Back to Journals » International Medical Case Reports Journal » Volume 9
Childhood neuroblastoma masquerading as pheochromocytoma: case report
Authors Moon S
Received 13 November 2015
Accepted for publication 7 January 2016
Published 17 March 2016 Volume 2016:9 Pages 65—67
DOI https://doi.org/10.2147/IMCRJ.S100479
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Ronald Prineas
Suk-Bae Moon
Department of Surgery, Kangwon National University Hospital, Kangwon National School of Medicine, Kangwon National University, Chuncheon, South Korea
Abstract: Neuroblastoma is the most common extracranial solid tumor in children. Mild hypertension is a frequent symptom, presumably an effect of catecholamines that tumors release. Reported herein is the rare occurrence of severe hypertension and subsequent heart failure attributable to adrenal gland neuroblastoma. A 3-year-old boy presented with anterior chest wall protrusion. Physical examination revealed severe hypertension, and left-sided cardiac failure was evident by echocardiography. Catecholamine metabolite (norepinephrine) levels were increased in serum (>2,000 pg/mL) and in urine (1,350.5 µg/day). Abdominal computed tomography scan showed a 7 cm solid mass arising from right adrenal gland. Oral phenoxybenzamine was given for hemodynamic stabilization, and right adrenalectomy was performed to remove an apparent pheochromocytoma. Ultimately, the pathologic diagnosis was ganglioneuroblastoma. Both hypertension and cardiac failure resolved postoperatively.
Keywords: hypertension, heart failure, adrenal gland, pheochromocytoma
Introduction
Neuroblastoma (NBL) is the most common extracranial solid tumor in childhood that derives from the neural crest and may vary in clinical symptoms, depending on location, distal spread, and secreted metabolites of tumors.1 Pheochromocytoma is also a neural crest-derived tumor that presents with headache, fever, palpitation, and weight loss, but the most common presentation is sustained hypertension.2 Hypertension secondary to catecholamine release is manifested in ~25% of patients with NBL, but it is generally not severe compared to that in pheochromocytoma.1 Described herein is the case of a 3-year-old boy who suffered severe hypertension and heart failure due to adrenal gland NBL.
Case report
This study was conducted in accord with all the guidelines stipulated by the Declaration of Helsinki. Because no interventional experimentation whatsoever was involved and the study was retrospective, written patient consent and ethics committee approval was not required by the Institutional Review Board of Kangwon National University Hospital.
A 3-year-old boy presented to our facility with anterior chest wall protrusion. His birth record and past medical history were unremarkable. On physical examination, a protruding anterior bony chest wall was obvious, and systolic blood pressure elevation was recorded (160–200 mmHg). In addition to marked cardiomegaly visible on plain chest radiograph, echocardiography revealed left-sided heart failure (ejection fraction, 27%) and mitral valve regurgitation. Laboratory testing disclosed elevated levels of catecholamines and their metabolites (Table 1). Abdominal computed tomography (CT) images revealed a heterogeneously enhanced, solid, and rounded tumor of right adrenal gland, measuring 7×6.5 cm (Figure 1). For what appeared to be pheochromocytoma, the patient was given a 2-week antihypertensive regimen of phenoxybenzamine, nitroprusside, and nitroglycerin. Open adrenalectomy was then performed, free of drastic blood pressure fluctuations during surgery. Postoperative hypotension was controlled by a 24-hour dopamine drip (5 μg/kg/h), tapered thereafter. The final pathologic examination was ganglioneuroblastoma intermixed type (Figure 2).
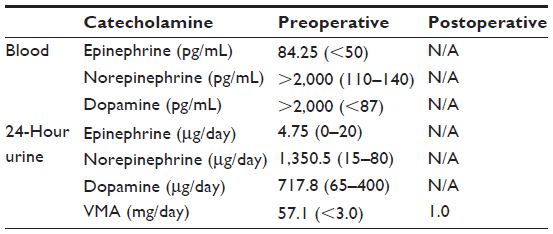 | Table 1 Blood and 24-hour urine catecholamine levels |
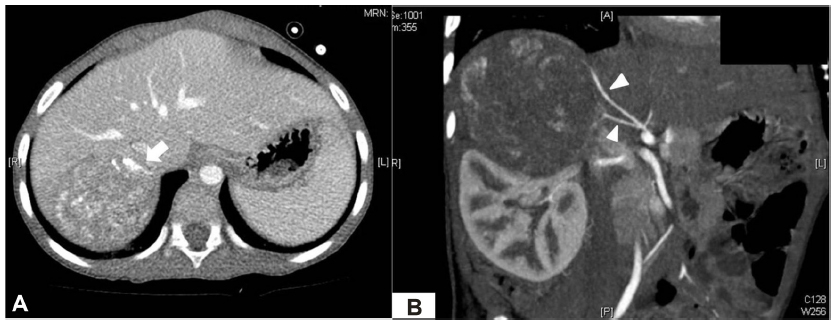 | Figure 1 Preoperative CT scan. |
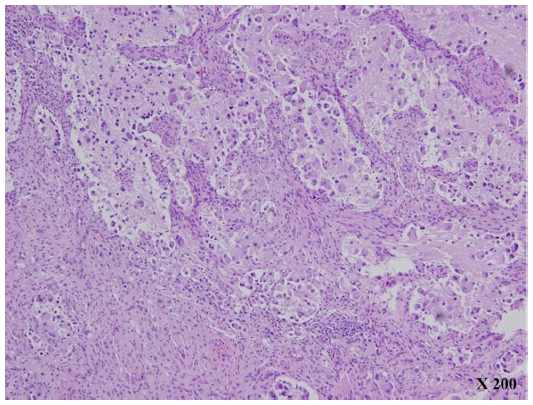 | Figure 2 Histologic features (H&E, ×200). |
This patient’s hospital course was uneventful, with abatement of both hypertension and cardiac failure following surgery. By International Neuroblastoma Staging System (INSS) criteria, a negative postoperative work-up for metastasis, including bone marrow examination, bone scan, and iodine-131-meta-iodobenzylguanidine whole-body scan, indicated stage I disease. His baseline vanillylmandelic acid level of 57.1 mg/day (normal for age, <18 mg/day) declined to 1.0 mg/day at postoperative Month 1. By postoperative Month 8, six cycles of chemotherapy were completed (without signs of NBL recurrence), his chest wall no longer protruded, and the cardiomegaly was resolving.
Discussion
NBL is a neuroendocrine tumor arising in sympathetic nerves. A significant proportion of patients are known to develop mild secondary hypertension from tumor-produced catecholamines.3,4 However, severe hypertension, culminating in heart failure, is rarely reported. In prior accounts, a 3-year-old boy5 and a 5-year-old girl6 who similarly developed severe hypertension due to adrenal tumors have been described. Likewise, these lesions were indistinguishable preoperatively from pheochromocytoma but proved to be NBL after adrenalectomy. Cho et al also reported a 7-year-old boy who experienced cardiac failure during the course of chemotherapy for NBL.7 The rarity of this symptom complex prompts a diagnosis of pheochromocytoma rather than NBL, based on clinical evidence.
A diagnosis of pheochromocytoma typically stems from hallmark features, including catecholamine metabolite elevations in blood and urine, using radiologic tests for localization. Usually, homogeneous enhancement is found on CT or magnetic resonance scans of pheochromocytomas, but heterogeneous enhancement is feasible (thus mimicking NBL) if a tumor necrosis occurs.8 In the absence of tissue confirmation, NBL with excessive catecholamine production and pheochromocytoma may thus be impossible to differentiate by blood and/or radiologic testing.9 Positron emission tomography may be a useful imaging study for pheochromocytoma in the near future, and if specific agent, such as 6-(18F)-fluorodopamine, becomes generally available, positron emission tomography scanning may prove to be the imaging method of choice.2 Our preliminary impression of pheochromocytoma was correctly diagnosed as NBL after surgery.
Activation of the renin–angiotensin system by renal artery compression is another known mechanism of hypertension due to adrenal NBL.10 Although plasma renin or aldosterone determinations were lacking in our patient, renovascular hypertension is excluded by CT evidence (ie, absence of vascular compression) and documented catecholamine elevations in blood and urine.
Use of alpha1-adrenergic blockers, such as phenoxybenzamine, for preoperative hemodynamic stabilization is of paramount importance in treating pheochromocytoma but has not been investigated in NBL-associated hypertension. However, this preoperative strategy did prove successful here, given that alpha1-adrenergic receptors are also implicated in the hemodynamic volatility of NBL. Patients with pheochromocytoma suffering long-term excessive catecholamine exposure often experience transient postoperative hypotension as well, requiring short-term inotropics. Indeed, our patient developed postoperative hypotension that responded to dopamine infusion within 24 hours.
Once completely resected, postoperative chemotherapy is not standard treatment for INSS stage I NBL.11 However, our patient’s atypical clinical manifestations prompted a course of adjuvant chemotherapy. More experience with such patients is truly needed to establish strict postoperative chemotherapeutic guidelines.
In summary, we encountered a child suffering severe hypertension and heart failure due to INSS stage I NBL, thus masquerading as pheochromocytoma. Although preoperative evaluation failed to suggest NBL, the patient was managed appropriately and successfully treated by complete tumor resection. Ultimately, a diagnosis of NBL resulted from postsurgical pathologic examination.
Disclosure
The author reports no conflicts of interest in this work.
References
Rich BS, La Quaglia MP. Neuroblastoma. In: Coran AG, Adzick NS, Krummel TM, et al, editors. Pediatric Surgery. 7th ed. Philadelphia: Elsevier; 2011:441–458. | |
Caty MG, Escobar MA. Adrenal tumors. In: Coran AG, Adzick NS, Krummel TM, et al, editors. Pediatric Surgery. 7th ed. Philadelphia: Elsevier; 2011:557–566. | |
Kedar A, Glassman M, Voorhess ML, et al. Severe hypertension in a child with ganglioneuroblastoma. Cancer. 1981;47(8):2077–2080. | |
Steinmetz JC. Neonatal hypertension and cardiomegaly associated with a congenital neuroblastoma. Pediatr Pathol. 1988;9(5):577–582. | |
Sendo D, Katsuura M, Akiba K, et al. Severe hypertension and cardiac failure associated with neuroblastoma: a case report. J Pediatr Surg. 1996;31(12):1688–1690. | |
Seefelder C, Sparks JW, Chirnomas D, Diller L, Shamberger RC. Perioperative management of a child with severe hypertension from a catecholamine secreting neuroblastoma. Paediatr Anaesth. 2005;15(7):606–610. | |
Cho HS, Kim HM, Park SW, et al. A case of severe hypertensive pulmonary edema associated with neuroblastoma during chemotherapy. J Korean Pediatr Soc. 2000;43(4):573–577. | |
Motta-Ramirez GA, Remer EM, Herts BR, Gill IS, Hamrahian AH. Comparison of CT findings in symptomatic and incidentally discovered pheochromocytomas. AJR Am J Roentgenol. 2005;185(3):684–688. | |
McHugh K. Renal and adrenal tumours in children. Cancer Imaging. 2007;7(1):41–51. | |
Weinblatt ME, Heisel MA, Siegel SE. Hypertension in children with neurogenic tumors. Pediatrics. 1983;71(6):947–951. | |
Kushner BH, Cohn SL, Matthay KK, et al. Treatment of neuroblastoma. In: Cheung NKV, Cohn SL, editors. Neuroblastoma. Berlin: Springer; 2005:123–192. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.