Back to Journals » International Journal of Nanomedicine » Volume 9 » Issue 1
Chemical modulation of M13 bacteriophage and its functional opportunities for nanomedicine
Authors Chung W, Lee D, Yoo SY ![]()
Received 7 September 2014
Accepted for publication 25 October 2014
Published 12 December 2014 Volume 2014:9(1) Pages 5825—5836
DOI https://doi.org/10.2147/IJN.S73883
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 5
Editor who approved publication: Dr Thomas Webster
Woo-Jae Chung,1 Doe-Young Lee,1 So Young Yoo2,3
1College of Biotechnology and Bioengineering, Sungkyunkwan University, Suwon, Republic of Korea; 2BIO-IT Foundry Technology Institute, Pusan National University, Busan, Republic of Korea; 3Research Institute for Convergence of Biomedical Science and Technology, Pusan National University Yangsan Hospital, Yangsan, Republic of Korea
Abstract: M13 bacteriophage (phage) has emerged as an attractive bionanomaterial owing to its genetically tunable surface chemistry and its potential to self-assemble into hierarchical structures. Furthermore, because of its unique nanoscopic structure, phage has been proposed as a model system in soft condensed physics and as a biomimetic building block for structured functional materials. Genetic engineering of phage provides great opportunities to develop novel nanomaterials with functional surface peptide motifs; however, this biological approach is generally limited to peptides containing the 20 natural amino acids. To extend the scope of phage applications, strategies involving chemical modification have been employed to incorporate a wider range of functional groups, including synthetic chemical compounds. In this review, we introduce the design of chemoselective phage functionalization and discuss how such a strategy is combined with genetic engineering for a variety of medical applications, as reported in recent literature.
Keywords: M13 bacteriophage, chemoselective modification, functionalization, biomimetic structure, bionanomaterial
Introduction
Phage display technology has opened up new avenues for utilizing biological materials as a useful sensor system to study biological interactions such as protein–protein, protein–peptide, and protein–DNA interactions, and even chemical reactions.1 Bacteriophages, viruses that infect bacteria, have been used to develop technology that links proteins with the genes that encode them, allowing for selection of antibodies and functional peptides against target antigens. Owing to their unique nanoscopic structures capable of displaying genetically programmable surface peptides, M13 bacteriophage (phage) has drawn attention as a powerful bionanomaterial with the advent of bionanotechnology.2 The unique advantages of phages over conventional bionanomaterials can be easily exploited for genetic information and biomimetic structure, which led to the development of various electronic and medical materials with precise molecular-level control. The biological advantages of phages such as evolution, specific recognition, and self-replication can be enhanced through genetic and chemical engineering.
By mimicking the natural evolutionary process, phage can be used as an information-mining tool to identify protein (or peptide) sequences that specifically recognize target materials at a molecular level.3These recognition elements have been used to design unprecedented materials such as template-synthesized organic–inorganic composite materials4,5 and sensory materials.6,7 The viral particles can self-assemble into various ordered structures with well-defined filamentous shapes, which can lead to novel materials for various functional applications, including energy generation,8–10 biosensors,11–14 semiconductors,4,5 and tissue-regenerating materials.13,15–18 In addition, there is growing interest in M13 phage as a model system for soft condensed physics19 and as a biomimetic building block for structured functional materials.16,20
Although genetic engineering approaches have been widely used to design novel bionanomaterials, there are two main motives for development of methods for chemical functionalization of phage. 1) The functional groups expressed by this genetically programmable bionanomaterial are limited to peptides composed of natural amino acids, which cannot incorporate the vast (bio)chemical diversity of natural or synthetic compounds. While genetic engineering is powerful in tuning every coat protein copy, excessive mutations diminish the packaging, replication, and assembly efficiency of the phage. 2) To expand the use of phage in novel functional applications such as (bio)chemical sensing, bioimaging, and tissue engineering, the chemical functionalization of M13 phage is essential. Incorporation of synthetic functional groups in a site-specific and quantitative manner is a challenging issue in chemical functionalization with further application in biomedical areas.
With the growing expectations for phage as a potential bionanomaterial in the development of next-generation functional materials, the functionalization of phage by combined biological and chemical methods is gaining enormous interest. The M13 phage is the most extensively developed system for peptide display and can be used in a wide range of applications in various fields by site-specific chemical modifications. In this study, we discuss recent study trends in chemical derivatization of M13 phage bionanomaterials and their potential applications in biomedical areas.
M13 phage
The M13 phage is a bacterial-virus composed of a single-stranded DNA encapsulated with major (pVIII) and minor (pIII, pVI, pVII, and pIX) coat proteins. Its long, rodlike shape is ~880 nm in length and 6.6 nm in width (Figure 1A).21,22 The M13 phage infects and grows only in the male strains (displaying F-pili) of Escherichia coli (E. coli). As with other lysogenic phages, M13 infects host cells by injecting its genetic material. The genetic material of the phage usurps the cellular host metabolism in order to replicate itself and its associated proteins. These protein products are transported to the host cell membranes, where new phages are packaged and released through a protein pore channel in the bacterial membrane,21,23,24 without disrupting the cell wall. Bacterial growth continues even after phage infection, providing an advantage for mass amplification of phages.25 The viral capsid is aligned along the shaft and is composed of 2,700 copies of pVIII and ~5 copies of minor coat proteins pIII, pVI, pIX, and pVII located at either end.21,22 The 50-residue pVIII (98% by mass) is composed of three distinct domains, namely, a negatively charged hydrophilic N-terminal domain (1–20), an intermediate hydrophobic domain (21–39), and a positively charged domain (40–50) that interacts electrostatically with phage genomic DNA (Figure 1B). Only the N-terminal domain is exposed to the media, allowing it to be targeted for genetic or chemical functionalization. The final five residues of pVIII are structurally unconstrained, thus providing an optimal target for genetic engineering. Additionally, the minor coat protein pIII, which resides on one tip of the phage, has been extensively exploited in phage display owing to its flexibility and the accessibility of its N-terminus,26 which allows for insertion of various peptide lengths, including larger proteins (>100 amino acids).27 This review will focus on the chemical functionalization of pVIII and pIII, which have more tolerance to genetic mutation for inserting specific amino acids, thereby leading to facile chemoselective modification.

|
Figure 1 Schematic structure of an M13 bacteriophage (A) and its major coat proteins (B). |
Chemical functionalization of M13 phage
The two most important challenges during functionalization of M13 phage with synthetic functional groups are 1) incorporation of functional groups in the desired active sites under mild reaction conditions where the phage structure is retained and 2) coupling synthetic functional groups with multiple copies of proteins that have abundant potential reactive groups. Therefore, the chemical conjugation strategy in phage modification requires mild and facile chemoselective reactions. Researchers have recently begun to understand the site-specific chemical accessibility of amino acids within the coat proteins. To achieve a controlled and orthogonal chemistry on the phage, several amino acids (cysteine, N-terminal alanine, lysine, N-terminal serine/threonine, aspartic acid/glutamic acid, and tyrosine) are often exploited as target residues (Table 1). Either wild-type or genetically engineered phages displaying specific amino acid(s) on the exposed coat protein domain were used for chemical functionalization. In the wild-type phage, amino groups in N-terminal alanine and in lysine (Lys8) and carboxylic acid groups in glutamic acid (Glu2) and aspartic acid (Asp4 and Asp5) on pVIII are viable targets for selective chemical functionalization such as amide bond formation (Figure 1B). In order to utilize a wide range of chemoselective modifications in phage, cysteine and other amino acids such as N-terminal serine/threonine or tyrosine, which do not appear in the solvent-exposed domain of the wild-type coat proteins, are used in genetic engineering. Several chemoselective modification methods have been employed for each amino acid residue incorporated (Table 1). This development of site-specific organic synthesis approaches28–31 enables phage surfaces to be modified with various chemicals such as fluorescent dyes, chromophores, enzymes, and synthetic oligomers (eg, poly(ethylene glycol) [PEG]) and allows them to be used in various applications, including bioimaging, biosensing, tissue engineering, and energy harvesting.29–31

|
Table 1 Selected reports on chemical functionalization of M13 bacteriophage |
Cysteine
Cysteine is one of the least abundant amino acids in phage coat proteins. The wild-type sequences of M13 major coat proteins do not contain cysteines, whereas pIII, pVII, and pIX have internal cysteines that play important roles in structure formation via disulfide bonds. Generally, cysteines are incorporated in the viral capsid by genetic engineering to create reactive handles. Cysteines on M13 phage have been used as alkylation sites for electrophilic halides or maleimides. Adjacent cysteine residues with the potential to form disulfide bridges must be reduced to create native thiol groups for cysteine-specific reactions. For the selective functionalization of incorporated cysteines, intrinsic cysteine residues in minor coat proteins are often removed by mutagenesis. However, if the application of the phage requires phage infectivity (ie, in phage display screening), reduction or removal of the intrinsic disulfide bonds might be problematic because it is reported to decrease phage infectivity.
To explore the accessibility and environment of cysteines, Khan et al32 genetically incorporated cysteines within the hydrophobic transmembrane domain of the M13 major coat protein (Figure 2A). The accessibility of Cys-sulfhydryl in the M13 phage was examined by its reaction with maleimido and iodoacetamido groups and 5,5′-dithiobis(2-nitrobenzoic acid). Briefly, genetically engineered phage (1 mg/mL) in Tris buffer (10 mM, pH 8.5) was mixed with the same volume of sulfhydryl-reactive reagents (1 mg/mL) at room temperature in the dark for 2 hours. After alkylation or disulfide bond formation, the accessibility of each reagent was deduced by labeling the unmodified sulfhydryl groups with [14C]iodoacetamide (14C-IAM). Excess 14C-IAM was incubated with phage solution (1 mg/mL in Tris buffer) at 37°C for 90 min. Labeled phage was isolated by PEG precipitation.33 The extent of sulfhydryl modification was measured by electron paramagnetic resonance spectroscopy. The reactivity of the Cys mutant phage showed potential for use as a versatile tool in probing virus structure and tracking conformational changes during the phage life cycle.
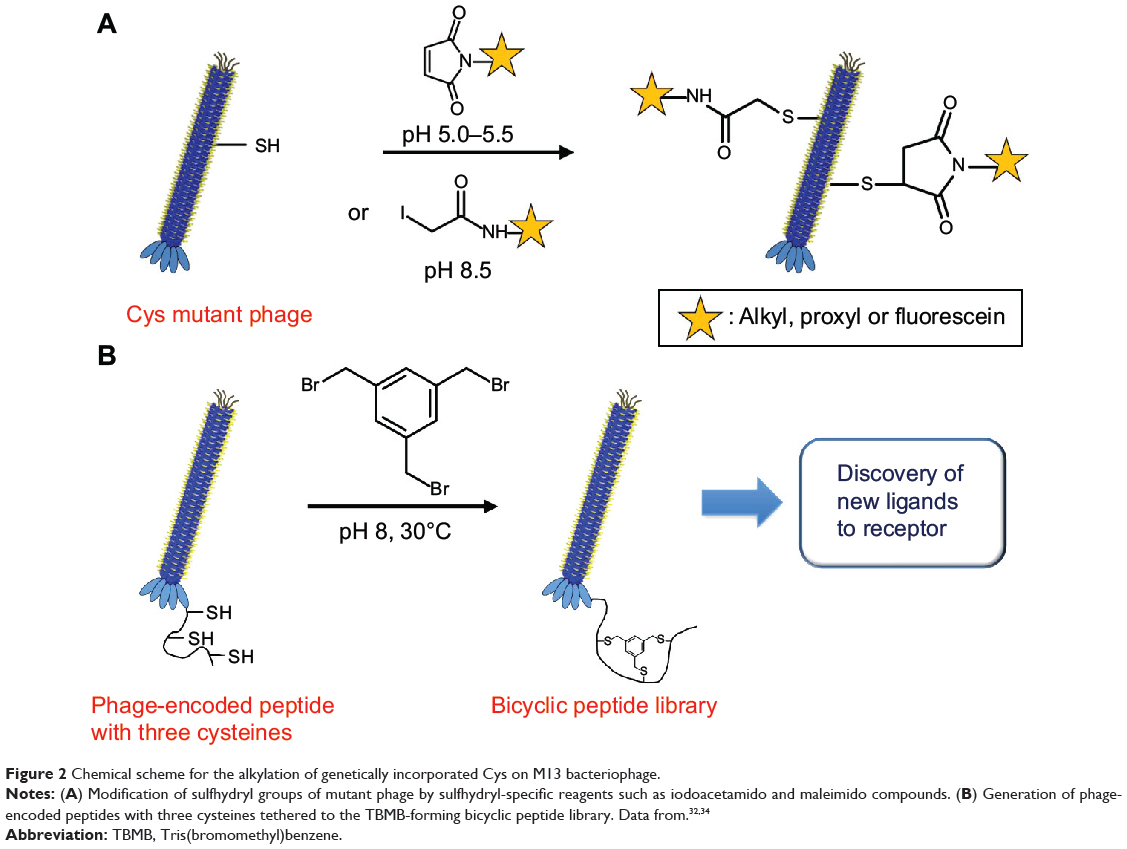
|
Figure 2 Chemical scheme for the alkylation of genetically incorporated Cys on M13 bacteriophage. |
Heinis et al34 fused a library of peptides containing three cysteine residues, each separated by six random amino acids, to the minor coat protein pIII (Figure 2B), which was genetically engineered to remove intrinsic disulfide bonds for the selective functionalization of the three newly introduced cysteine residues. The reactive cysteines were conjugated with Tris(bromomethyl)benzene (TBMB) to generate a library of bicyclic peptides at the N-terminus of pIII. Briefly, the phage (1011–1012 transducing units) in 20 mL of ammonium carbonate buffer (20 mM, pH 8) was pretreated with Tris(2-carboxyethyl)phosphine (TCEP) (1 mM, 1 h). After concentrating and washing the phage, it was resuspended in the reaction buffer (32 mL), and TBMB solution in acetonitrile (8 mL, 50 μM) was added to the mixture. The mixture was incubated at 30°C for 1 hour, and the phage containing the peptide loops was isolated using PEG precipitation. These phage-encoded combinatorial chemical libraries showed the potential to identify novel bicyclic peptide drugs (eg, enzyme inhibitors). However, the improved systemic approach should be addressed since disulfide-free pIII caused phage instability, leading to reduced infectivity. The chemical conjugation reaction often led to loss of phage infectivity (5-fold or greater).
Spruijt et al35 employed site-directed labeling in the phage system by incorporating single cysteines in M13 pVIII.36 The several mutants were subsequently spin-labeled for electron spin resonance spectroscopy or fluorescently labeled. This study provided insight into the location and incorporation of the hydrophobic domain of pVIII in the phospholipid bilayer.
N-terminal alanine
Amide bond formation between amino groups on coat proteins and acylating reagents is the most widely used bioconjugation strategy. N-terminal amines and lysine residues (Lys8 in wild-type pVIII) have been used for acylation sites to incorporate either a final functional group or a chemical linker (eg, alkynyl group) for further chemical modification (eg, azido-alkyne cycloaddition). N-terminal groups in the minor coat proteins can also participate in the acylation reaction, although they are far less abundant than the accessible amines on major coat proteins.
Rong et al37 (Figure 3A) used M13 phages as building blocks to generate thin films with aligned topography for directing oriented cell growth. In order to incorporate cell-binding motifs to the phage coat protein, two-step chemical modification was carried out on the N-terminal amines. M13 phage dispersion (10.2 mg/mL, 4 mL) in phosphate-buffered saline (PBS; 10 mM, pH 7) was mixed with alkyne-derivatized N-hydroxysuccinimide ester (0.21 M, 320 μL) and incubated overnight at 4°C. The phage mixture was purified by dialysis against PBS buffer (MWCO; 100 kDa). Thereafter, azido-RGDS (arginyl-glycyl-aspartyl-serine)-peptide was coupled to the phage via the copper(I)-catalyzed azido-alkyne cycloaddition reaction (click chemistry). The alkyne-modified phage (4.3 mg/mL, 925 μL) in PBS was mixed with azido-RGDS-peptide (100 mM, 55 μL), copper sulfate (100 mM, 10 μL), and sodium ascorbate (200 mM, 10 μL) and incubated overnight at room temperature. The phage was purified as described previously.
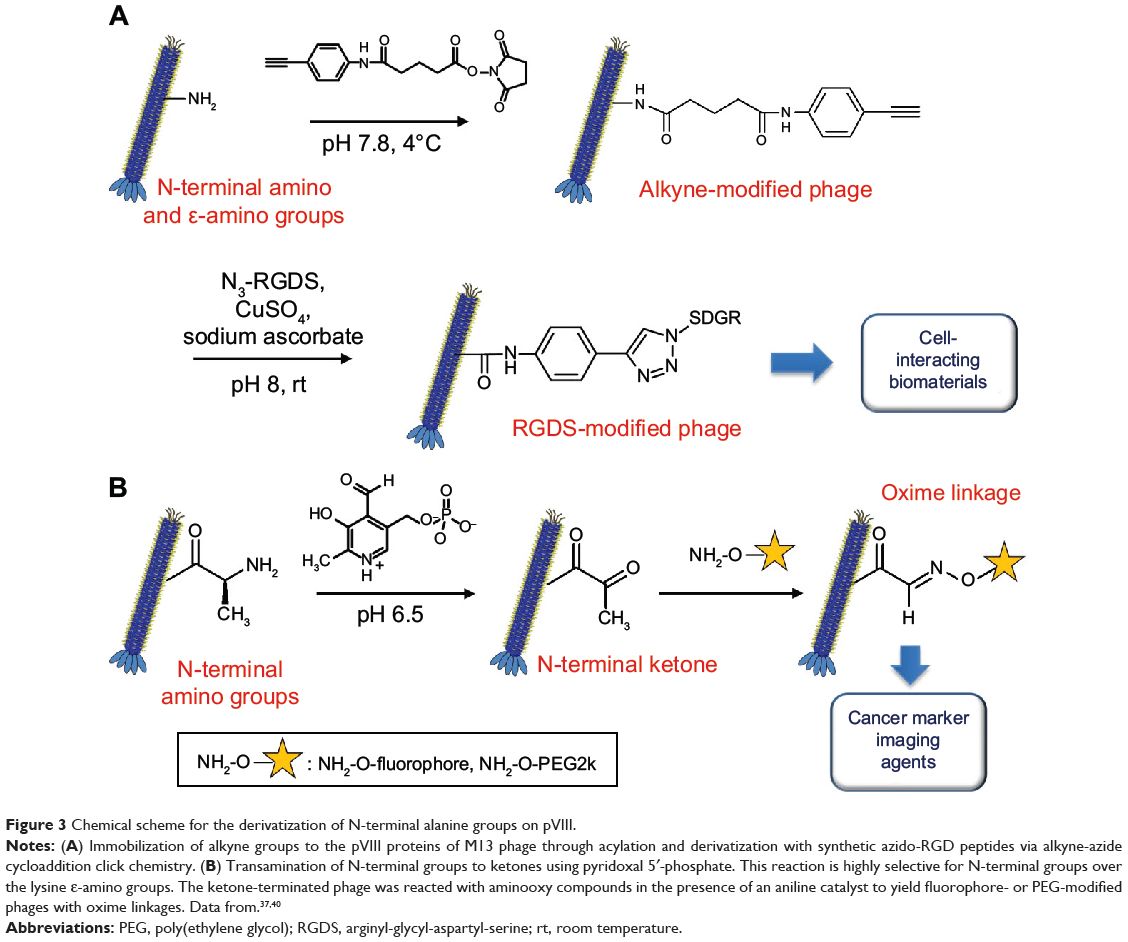
|
Figure 3 Chemical scheme for the derivatization of N-terminal alanine groups on pVIII. |
Chung et al15 biotinylated the amino groups (N-terminal or lysine residue) of M13 phage major coat proteins genetically engineered to display RGD peptides on every copy of pVIII. Briefly, RGD-phage dispersion (1 mg/mL, 1 mL) in PBS (10 mM, pH 7) was mixed with 6-((biotinoyl)amino)hexanoic acid, succinimidyl ester in dimethyl sulfoxide (DMSO) (0.2 M, 200 μL), and incubated overnight at room temperature. The phage mixture was purified by dialysis against PBS buffer (MWCO 10 kDa). Biotinylated RGD-phages (b-RGD-phage) were used for observing and quantifying the diffusion of fluorescent dye-labeled streptavidin into the fibers made of b-RGD-phage and polylysine.
Li et al38 examined the chemical reactivity of phage N-terminal and lysine amino groups by conjugating them to fluorescent dye molecules. The authors used an N-hydroxysuccinimidyl (NHS) ester derivative of tetramethylrhodamine (TMR-NHS), which reacts with amines via amide bond formation. M13 phage (1.0 mg/mL) was incubated in TMR-NHS (40 μM to 8 mM) in a mixed solution containing phosphate buffer (pH 7.8, 10 mM) and DMSO (v/v 80:20) at 4°C for 24 hours. The modified phage was purified by dialysis with the phosphate buffer (pH 7.8, 10 mM). The degree of coupling was assessed by measuring the visible light absorbance of the fluorescent dye attached to the phage (up to ~1,600 dyes/phage). MALDI-TOF MS and HPLC-MS/MS were used to identify the modified pVIII proteins and their regioselectivities. Only the N-terminus and Lys8 were reactive toward the TMR-NHS, whereas Lys40, 43, 44, and 48 (associated with the phage DNA) were not reactive. The fluorescence intensity of the dye-modified phage was the highest without significant self-quenching for about 400 coupled dye molecules per M13 phage. The fluorescent M13 phages were conjugated with cancer-targeting motifs (folic acid) using an orthogonal reaction, which will be described in detail later in this review. Such dual-modified phages showed the potential of M13 phage in drug delivery and bioimaging applications.
Zhang et al39 grafted PEG to the amino groups of pVIII to tune fd phage surface properties. The overall structures of fd and M13 phages are identical, and the fd phage differs from M13 by a single amino acid residue: an asparagine at position 12 in M13 is replaced by an aspartic acid in fd. The reaction between the NHS ester derivative of PEG (MW =5 kDa) (3.2 mM) and fd phage (5 mL, 2 mg/mL) was conducted in phosphate buffer (100 mM, pH 8.2) in a shaker for 12 hours at room temperature. The PEG-grafted phage was purified by ultracentrifugation (100,000× g, 6 hours) and resuspended in Tris buffer (pH 8.2, 20 mM). This modified phage can be used as a model system in soft condensed matter physics.
Carrico et al40 employed a transamination/oxime formation strategy for selective labeling of fd phage pVIII N-terminal groups. This strategy led to the transamination of only the N-terminal amino groups, leaving the ε-amino group of Lys8 intact (Figure 3B).40 The phage (75–128 nM) was transaminated with pyridoxal 5′-phosphate (100 mM) in phosphate buffer (25 mM, pH 6.5) for 13 hours at room temperature. Excess reagent was removed using the standard PEG precipitation method. The resulting N-terminal ketones were subject to reaction with various alkoxyamine compounds such as aminooxy biotin, aminooxy PEG2000, and aminooxy fluorophore (10–20 mM) in the phosphate buffer (10–20 mM, pH 6.2) for 15–21 hours at room temperature. Aniline was used as a catalyst to accelerate the rate of reaction. The excess reagent was removed by either PEG precipitation or gel filtration. The authors adopted this chemoselective modification to the fd phage displaying antibody fragments targeting EGFR and HER2 (epidermal growth factor receptors) on pIII minor coat proteins. Such modified phages can be used for bioimaging modality with active targeting capability.
Adhikari et al41 introduced sulfhydryl groups as reactive handles in the viral capsid of M13 phage for chemoselective protein immobilization. M13 phage (1010 plaque forming units [pfu]) in PBS buffer (800 μL, including 3 mM ethylenediaminetetraacetic acid) was added to Traut’s reagent (2-iminothiolane-HCl, 7 μM) and incubated for 90 minutes at 25°C. After purifying the resultant thiolated phage from excess reagents using a spin filter, the phage suspension was mixed with maleimide-functionalized horseradish peroxidase solution in PBS (22 μM to 1.14 mM) for 90 minutes at 25°C. The enzyme-immobilized phage was purified similarly and it can be used for immunochromatographic assays to detect viral pathogens after incorporating specific antibodies on the phage.
Phages have been labeled with a number of different fluorescent dyes for in vivo bioimaging by Kelly et al.42 The phages were first genetically engineered to display SPARC (secreted protein, acidic and rich in cysteine) or vascular cell adhesion molecule-1-binding ligands, which were then labeled with fluorescent dyes using standard NHS or isothiocyanate chemistry.
The pVIII N-terminal amino groups and lysine residues are often used for incorporating fluorescent dyes into the phage body because the amino group functionalization is straightforward and efficient. Using the same chemical conjugation method, imaging agents such as gadolinium (Gd) complexes and quantum dots or metallic nanoparticles can be incorporated to the phage particle for magnetic resonance imaging (MRI) and fluorescence imaging or plasmonic detection, respectively. By incorporating targeting ligands such as folates and RGD motifs with the imaging agents on phage particles through orthogonal chemical conjugation or genetic engineering, the modified phages have been demonstrated as a useful tool for disease-specific imaging or therapeutics.37,41,43 In addition, application of phage as targeted drug carriers for the treatment of pathogenic bacteria has been reported. In this study, the bactericidal drug was linked to the phage surface using an acylation method similar to that described earlier in which drugs were designed to be released by enzymatic cleavage.44
Aspartic acid and glutamic acid
Carboxylic acid groups are often exploited in protein derivatization using a mild carboxylic acid activation method. Wild-type M13 phage pVIII contains surface carboxylic acid-containing amino acids (Glu2, Glu20, Asp4, and Asp5). Generally, the acylating reagent O-acylurea is generated in situ in the presence of the amino-containing chemical groups by adding water-soluble carbodiimide to phage carboxyl groups. However, the intermediate O-acylurea is vulnerable to hydrolysis and may undergo side reactions involving the rearrangement of the O-acylisourea to the unreactive N-acylurea, which may lower the reaction yield. Converting the O-acylurea to an active ester by adding NHS or sulfo-NHS to carbodiimide reactions often increases efficiency and enables the carboxyl groups to be activated for storage and later use. Aspartic acids and glutamic acids in major coat proteins also provide negatively charged surface regions that can be coated with positively charged oligomers such as oligolysines.
Zhang et al39 utilized carbodiimide chemistry in fd phage to modify abundant carboxyl groups (Glu and Asp) with organic amine compounds such as ethylenediamine, N,N-dimethylethylenediamine, and 1-(2-aminoethyl)piperazine (Figure 4). Water-soluble carbodiimide, 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (EDAC), was used to activate the carboxyl groups of Glu2, Glu20, Asp4, Asp5, and Asp12 into O-acylurea, which reacts with amino groups to form amide bonds. Diamine solution (pH 5, 200 mM, 4.5 mL) was mixed with an fd phage dispersion (20 mg/mL, 0.5 mL). While the pH of the mixture was adjusted to 5.0–5.5, EDAC (0.4 mmol) was added to the mixture and incubated at room temperature for 5 hours. The modified phage was purified by dialysis in deionized water. The charge-reversed phages have been demonstrated to interact with negatively charged phages, which can be useful for studying colloidal soft matter physics.
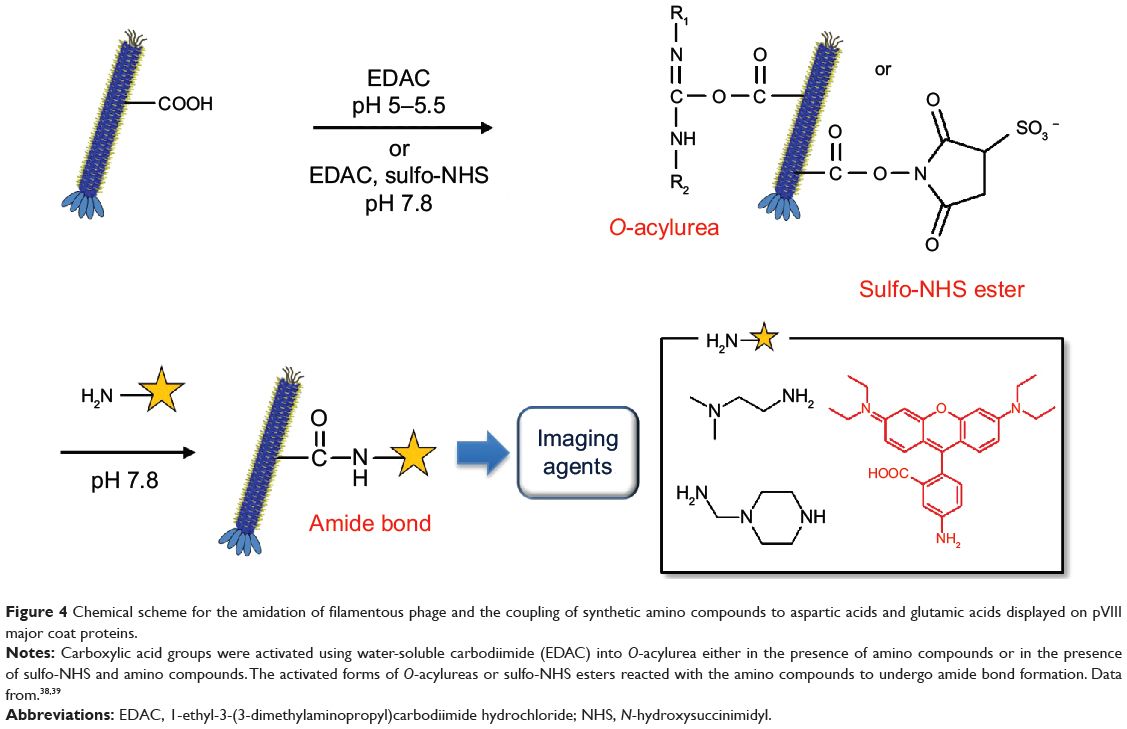
|
Figure 4 Chemical scheme for the amidation of filamentous phage and the coupling of synthetic amino compounds to aspartic acids and glutamic acids displayed on pVIII major coat proteins. |
Li et al38 identified the chemical reactivity of Glu2, Glu20, Asp4, and Asp5 carboxylic acid groups on pVIII by conjugating them with several concentrations of amino-functionalized fluorescent dye (Rhodamine B amine). M13 phage was incubated with Rhodamine B amine, EDAC (10 mM), and N-hydroxysulfosuccinimide (30 mM) in phosphate buffer (10 mM, pH 7.8) at room temperature for 12 hours. After the modified phage was purified by dialysis, the degree of coupling was assessed by measuring the visible light absorbance of fluorescent dyes attached to the phage (up to ~150 dyes/phage). HPLC-MS/MS was used to identify reactive residues. Glu2, Asp4, and Asp5 were found to react with carbodiimide activation-based amidation, whereas Glu20 was not reactive, possibly due to its poor accessibility. This is not surprising because Glu20 is buried compared to Glu2, Asp4, and Asp5.
Lamboy et al45 attempted to reduce nonspecific binding of positively charged proteins to the native negatively charged surface of phage for improved phage display system development. In order to neutralize the phage surface, the authors had either genetically engineered the phage to display charge-neutralizing peptides on the solvent-exposed surface or blocked the surface with oligolysine (eg, Lys8) against highly basic proteins in the proteome. In both cases, the infectivity of phage diminished because the neutralized phage interacts less with F-pili, which are composed of the highly positively charged F-pilin, on E. coli.
N-terminal serine and threonine
N-terminal serine and threonine yield aldehyde functional groups upon oxidative cleavage by NaIO4.46 These N-terminal amino acids are not found in native M13 phage and therefore can act as chemoselective modification sites. The pIII minor coat proteins are common targets for incorporating N-terminal serine/threonine.
Ng et al47 synthesized genetically encoded glycosylated peptide libraries displayed on M13 phage using N-terminal serines/threonines (Figure 5A). Among the peptide libraries, the phages carrying N-terminal serine and threonine on pIII were oxidized to form N-terminal glyoxals, which can act as reactive handles for subsequent immobilization of glycans. The phage (1011 pfu/mL) in PBS (pH 7.4) was oxidized with NaIO4 (6 mM) for 5 minutes on ice and quenched with glutathione for 10 minutes at room temperature. An equal volume of aminooxy compounds (aminooxy-biotin or aminooxy-mannose, 2 mM) in anilinium acetate buffer (pH 4.7) was added to the phage mixture and incubated for 1 hour at room temperature. Such oxime ligation on phage-displayed peptides can have broad applications in the synthesis of custom-designed chemically modified phage libraries.47,48 Although no detailed results of phage stability were reported, the authors claimed that this chemical modification had minimal interference on phage infectivity.
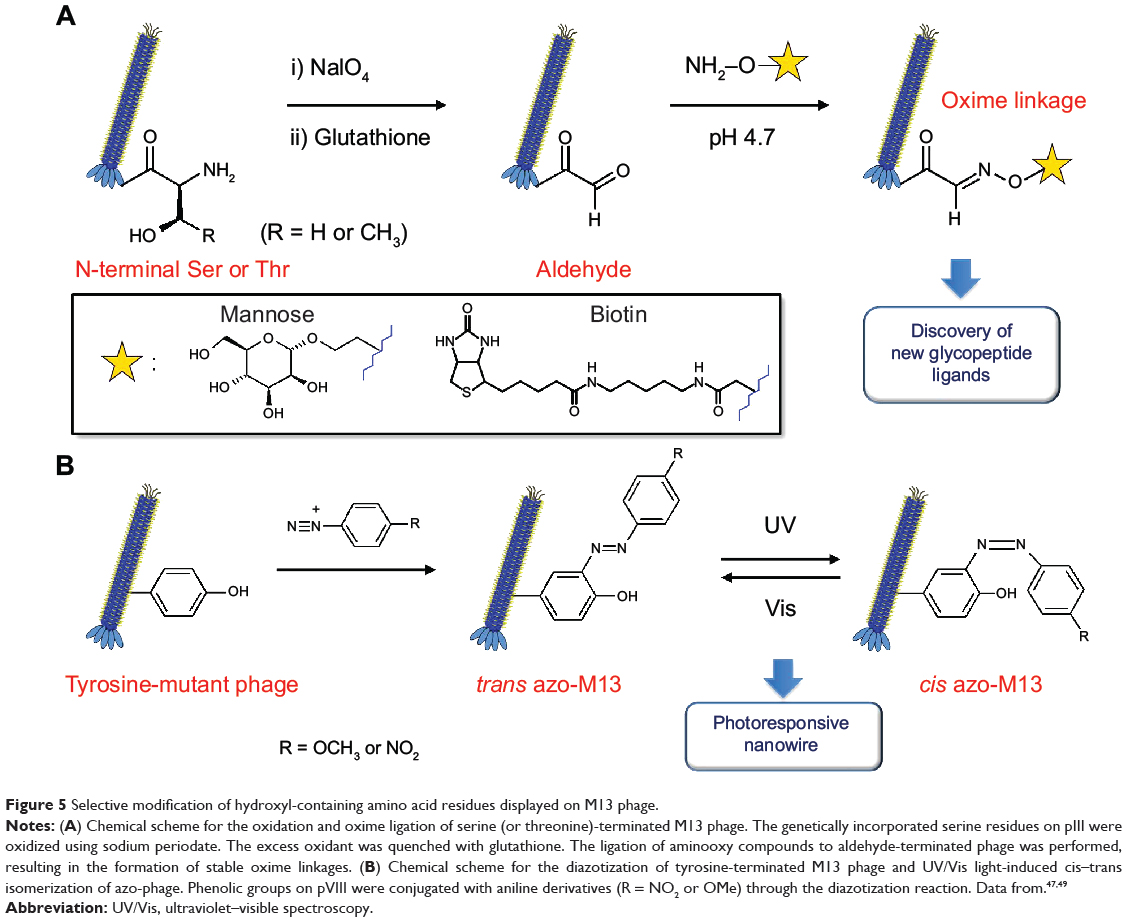
|
Figure 5 Selective modification of hydroxyl-containing amino acid residues displayed on M13 phage. |
Tyrosine
Among the aromatic groups in proteins, the phenol groups of tyrosine residues are the most susceptible to diazotization reactions. Tryptophan and phenylalanine in pVIII are within the hydrophobic domain and therefore not exposed to solvents. The use of chemoselective modification of tyrosine residues requires incorporation within the N-terminal hydrophilic domain for chemical access. The chemoselective modification of tyrosine residues on coat proteins is orthogonal to the other chemical modification methods for preparing dual or multimodified phage particles.
Murugesan et al49 devised photoresponsive nanowires out of genetically engineered M13 phage through diazotization of tyrosine residues displayed on pVIII major coat proteins (Figure 5B). Tyrosine-rich decapeptides (amino acid sequence: Y-Y-G-Y-Y-G-Y-Y-G-Y) were fused with the N-terminus of major coat protein by the phage display technique. Coupling phenolic tyrosine residues to the side wall of M13 phage with different aniline derivatives (4-methoxyaniline, 4-nitroaniline) was completed as follows. An excess of diazonium salts was prepared by mixing aniline derivatives with sodium nitrite and aqueous HCl solution at 0°C for 1 hour. Tyrosine peptide-fused M13 phage solution in phosphate buffer (500 μL, 10 mM) with borate buffer/NaCl aqueous solution (500 μL, 0.1 M NaCl) was added to sufficiently cooled mixtures of diazonium reaction. Color change (colorless to yellow) indicated the conjugation of diazonium salts with genetically engineered M13 phages. The mixture was incubated with occasional shaking at 5°C for 3 hours, and the modified phage was purified by dialysis against the phosphate buffer. The resulting azo-phage exhibited reversible photoresponsive properties through cis–trans isomerization of the azo moieties. This property of modified phage can be potentially used in light-controllable smart devices.
One of the weak points of this chemical modification strategy is that it is not applicable to phages carrying aromatic amino acid residues as part of the additional active sites other than the photoreaction sites. The accessibility of the amino acid residues in the buried hydrophobic domain toward the diazotization reaction has not been fully studied.
Challenges and next-generation functional phage
The various chemical conjugation strategies have shown a great potential for use in the development of novel nanomedicines for tissue engineering, bioimaging, drug discovery, and targeted drug delivery even though the chemical modification strategies must be carefully devised to ensure chemical/physical integrity and phage infectivity. This review focused on the chemical modification of pIII and pVIII and their further functional opportunities; however, we also note that pVII and pIX are currently studied as new targets for genetic engineering and chemoselective modification, and an enzymatic conjugation strategy was recently reported. The minor coat protein pIII is the critical component for E. coli infection through interaction with F-pili. Consequently, genetic engineering and chemical modification of pIII always bear the possible effect of reducing phage infectivity and leading to a low production yield and inefficient combinatorial phage display screening. Recently, interest in two alternative coat proteins, pVII and pIX, which are located at the opposite end, has increased both for genetic engineering and for subsequent chemical modification because they are relatively less involved in phage stability and infectivity.50 An enzymatic approach for conjugating synthetic molecules to the phage particle in a highly efficient, site-specific manner has also been reported.51 The authors showed that pIII, pVIII, and pIX can be functionalized with a broad range of molecules from small molecules to folded proteins in a site-specific manner with higher yields than conventional genetic engineering approaches. This novel enzyme-mediated functionalization can be done under very mild biocompatible conditions, which may expand the use of M13 phage in nanomedicine.
Conclusion
The M13 phage is one of the most extensively developed systems for site-specific peptide display, which endows it with great potential for development of novel multifunctional bionanomaterials. This nanomaterial with genetically programmable peptide functionalities can be further modified with synthetic compounds using well-established chemoselective chemical modifications. The N-terminal domains of pIII and pVIII, which are exposed to the media, are targets for chemical functionalization. The target reaction sites span N-terminal amino groups for acylation and transamination or thiourea formation, N-terminal serine/threonine for oxidation to aldehyde groups, cysteines for alkylation, tyrosines for diazotization, and carboxylic acid groups (from Asp and Glu) for amidation via carbodiimide activation. Synthetic compounds that have been chemically attached to the filamentous phage encompass a wide range of possible compounds, including fluorophores, PEG, synthetic peptides, enzymes, sugars, biotin, photoresponsive chemicals, positively charged compounds, and isotope-labeled compounds.
The combined strategy using genetic engineering and chemoselective modification is expanding the scope of potential applications of M13 phage in bioimaging, biosensing, and tissue engineering. Recently, phages have shown a new possibility of use as building blocks for hierarchically assembled biomimetic structures in addition to their usefulness as a filamentous nanomaterial.16 The biomimetic structures based on hierarchical phage assembly have demonstrated usefulness in tissue engineering materials,13,17,18 therapeutic delivery systems,43,52 colorimetric sensors,6 and piezoelectric energy-generating biomaterials.8–10 The aforementioned functionalization strategy is expected to extend its utility in the hierarchically assembled structures, which will lead to future advanced material design for a wide range of applications, including biomedical, energy, and sensing devices. Recently, many research groups have begun to use pVII and pIX minor coat proteins as alternative targets for phage display and subsequent chemoselective modification, which will be advantageous for the development of combinatorial library systems, including peptides conjugated with synthetic materials. Although this review focused on the chemical functionalization of M13 phage, the chemoenzymatic strategy can be an alternative or more efficient method for modifying phages if there are stability or infectivity issues in chemical modification.
Acknowledgments
This research was supported by the Basic Research Promotion Fund from Samsung (S-2013-0902-000) and by the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Science, ICT and Future Planning and the Korean government (2013R1A1A3008484) and by the Korean government (NRF-2014S1A2A2027641).
Disclosure
The authors report no conflicts of interest in this work.
References
Jaworski JW, Raorane D, Huh JH, Majumdar A, Lee S-W. Evolutionary screening of biomimetic coatings for selective detection of explosives. Langmuir. 2008;24(9):4938–4943. | ||
Merzlyak A, Lee S-W. Phage as templates for hybrid materials and mediators for nanomaterial synthesis. Curr Opin Chem Biol. 2006;10(3):246–252. | ||
Whaley SR, English DS, Hu EL, Barbara PF, Belcher AM. Selection of peptides with semiconductor binding specificity for directed nanocrystal assembly. Nature. 2000;405:665–668. | ||
Lee SW, Mao CB, Flynn CE, Belcher AM. Ordering of quantum dots using genetically engineered viruses. Science. 2002;296:892–895. | ||
Mao C, Solis DJ, Reiss BD, et al. Virus-based toolkit for the directed synthesis of magnetic and semiconducting nanowires. Science. 2004;303(5655):213–217. | ||
Oh J-W, Chung W-J, Heo K, et al. Biomimetic virus-based colourimetric sensors. Nat Commun. 2014;5:3043. | ||
Lee JH, Xu PF, Domaille DW, Choi C, Jin S, Cha JN. M13 bacteriophage as materials for amplified surface enhanced Raman scattering protein sensing. Adv Funct Mater. 2014;24(14):2079–2084. | ||
Lee YJ, Yi H, Kim W-J, et al. Fabricating genetically engineered high-power lithium-ion batteries using multiple virus genes. Science. 2009;324(5930):1051–1055. | ||
Nam KT, Kim D-W, Yoo PJ, et al. Virus-enabled synthesis and assembly of nanowires for lithium ion battery electrodes. Science. 2006;312(5775):885–888. | ||
Lee BY, Zhang J, Zueger C, et al. Virus-based piezoelectric energy generation. Nat Nano. 2012;7(6):351–356. | ||
Cerruti M, Jaworski J, Raorane D, et al. Polymer-oligopeptide composite coating for selective detection of explosives in water. Anal Chem. 2009;81(11):4192–4199. | ||
Jaworski JW, Raorane D, Huh JH, Majumdar A, Lee SW. Evolutionary screening of biomimetic coatings for selective detection of explosives. Langmuir. 2008;24(9):4938–4943. | ||
Yoo SY, Oh J-W, Lee S-W. Phage-chips for novel optically readable tissue engineering assays. Langmuir. 2011;28(4):2166–2172. | ||
Lee JW, Song J, Hwang MP, Lee KH. Nanoscale bacteriophage biosensors beyond phage display. Int J Nanomed. 2013;8(1):3917–3925. | ||
Chung W-J, Merzlyak A, Lee S-W. Fabrication of engineered M13 bacteriophages into liquid crystalline films and fibers for directional growth and encapsulation of fibroblasts. Soft Matter. 2010;6(18):4454–4459. | ||
Chung W-J, Oh J-W, Kwak K, et al. Biomimetic self-templating supramolecular structures. Nature. 2011;478(7369):364–368. | ||
Yoo SY, Merzlyak A, Lee S-W. Facile growth factor immobilization platform based on engineered phage matrices. Soft Matter. 2011;7(5):1660–1666. | ||
Yoo SY, Merzlyak A, Lee S-W. Synthetic phage for tissue regeneration. Mediators Inflamm. 2014;2014:11. | ||
Adams M, Dogic Z, Keller SL, Fraden S. Entropically driven microphase transitions in mixtures of colloidal rods and spheres. Nature. 1998;393(6683):349–352. | ||
Yang SH, Chung W-J, McFarland S, Lee S-W. Assembly of bacteriophage into functional materials. Chem Rec. 2013;13(1):43–59. | ||
Marvin DA, Welsh LC, Symmons MF, Scott WR, Straus SK. Molecular structure of fd (f1, M13) filamentous bacteriophage refined with respect to X-ray fibre diffraction and solid-state NMR data supports specific models of phage assembly at the bacterial membrane. J Mol Biol. 2006;355(2):294–309. | ||
Rodi DJ, Mandova S, Makowski L. Filamentous bacteriophage structure and biology. In: Sidhu SS, editor. Phage Display in Biotechnology and Drug Discovery. Boca Raton, FL: CRC Press Taylor and Francis Group; 2005:748. | ||
Opalka N, Beckmann R, Boisset N, Simon MN, Russel M, Darst SA. Structure of the filamentous phage pIV multimer by cryo-electron microscopy. J Mol Biol. 2003;325(3):461–470. | ||
Russel M, Linderoth NA, Sali A. Filamentous phage assembly: variation on a protein export theme. Gene. 1997;192(1):23–32. | ||
Hohn B, Lechner H, Marvin DA. Filamentous bacterial viruses. I. DNA synthesis during early stages of infection with fd. J Mol Biol. 1971;56(1):143–154. | ||
Lubkowski J, Hennecke F, Pluckthun A, Wlodawer A. The structural basis of phage display elucidated by the crystal structure of the N-terminal domains of g3p. Nat Struct Mol Biol. 1998;5(2):140–147. | ||
Sidhu SS, Weiss GA, Wells JA. High copy display of large proteins on phage for functional selections. J Mol Biol. 2000;296(2):487–495. | ||
Carrico ZM, Romanini DW, Mehl RA, Francis MB. Oxidative coupling of peptides to a virus capsid containing unnatural amino acids. Chem Commun. 2008(10):1205–1207. | ||
Miller RA, Presley AD, Francis MB. Self-assembling light-harvesting systems from synthetically modified tobacco mosaic virus coat proteins. J Am Chem Soc. 2007;129(11):3104–3109. | ||
Nicholas S, Zachary C, Matthew F. Nanoscale integration of sensitizing chromophores and porphyrins with bacteriophage MS213. Angew Chem Int Ed Engl. 2009;48(50):9498–9502. | ||
Schlick TL, Ding Z, Kovacs EW, Francis MB. Dual-surface modification of the tobacco mosaic virus. J Am Chem Soc. 2005;127(11):3718–3723. | ||
Khan AR, Williams KA, Boggs JM, Deber CM. Accessibility and dynamics of Cys residues in bacteriophage IKe and M13 major coat protein mutants. Biochemistry. 1995;34(38):12388–12397. | ||
Maniatis T, Fritsch EF, Sambrook J. Molecular Cloning: A Laboratory Manual. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory; 1982. | ||
Heinis C, Rutherford T, Freund S, Winter G. Phage-encoded combinatorial chemical libraries based on bicyclic peptides. Nat Chem Biol. 2009;5(7):502–507. | ||
Spruijt RB, Wolfs CJAM, Verver JWG, Hemminga MA. Accessibility and environment probing using cysteine residues introduced along the putative transmembrane domain of the major coat protein of bacteriophage M13. Biochemistry. 1996;35(32):10383–10391. | ||
Hemminga M, Vos W, Nazarov P, et al. Viruses: incredible nanomachines. New advances with filamentous phages. Eur Biophys J. 2010;39(4):541–550. | ||
Rong J, Lee LA, Li K, et al. Oriented cell growth on self-assembled bacteriophage M13 thin films. Chem Commun. 2008(41):5185–5187. | ||
Li K, Chen Y, Li S, et al. Chemical modification of M13 bacteriophage and its application in cancer cell imaging. Bioconjug Chem. 2010;21(7):1369–1377. | ||
Zhang Z, Buitenhuis J, Cukkemane A, Brocker M, Bott M, Dhont JKG. Charge reversal of the rodlike colloidal fd virus through surface chemical modification. Langmuir. 2010;26(13):10593–10599. | ||
Carrico ZM, Farkas ME, Zhou Y, et al. N-terminal labeling of filamentous phage to create cancer marker imaging agents. ACS Nano. 2012;6(8):6675–6680. | ||
Adhikari M, Dhamane S, Hagstrom AEV, et al. Functionalized viral nanoparticles as ultrasensitive reporters in lateral-flow assays. Analyst. 2013;138(19):5584–5587. | ||
Kelly KA, Waterman P, Weissleder R. In vivo imaging of molecularly targeted phage. Neoplasia. 2006;8(12):1011–1018. | ||
Bhattarai SR, Yoo SY, Lee S-W, Dean D. Engineered phage-based therapeutic materials inhibit Chlamydia trachomatis intracellular infection. Biomaterials. 2012;33(20):5166–5174. | ||
Yacoby I, Shamis M, Bar H, Shabat D, Benhar I. Targeting antibacterial agents by using drug-carrying filamentous bacteriophages. Antimicrob Agents Chemother. 2006;50(6):2087–2097. | ||
Lamboy JA, Tam PY, Lee LS, et al. Chemical and genetic wrappers for improved phage and RNA display. Chembiochem. 2008;9(17):2846–2852. | ||
Geoghegan KF, Stroh JG. Site-directed conjugation of nonpeptide groups to peptides and proteins via periodate oxidation of a 2-amino alcohol. Application to modification at N-terminal serine. Bioconjug Chem. 1992;3(2):138–146. | ||
Ng S, Jafari MR, Matochko WL, Derda R. Quantitative synthesis of genetically encoded glycopeptide libraries displayed on M13 phage. ACS Chem Biol. 2012;7(9):1482–1487. | ||
Kitov PI, Vinals DF, Ng S, Tjhung KF, Derda R. Rapid, hydrolytically stable modification of aldehyde-terminated proteins and phage libraries. J Am Chem Soc. 2014;136(23):8149–8152. | ||
Murugesan M, Abbineni G, Nimmo SL, Cao B, Mao C. Virus-based photo-responsive nanowires formed by linking site-directed mutagenesis and chemical reaction. Sci Rep. 2013;3:1820. | ||
Løset GÅ, Sandlie I. Next generation phage display by use of pVII and pIX as display scaffolds. Methods. 2012;58(1):40–46. | ||
Hess GT, Cragnolini JJ, Popp MW, et al. M13 bacteriophage display framework that allows sortase-mediated modification of surface-accessible phage proteins. Bioconjug Chem. 2012;23(7):1478–1487. | ||
Choi DS, Jin H-E, Yoo SY, Lee S-W. Cyclic RGD peptide incorporation on phage major coat proteins for improved internalization by HeLa cells. Bioconjug Chem. 2013;25(2):216–223. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.