Back to Journals » International Journal of Nanomedicine » Volume 14
Characterization and bioactivity of self-assembled anti-angiogenic chondroitin sulphate-ES2-AF nanoparticle conjugate
Authors Xing L, Sun F, Wang Z, Li Y, Yang Z, Wang F ![]() , Zhai G, Tan H
, Zhai G, Tan H
Received 25 November 2018
Accepted for publication 26 February 2019
Published 10 April 2019 Volume 2019:14 Pages 2573—2589
DOI https://doi.org/10.2147/IJN.S195934
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Linlin Sun
Liang Xing,1,2,* Feng Sun,1,2,* Zhendong Wang,1,2 Yan Li,1,2 Zhifang Yang,1,2 Fengshan Wang,1,3 Guangxi Zhai,3 Haining Tan1,2
1National Glycoengineering Research Center, School of Pharmaceutical Sciences, Shandong University, Jinan 250012, Shandong, People’s Republic of China; 2Shandong Provincial Key Laboratory of Carbohydrate Chemistry and Glycobiology, Shandong University, Jinan 250012, Shandong, People’s Republic of China; 3School of Pharmaceutical Sciences, Shandong University, Jinan 250012, Shandong, People’s Republic of China
*These authors contributed equally to this work
Background: In the past few years, significant progress has been made in inhibiting neovascularization at the tumor site, cutting off the nutrient supply of the tumor, and inhibiting tumor growth and metastasis. However, many proteins/peptides have the disadvantage of poor stability, short half-life, and uncertain targeting ability. Chemical modification can be used to overcome these disadvantages; many polyethylene glycol-modified proteins/peptides have been approved by US FDA. The purpose of this study was to obtain a novel anti-angiogenic chondroitin sulfate (CS)-peptide nanoparticle conjugate with efficient anti-neovascularization and tumor targeting ability and an acceptable half-life.
Materials and methods: The CS-ES2-AF nanoparticle conjugate was synthesized and characterized using 1H-nuclear magnetic resonance spectroscopy, transmission electron microscopy, and particle size and zeta potential analyzer. The anti-angiogenic ability was studied using MTT, migration, tube formation, and chick chorioallantoic membrane assays. The targeting ability of CS-ES2-AF was studied by ELISA, surface plasmon resonance, and bioimaging. The pharmacokinetics was also studied.
Results: The CS-ES2-AF could self-assemble into stable nanoparticles in aqueous solution, which significantly enhances its anti-neovascularization activity, tumor targeting more explicit, and prolongs its half-life.
Conclusion: CS is an effective protein/peptide modifier, and CS-ES2-AF displayed good potential in tumor targeting therapy.
Keywords: chondroitin sulfate, ES2-AF, nanoparticles, anti-angiogenesis, targeting
Introduction
Chondroitin sulfate (CS) is a glycosaminoglycan composed of alternating D-glucuronic acid and N-acetyl-D-galactosamine residues and is widely present in the extracellular matrix and the cell surface of animal tissues, particularly in abundance in cartilage tissue.1,2 It is an endogenous mucopolysaccharide with good histocompatibility, which is often used for drug modification and the construction of drug carriers. CS-modified cisplatin displayed improved anti-cancer activity and reduced drug toxicity.3 CS-modified glucocorticoids can prolong the release time of the drug in vivo and enhance the efficacy, without toxicity or side effects.4 In addition, CS is also a ligand of CD44, which can specifically target the CD44-overexpressed tumor cells.5–8
A short peptide, ES2, containing eleven amino acids (IVRRADRAAVP) is the main active fragment of endostatin (ES), which has anti-angiogenic properties. We have studied the bioactivity of ES2 and found that it could effectively inhibit the proliferation and migration of endothelial cells.9 A VEGFR1-selective hexapeptide (GNQWFI, AF) was selected from a Position Scanning Synthetic Peptide Library, which can specifically bind to vascular endothelial growth factor (VEGF), thereby inhibiting the interaction of VEGF with a series of ligands. Although the AF peptide cannot directly inhibit the proliferation and migration of endothelial cells, it can significantly inhibit the binding of VEGF to the receptor, thereby blocking the pathway of neovascularization.6,7,10 However, the AF peptide contains three hydrophobic amino acids (tryptophan, phenylalanine, and isoleucine) that lead to poor water solubility, thus restricting their applicability.11–13
To obtain a more biologically active and effective target for the inhibition of angiogenic peptides, a synthetic peptide anti-Flt1-ES2 (IVRRADRAAVPGGGGGNQWFI, ES2-AF) was prepared by solid-phase synthesis. Polypeptides and protein drugs are easily degraded in plasma, tissues, and cells by various metabolic enzymes in the body, resulting in their poor stability and a short half-life in vivo. Therefore, we used CS to chemically modify ES2-AF to improve the shortcomings of peptide drugs in this study. We hoped that the addition of CS could increase the tumor tissue-targeting ability of these drugs and increase their efficacy in vivo.14–16 The structural confirmation of the ES2-AF derivative (CS-ES2-AF) was performed using 1H-nuclear magnetic resonance spectroscopy (1H-NMR), and the morphology and stability of the conjugates were studied by transmission electron microscopy (TEM) and particle size and zeta potential analyzer. The anti-angiogenic ability of CS-ES2-AF on endothelial cells was studied using MTT, migration, tube formation, and chick chorioallantoic membrane (CAM) assays in vivo and in vitro. Targeting studies with CS-ES2-AF were performed by ELISA, surface plasmon resonance (SPR), and in vivo bioimaging. The pharmacokinetics of the drugs was also studied.
Materials and methods
Materials
Synthetic short peptides AF, ES2, and AF-ES2 were purchased from China Peptides Co., Ltd. (Shanghai, China). CS, average molecular weight 20 kDa, was purchased from Shanghai Yuanye Bio-Technology Co., Ltd (Shanghai, China). EA.hy926 endothelial cells (ATCC Number: CRL-2922) and B16 cells (ATCC Number: FS-0467) were obtained from Shanghai Cell Bank, the Institute of Cell Biology, China Academy of Sciences (Shanghai, China). PBS, trypsin, and MTT were purchased from Solarbio Science & Technology Co., Ltd. (Shanghai, China). DMEM was purchased from Gibco®, Life Technologies (Carlsbad, CA, USA). FBS was purchased from Hangzhou Sijiqing Biological Engineering Co., Ltd. (Hangzhou, China). Matrigel matrix, 96-well plates, 48-well plates, 25-mL cell culture bottles, and 15-mL centrifuge tubes were purchased from Corning INC (New York, NY, USA). Dimethyl sulphoxide (DMSO) and tetrabutylammonium hydroxide (TBA) were purchased from Sinopharm Chemical Reagent Co., Ltd. (Shanghai, China). (Benzotriazol-1-yloxy)tris(dimethylamino)phosphonium hexafluorophosphate (BOP) was purchased from TCI Co., Ltd. (Shanghai, China). Fluorescein isothiocyanate (FITC) was purchased from Shanghai Purple One Reagent Factory (Shanghai, China). VEGF165 and basic fibroblast growth factor (bFGF) were purchased from Peprotech (Rocky Hill, NJ, USA). Tween 20 and 3,3’,5,5’-Tetramethylbenzidine color liquid were purchased from Beyotime Biological Technology Co., Ltd. (Shanghai, China). Anti-human IgG-HRP-Fc was purchased from Cusabio Biological Engineering Co., Ltd. (Wuhan, China). Recombinant CD44 protein and Flt-Fc were purchased from R&D Systems (Minneapolis, MN, USA). All other chemicals and reagents were of the highest commercial grade available.
Synthesis of the CS-ES2-AF conjugate
The CS-TBA was prepared according to previously described protocols.10,17,18 Then, the CS-TBA was dissolved in DMSO and BOP (1 g) was added and mixed for 30 minutes to activate the carboxyl groups of CS-TBA. The ES2-AF peptide (11 mg), dissolved in DMSO, was added to the reaction solution. Finally, DIPEA (50 μL) was added to the reaction solution and mixed at 37°C. After 24 hours, an equal volume of 1 M NaCl aqueous solution was added to the reaction solution, the pH was adjusted to 3.0 with 1 M HCl to terminate the reaction, and then the pH was raised to 7.0 with 1 M NaOH. The resulting solution was dialyzed in a dialysis bag (5 kDa molecular weight cutoff) and subsequently freeze-dried. The structure of the obtained CS-ES2-AF was determined by 1H-NMR and stored at 2°C–8°C until further analysis.
Characterization of CS-ES2-AF
The morphology of ES2-AF and CS-ES2-AF was observed under TEM. The CS-ES2-AF sample (10 mg) was dissolved in 1 mL distilled water and then stained with a 2% aqueous solution of phosphotungstic acid and allowed to dry before observation under the microscope.
ES2-AF and CS-ES2-AF solutions (1 mg/mL) were prepared with deionized water. The size distribution and zeta potential of ES2-AF and CS-ES2-AF in the resultant solutions were determined using particle size and zeta potential analyzer.
Cells and culture conditions
All experiments were performed with EA.hy926 or B16 cells obtained from the Shanghai Cell Bank, the Institute of Cell Biology, China Academy of Sciences (Shanghai, China). The EA.hy926 cells were cultivated in DMEM supplemented with 10% FBS and 1% penicillin/streptomycin, and incubated at 37°C in a 5% CO2-humidified atmosphere.19,20 The B16 cells were cultured under the same conditions, in RPMI 1640 medium. Cells were subcultured at ~80% confluence.
Cytotoxicity assay
The toxicities of CS-ES2-AF and ES2-AF were evaluated using the MTT assay.9,21,22 Dehydrogenase present in the mitochondria of living cells can reduce MTT. EA.hy926 cells were seeded in 96-well plates at a density of 5.0×103 cells per well and treated with various concentrations of CS-ES2-AF and ES2-AF (5 μg/mL, 25 μg/mL, 50 μg/mL, 100 μg/mL, or 200 μg/mL; concentrations refer to the ES2-AF portion only). After 48 hours of incubation, the media was removed and 20 μL MTT solution was added to each well, followed by incubation for 4 hours in the dark. Then, the supernatants were removed and 150 μL DMSO was added to each well and gently shaken for 15 minutes to completely dissolve the precipitate. A microplate reader was used to measure the absorbance of the dissolved precipitate at 490 nm. The inhibition rate (IR) was calculated according to the formula below:
|

Wound-healing assay
EA.hy926 cells were seeded onto six-well plates and treated with different concentrations of CS-ES2-AF or ES2-AF (50 μg/mL, 100 μg/mL, and 200 μg/mL; concentrations refer to the ES2-AF portion only) for 48 hours. Three parallel cell-free gaps were created in each well with a pipette tip. Images were taken using an inverted fluorescence microscope, and the cells that migrated into the wound area were calculated.23,24
Tube formation assay
EA.hy926 cells were propagated in DMEM supplemented with 10% FBS. The cells (4×104/well) were seeded onto 48-well plates coated with Matrigel, followed by treatment with DMEM containing different concentrations of CS-ES2-AF or ES2-AF (50 μg/mL, 100 μg/mL, or 200 μg/mL; concentrations refer to the ES2-AF portion only). Then, bFGF was added to each well and made up to a final concentration of 5 ng/mL. Peak tube formation was observed after 8 hours of culture, and five random fields were observed under a microscope. The number of branches per field of each tube for each treatment was quantified. This experiment was independently repeated five times.23–27
CAM assay
The chicken embryo is a commonly used model to detect anti-angiogenic inhibitors; therefore, we used the CAM assay to determine the effect of CS-ES2-AF on neovascularization.28,29 Forty well-developed 6-day-old chicken embryos were placed in a sterile incubator at 37°C and 70% relative humidity. In a sterile environment, the large end of the chicken embryo was wiped with iodine and 75% alcohol. A small hole was made on the top of the chicken embryo, the upper air chamber membrane was peeled off, and the lower CAM membrane was exposed and placed in an incubator. After incubation for 24 hours, a gelatin sponge (0.5 cm diameter) was used as a loading carrier and placed on the chicken embryo allantoic membrane. CS-ES2-AF and ES2-AF (10 μg/mL, 25 μg/mL, or 50 μg/mL; concentrations refer to the ES2-AF portion only) were separately added to the gelatin sponges. The blank control was saline. The volume of each group was 20 μL, and each group had three chicken embryos.
ELISA
The PBS-dissolved VEGF165 solution was added to each well of a 96-well plate and kept overnight at 4°C and then washed with PBS to remove the free, unbound VEGF165. Then, 3% BSA in PBS was added to each well and the plate was incubated for 2 h at 37°C. This step was followed by treatment with the primary antibody positive control (1% BSA in PBS solution containing 500 ng/mL Flt-Fc) or different concentrations of CS-ES2-AF or ES2-AF (50 μg/mL, 100 μg/mL, 200 μg/mL, or 500 μg/mL; concentrations refer to the ES2-AF portion only). After incubation for 1 hour at room temperature, the plate was washed with PBS buffer containing 0.05% Tween 20. Secondary antibodies (anti-human IgG-HRP-Fc) were added to each well and incubated for 1 hour at room temperature. The plate was washed and dried. After incubation with 3,3’,5,5’-Tetramethylbenzidine solution for 2–5 minutes in the dark, the reaction was stopped by the addition of dilute H2SO4 to each well. The absorbance at 450 nm was measured using a microplate reader.30
SPR assay
According to the preconcentration experiment of the ligand protein CD44, the optimum pH of the coupling reaction was selected to be 3.8.18,31 The CD44 protein was covalently bound to the CM5 chip using a classical amino coupling method. The carboxyl group on the surface of the CM5 chip was activated with the activating reagent 1,2-Dichloroethane/N-Hydroxysuccinimide, then CD44 protein (10 μg/mL) was added, and finally the chip was blocked by an ethanolamine-HCl solution. Finally, affinity kinetic data between CS-ES2-AF and ES2-AF to CD44 were tested by a single-loop experiment according to the wizard program in the operating system, and the affinity of the two drugs for CD44 was compared.19,32–34
Bioimaging of FITC-labeled CS-ES2-AF conjugate
To study the targeting properties, the modified product and ES2-AF were labeled with FITC according to a previous protocol.9 The tumor model was established by subcutaneous injection of B16 cells (5×106/mL) into the right armpit of each nude mouse (approval number by ethical committee: 2017-D040). When the tumor volume reached approximately 200 mm3, the nude mice were injected with the FITC-labeled compounds FITC-ES2-AF, FITC-CS-ES2-AF, and CS&FITC-ES2-AF via tail vein. All animal experiments in this study were approved by the Ethics Committee of School of Nursing of Shandong University and performed following ethical guidelines for the care and use of animals. Saline was used as the control and the dose of the experimental group was 25 mg/kg (concentration refers to the ES2-AF portion only). The In Vivo Imaging System kinetics system was used to visualize the nude mice at 0.5, 1, 2, and 4 hours after administration, and the distribution of the drugs was observed in the mice at each time point.22,35,36
Pharmacokinetic study
BALB/c mice (~20 g) that were fasted for 12 hours were randomly divided into three groups. Then, 200 μL of FITC-ES2-AF or FITC-CS-ES2-AF was injected via the tail vein at a peptide dose of 20 mg/kg (concentration refers to the ES2-AF portion only). Blood samples were collected through the fundus venous plexus, 0.5–24 hours after administration. The samples were allowed to stand for 2 hours and the supernatants were collected by centrifugation at 4°C for 10 minutes. Then, an equal volume of 0.1% SDS solution was added to the supernatants as a fluorescent sensitizer. Finally, the fluorescence intensity of the samples was measured by a fluorescence spectrophotometer (excitation at 280 nm and emission at 350 nm).37,38
Results
Synthesis of CS-ES2-AF conjugate
The polypeptide ES2-AF, which was synthesized by solid-phase peptide synthesis, was analyzed by high-performance liquid chromatography. The purity of polypeptide ES2-AF was 95.41% and the retention time was 7.750 min. The molecular weight of polypeptide ES2-AF, measured by mass spectrometry (MS), was 2,198.12. To improve the solubility of CS in organic solvents, CS was modified using TBA-OH to form the DMSO-soluble conjugate CS-TBA.10,17 The yield of CS-TBA was 89.7%. The 1H-NMR spectrum of CS-TBA shown in Figure 1C, compared with the 1H-NMR spectra of CS (Figure 1B), shows four signal peaks with chemical shifts of δ=0.97 ppm, 1.40 ppm, 1.68 ppm, and 3.20 ppm, which are characteristic peaks of the butyl group on the reactant TBA-OH. The signal peak in the range δ=1.85–1.95 ppm corresponds to the characteristic methyl group peak of the acetylamino group on CS. The 1H-NMR spectra of the products showed that the conjugate CS-TBA was successfully prepared by ion exchange and it had good solubility in DMSO.
 | Figure 1 Synthesis and 1H-NMR spectra of CS-ES2-AF. (A) Synthetic route of CS-ES2-AF. 1H-NMR spectra of (B) CS, (C) CS-TBA, and (D) CS-ES2-AF. |
The CS-ES2-AF conjugate was prepared and the yield was approximately 88%–93% (Figure 1A). The 1H-NMR spectrum is shown in Figure 1D. Comparing Figure 1D with Figure 1B, it was found that besides CS signal peak, ES2-AF signal peak was also found in Figure 1D. From the obtained spectra, peaks at 7.00–8.00 ppm corresponded to the aromatic rings of phenylalanine and tryptophan and peaks at 1.85–1.95 ppm corresponded to the methyl resonance of the acetamido moiety of CS. By comparing peaks at 1.85–1.95 ppm and peaks at 7.00–8.00 ppm, we determined the peptide content. The successful conjugation of CS and the novel angiogenic peptide ES2-AF was confirmed by 1H-NMR spectroscopy, and the prepared ES2-AF contained two peptide molecules per CS chain.
Physicochemical characterization of CS-ES2-AF
The TEM images of CS-ES2-AF and ES2-AF (Figure 2A) show that all the particles are uniform, nanosized spheres. The average sizes of the CS-ES2-AF and ES2-AF particles were 144.20 nm and 130.20 nm, respectively (Figure 2B). The small particle size, as well as the uniform size distribution of the CS-ES2-AF nanoparticles, was suitable for the development of nanomedicines. The zeta potentials of CS-ES2-AF and ES2-AF were −34.00±4.53 mV and −7.50±4.07 mV, respectively, indicating that the conjugation with CS enhanced the stability of ES2-AF in the system. Based on this result, we can conclude that CS-ES2-AF can self-assemble into well-stabilized nanoparticles in aqueous solution. This structure enables the peptide to be encapsulated in the CS-formed nanoparticles, avoiding the loss of activity caused by protease hydrolysis.39–41 Studies have shown that CS can specifically bind to CD44 on the surface of tumor cells; the presence of CS-ES2-AF nanoparticles enables CS to act as a targeting vector to carry ES2-AF to the surface of tumor cells, thereby indicating that ES2-AF can play a better role in anti-angiogenesis.5
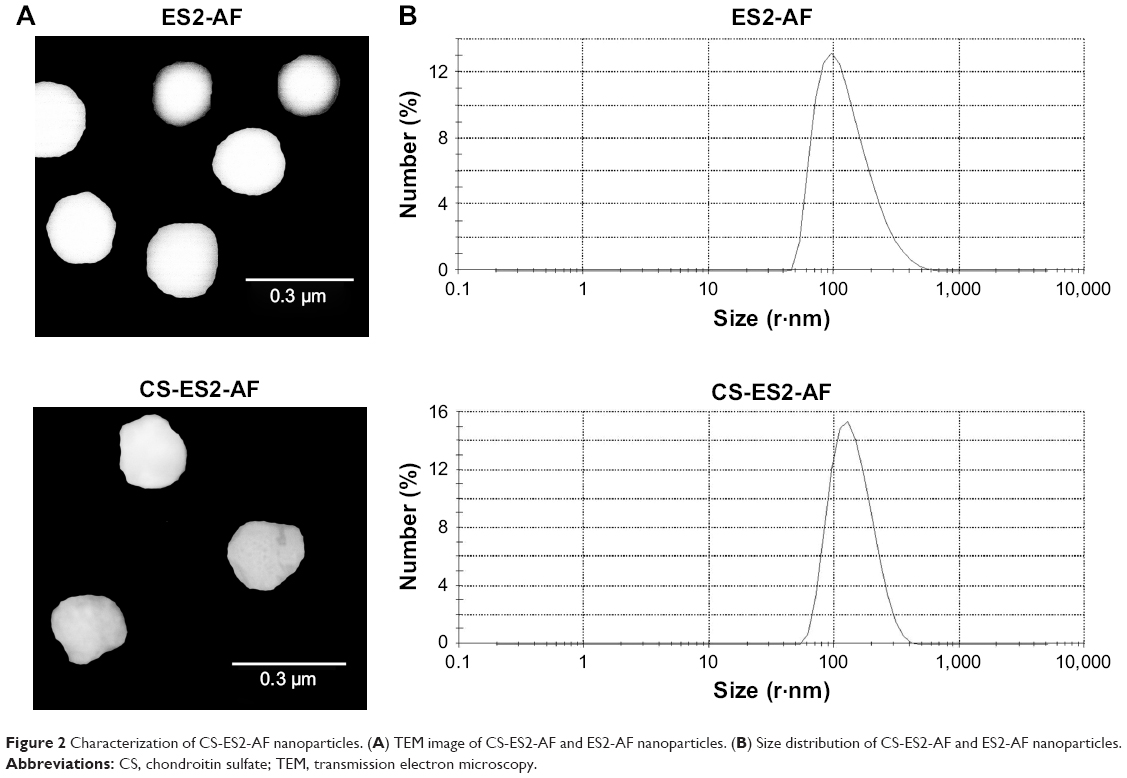 | Figure 2 Characterization of CS-ES2-AF nanoparticles. (A) TEM image of CS-ES2-AF and ES2-AF nanoparticles. (B) Size distribution of CS-ES2-AF and ES2-AF nanoparticles. |
MTT assay
The inhibitory effect of ES2-AF and CS-ES2-AF on the proliferation of the endothelial cells was measured by MTT assay.9,21,22,42 As shown in Figure 3A, when the concentrations of ES2-AF were 5, 25, 50, 100, and 200 μg/mL, the IRs of EA.hy926 endothelial cells were 8.38%±1.29%, 13.38%±1.46%, 17.16%±2.14%, 21.81%±1.59%, and 25.91%±200%, respectively. At the same concentrations of CS-ES2-AF, the IRs of EA.hy926 endothelial cells were 9.72%±0.51%, 12.75%±1.20%, 18.66%±2.26%, 25.72%±3.55%, and 33.01%±3.63%, respectively. From these results, it can be seen that the IRs of ES2-AF and CS-ES2-AF on EA.hy926 cells showed concentration-dependent effect ranging from 5 μg/mL to 200 μg/mL. The inhibitory rates of ES2-AF and CS-ES2-AF were similar (5 μg/mL–100 μg/mL), which indicated that the activity of ES2-AF was similar to CS-ES2-AF at relatively low concentrations. When the concentration increased, the IR of CS-ES2-AF was higher than that of ES2-AF at the same concentration, which indicated that the activity of CS-ES2-AF was significantly better than that of ES2-AF at high concentrations.
 | Figure 3 (A) Inhibitory effects of peptide ES2-AF and conjugate CS-ES2-AF on the proliferation of EA.hy926 endothelial cells after incubation for 48 hours. (B) Comparison of the effects of ES2-AF, the mixture of CS&ES2-AF, and CS-ES2-AF conjugation based on in vitro ELISA assay. Data represent mean ± SD (n=5). **P<0.01 is considered significantly different compared with the ES2-AF group. |
The activity of polypeptide drugs is often related to their structure. Compared to small molecule drugs, polypeptide drugs often have poor aqueous stability due to their spatial structure. When the anti-angiogenic peptide ES2-AF was modified with CS, the stability of the conjugate in aqueous solution improved. This is due to the fact that the CS group in the aqueous solution forces the water molecules around the ES2-AF polypeptide to enhance the stability of ES2-AF. Therefore, it can be inferred that the higher the concentration of CS, the better the stability of ES2-AF. In addition, the results of the MTT assay are also related to the state of endothelial cells and the degree of differentiation. Therefore, when the MTT assay is used to evaluate the inhibitory effect of drugs on endothelial cell proliferation, cells with similar passage numbers should be selected for experiments.
Cell migration assay
EA.hy926 cells were co-cultured in cell culture medium containing different concentrations of ES2-AF, CS&ES2-AF, and CS-ES2-AF for 48 hours and photographed under a fluorescence microscope to obtain a graph of the cell migration. The initial position of the scratch was marked with a black solid line (Figure 4A). As shown in Figure 4D, inhibition of cell migration occurred in a concentration-dependent manner. When the concentration increased, the inhibitory effect on the endothelial cell migration increased. When the concentrations were 50 μg/mL, 100 μg/mL, and 200 μg/mL, the numbers of migrated cells in the CS-ES2-AF group were 164.33±8.89, 125.17±7.03, and 100.67±15.69, respectively. At the same concentrations, the numbers of migrated cells were 210.67±8.50, 156.50±12.16, and 108.00±7.85, respectively, in the ES2-AF group and the numbers of migrated cells in the mixture group were 201.33±19.61, 162.50±11.90, and 125.67±5.65, respectively.
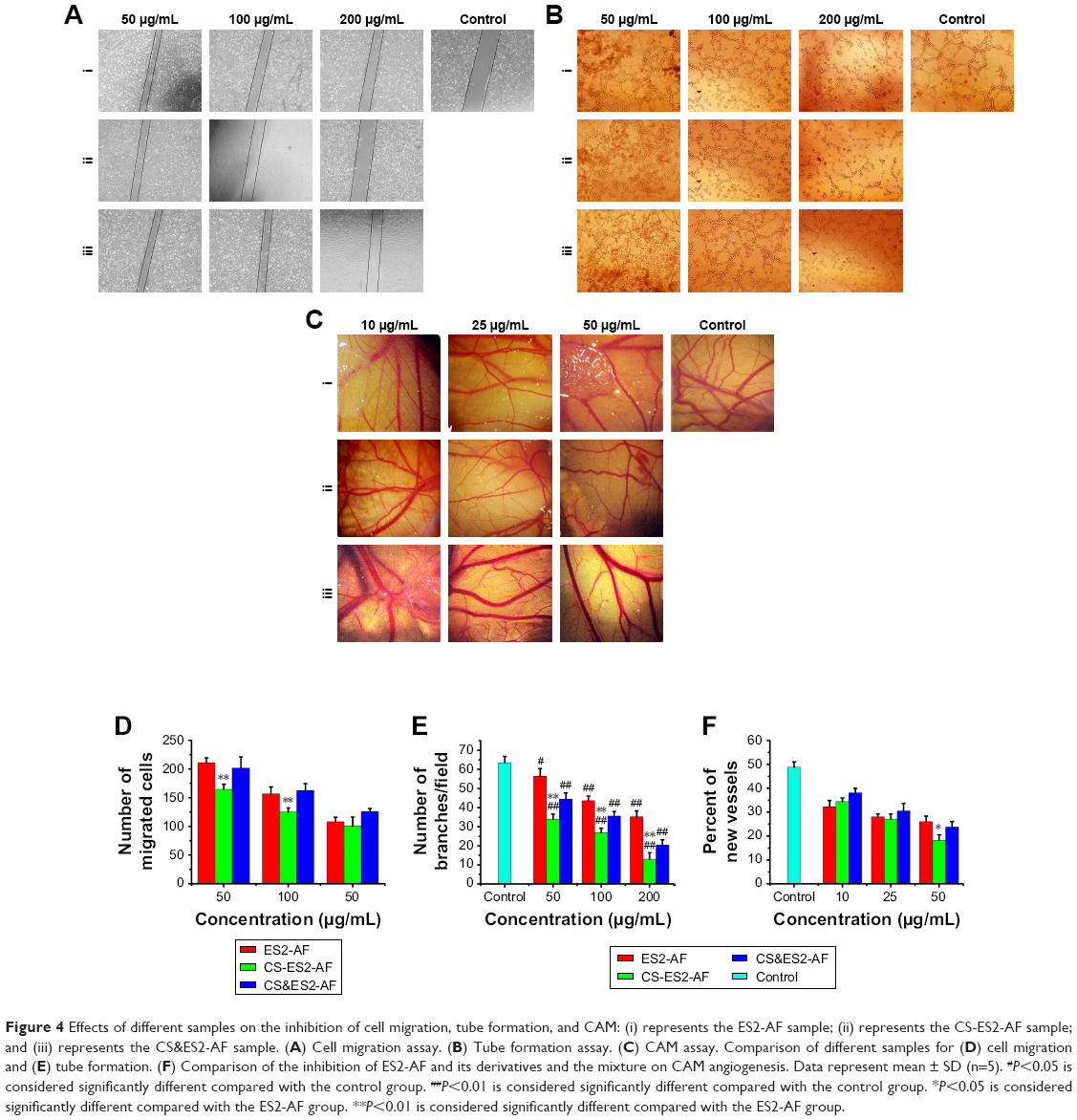 | Figure 4 Effects of different samples on the inhibition of cell migration, tube formation, and CAM: (i) represents the ES2-AF sample; (ii) represents the CS-ES2-AF sample; and (iii) represents the CS&ES2-AF sample. (A) Cell migration assay. (B) Tube formation assay. (C) CAM assay. Comparison of different samples for (D) cell migration and (E) tube formation. (F) Comparison of the inhibition of ES2-AF and its derivatives and the mixture on CAM angiogenesis. Data represent mean ± SD (n=5). #P<0.05 is considered significantly different compared with the control group. ##P<0.01 is considered significantly different compared with the control group. *P<0.05 is considered significantly different compared with the ES2-AF group. **P<0.01 is considered significantly different compared with the ES2-AF group. |
For different samples at the same concentration, the activity of CS-ES2-AF was higher than the activity of ES2-AF, and when the concentrations were 50 μg/mL and 100 μg/mL, the effect of CS-ES2-AF on the endothelial cell migration was significantly better than that of ES2-AF. Like the MTT assay results, even when the concentration was relatively low, CS could improve the solubility of ES2-AF. CS could increase the number of water molecules near ES2-AF and increase the stability of ES2-AF. Thus, CS-ES2-AF could inhibit the endothelial cell metastasis more efficiently than ES2-AF. When the concentration was increased to 200 μg/mL, the inhibitory effect of CS-ES2-AF on the endothelial cells was similar to that of ES2-AF. Thus, CS could promote the adhesion and growth of endothelial cells, and, to a certain extent, counteract the inhibitory effect of ES2-AF.
Tube formation assay
The tube formation assay was used to investigate the effects of ES2-AF, CS-ES2-AF, and CS&ES2-AF on blood vessel formation in EA.hy926 cells.23,24 The number of tubes is represented by the number of branches/fields. Different concentrations of the drugs were added to 48-well plates and incubated for 8 hours. The plates were photographed under a fluorescence microscope (Figure 4B); the tube formation of the endothelial cells could be observed from the figure. A large number of tubes were formed in the control group after incubation for 8 hours. Compared with the control group, the number of tubes formed in the low-concentration experimental group was less, indicating that the low concentration of the drug could also inhibit the endothelial cell tube formation. When the concentration was increased to 200 μg/mL, the number of tube branches/field in the CS-ES2-AF group was significantly reduced, and most cells were isolated, while more tube branches/field were still visible in the ES2-AF group.
After processing the data (Figure 4E), it was found that the number of tubes formed in the control group was 63.40±3.36. When the concentrations were 50 μg/mL, 100 μg/mL, and 200 μg/mL, the numbers of branches/fields in the CS-ES2-AF group were 33.80±2.77, 26.80±2.39, and 13.00±3.39, respectively. At the same concentrations, the numbers of branches/fields in the ES2-AF and CS&ES2-AF mixture groups were 56.40±4.03, 43.60±2.41, and 35.20±3.03; and 44.40±3.29, 35.60±2.30, and 20.40±2.70, respectively. These results could be verified with intuitive observations. Thus, at concentrations of 50–200 μg/mL, the effect of CS-ES2-AF on the inhibition of tube formation in endothelial cells is superior to that of ES2-AF.
CAM assay
The traditional CAM test was used to study the effects of the drugs on angiogenesis in vivo. The anti-angiogenic ability of the drug is expressed as a percentage of neovascularization.28,29 Under the stereoscopic microscope, the CAM vascular network of the control group showed a clear vein-like distribution after the addition of physiological saline and bFGF (Figure 4C). The number of small vessels generated around the main vessel increased, the small blood vessels grew exuberantly, and then, produced more branches, thereby increasing the density of the whole blood vessels in the field. Within the same drug group, the density of the blood vessels decreased significantly when the concentration of the drug increased, which showed that the activity of the CAM was positively correlated with the concentration. At the same concentrations, the activity of the CS&ES2-AF mixture group was slightly less than the other two groups. The chemically modified product CS-ES2-AF had the smallest number of small blood vessels around the main blood vessels, and almost no visible small blood vessels existed in the field of view under the stereoscope.
To corroborate the above results, the newly generated vessels in each group were statistically analyzed (Figure 4F). After drug treatment for 24 hours, the neovascularization rate of the control group was 48.75%±2.27%. When the concentrations of drug were 10 μg/mL, 25 μg/mL, and 50 μg/mL, the neovascularization rates in the CS-ES2-AF group were 34.40%±1.54%, 27.14%±2.08%, and 18.25%±2.62%, respectively. At these same concentrations, the proportions of new vessels in the ES2-AF and CS&ES2-AF groups were 32.29%±2.54%, 28.02%±1.23%, and 26.02%±2.28%; and 38.11%±1.81%, 30.51%±3.13%, and 23.79%±2.29%, respectively. Comparing the groups, it was found that when the concentration was 10 μg/mL–25 μg/mL, there was no significant difference in the activity of ES2-AF and the modified product CS-ES2-AF. When the concentration was 10 μg/mL, the activity of CS-ES2-AF was lower than that of the unmodified polypeptide ES2-AF. This might be due to the water-soluble CS forming a gel-like barrier near ES2-AF at low concentrations, affecting the contact between ES2-AF and the chicken embryos, thereby reducing the effect of ES2-AF, and making the conjugates less active at low concentration. When the concentration was 50 μg/mL, the CS-ES2-AF activity was higher than the unmodified polypeptide ES2-AF. This might be due to the increase in stability of CS-ES2-AF in vivo, thereby improving the activity.
ELISA
The inhibitory effect of CS-ES2-AF on the binding capacity of VEGF to its receptor VEGFR1 (Flt-1) was determined by ELISA.10,30 The absorbance of the positive control well was set to 100%, and the ratio of the experimental well and the contrast well absorption values was used to express the inhibitory ability of the different experimental samples. As shown in Figure 3B, the inhibitory ability of the three experimental groups increased significantly with increasing concentrations in a range of 5–500 μg/mL, indicating that the inhibition occurred in a concentration-dependent manner. At the same concentration, the inhibition of CS-ES2-AF was significantly better than both ES2-AF and CS&ES2-AF, indicating that the glycosylation (modification) significantly enhanced the ability of ES2-AF to inhibit the binding of VEGF to VEGFR1 (Flt-1). This was consistent with the cell proliferation experimental results mentioned earlier.
In this study, the active fragment AF inhibited the binding of VEGF to its receptor. Because the AF peptide contains several aromatic amino acids, it has poor water solubility. ES2 peptide and water-soluble mucopolysaccharide CS were used to modify AF, which resulted in a modified product CS-ES2-AF with improved water solubility; thus, CS-ES2-AF inhibited the binding of VEGF to its receptor VEGFR1 better than ES2-AF. In addition, glycosylation could also increase the stability of ES2-AF in aqueous solution.5 Compared to unmodified ES2-AF, the modified CS-ES2-AF was more stable at 37°C and, thus, showed better inhibition than ES2-AF, a result that could also be verified by CAM experiments.
SPR assay
The pH value of the most suitable coupling reaction was screened by pre-enrichment experiments to ensure that sufficient CD44 protein was coupled onto the CM5 chip to facilitate the next screening of drugs. The experimental results showed that when the pH value of the CD44 protein solution was 3.8, the response value of the protein-chip binding was the highest; pH 3.8 was thus taken as the ideal ligand coupling condition (Figure 5A). Ligand conjugation was performed under these conditions with a 10 μg/mL concentration of the coupled protein CD44 solution, and the final protein coupling amount was 394.8 RU.
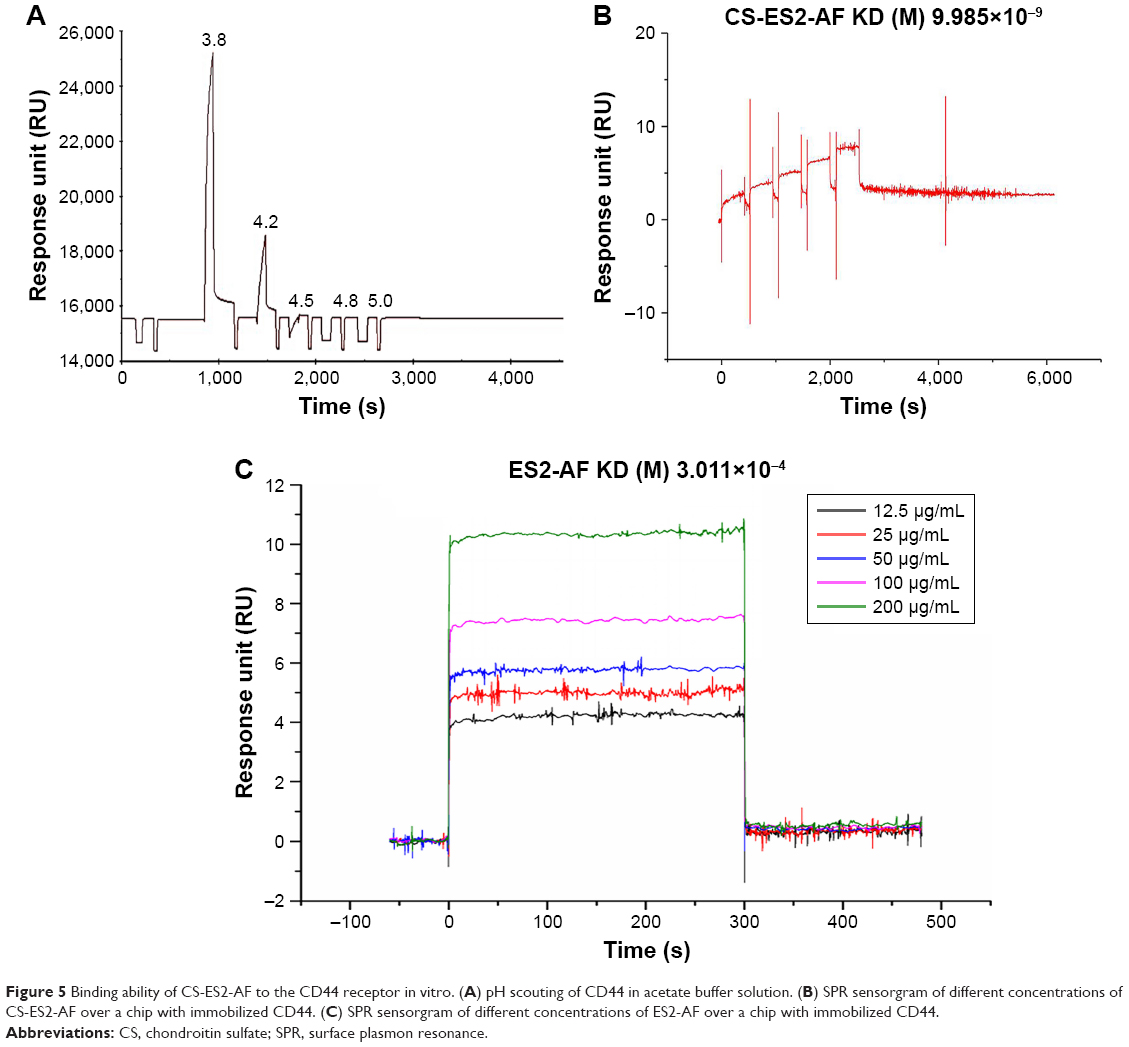 | Figure 5 Binding ability of CS-ES2-AF to the CD44 receptor in vitro. (A) pH scouting of CD44 in acetate buffer solution. (B) SPR sensorgram of different concentrations of CS-ES2-AF over a chip with immobilized CD44. (C) SPR sensorgram of different concentrations of ES2-AF over a chip with immobilized CD44. |
CS-ES2-AF flowed through the CM5 chip to generate dynamic sensing fitting plots. The affinity kinetic parameters Ka (L/(mol × s)) and Kd (1/s) of the interaction of CD44 protein with CS-ES2-AF and ES2-AF were 6,846, 6.863×10−5 and 0, 0, respectively. Here, Ka is the binding constant between the sample and CD44, Kd is the dissociation constant of the sample and CD44, and KD (Kd/Ka) is the equilibrium dissociation constant, which was used to measure the ability of CD44 to interact with the sample; the smaller the KD value, the stronger the affinity of CD44 to the sample. The KD value of ES2-AF and CD44 was 3.011×10−4 (Figure 5C), while the KD value of CS-ES2-AF and CD44 was much smaller, 9.985×10−9 (Figure 5B). This indicated that the binding affinity of the CS-modified glycosylated polypeptide ES2-AF to the CD44 receptor was better than the affinity of the unmodified polypeptide ES2-AF.
In general, a Kd value in the range of 10−5–10−12 indicates a strong sample-to-ligand affinity.43 According to the results presented here, the affinity of the polypeptide ES2-AF to the CD44 receptor was weak, and CS, a ligand of the CD44 receptor, can bind specifically to the CD44 receptor. Therefore, the affinity of CS to CD44 receptors was theoretically stronger than the unmodified ES2-AF, and it could be concluded that CS could increase the targeting of drugs to tumor tissues.
Tissue distribution and targeting
The nude mice bearing melanoma B16 were randomly divided into two groups. When the tumor volume reached 30 mm3, ES2-AF-FITC and CS-ES2-AF-FITC were administered by tail vein. The nude mice were anesthetized by intraperitoneal injection of chloral hydrate. When the nude mice were stable under anesthesia, they were photographed in a live imager to record the in vivo bioimaging at different time points.
As shown in Figure 6, no fluorescence was observed in both the groups of mice at 0 hour, indicating that the mice had no fluorescence interference, and the fluorescence captured by the in vivo bioimaging was from the drug distributed within the mice. The distribution and metabolism of the drug ES2-AF-FITC in nude mice were as follows. At 0.5 hour, ES2-AF-FITC was distributed throughout the whole body of the nude mice. Fluorescence appeared in the tumor; there was no significant difference between the fluorescence intensity in the tumor cells and in the other parts of the mice. The image showed that ES2-AF-FITC had entered the tumor tissue. At 1 hour, the fluorescence intensity within the tumor weakened, indicating that some ES2-AF-FITC had been metabolized. At 2 hours and 4 hours, there was a strong fluorescent signal in the urethral opening of the nude mice, indicating that the ES2-AF-FITC had circulated in the body and entered the bladder from the kidneys, which was excreted in the form of urine.
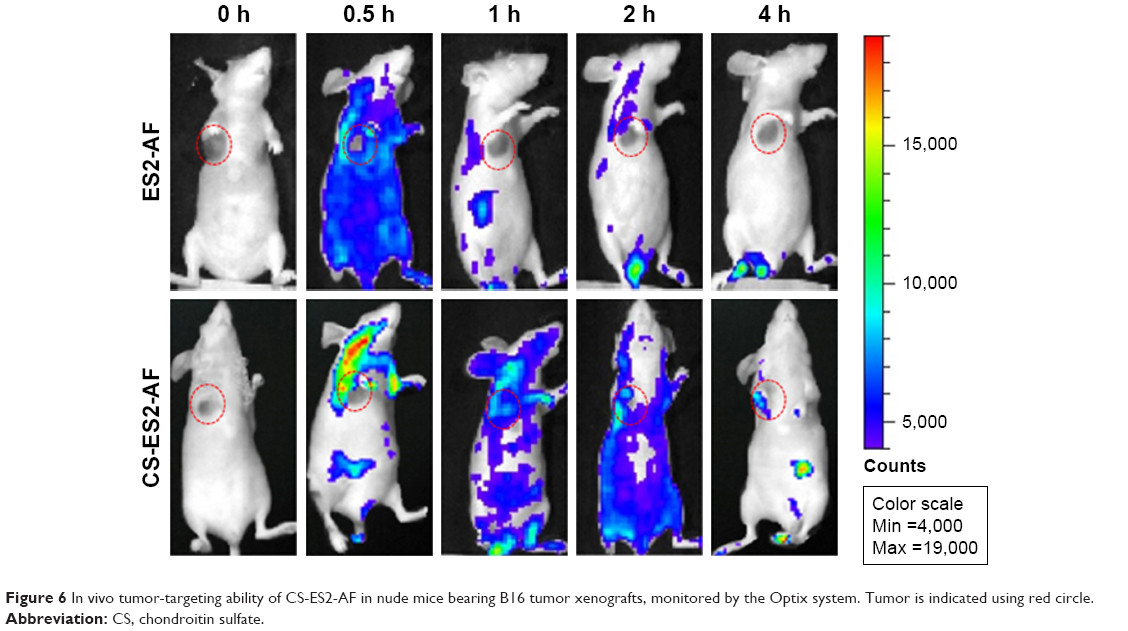 | Figure 6 In vivo tumor-targeting ability of CS-ES2-AF in nude mice bearing B16 tumor xenografts, monitored by the Optix system. Tumor is indicated using red circle. |
When CS-ES2-AF-FITC was used, a strong fluorescence signal was observed at the tumor site and in the cervical lymph nodes near the tumor at 0.5 hours, indicating that there was a large CS-ES2-AF-FITC distribution. The fluorescence signals in the other parts of the mice were weak, indicating that the distribution of CS-ES2-AF-FITC in the tumor-bearing nude mice was regular. At 1, 2, and 4 hours, the fluorescence signal still appeared in the tumor site, and the fluorescence intensity was stronger than in the other tissues in the body. At the same time, almost no fluorescence appeared at the tumor site of the ES2-AF-FITC group, indicating that the metabolism of CS-ES2-AF-FITC was slower than ES2-AF-FITC. This is because glycosylation induces structural changes in the conjugates and polysaccharide chain increases the steric hindrance of the conjugate. This improves the drug’s ability to resist hydrolysis by hydrolase enzymes, making it stable in the blood, thereby demonstrating that a glycosylation modification can prolong the stability of a drug in the body. The fluorescence intensity of the glycosylated compound at the tumor site was stronger than the polypeptide group, demonstrating that CS had a certain ability to target tumor tissue in B16-bearing nude mice, which was consistent with the SPR experimental results.
Pharmacokinetics study
Blood samples were collected at the predetermined time points from BALB/c mice and processed, the fluorescence intensity was measured, and the concentration of drug in the plasma was calculated. The pharmacokinetic time curves of the mice treated with CS-ES2-AF and ES2-AF are presented in Figure 7. The plasma concentration of ES2-AF, which had not been modified by glycosylation, was lower than the lowest limit of quantitation after 12 hours of administration, and ES2-AF was not detected in plasma after 24 hours. While the plasma concentration of the glycosylated product CS-ES2-AF was lower than the ES2-AF group at 0.5 hour, the plasma concentrations were higher at the other time points.
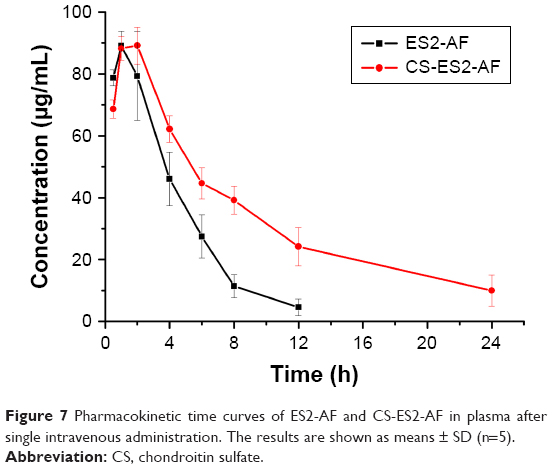 | Figure 7 Pharmacokinetic time curves of ES2-AF and CS-ES2-AF in plasma after single intravenous administration. The results are shown as means ± SD (n=5). |
The data were simulated by DAS2.0 software. The pharmacokinetic parameters of CS-ES2-AF and ES2-AF are shown in Table 1. Compared with the unmodified ES2-AF, the half-life and average residence time of the glycosylation-modified product CS-ES2-AF was significantly prolonged, and the clearance rate was decreased in vivo. It was also found that the peak concentrations of the two drugs were similar. Thus, the plasma concentration of CS-ES2-AF in mice increased; maintenance of a high plasma concentration for a long time is expected to reduce the dosage and frequency of administration in clinical applications.
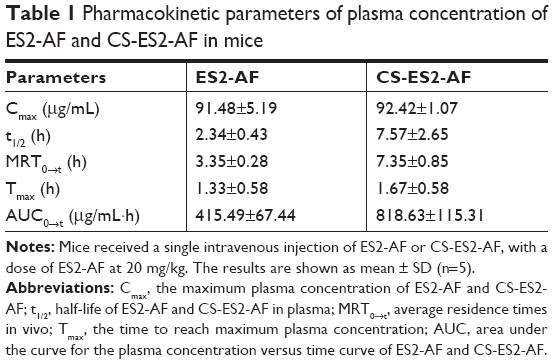 | Table 1 Pharmacokinetic parameters of plasma concentration of ES2-AF and CS-ES2-AF in mice |
Discussion
Solid-phase synthesis method has been extensively used in the field of protein/peptide drugs research to synthesize polypeptides. This method can quickly and accurately synthesize the designed target peptide chain, and the products synthesized can be quickly and accurately purified and identified by HPLC and MS. In this study, polypeptide ES2-AF was successfully synthesized by this method, with 95.41% purity and a molecular weight of 298.12. The results indicated that polypeptide ES2-AF can be synthesized by solid-phase method and the peptide can be used in future studies.
The active peptide that inhibits endothelial cell proliferation in the ES2-AF peptide is ES2, which is derived from the ES sequence (amino acid position 60–70) and is about three times more active than the ES sequence.28 The ES2 can directly block tyrosine phosphorylation of VEGF receptor on the endothelial cell surface, thereby blocking the transmission of VEGF-mediated signaling pathway.44 In addition, studies have speculated that nucleolin may be a potential receptor for ES2 in the nucleus of endothelial cells. When ES2-AF enters the endothelial cells, the ES2 fragment can inhibit the phosphorylation process of the nucleolus, directly affecting the biosynthesis and maturation of ribosomes and indirectly influencing the process of chromatin replication and nucleogenesis. Then, the mitotic process of the endothelial cells is arrested, inhibiting the endothelial cell proliferation.45
The MTT assay was used to evaluate the inhibitory effects of ES2-AF and its glycosylation modification products on the proliferation of EA.hy926 cells. It was found that the inhibitory activities of both the drugs were dose-dependent. The activity of polypeptide drugs is often related to their structure. Compared with small molecule drugs, polypeptide drugs tend to have poor stability in aqueous solution due to their spatial structure. The peptide ES2-AF was modified with CS, and the stability of the modified product CS-ES2-AF was improved in aqueous solution. The CS can increase the solubility of ES2-AF in aqueous solution as the CS group forces water molecules around the polypeptide ES2-AF to improve the stability of ES2-AF. Moreover, the higher the concentration of ES2-AF, the more the stability of ES2-AF. Previous research also supports this conclusion.46 In addition, MTT results are also related to cell status and degree of differentiation, so cells with better status should be selected for experiments.
Studies have shown that ES could stably bind to the precursor protein of matrix metalloproteinase to form a complex,47 blocking the activation and catalytic ability of matrix metalloproteinases, whereas matrix metalloproteinases could create necessary conditions for cell migration by dissolving the extravascular matrix. ES2, as the main active fragment of ES, also has the ability to stably bind to the precursor proteins of matrix metalloproteinase. The results of cell metastasis showed that the activity of CS-ES2-AF in inhibiting cell metastasis was significantly better than unmodified ES2-AF at low concentrations, and there was no significant difference between CS-ES2-AF and ES2-AF at high concentration. In agreement with the results of MTT, the efficiency of CS in improving the solubility of ES2-AF increased at low drug concentration. Therefore, CS-ES2-AF was more efficient in inhibiting endothelial cell metastasis than ES2-AF. When the concentration was increased, the activity of the two drugs was equivalent, which might be due to CS promoting the adhesion and growth of endothelial cells to counteract the inhibition of ES2-AF at a certain degree.48
Endothelial cell proliferation and differentiation are the vital parts in neovascularization. Matrigel is a soluble basement membrane matrix that is similar to mammalian cell basement membrane components.49 Related studies have shown that the matrigel, as an extracellular mechanism complex, added to the cell culture environment could promote cell proliferation, differentiation, and collagen secretion.50 The mechanism of ES inhibiting endothelial cells tube formation is similar to that of inhibiting endothelial cell migration. At the same time, endothelial cell migration and tube formation is a continuous process. ES can disrupt the integrity of microfilaments, thereby disrupting normal adhesion between these endothelial cells and between the endothelial cells and the matrix. Ultimately, the growth of endothelial cells is limited and the formation of vessels is blocked.51
The CAM model is a widely used angiogenic model due to its many advantages: cost effectiveness, short experiment period, high reproducibility, and easy handling. In the experiment, normal saline was used as the control group and a small amount of bFGF was added to each group. bFGF is an inducing factor for vascular growth, which makes the results of neovascularization more obvious. The result showed that the activity of CS-ES2-AF in the high-concentration group was better than ES2-AF, which could be because the stability of CS-ES2-AF was better than that of ES2-AF. However, comparing the results of mixture group with that of peptide group at a low concentration condition, it was found that adding CS in mixture had an effect on the activity of peptide and could increase the activity of mixture. It was also found that the activity of conjugated group was slightly lower than that of peptide group at this concentration. This phenomenon may be due to the fact that CS in the conjugate could form a gel-like barrier structure near ES2-AF at a lower concentration. Therefore, the conjugate and mixture groups were inferior in activity at low concentrations.
The VEGF/VEGFR1 (Flt-1) pathway is an important signaling pathway that can regulate the proliferation, division, and migration of vascular endothelial cells.52 Endothelial cells can be regulated from the beginning of the signal by inhibiting the binding of VEGF to its receptor, thereby inducing endothelial cell apoptosis and migration inhibition, thus reducing the formation of vessels in tumor tissue. In this study, the main active fragment that inhibited the binding of VEGF to its receptor was short peptide AF section. The water solubility of AF is poor due to its high content of aromatic amino acids. Therefore, AF was modified with ES2 peptide by solid-phase synthesis to improve its water solubility. Then, ES2-AF was modified with CS, a better water-soluble mucopolysaccharide, to further improve the water-solubility of ES2-AF. The water solubility of AF was significantly improved after modification, so the ability of CS-ES2-AF to inhibit the binding of VEGF to its receptor, VEGFR1, was stronger than that of ES2-AF.
At present, the methods commonly used to detect drug activity include cell screening methods and biochemical screening methods. Compared with cell screening methods, biochemical screening methods are widely used because they are rapid, simple, and efficient.53 The SPR method is one of the widely used biochemical screening methods because it is a real-time, fast, high-flux method and does not require special marking. The technique utilizes the principle of physical optics. When studying the interaction of two molecules, one molecule is fixed on the surface of the sensor sheet, and the solution of another molecule flows through the surface. The combination of the two molecules will change the refractive index of the sensor surface, so the interaction between the two molecules can be detected.
CD44 protein is often expressed in some malignant and highly metastatic tumor cells, and the prognosis of these tumors is not satisfactory after chemotherapy. Related studies have shown that CD44 was closely related to the invasion and metastasis of many malignant tumor cells.54 CD44 protein could be detected in various tumor cells, such as gastric carcinoma,55 glioma,56 breast cancer,57,58 and pancreatic cancer.59,60 Thus, CD44 can be used for targeted tumor therapy, and the therapeutic efficacy of the tumor can be improved by designing a drug that targets CD44 receptor.
In SPR study, the pH value of the suitable CD44 protein solution was screened to ensure the coupling of more CD44 protein to the CM5 chip, which facilitated the screening of the drug in the next step. When the ligand protein CD44 is coupled to the chip surface, it is necessary to dissolve the protein in a buffer solution with pH lower than its isoelectric point and higher than pH 3.5, so that the protein surface is positively charged, and the carboxyl groups with negative charges on the chip surface are combined by electrostatic adsorption. The isoelectric point of CD44 was pH 5.15, so the optimum pH value of CD44 protein solution was 3.8 after screening. The samples were dissolved in PBS buffer and diluted with running buffer (50 mM phosphate, 100 mM sodium chloride, and 0.01% vol/vol Tween 20, pH 7.4). Then, different concentrations of the sample were flowed through the surface of the chip immobilized with the CD44 receptor protein and the Kd values were obtained by Biacore T2000. In general, when the value of KD was in the range of 10−5–10−12, it meant that the sample had a strong affinity with the ligand protein.33 Because CS is the ligand of CD44 receptor and can specifically bind to CD44 receptor, it had a stronger affinity toward CD44 receptor than the unmodified ES2-AF. Therefore, CS was used to modify ES2-AF to improve the targeting of drugs to tumor tissues.
In vivo imaging is a new technology that qualitatively or quantitatively analyzes the distribution and metabolism of drugs in the living body by using various imaging modes at the cellular and molecular level. Compared with traditional in vitro imaging techniques or slice observation, in vivo imaging is better. First, it can obtain multiple data on the same animal, which helps to eliminate individual differences. Second, it can be continuously observed for a period of time, and dynamic experimental results and intuitive drug distribution images can be obtained, so that the experimental results can be seen at a glance. Finally, it is a non-invasive and painless detection technology of biological behavior in vivo, which complies with the requirements of experimental ethics. Therefore, small animal in vivo imaging technology is widely used in the field of oncology research.
As many substances in organisms can produce fluorescence, such as skin, hair, and rat food, nude mice were selected as experimental animals and fasted 12 h before the experiment. The background noise was removed or attenuated by photographing nude mice before administration.
Comparing the distribution and metabolism of peptides and their modifications in tumor-bearing nude mice, it was found that the glycosylation-modified products had a longer metabolism time in nude mice. This might be because glycosylation modification changed the structure of the conjugates, increasing their steric hindrance and thus improving the ability to resist hydrolysis by hydrolases. Glycosylation also increases the stability of the conjugates in blood. The fluorescence intensity of the glycosylation-modified product at the tumor site was stronger than that of polypeptide group, demonstrating the ability of CS to target tumor tissues, which was consistent with the SPR results.
Protein or peptide drugs are affected by many factors in pharmacokinetic studies compared to small molecule chemicals. On one hand, the sensitivity of the analytical method is an issue since the dosage of the polypeptide drug is small, and, on the other hand, the amino acids of the polypeptide drugs are susceptible to the endogenous substances in organisms as most often they are natural amino acids. Therefore, it is required to distinguish the amino acids and endogenous substances of exogenous polypeptide drugs in the analysis process.
In the pharmacokinetic study of peptide drugs, several commonly used methods include isotopic tracer method,61 ELISA,10 liquid chromatography-mass spectrometry (LC-MS),62,63 and fluorescence labeling. At present, the US FDA uses the pharmacokinetic data obtained by radioisotope determination of peptide drugs for the effective evaluation of the safety of the drugs. However, polypeptides labeled with radioisotopes, such as 125I are prone to be labeled off-label during the experiment, as they are unable to accurately reflect the concentration of the polypeptide drugs from the measured radioactivity. This method also has the shortcomings of radioactive contamination, thus limiting its application in clinical pharmacokinetics research. Although ELISA method is simple and convenient to operate, it is necessary to develop related antibody-coated kits. The preparation period of the kit is not long and it is not susceptible to interference by biological endogenous and exogenous substances during the measurement process. The LC/MS has the advantages of good specificity and high sensitivity. However, plasma samples contain a large amount of salts and endogenous proteins, which may interfere with the measurement process. Fluorescence labeling has been used by increasing number of people because of its mild reaction conditions and simple detection method.
ES2-AF, as a new anti-angiogenic peptide prepared by solid-phase synthesis technology, has no light absorption and chromogenic groups, so it cannot be directly determined by spectroscopy or chromatography. As a common fluorescent marker, FITC, due to its mild labeling reaction conditions, is often used to label polysaccharides and polypeptides to study their pharmacokinetic process in animals. In this study, FITC was used to label ES2-AF. FITC could be linked to several amino acids in the ES2-AF peptide chain. After dialysis, free FITC was removed and ES2-AF-FITC and CS-ES2-AF-FITC samples were prepared.
To enhance the fluorescence intensity of the sample, 0.1% SDS solution was used as fluorescence sensitizer. The addition of SDS could produce a large amount of pre-micelles in the sample system due to which the FITC-labeled molecules dispersed and adhered to the micelle system. The collision energy loss between the fluorescent molecules was reduced, the quenching effect of oxygen was reduced, and the absorption interface of fluorescent groups was increased. The fluorescence sensitization by the SDS solution brought order to the fluorophore microenvironment in biological samples. This reduced the competition with other non-radiative deactivators and fluorescence quenching processes in the system, and improved the quantum yield of fluorescence. This was manifested by an increase in the fluorescence intensity of the sample.
Among the pharmacokinetic parameters obtained in this study, the clearance rate of CS-ES2-AF in vivo was about 50% of that of ES2-AF, and the half-life of CS-ES2-AF in mice was about three times that of ES2-AF. The reason for this phenomenon might be because the fluorescence intensity of FITC represents the concentration of ES2-AF in this study, but the metabolism of FITC and ES2-AF was not completely synchronized in vivo, and ES2-AF-FITC and free FITC might coexist in mouse plasma, which affected the determination of the results. Blood samples were taken from several mice at each time point, and there were some individual differences between the mice, which led to some inconsistencies in pharmacokinetic parameters.
Conclusion
To conclude, ES2-AF was synthesized by a solid-phase method in this study and CS-ES2-AF was obtained by chemical modification of a new peptide ES2-AF with CS. First, 1H-NMR spectrum showed that CS successfully bound to ES2-AF and that there were two peptides per one CS chain. Then, the in vitro activities of ES2-AF and CS-ES2-AF were studied. The results showed that CS-ES2-AF had better ability to inhibit the proliferation and metastasis of endothelial cells than ES2-AF. Then, the binding ability of ES2-AF and CS-ES2-AF in vitro and in vivo was studied. ELISA results showed that the glycosylation-modified product CS-ES2-AF inhibited the binding ability of VEGF to its receptor, better than the peptide ES2-AF; SPR results showed that the binding ability of CS-ES2-AF to CD44 receptor was better than that of ES2-AF; in vivo imaging experiments showed that the distribution of CS-ES2-AF in tumor-bearing nude mice was targeted. In addition, bioimaging results showed that CS-ES2-AF had a longer metabolic time compared to ES2-AF in vivo, which may be due to the increased stability of CS-ES2-AF in plasma. That was consistent with the in vivo half-life experiments. The half-life of CS-ES2-AF was approximately threefold longer than that of ES2-AF. These data strongly suggest that CS has the desired effect of extending the half-life of this peptide drug.
In conclusion, ES2-AF that has been modified by CS has improved anti-neovascularization activity in vitro and in vivo, enhanced binding ability to different receptors in vitro and in vivo, targeted the tumor tissues, and significantly prolonged the half-life in mice. The results of this study have laid an experimental foundation for further development of targeted anti-tumor polypeptide drugs.
Acknowledgments
This work was supported by National Natural Science Foundation of China (81473129), Shandong Provincial Key Research and Development Program (2015GSF118099), Shandong Provincial Natural Science Foundation (ZR2015HM043), and the Fundamental Research Funds of Shandong University (2014JC044).
Disclosure
The authors report no conflicts of interest in this work.
References
Novoa-Carballal R, Perez-Martin R, Blanco M, et al. By-products of Scyliorhinus canicula, Prionace glauca and Raja clavata: A valuable source of predominantly 6S sulfated chondroitin sulfate. Carbohydr Polym. 2017;157:31–37. doi:10.1016/j.carbpol.2016.09.050 | ||
Yamada S, Sugahara K. Potential therapeutic application of chondroitin sulfate/dermatan sulfate. Curr Drug Discov Technol. 2008;5(4):289–301. | ||
Zhang JS, Imai T, Suenaga A, Otagiri M. Molecular-weight-dependent pharmacokinetics and cytotoxic properties of cisplatin complexes prepared with chondroitin sulfate A and C. Int J Pharm. 2002;240(1–2):23–31. | ||
Onishi H, Matsuyama M. Conjugate between chondroitin sulfate and prednisolone with a glycine linker: preparation and in vitro conversion analysis. Chem Pharm Bull (Tokyo). 2013;61(9):902–912. | ||
Lo YL, Sung KH, Chiu CC, Wang LF. Chemically conjugating polyethylenimine with chondroitin sulfate to promote CD44-mediated endocytosis for gene delivery. Mol Pharm. 2013;10(2):664–676. doi:10.1021/mp300432s | ||
Tjin Tham Sjin RM, Satchi-Fainaro R, Birsner AE, Ramanujam VM, Folkman J, Javaherian K. A 27-amino-acid synthetic peptide corresponding to the NH2-terminal zinc-binding domain of endostatin is responsible for its antitumor activity. Cancer Res. 2005;65(9):3656–3663. doi:10.1158/0008-5472.CAN-04-1833 | ||
Wickstrom SA, Alitalo K, Keski-Oja J. An endostatin-derived peptide interacts with integrins and regulates actin cytoskeleton and migration of endothelial cells. J Biol Chem. 2004;279(19):20178–20185. doi:10.1074/jbc.M312921200 | ||
Xu HL, Tan HN, Wang FS, Tang W. Research advances of endostatin and its short internal fragments. Curr Protein Pept Sci. 2008;9(3):275–283. | ||
Yu Y, Sun F, Zhang C, Wang Z, Liu J, Tan H. Study on glyco-modification of endostatin-derived synthetic peptide endostatin2 (ES2) by soluble chitooligosaccharide. Carbohydr Polym. 2016;154:204–213. doi:10.1016/j.carbpol.2016.08.043 | ||
Oh EJ, Choi JS, Kim H, Joo CK, Hahn SK. Anti-Flt1 peptide – hyaluronate conjugate for the treatment of retinal neovascularization and diabetic retinopathy. Biomaterials. 2011;32(11):3115–3123. doi:10.1016/j.biomaterials.2011.01.003 | ||
Pasut G, Veronese FM. State of the art in PEGylation: the great versatility achieved after forty years of research. J Control Release. 2012;161(2):461–472. doi:10.1016/j.jconrel.2011.10.037 | ||
Schumacher D, Hackenberger CP. More than add-on: chemoselective reactions for the synthesis of functional peptides and proteins. Curr Opin Chem Biol. 2014;22:62–69. doi:10.1016/j.cbpa.2014.09.018 | ||
Suganuma T, Ino K, Shibata K, et al. Functional expression of the angiotensin II type 1 receptor in human ovarian carcinoma cells and its blockade therapy resulting in suppression of tumor invasion, angiogenesis, and peritoneal dissemination. Clin Cancer Res. 2005;11(7):2686–2694. doi:10.1158/1078-0432.CCR-04-1946 | ||
Armstrong JK, Hempel G, Koling S, et al. Antibody against poly(ethylene glycol) adversely affects PEG-asparaginase therapy in acute lymphoblastic leukemia patients. Cancer. 2007;110(1):103–111. doi:10.1002/cncr.22739 | ||
Slevin M, Krupinski J, Gaffney J, et al. Hyaluronan-mediated angiogenesis in vascular disease: uncovering RHAMM and CD44 receptor signaling pathways. Matrix Biol. 2007;26(1):58–68. doi:10.1016/j.matbio.2006.08.261 | ||
Stern R, Asari AA, Sugahara KN. Hyaluronan fragments: an information-rich system. Eur J Cell Biol. 2006;85(8):699–715. doi:10.1016/j.ejcb.2006.05.009 | ||
Oh EJ, Park K, Choi JS, Joo CK, Hahn SK. Synthesis, characterization, and preliminary assessment of anti-Flt1 peptide-hyaluronate conjugate for the treatment of corneal neovascularization. Biomaterials. 2009;30(30):6026–6034. doi:10.1016/j.biomaterials.2009.07.024 | ||
Sun F, Yu Y, Yang Z, et al. Hyaluronic acid-endostatin2-alft1 (HA-ES2-AF) nanoparticle-like conjugate for the target treatment of diseases. J Control Release. 2018;288:1–13. doi:10.1016/j.jconrel.2018.08.038 | ||
Chen FF, Liu Y, Wang F, et al. Effects of upregulation of Id3 in human lung adenocarcinoma cells on proliferation, apoptosis, mobility and tumorigenicity. Cancer Gene Ther. 2015;22(9):431–437. doi:10.1038/cgt.2015.38 | ||
Kim H, Choi JS, Kim KS, Yang JA, Joo CK, Hahn SK. Flt1 peptide-hyaluronate conjugate micelle-like nanoparticles encapsulating genistein for the treatment of ocular neovascularization. Acta Biomater. 2012;8(11):3932–3940. doi:10.1016/j.actbio.2012.07.016 | ||
Huang XY, Zhang XM, Chen FH, et al. Anti-proliferative effect of recombinant human endostatin on synovial fibroblasts in rats with adjuvant arthritis. Eur J Pharmacol. 2014;723:7–14. doi:10.1016/j.ejphar.2013.10.068 | ||
Liang DS, Su HT, Liu YJ, Wang AT, Qi XR. Tumor-specific penetrating peptides-functionalized hyaluronic acid-d-alpha-tocopheryl succinate based nanoparticles for multi-task delivery to invasive cancers. Biomaterials. 2015;71:11–23. doi:10.1016/j.biomaterials.2015.08.035 | ||
Bai Y, Zhao M, Zhang C, et al. Anti-angiogenic effects of a mutant endostatin: a new prospect for treating retinal and choroidal neovascularization. PLoS One. 2014;9(11):e112448. doi:10.1371/journal.pone.0112448 | ||
Xu X, Mao W, Chen Q, et al. Endostar, a modified recombinant human endostatin, suppresses angiogenesis through inhibition of Wnt/beta-catenin signaling pathway. PLoS One. 2014;9(9):e107463. doi:10.1371/journal.pone.0107463 | ||
Bai YJ, Huang LZ, Zhou AY, Zhao M, Yu WZ, Li XX. Antiangiogenesis effects of endostatin in retinal neovascularization. J Ocul Pharmacol Ther. 2013;29(7):619–626. doi:10.1089/jop.2012.0225 | ||
Wang F, Bai Y, Yu W, et al. Anti-angiogenic effect of KH902 on retinal neovascularization. Graefes Arch Clin Exp Ophthalmol. 2013;251(9):2131–2139. doi:10.1007/s00417-013-2392-6 | ||
Yu WZ, Li XX, She HC, et al. Gene transfer of kringle 5 of plasminogen by electroporation inhibits corneal neovascularization. Ophthalmic Res. 2003;35(5):239–246. doi:10.1159/000072143 | ||
Qiu Z, Hu J, Xu H, Wang W, Nie C, Wang X. Generation of antitumor peptides by connection of matrix metalloproteinase-9 peptide inhibitor to an endostatin fragment. Anticancer Drugs. 2013;24(7):677–689. doi:10.1097/CAD.0b013e328361b7ad | ||
Ren Z, Wang Y, Jiang W, Dai W, Jiang Y. Anti-tumor effect of a novel soluble recombinant human endostatin: administered as a single agent or in combination with chemotherapy agents in mouse tumor models. PLoS One. 2014;9(9):e107823. doi:10.1371/journal.pone.0107823 | ||
Guo L, Song N, He T, et al. Endostatin inhibits the tumorigenesis of hemangioendothelioma via downregulation of CXCL1. Mol Carcinog. 2015;54(11):1340–1353. doi:10.1002/mc.22210 | ||
Rooney P, Kumar S, Ponting J, Wang M. The role of hyaluronan in tumour neovascularization (review). Int J Cancer. 1995;60(5):632–636. | ||
Liu LK, Finzel BC. Fragment-based identification of an inducible binding site on cell surface receptor CD44 for the design of protein-carbohydrate interaction inhibitors. J Med Chem. 2014;57(6):2714–2725. doi:10.1021/jm5000276 | ||
Park KC, Choi SH. Effects of endostatin and a new drug terpestacin against human neuroblastoma xenograft and cell lines. Pediatr Surg Int. 2013;29(12):1327–1340. doi:10.1007/s00383-013-3398-1 | ||
Ying T, Prabakaran P, Du L, et al. Junctional and allele-specific residues are critical for MERS-CoV neutralization by an exceptionally potent germline-like antibody. Nat Commun. 2015;6:8223. doi:10.1038/ncomms9223 | ||
Oh EJ, Park K, Kim KS, et al. Target specific and long-acting delivery of protein, peptide, and nucleotide therapeutics using hyaluronic acid derivatives. J Control Release. 2010;141(1):2–12. doi:10.1016/j.jconrel.2009.09.010 | ||
Yang JA, Kong WH, Sung DK, et al. Hyaluronic acid-tumor necrosis factor-related apoptosis-inducing ligand conjugate for targeted treatment of liver fibrosis. Acta Biomater. 2015;12:174–182. doi:10.1016/j.actbio.2014.10.002 | ||
Zhang P, Jain P, Tsao C, et al. Butyrylcholinesterase nanocapsule as a long circulating bioscavenger with reduced immune response. J Control Release. 2016;230:73–78. doi:10.1016/j.jconrel.2016.04.008 | ||
Lin X, Wang S, Jiang Y, et al. Poly(ethylene glycol)-Radix ophiopogonis polysaccharide conjugates: preparation, characterization, pharmacokinetics and in vitro bioactivity. Eur J Pharm Biopharm. 2010;76:230–237. doi:10.1016/j.ejpb.2010.07.003 | ||
Pang X, Yang X, Zhai G. Polymer-drug conjugates: recent progress on administration routes. Expert Opin Drug Deliv. 2014;11(7):1075–1086. doi:10.1517/17425247.2014.912779 | ||
Pelegri-O’day EM, Lin EW, Maynard HD. Therapeutic protein-polymer conjugates: advancing beyond PEGylation. J Am Chem Soc. 2014;136(41):14323–14332. doi:10.1021/ja504390x | ||
Saravanakumar G, Deepagan VG, Jayakumar R, Park JH. Hyaluronic acid-based conjugates for tumor-targeted drug delivery and imaging. J Biomed Nanotechnol. 2014;10(1):17–31. | ||
West DC, Kumar S. The effect of hyaluronate and its oligosaccharides on endothelial cell proliferation and monolayer integrity. Exp Cell Res. 1989;183(1):179–196. | ||
Nguyen HH, Park J, Kang S, Kim M. Surface plasmon resonance: a versatile technique for biosensor applications. Sensors (Basel). 2015;15(5):10481–10510. doi:10.3390/s150510481 | ||
Kim YM, Hwang S, Kim YM, et al. Endostatin blocks vascular endothelial growth factor-mediated signaling via direct interaction with KDR/Flk-1. J Biol Chem. 2002;277(31):27872–27879. doi:10.1074/jbc.M202771200 | ||
Shi H, Huang Y, Zhou H, et al. Nucleolin is a receptor that mediates antiangiogenic and anti-tumor activity of endostatin. Blood. 2007;110(8):2899–2906. doi:10.1182/blood-2007-01-064428 | ||
Tan H, Yang S, Feng Y, et al. Characterization and secondary structure analysis of endostatin covalently modified by polyethylene glycol and low molecular weight heparin. J Biochem. 2008;144(2):207–213. doi:10.1093/jb/mvn060 | ||
Lee SJ, Jang JW, Kim YM, et al. Endostatin binds to the catalytic domain of matrix metalloproteinase-2. FEBS Lett. 2002;519(1–3):147–152. | ||
Wight TN, Kinsella MG, Qwarnström EE. The role of proteoglycans in cell adhesion, migration and proliferation. Curr Opin Cell Biol. 1992;4:793–801. doi:10.1016/0955-0674(92)90102-I | ||
Kleinman HK, Martin GR. Matrigel: basement membrane matrix with biological activity. Semin Cancer Biol. 2005;15(5):378–386. doi:10.1016/j.semcancer.2005.05.004 | ||
Mondrinos MJ, Koutzaki S, Jiwanmall E, et al. Engineering three-dimensional pulmonary tissue constructs. Neuroimage. 2006;12(4):717–728. | ||
Wickstrom SA, Veikkola T, Rehn M, et al. Endostatin-induced modulation of plasminogen activation with concomitant loss of focal adhesion and actin stess fibers in cultured human endothelial cells. Cancer Res. 2001;61(17):6511–6516. | ||
Lee WS, Pyun BJ, Kim SW, et al. TTAC-0001, a human monoclonal antibody targeting VEGFR-2/KDR, blocks tumor angiogenesis. Mabs. 2015;7(5):957–968. doi:10.1080/19420862.2015.1045168 | ||
Zhao H, Chen Z. Screening of neuraminidase inhibitors from traditional Chinese medicines by integrating capillary electrophoresis with immobilized enzyme microreactor. J Chromatogr A. 2014;1340(8):139–145. doi:10.1016/j.chroma.2014.03.028 | ||
Ishida T. Immunohistochemical expression of the CD44 variant 6 in colorectal adenocarcinoma. Surg Today. 2000;30(1):28–32. doi:10.1007/PL00010042 | ||
Wang C, Xie J, Guo J, Manning HC, Gore JC, Guo N. Evaluation of CD44 and CD133 as cancer stem cell markers for colorectal, cancer. Oncol Rep. 2012;28(4):1301–1308. doi:10.3892/or.2012.1951 | ||
Pietras A, Katz AM, Ekström EJ, et al. Osteopontin-CD44 signaling in the glioma perivascular niche enhances cancer stem cell phenotypes and promotes aggressive tumor growth. Cell Stem Cell. 2014;14(3):357–369. doi:10.1016/j.stem.2014.01.005 | ||
Berner HS, Suo Z, Risberg B, Villman K, Karlsson MG, Nesland JM. Clinicopathological associations of CD44 mRNA and protein expression in primary breast carcinomas. Histopathology. 2003;42(6):546–554. | ||
Diaz LK, Zhou X, Wright ET, et al. CD44 expression is associated with increased survival in node-negative invasive breast carcinoma. Breast Dis. 2006;16(4):359. | ||
Hermann PC, Huber SL, Herrler T, et al. Distinct populations of cancer stem cells determine tumor growth and metastatic activity in human pancreatic cancer. Cell Stem Cell. 2007;1(3):313–323. doi:10.1016/j.stem.2007.06.002 | ||
Li C, Lee CJ, Simeone DM. Identification of human pancreatic cancer stem cells. Methods Mol Biol. 2009;568:161–173. doi:10.1007/978-1-59745-280-9_10 | ||
Setchell KD, Zhao XH, Jha P, Heubi JE, Brown NM. The pharmacokinetic behavior of the soy isoflavone metabolite S-(-)equol and its diastereoisomer R-(+)equol in healthy adults determined by using stable-isotope-labeled tracers. Am J Clin Nutr. 2009;90:1029–1037. doi:10.3945/ajcn.2009.27981 | ||
Zhu Y, D’Arienzo C, Lou Z, et al. LC-MS/MS multiplexed assay for the quantitation of a therapeutic protein BMS-986089 and the target protein Myostatin. Bioanalysis. 2016;8(3):193–204. doi:10.4155/bio.15.238 | ||
Jenkins R, Duggan JX, Aubry AF, et al. Recommendations for validation of LC-MS/MS bioanalytical methods for protein biotherapeutics. Aaps J. 2015;17(1):1–16. doi:10.1208/s12248-014-9685-5 |
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.