boer H, van der Veen EA. Why retest young adults with childhood-onset growth hormone deficiency? J Clin Endocrinol Metab. 1997;82(7):2032–2036. | ||
Gasco V, Corneli G, Beccuti G, et al. Retesting the childhood-onset GH-deficient patient. Eur J Endocrinol. 2008;159 Suppl 1(1):S45–S52. | ||
Maghnie M, Aimaretti G, Bellone S, et al. Diagnosis of GH deficiency in the transition period: accuracy of insulin tolerance test and insulin-like growth factor-I measurement. Eur J Endocrinol. 2005;152(4):589–596. | ||
Secco A, di Iorgi N, Napoli F, et al. Reassessment of the growth hormone status in young adults with childhood-onset growth hormone deficiency: reappraisal of insulin tolerance testing. J Clin Endocrinol Metab. 2009;94(11):4195–4204. | ||
Clayton P, Gleeson H, Monson J, Popovic V, Shalet SM, Christiansen JS. Growth hormone replacement throughout life: insights into age-related responses to treatment. Growth Horm IGF Res. 2007;17(5):369–382. | ||
Attanasio AF, Shavrikova EP, Blum WF, Shalet SM. Quality of life in childhood onset growth hormone-deficient patients in the transition phase from childhood to adulthood. J Clin Endocrinol Metab. 2005;90(8):4525–4529. | ||
Boot AM, van der Sluis IM, Krenning EP, de Muinck Keizer-Schrama SM. Bone mineral density and body composition in adolescents with childhood-onset growth hormone deficiency. Horm Res. 2009;71(6):364–371. | ||
Drake WM, Carroll PV, Maher KT, et al. The effect of cessation of growth hormone (GH) therapy on bone mineral accretion in GH-deficient adolescents at the completion of linear growth. J Clin Endocrinol Metab. 2003;88(4):1658–1663. | ||
Attanasio AF, Howell S, Bates PC, et al. Body composition, IGF-I and IGFBP-3 concentrations as outcome measures in severely GH-deficient (GHD) patients after childhood GH treatment: a comparison with adult onset GHD patients. J Clin Endocrinol Metab. 2002;87(7):3368–3372. | ||
Boot AM, Engels MA, Boerma GJ, Krenning EP, de Muinck Keizer-Schrama SM, Keizerschrama S. Changes in bone mineral density, body composition, and lipid metabolism during growth hormone (GH) treatment in children with GH deficiency. J Clin Endocrinol Metab. 1997;82(8):2423–2428. | ||
Schweizer R, Martin DD, Haase M, et al. Similar effects of long-term exogenous growth hormone (GH) on bone and muscle parameters: a pQCT study of GH-deficient and small-for-gestational-age (SGA) children. Bone. 2007;41(5):875–881. | ||
Aimaretti G, Corneli G, Rovere S, Croce CG, Ghigo E, Procopio M. Is GH therapy useful to preserve bone mass in transition-phase patients with GH deficiency? J Endocrinol Invest. 2005;28(10 Suppl):28–32. | ||
Conway GS, Szarras-Czapnik M, Racz K, et al. Treatment for 24 months with recombinant human GH has a beneficial effect on bone mineral density in young adults with childhood-onset GH deficiency. Eur J Endocrinol. 2009;160(6):899–907. | ||
Underwood LE, Attie KM, Baptista J; Genentech Collaborative Study Group. Growth hormone (GH) dose-response in young adults with childhood-onset GH deficiency: a two-year, multicenter, multiple-dose, placebo-controlled study. J Clin Endocrinol Metab. 2003;88(11):5273–5280. | ||
Shalet SM, Shavrikova E, Cromer M, et al. Effect of growth hormone (GH) treatment on bone in postpubertal GH-deficient patients: a 2-year randomized, controlled, dose-ranging study. J Clin Endocrinol Metab. 2003;88(9):4124–4129. | ||
Fors H, Bjarnason R, Wirén L, et al. Currently used growth-promoting treatment of children results in normal bone mass and density. A prospective trial of discontinuing growth hormone treatment in adolescents. Clin Endocrinol. 2001;55(5):617–624. | ||
Mauras N, Pescovitz OH, Allada V, Messig M, Wajnrajch MP, Lippe B; Transition Study Group. Limited efficacy of growth hormone (GH) during transition of GH-deficient patients from adolescence to adulthood: a phase III multicenter, double-blind, randomized two-year trial. J Clin Endocrinol Metab. 2005;90(7):3946–3955. | ||
Wüster C, Abs R, Bengtsson BA, et al; KIMS Study Group and the KIMS International Board. Pharmacia & Upjohn International Metabolic Database. The influence of growth hormone deficiency, growth hormone replacement therapy, and other aspects of hypopituitarism on fracture rate and bone mineral density. J Bone Miner Res. 2001;16(2):398–405. | ||
Rosen T, Wilhelmsen L, Landin-Wilhelmsen K, Lappas G, Bengtsson B. Increased fracture frequency in adult patients with hypopituitarism and GH deficiency. Eur J Endocrinol. 1997;137(3):240–245. | ||
Bouillon R, Koledova E, Bezlepkina O, et al. Bone status and fracture prevalence in Russian adults with childhood-onset growth hormone deficiency. J Clin Endocrinol Metab. 2004;89(10):4993–4998. | ||
Abs R, Mattsson AF, Bengtsson BA, et al; KIMS Study Group. Isolated growth hormone (GH) deficiency in adult patients: baseline clinical characteristics and responses to GH replacement in comparison with hypopituitary patients. A sub-analysis of the KIMS database. Growth Horm IGF Res. 2005;15(5):349–359. | ||
Holmer H, Svensson J, Rylander L, et al. Fracture incidence in GH-deficient patients on complete hormone replacement including GH. J Bone Miner Res. 2007;22(12):1842–1850. | ||
Baroncelli GI, Bertelloni S, Sodini F, Saggese G. Lumbar bone mineral density at final height and prevalence of fractures in treated children with GH deficiency. J Clin Endocrinol Metab. 2002;87(8):3624–3631. | ||
Carroll PV, Drake WM, Maher KT, et al. Comparison of continuation or cessation of growth hormone (GH) therapy on body composition and metabolic status in adolescents with severe GH deficiency at completion of linear growth. J Clin Endocrinol Metab. 2004;89(8):3890–3895. | ||
Bechtold S, Bachmann S, Putzker S, Dalla Pozza R, Schwarz HP. Early changes in body composition after cessation of growth hormone therapy in childhood-onset growth hormone deficiency. J Clin Densitom. 2011;14(4):471–477. | ||
Sartorio A, Narici M, Conti A, Monzani M, Faglia G. Quadriceps and hand-grip strength in adults with childhood-onset growth hormone deficiency. Eur J Endocrinol. 1995;132(1):37–41. | ||
Sartorio A, Agosti F, de Col A, et al. Muscle Strength and Power, Maximum Oxygen Consumption, and Body Composition in Middle-Aged Short-stature Adults with Childhood-onset Growth Hormone Deficiency. Arch Med Res. 2008;39(1):78–83. | ||
Modesto MJ, Amer NM, Erichsen O, et al. Muscle strength and body composition during the transition phase in patients treated with recombinant GH to final height. J Pediatr Endocrinol Metab. 2014;27(9–10):813–820. | ||
Chikani V, Ho KKY, Kk H. Action of GH on skeletal muscle function: molecular and metabolic mechanisms. J Mol Endocrinol. 2014;52(1):R107–R123. | ||
Högler W, Shaw N. Childhood growth hormone deficiency, bone density, structures and fractures: scrutinizing the evidence. Clin Endocrinol. 2010;72(3):281–289. | ||
Klefter O, Feldt-Rasmussen U. Is increase in bone mineral content caused by increase in skeletal muscle mass/strength in adult patients with GH-treated GH deficiency? A systematic literature analysis. Eur J Endocrinol. 2009;161(2):213–221. | ||
Capalbo D, Esposito A, di Mase R, et al. Update on early cardiovascular and metabolic risk factors in children and adolescents affected with growth hormone deficiency. Minerva Endocrinol. 2012;37(4):379–389. | ||
Attanasio AF, Howell S, Bates PC, et al. Confirmation of severe GH deficiency after final height in patients diagnosed as GH deficient during childhood. Clin Endocrinol. 2002;56(4):503–507. | ||
Meazza C, Elsedfy HH, Pagani S, Bozzola E, El Kholy M, Bozzola M. Metabolic parameters and adipokine profile in growth hormone deficient (GHD) children before and after 12-month GH treatment. Horm Metab Res. 2014;46(3):219–223. | ||
Capalbo D, Mattace Raso G, Esposito A, et al. Cluster of cardiometabolic risk factors in children with GH deficiency: a prospective, case-control study. Clin Endocrinol. 2014;80(6):856–862. | ||
Colao A, di Somma C, Pivonello R, et al. The cardiovascular risk of adult GH deficiency (GHD) improved after GH replacement and worsened in untreated GHD: a 12-month prospective study. J Clin Endocrinol Metab. 2002;87(3):1088–1093. | ||
Kołtowska-Häggström M, Geffner ME, Jönsson P, et al. Discontinuation of growth hormone (GH) treatment during the transition phase is an important factor determining the phenotype of young adults with nonidiopathic childhood-onset GH deficiency. J Clin Endocrinol Metab. 2010;95(6):2646–2654. | ||
Prodam F, Savastio S, Genoni G, et al. Effects of growth hormone (GH) therapy withdrawal on glucose metabolism in not confirmed GH deficient adolescents at final height. PLoS One. 2014;9(1):e87157. | ||
Maison P, Griffin S, Nicoue-Beglah M, Haddad N, Balkau B, Chanson P. Metaanalysis of Blinded, Randomized, Placebo-Controlled Trials. Impact of growth hormone (GH) treatment on cardiovascular risk factors in GH-deficient adults: a Metaanalysis of Blinded, Randomized, Placebo-Controlled Trials. J Clin Endocrinol Metab. 2004;89(5):2192–2199. | ||
Gazzaruso C, Gola M, Karamouzis I, Giubbini R, Giustina A. Cardiovascular risk in adult patients with growth hormone (GH) deficiency and following substitution with GH – an update. J Clin Endocrinol Metab. 2014;99(1):18–29. | ||
Feinberg MS, Scheinowitz M, Laron Z. Cardiac dimension and function in patients with childhood onset growth hormone deficiency, before and after growth hormone retreatment in adult age. Am Heart J. 2003;145(3):549–553. | ||
Amato G, Carella C, Fazio S, et al. Body composition, bone metabolism, and heart structure and function in growth hormone (GH)-deficient adults before and after GH replacement therapy at low doses. J Clin Endocrinol Metab. 1993;77:1671–1676. | ||
Husbands S, Ong KK, Gilbert J, Wass JA, Dunger DB. Increased insulin sensitivity in young, growth hormone deficient children. Clin Endocrinol. 2001;55(1):87–92. | ||
Nørrelund H, Vahl N, Juul A, et al. Continuation of growth hormone (GH) therapy in GH-deficient patients during transition from childhood to adulthood: impact on insulin sensitivity and substrate metabolism. J Clin Endocrinol Metab. 2000;85(5):1912–1917. | ||
Giavoli C, Porretti S, Ronchi CL, et al. Long-term monitoring of insulin sensitivity in growth hormone-deficient adults on substitutive recombinant human growth hormone therapy. Metabolism. 2004;53(6):740–743. | ||
Child CJ, Zimmermann AG, Scott RS, Cutler GB, Battelino T, Blum WF; GeNeSIS International Advisory Board. Prevalence and incidence of diabetes mellitus in GH-treated children and adolescents: analysis from the GeNeSIS observational research program. J Clin Endocrinol Metab. 2011;96(6):E1025–E1034. | ||
Cutfield WS, Wilton P, Bennmarker H, et al. Incidence of diabetes mellitus and impaired glucose tolerance in children and adolescents receiving growth-hormone treatment. Lancet. 2000;355(9204):610–613. | ||
Nyberg F, Hallberg M. Growth hormone and cognitive function. Nat Rev Endocrinol. 2013;9(6):357–365. | ||
Schneider-Rivas S, Rivas-Arancibia S, Vázquez-Pereyra F, Vázquez-Sandoval R, Borgonio-Pérez G. Modulation of long-term memory and extinction responses induced by growth hormone (GH) and growth hormone releasing hormone (GHRH) in rats. Life Sci. 1995;56(22):PL433–PL441. | ||
Webb EA, O’Reilly MA, Clayden JD, et al. Effect of growth hormone deficiency on brain structure, motor function and cognition. Brain. 2012;135(1):216–227. | ||
Bullinger M, Kołtowska-Häggström M, Sandberg D, et al. Health-related quality of life of children and adolescents with growth hormone deficiency or idiopathic short stature – part 2: available results and future directions. Horm Res. 2009;72(2):74–81. | ||
Eiser C, Morse R. Quality-of-life measures in chronic diseases of childhood. Health Technol Assess. 2001;5(4):1–157. | ||
Wirén L, Johannsson G, Bengtsson BA. A prospective investigation of quality of life and psychological well-being after the discontinuation of GH treatment in adolescent patients who had GH deficiency during childhood. J Clin Endocrinol Metab. 2001;86(8):3494–3498. | ||
Stouthart PJ, Deijen JB, Roffel M, Delemarre-van de Waal HA. Quality of life of growth hormone (GH) deficient young adults during discontinuation and restart of GH therapy. Psychoneuroendocrinology. 2003;28(5):612–626. | ||
van Nieuwpoort IC, Drent ML. Cognition in the adult with childhood-onset GH deficiency. Eur J Endocrinol. 2008;159(Suppl 1):S53–S57. | ||
Geisler A, Lass N, Reinsch N, et al. Quality of life in children and adolescents with growth hormone deficiency: association with growth hormone treatment. Horm Res Paediatr. 2012;78(2):94–99. | ||
Chaplin JE, Kriström B, Jonsson B, et al. Improvements in behaviour and self-esteem following growth hormone treatment in short prepubertal children. Horm Res Paediatr. 2011;75(4):291–303. | ||
Spielhagen C, Schwahn C, Möller K, et al. The benefit of long-term growth hormone (GH) replacement therapy in hypopituitary adults with GH deficiency: results of the German KIMS database. Growth Horm IGF Res. 2011;21(1):1–10. | ||
Gleeson H, Davis J, Jones J, O’Shea E, Clayton PE. The challenge of delivering endocrine care and successful transition to adult services in adolescents with congenital adrenal hyperplasia: experience in a single centre over 18 years. Clin Endocrinol. 2013;78(1):23–28. | ||
Arlt W, Willis DS, Wild SH, et al; United Kingdom Congenital Adrenal Hyperplasia Adult Study Executive (CaHASE). Health status of adults with congenital adrenal hyperplasia: a cohort study of 203 patients. J Clin Endocrinol Metab. 2010;95(11):5110–5121. | ||
Hokken-Koelega A, van der Lely A-J, Hauffa B, et al. Bridging the gap: metabolic and endocrine care of patients during transition. Endocr Connect. 2016;5(6):R44–R54. | ||
Gleeson H. ‘Part of the problem, part of the solution’ – adult physicians’ role in adolescent and young adult health. Clin Med. 2015;15(5):413–414. | ||
Peters A, Laffel L. American Diabetes Association Transitions Working Group. Diabetes Care. 2011;34(11):2477–2485. | ||
Lucaccioni L, Wong SC, Smyth A, et al. Turner syndrome – issues to consider for transition to adulthood. Br Med Bull. 2015;113(1):45–58. | ||
Dwyer AA, Phan-Hug F, Hauschild M, Elowe-Gruau E, Pitteloud N. Transition in endocrinology: Hypogonadism in adolescence. Eur J Endocrinol. 2015;173(1):R15–R24. | ||
Crowley R, Wolfe I, Lock K, Mckee M. Improving the transition between paediatric and adult healthcare: a systematic review. Arch Dis Child. 2011;96(6):548–553. | ||
Downing J, Gleeson HK, Clayton PE, Davis JR, Wales JK, Callery P. Transition in endocrinology: the challenge of maintaining continuity. Clin Endocrinol. 2013;78(1):29–35. | ||
Gleeson H, Mccartney S, Lidstone V. ‘Everybody’s business’: transition and the role of adult physicians. Clin Med. 2012;12(6):561–567. | ||
Kennedy A, Sawyer S. Transition from pediatric to adult services: are we getting it right? Curr Opin Pediatr. 2008;20(4):403–409. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.