Back to Journals » Therapeutics and Clinical Risk Management » Volume 16
Changing Times for CLN2 Disease: The Era of Enzyme Replacement Therapy
Authors Specchio N ![]() , Pietrafusa N
, Pietrafusa N ![]() , Trivisano M
, Trivisano M
Received 29 January 2020
Accepted for publication 18 March 2020
Published 30 March 2020 Volume 2020:16 Pages 213—222
DOI https://doi.org/10.2147/TCRM.S241048
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Garry Walsh
Nicola Specchio, Nicola Pietrafusa, Marina Trivisano
Rare and Complex Epilepsy Unit, Department of Neuroscience, Bambino Gesù Children’s Hospital, IRCCS, Rome, Italy
Correspondence: Nicola Specchio
Department of Neuroscience, Bambino Gesù Children’s Hospital, IRCCS, Rome, Italy
Tel +39 0669592645
Fax +39 0668592463
Email [email protected]
Abstract: Neuronal ceroid lipofuscinosis type 2 (CLN2 disease) is a progressive neurodegenerative disease that results in early-onset, severe, progressive, neurological disabilities, leading to death in late childhood or early adolescence. Management has relied on symptomatic care, and supportive and palliative strategies, but the approval of the enzyme replacement therapy cerliponase alfa in the USA and Europe in 2017 brought different treatment opportunities. We describe the natural history of CLN2 disease, its diagnosis and management, and the preclinical and clinical development of cerliponase alfa. A PubMed search was undertaken for cerliponase alfa and rhTPP1 to identify preclinical and clinical studies. The hallmark-presenting symptoms of CLN2 disease are unprovoked seizures and a history of language delay, and progression involves motor dysfunction, and cognitive and visual decline. Cerliponase alfa has shown efficacy and tolerability in mouse and canine models of CLN2 disease when delivered intracerebroventricularly. Administration of cerliponase alfa in patients with CLN2 disease has led to significant reductions in the rate of decline of motor and language functions in comparison with a natural history population. The approval of cerliponase alfa has brought a new era for CLN2 disease, highlighting the need to understand different patterns of disease progression and clinical needs in treated patients.
Keywords: neuronal ceroid lipofuscinosis type 2, TPP1, cerliponase alfa, late infantile, seizures
Introduction
Neuronal ceroid lipofuscinosis type 2 (CLN2 disease), is a rare neurodegenerative pediatric disorder in the neuronal ceroid lipofuscinosis (NCL) family of lysosomal storage disorders.1,2 Although the NCLs are the most common cause of childhood neurodegeneration, even as a combined group they are rare. CLN2 disease is one of the most commonly reported NCLs, but it still affects fewer than 1 in 100,000 live births in most studied populations.1–7 CLN2 disease is caused by autosomal recessive mutations in the TPP1 gene resulting in a deficiency of the lysosomal enzyme tripeptidyl peptidase 1 (TPP1). TPP1 enzyme is produced as a proenzyme, which is processed in lysosomes into the active enzyme that cleaves proteins into tripeptides during protein degradation.8,9 While the function of TPP1 is not yet fully characterized, in humans, its deficiency leads to the accumulation of intracellular autofluorescent ceroid lipofuscin, which is followed by neuronal dysfunction and death, resulting in brain and retinal degeneration by an as-yet-undetermined mechanism.1,2,10
TPP1 is conserved between non-human primates, other mammals, amphibians, fish, amoeba, and humans,9 and an understanding of its cellular function is being developed through studies of knockout and loss-of-function mutations in these model organisms. Although the substrates of TPP1 are not fully described, TPP1 has been implicated in a range of cellular processes. In the amoeba Dictyostelium discoideum roles in growth and development have been shown, as well as inhibition of autophagy – one of the processes by which materials are delivered to the lysosome for degradation.11,12 The TOR pathway and regulation of the Golgi body are further mechanisms by which TPP1 deficiency may impart its effects.13,14 Zebrafish models of TPP1 deficiency reiterate some aspects of the human phenotype, and show early-onset progressive neurodegeneration, with apoptosis affecting the retina and brain.15 These zebrafish models display enlarged lysosomes and show developmental abnormalities of the brain, retina, optic nerve and spinal cord.15 Murine models show lysosomal accumulations of ceroid lipofuscin throughout the brain, such that the neuronal cell body appears “full” of accumulated storage material.16,17 Further, the TPP1 Dachshund displays autofluorescent storage material throughout the central nervous system.18 The studies in these models, from simple eukaryotes to mammals, reinforce the importance of TPP1’s role within the lysosome, and within the more complex models, it is clear that the nervous system is the area targeted by lysosomal accumulations. Our growing understanding of how to treat CLN2 disease is based not only on managing clinical signs and symptoms but also on its pathological basis within the lysosome.
The Natural History of CLN2 Disease
CLN2 disease typically presents between the age of 2 and 4 years, although a small number of patients with later symptom onset have been reported.19 Although seizures are frequently reported at an early disease stage, language delay or deficits are becoming increasingly noted as an early, if not the first, symptom.3,20–23 Motor dysfunction may also occur in early disease stages, in the form of clumsiness or ataxia,20,21 and a low proportion of patients may present with behavioral problems or dementia.20 The majority of patients (approximately 85%) show a classic late-infantile phenotype.19 Beyond the early stages of the disease (during which there may be some variation in presenting symptoms), an almost universal, rapid progression occurs, including further regression of motor and language skills, development of myoclonus, decline in vision, deterioration of cognitive function and death by early adolescence.1,2,21,24–28 Seizures tend to continue with disease progression; these are usually polymorphic, including myoclonic, tonic, tonic–clonic, atonic, and absence,3,21,23 and are difficult to treat, becoming resistant to multiple antiepileptic drugs over time.3,24 A large observational study of the natural history of CLN2 disease in patients in the DEM-CHILD registry and Weill Cornell Medical College data set has shown that this period of rapid progression occurs in most patients between the ages of approximately 3 and 6 years, in very quick succession after the onset of symptoms.20 During this time, children with CLN2 disease can become wheelchair-bound and dependent upon caregivers, and commonly have respiratory infections, and feeding and sleep disturbances, before CLN2 disease is fatal.2,20,21,23,24 The early-stage symptoms and progressive symptoms of CLN2 disease are shown in Figure 1.1,20,23–26,29
|
Figure 1 The presenting symptoms and progression of CLN2 disease. Reprinted from Lancet Child Adolesc Health; 2(8); Nickel M, Simonati A, Jacoby D, Lezius S, Kilian D, van de Graaf B, Pagovich OE, Kosofsky B, Yohay K, Downs M, Slasor P, Ajayi T, Crystal RG, Kohlschütter A, Sondhi D, Schulz A; Disease characteristics and progression in patients with late-infantile neuronal ceroid lipofuscinosis type 2 (CLN2) disease: An observational cohort study; 582–590; Copyright © 2018 Elsevier Ltd. All rights reserved, with permission from Elsevier.20 Age ranges represent those typical of the classic late-infantile phenotype. The first circle indicates the age at which the period of rapid progression typically begins (3 years), and the second circle indicates the typical age at diagnosis (approximately 5 years). Atypical phenotypes of CLN2 disease can vary in age of onset, rate of progression, and disease manifestation. Abbreviation: CI, confidence interval. |
Monitoring Progression of CLN2 Disease
As CLN2 disease progresses, functional decline is usually measured using either the Hamburg scale (focusing on seizures, motor function, language, and vision) or the Weill Cornell scale (assessing gait, language, feeding or swallowing, and myoclonus).23,25 The CLN2 Disease Clinical Rating Scale, a modified version of the Hamburg scale, is used in clinical trials to understand how treatments affect motor, language, and visual function, and seizures. The CLN2 Disease Clinical Rating Scale has been shown to be comparable with the Hamburg scale and allows comparison of trial data and historically collected data.30 Clinical trial endpoints have focused on the motor and language aspects of the CLN2 Disease Clinical Rating Scale as they capture early-stage disease features and allow a comparison of treatment effect with natural history progression. In this scale, patients with the motor or language function of a person from the general population are given a rating of 3, and patients who are no longer able to walk or crawl, or produce intelligible words, are given a rating of 0 (Table 1).30
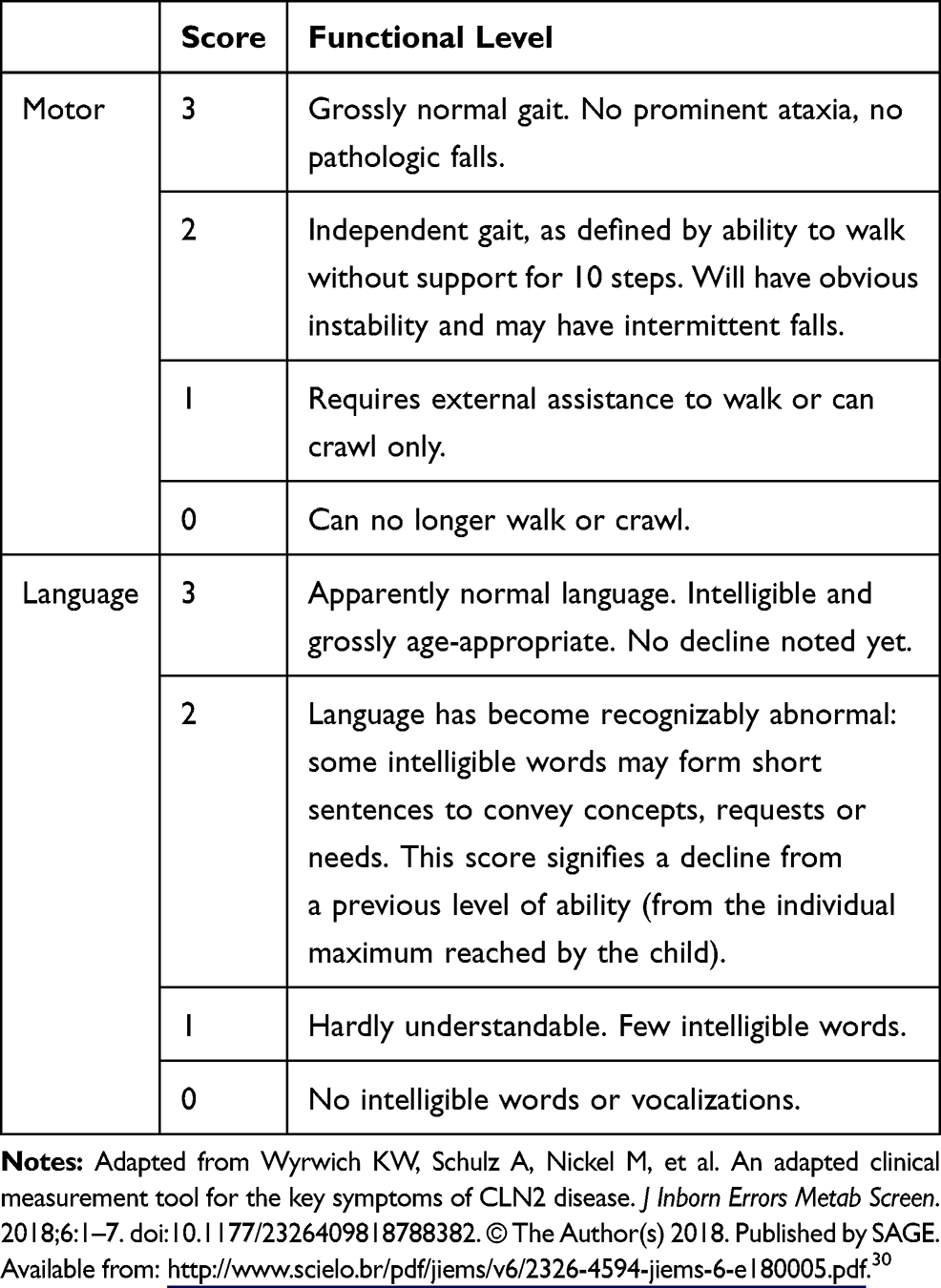 |
Table 1 Motor and Language Scores in the CLN2 Disease Clinical Rating Scale |
With the use of these clinical scores and the relatively uniform progression of the disease, natural history populations have been used as the comparator group in clinical trials.
Diagnosis of CLN2 Disease
Diagnostic delays are common in patients without a history of CLN2 disease [1], and the delay between the onset of symptoms and definitive diagnosis may span from 2 to 3 years.3,20
Other subtle clinical features that can be key in raising suspicion of CLN2 disease may be recorded in patients in the early stages of the disease. For example, cerebellar or cerebral atrophy, cortical thinning, or white matter changes may be detectable on brain magnetic resonance imaging (MRI).23,26,31–33 Indeed, in one study of 14 patients with CLN2 disease, cerebellar atrophy was identified in 100% of patients at first brain MRI and periventricular white matter changes were found in 79% of patients.31 MRI is frequently used in the early stages of assessing patients with epileptic seizures, and ensuring that clinicians are aware that subtle atrophy could be detected in CLN2 disease may increase the likelihood of early diagnosis. Patients with CLN2 disease may also show a characteristic photoparoxysmal response during electroencephalogram (EEG) with low-frequency (1–3 Hz) intermittent photic stimulation (IPS).31,34,35 At first EEG, this response has been identified in over 78% of patients in some studies,31,34 although it should be noted that another study reported that only 22% of patients showed this response at first EEG.36 EEGs have a key role in the diagnostic process for patients with seizures, and the use of low-frequency IPS could raise suspicion of CLN2 disease, providing opportunities for early diagnosis.
Aside from limited awareness of CLN2 disease, there are several other factors that may delay its diagnosis. The early symptoms, such as seizures and language delays, can also occur in many other more common diseases, and as a result, non-specific language disorders or epilepsy syndromes are common misdiagnoses. Given the severity of the seizures; the potentially subtle, variable language deficits that may be present;23 and the wide range of language skills in early childhood in the general population, language deficits may be overlooked as a key presenting symptom in some patients and the focus may be on seizure management.3 In addition, antiepileptic drugs may be associated with adverse events that affect motor function, so any subtle motor deficits that are present may be attributed to drugs instead of being recognized as a distinct symptom that should be considered in the differential diagnosis.3
Diagnosis should be carried out through both genetic testing for mutations in TPP1 and enzyme activity testing of TPP1 protein.29 In practice, requesting appropriate tests requires clinicians to have an awareness of CLN2 disease. Indeed, even if a broad metabolic screen is requested for diagnostic purposes, the number of screens that include either genetic or enzymatic activity testing of TPP1 is not known. Clinician awareness and availability of TPP1 testing have the potential to cause large regional differences in time to diagnosis, and ensuring that clinicians keep CLN2 disease in mind during the differential diagnosis, it is essential that gene panels or metabolic screens utilized within hospitals include the TPP1 gene and TPP1 enzyme activity, so the diagnosis of CLN2 disease is not missed. An understanding of these regional variations can be used to target awareness campaigns, not only on CLN2 disease itself but also on the availability of appropriate genetic and enzyme activity screens. It should be ensured that CLN2 disease is excluded during any differential diagnosis of a patient presenting with early language delay and unprovoked epileptic seizures starting between the ages of 2 and 4 years, and other symptoms of CLN2 disease, so that opportunities for early treatment are not missed. As large-scale gene panels become increasingly available, the key to early diagnosis may lie in prompt use of these next-generation techniques in patients with unexplained epilepsies or other symptoms of developmental regression, rather than waiting for a clear-cut clinical picture suggesting CLN2 disease.
Treatment and Management of CLN2 Disease
No international guidelines exist for the management of CLN2 disease. However, strategies commonly focus on a 2017 publication written by a team of CLN2 disease experts with specialties including neurology, palliative care, genetics, and pediatrics.3 Given the wide range of symptoms experienced by patients with CLN2 disease and the complexity of clinical needs as the disease rapidly progresses, management requires a diverse multidisciplinary team. Alongside symptom management involving pediatric neurologists, epileptologists, ophthalmologists, physiotherapists, and pediatricians, the progressive disease requires increasing input from respiratory clinicians, dieticians, and pain and sleep specialists, and provision of palliative support to optimize the quality of life and minimize complications in later disease stages. Management plans also need to incorporate support for families, not only in helping them to care for their severely affected child but also in considering new treatments and entering clinical trials, and providing them with family planning options.
Adding further complexity, several commonly used antiepileptic treatments, such as carbamazepine and phenytoin, may be associated with adverse events in CLN2 disease and should be avoided.3,21 As the disease progresses and as language and communication skills continue to deteriorate, it can become increasingly difficult to understand patients’ needs; maintaining communication becomes even more important, as patients may be very uncomfortable because of motor dysfunction, myoclonus, and pain, and be totally dependent on caregivers.21
Several strategies have been examined in the development of treatments for CLN2 disease. Three studies of gene therapy using AAV vectors for CLN2 disease are currently completed or active,37 and studies in the canine model of CLN2 disease have shown that gene therapy can deliver TPP1 enzyme throughout the brain, and reduce lysosomal storage and neurological symptoms.38 In a mouse model of CLN2 disease carrying knockout mutations, injections of either an AAV2 or AAV5 vector containing human TPP1 resulted in reduced autofluorescent storage material.39 In a Phase I study of 10 children with CLN2 disease, administration of AAV2-delivered TPP1 was not reported to have a clinical benefit; even though there were some reductions in progression as measured by MRI, this technique was also associated with serious adverse events.40 A study of stem cell transplantation into the cerebral hemispheres and lateral ventricles has not shown efficacy in CLN2 disease, although this study was undertaken in a small number of patients in later disease stages.41 The use of drugs that upregulate lysosomal function, such as gemfibrozil and fenofibrate, have been examined in neural progenitor cells from patients with CLN2 disease, but no increases in TPP1 messenger RNA were induced.42
A further strategy has been the development of an enzyme replacement therapy and in 2017, enzyme replacement therapy with cerliponase alfa (recombinant human TPP1 [rhTPP1]; Brineura) was granted regulatory approval in the USA and Europe for the treatment of CLN2 disease (specifically for slowing the loss of ambulation in symptomatic children aged ≥3 years in the USA), [28] marking the first disease-specific therapeutic advance for the NCLs.3 Aside from allowing an appropriate multidisciplinary care strategy to be put in place in early disease3 and enabling families to be provided with support and family planning advice, prompt diagnosis is now key in providing opportunities to treat patients with cerliponase alfa from an early stage.
The timeline of the development of cerliponase alfa is shown in Figure 2.
|
Figure 2 Timeline showing the preclinical and clinical development of cerliponase alfa and the year of its approval in the USA and Europe. Abbreviations: CNS, central nervous system; ICV, intracerebroventricular; IT-L, intrathecal–lumbar. |
Cerliponase Alfa
Cerliponase alfa is a recombinant human TPP1 (rhTPP1) in its proenzyme form and is manufactured in a Chinese hamster ovary cell line.43 The drug is processed in lysosomes into mature, active TPP1.44,45
Preclinical Development of Cerliponase Alfa
One of the first challenges in the development of a treatment for CLN2 disease was to determine how it could be most effectively delivered to degenerating neurons. Preclinical studies have been supported by mouse and canine models of CLN2 disease.16,18,46,47
The utilized mouse model of CLN2 disease carries a missense mutation (p.Arg447His) and an intronic insertion, which results in undetectable TPP1, motor dysfunction (tremor and ataxia) by 7 weeks of age, and extensive neuronal pathology (including accumulation of lysosomal storage material).16 Administration of rhTPP1 proenzyme into the lateral ventricles of these mice at Day 60 resulted in the presence of mature TPP1 throughout the brain, indicating that the proenzyme had entered neurons and been correctly processed within lysosomes.48 Lysosomal storage was decreased, and tremor amplitude was lower than in vehicle-treated mice and neuronal integrity in the deep cerebellar nuclei was improved.
The canine model of CLN2 disease, the TPP1-null dachshund, exhibits loss of cognitive function, ataxia, visual decline, and myoclonic seizures from the age of 9 months, and death by the age of 12 months, which mirrors the pattern of neurodegenerative decline seen in patients.18,46,47 Monthly intrathecal–lumbar (IT-L) administration of cerliponase alfa into one wild-type and one TPP1-null dachshund over a 4–7 month period resulted in expression of TPP1 throughout the central nervous system and reductions in lysosomal storage material in the TPP1-null dachshund versus the wild-type.49 However, no functional improvements were reported, and several infusion-associated reactions occurred, including hypersensitivity, swelling, urticaria, vomiting, hypotension, and tachycardia.49
Repeated intracerebroventricular (ICV) infusions of 4 mg or 16 mg rhTPP1 every other week into wild-type and TPP1-null animals at 2.5 months of age were used to assay distribution, pharmacology, efficacy, and safety.50,51 This treatment regimen was associated with reductions in lysosomal storage in contrast to monthly IT-L infusions. Neuronal morphology was maintained in comparison with vehicle-treated animals.50 Survival was 39–47 weeks in vehicle-treated animals, but was extended to 57–67 weeks in animals treated with 16 mg rhTPP1.51 All animals were assessed on a weekly basis, and treatment with rhTPP1 delayed onset of neurological symptoms such as ataxia, tremors, myoclonus, and visual deficits, although all animals eventually progressed to end-stage disease.51 Cognitive function was also preserved, as assessed through T-maze testing. Importantly, enlargement of the ventricles, which was used to assess brain atrophy, was found to be reduced in rhTPP1-treated animals, and this trend was higher in animals receiving higher doses.51 Placement of the ICV catheters caused inflammation, but this was not associated with dose. TPP1 enzyme was found throughout the brain, including in the frontal and occipital cortices, striatum, thalamus, hypothalamus, midbrain, cerebellum, pons, and medulla. The higher dose (16 mg) was associated with mild to moderate infusion-associated reactions, including facial edema, erythema, urticaria, hypotension, diarrhea, and vomiting. These reactions were managed with antihistamine and could be eliminated by extending the infusion time from 2 hrs to 4 hrs. Repeated administration resulted in the development of anti-rhTPP1 antibodies in the plasma and cerebrospinal fluid (CSF) of all wild-type and TPP1-null animals, although levels were approximately 10- to 100-fold higher in TPP1-null animals. Maintaining patency of the ICV catheters was a challenge because of debris within the catheter or dislocation of the catheter from the ventricle due to the growth of the skull. This provided valuable information for trials in humans, in terms of determining the time at which to begin treatment in growing patients, although it is important to consider that these challenges may be associated with the small size of animal ventricles in comparison with those in humans.
To understand the pharmacological distribution of cerliponase alfa with different techniques, a single dose of rhTPP1 was injected via an ICV or IT-L route in healthy cynomolgus monkeys, and neurological samples were collected at 3, 7, and 14 days after infusion. Although some lesions developed in association with the placement of the infusion devices, no serious adverse events were noted and there were no effects on blood, serum, or urinary chemistry.52 Both ICV and IT-L infusions resulted in TPP1 distribution in most areas of the brain, including the frontal and occipital cortices and the cerebellum, but ICV infusions also resulted in detectable TPP1 in deep brain structures, such as the thalamus, midbrain, and striatum. This is unsurprising given that administration into the ventricles results in drug entry closer to the site of CSF production compared with IT-L administration.52 No antibodies to rhTPP1 were detected in either CSF or plasma, although because cynomolgus monkeys produce TPP1 that is highly homologous to rhTPP1,9 it was unclear if this would provide useful immunogenicity information for studies in patients with CLN2 disease who produce very little or no TPP1.
The overall results of these studies in healthy animals and CLN2 disease models have informed the design of clinical trials, in terms of administration, infusion times and frequencies, and dosages. The feasibility of administering cerliponase alfa via ICV devices was established, in terms of causing fewer adverse events and resulting in the higher distribution of the drug in deep brain structures compared with IT-L administration. The need for management of hypersensitivity reactions through the use of slow infusion times and the administration of antihistamine has also been carried forward into studies in patients.
Clinical Trials of Cerliponase Alfa
Safety and efficacy of cerliponase alfa has been studied in a multicenter, international, open-label study in patients with CLN2 disease aged between 3 and 16 years.53 In the Phase I/II dose-escalation stage (NCT01907087), doses were increased every 2 weeks from 30 mg to 100 mg, and then to 300 mg to determine the side effect profile at each dose. Patients received at least two infusions of a particular dose before the dose was increased, and all patients received the 300 mg dose for 48 weeks. A total of 23 patients provided efficacy data and 24 provided safety data. In the 240-week extension stage (NCT02485899), patients received 300 mg doses every 2 weeks for at least 96 weeks. The primary endpoint for each stage was the time until an unreversed 2-point reduction in motor and language scores of the CLN2 Disease Clinical Rating Scale (a modified version of the Hamburg scale) as compared with a group of 42 natural history patients. As patients with CLN2 disease show a relatively uniform disease progression, natural history populations are used as the comparator group in clinical trials, instead of placebo controls. The use of placebo controls, when an ICV device would need to be implanted, would raise some ethical issues in this rare disease population.
At study entry, patients were required to have a combined motor and language score between 3 and 6 inclusive, with a score of at least 1 in each domain. At screening, most patients had a score of 3 or 4, although some patients had scores of 1 by the time the 300 mg dose regimen was initiated. The mean (± standard deviation [SD]) duration of treatment at 300 mg was 115 ± 30 weeks (range: 1–145 weeks), with patients receiving 99% of planned infusions.
Administration of cerliponase alfa resulted in treated patients being less likely to experience an unreversed decline in combined motor and language score (hazard ratio [HR]: 0.08; 95% confidence interval [CI]: 0.02–0.23; P < 0.001). Both motor and language scores were also affected independently ([motor: HR: 0.04; 95% CI: 0.00–0.29; P = 0.002] [language: HR: 0.15; 95% CI: 0.04–0.52; P < 0.003]) [38]. The mean rate of decline in the motor–language score in treated patients was reduced to 0.27 ± 0.35 points compared with 2.12 ± 0.98 in the natural history population – a difference of 1.85 ± 0.21 points (95% CI: 1.51–2.18; P < 0.001).[38] Changes from baseline in combined motor-language scores in 17 matched pairs from the treated and natural history populations are shown in Figure 3.
|
Figure 3 Change from baseline in motor and language scores of the CLN2 Disease Clinical Rating Scale in matched pairs of patients treated with cerliponase alfa and historical controls. From N Engl J Med; Schulz A, Ajayi T, Specchio N, de Los Reyes E, Gissen P, Ballon D, Dyke JP, Cahan H, Slasor P, Jacoby D, Kohlschütter A; CLN2 Study Group; Study of intraventricular cerliponase alfa for CLN2 disease; 378(20); 1898–1907. Copyright © 2018 Massachusetts Medical Society. Reprinted with permission from Massachusetts Medical Society.53 |
If seizures and visual decline were also incorporated into the clinical scores (each using a scale of 0–3 encompassing no function to normal function) at Week 48, treated patients showed a mean increase of 0.30 ± 1.70 points compared with a decrease of 2.80 ± 2.04 in the natural history population. This difference had become even more apparent by Week 96, with an increase of 0.40 ± 2.08 points compared with a decrease of 4.30 ± 2.26 in the natural history population. Brain atrophy, as assessed by MRI, was ongoing throughout the trial, occurring at an annualized rate of 10.5% in Year 1 and 3.3% in Year 2.
In terms of safety, all patients experienced at least one adverse event, although these were usually mild and resolved by clinical management. The most common were reported as convulsions (including seizures; 96%), pyrexia (71%), vomiting (63%), hypersensitivity reactions (63%), upper respiratory tract infections (54%), nasopharyngitis (42%), and rhinitis (42%). Fifty-five serious adverse events occurred in 83% of patients, and included hypersensitivity (29%), upper respiratory tract infections (21%), epilepsy (including seizures; 17%), pharyngitis (17%), gastroenteritis (13%), pyrexia (8%), and device-related infections (8%). Seizures, epilepsy, and generalized tonic–clonic seizures were mapped to the Convulsions Standardised MedDRA (Medical Dictionary for Regulatory Activities) Query term.[39] Although experienced by the vast majority of patients, seizures are not unexpected given the natural history of CLN2 disease, and only 6% of all convulsion events were considered to be associated with cerliponase alfa treatment.44 There were no deaths or treatment discontinuations caused by an adverse event, although two patients (8%) experienced device-related infections that required replacement of the ICV device, resulting in treatment delays while these infections were managed.53 Antibodies to cerliponase alfa were detected in serum in 79% of patients and in CSF in 33%, but these were not associated with the frequency or severity of hypersensitivity events.44,45
In an ongoing study assessing the safety and efficacy of cerliponase alfa in 11 patients under the age of 3 years, the most common adverse events have been mild or moderate, including pyrexia (55%), generalized tonic–clonic seizures (36%), upper respiratory tract infections (36%), and vomiting (36%), similar to those in the previous trial in patients over the age of 3 years.54 Two patients experienced hypersensitivity reactions. A total of 19 serious adverse events occurred in 9 patients, with pyrexia being the most common (occurring in 27% of patients). After a median treatment duration of 44.1 weeks (range: 42.6–114.0), no deaths or treatment discontinuations had occurred. Over the course of treatment, seven patients maintained stable combined motor and language scores, two gained a point, and one lost a point. The remaining patient maintained baseline motor score; language was not assessed, as the patient had an autism spectrum disorder. This ongoing trial is important because the inclusion of patients under the age of 3 years incorporates a population that may not yet be showing overt presenting symptoms and have been diagnosed based on a family history of CLN2 disease.
An expanded access program trial (NCT02963350) giving patients over the age of 2 years who cannot participate in clinical trials access to cerliponase alfa is also underway.
Cerliponase Alfa in Patients
Based on the aforementioned clinical trial results, cerliponase alfa is available in Europe in patients of all ages and in the USA for patients aged 3 years or over. Treatment is required every 2 weeks via an ICV Ommaya or Rickham reservoir device, which is implanted under the scalp of patients using MRI guidance.44,45,53
Based on information from preclinical studies, cerliponase alfa is infused after antihistamine administration to reduce hypersensitivity reactions. The procedure has been accepted by patients and families, and as yet, no patients have withdrawn from treatment for efficacy or safety reasons.
Conclusion
CLN2 disease is a severe progressive neurodegenerative disorder, which in its classic late-infantile form results in death in late childhood or early adolescence. The hallmark-presenting symptoms of unprovoked seizures between the ages of 2 and 4 years and a history of early language delay are frequently highlighted as being key in raising suspicion of CLN2 disease. Because of improvements in, and clarification of, diagnostic processes, the identification of patients in early disease stages (particularly those with a family history) provides valuable opportunities to initiate treatment prior to the onset of rapid progression. The early diagnosis of patients also provides opportunities to understand the early clinical features detected by MRI or EEG in more depth, and enables large-scale biomarker analyses to be carried out to potentially identify patients with differing rates of progression or responses to treatment.
With the approval of cerliponase alfa in the USA and Europe in 2017, the need for early diagnosis has become even more important, given the clinical benefit of this therapy in reducing the rate of decline of motor and language function. Further investigations are required for a more in-depth understanding of longer-term effects and the impact of cerliponase alfa on seizures and vision loss, and indeed prolongation of patient lives. Several other strategies for treating CLN2 disease and the other NCLs have been studied or are under development.22,37–42 These have included gene therapy, stem cell therapy, immunomodulation, and the use of drugs that upregulate lysosome function across a range of NCL animal models and cell lines and in patients in clinical trials.
With the potential for treatment options to be developed in the coming years, the new era for patients with CLN2 disease, and potentially other NCLs, means that clinicians will need to understand a different disease profile for treated patients. Longer-term follow-up of patients treated at an early stage is required to understand the potential for functional development; the combination of early diagnosis and treatment with cerliponase alfa may lead to patients continuing to develop in terms of, for example, motor or language skills, instead of regressing according to the expected patterns seen in the natural history population. Beyond understanding how disease progression changes, multidisciplinary approaches may need to be adapted for patients with longer survival times and different clinical needs. The established multidisciplinary team may need to be expanded to include, for example, specialists who can provide educational support as children continue to progress or maintain function beyond currently expected ages.
The approval of cerliponase alfa also marks a changing time for providing support to families; when patients are diagnosed, families would previously have needed to come to terms with the expected progression of the disease. The benefits of treatment with cerliponase alfa must be combined with continued support and guidance for families.
Acknowledgments
Writing support was provided by Emma Conran, Porterhouse Medical, Reading, UK, and funded by BioMarin Pharmaceutical Inc.
Disclosure
Nicola Specchio has been a consultant for and received grant/research support from BioMarin Pharmaceutical Inc., and reports non-financial support from them during the conduct of the study and grants and personal fees from them outside the submitted work. Nicola Pietrafusa has been a consultant for BioMarin Pharmaceutical Inc. Marina Trivisano has been a consultant for BioMarin Pharmaceutical Inc.
References
1. Chang M, Cooper JD, Davidson BL, et al. CLN2. In: Mole S, Williams R, Goebel H, editors. The Neuronal Ceroid Lipofuscinoses (Batten Disease). Oxford: Oxford University Press; 2011:80–109.
2. Bennett MJ, Rakheja D. The neuronal ceroid-lipofuscinoses. Dev Disabil Res Rev. 2013;17(3):254–259. doi:10.1002/ddrr.v17.3
3. Williams RE, Adams HR, Blohm M, et al. Management strategies for CLN2 disease. Pediatr Neurol. 2017;69:102–112. doi:10.1016/j.pediatrneurol.2017.01.034
4. Williams RE, Mole SE. New nomenclature and classification scheme for the neuronal ceroid lipofuscinoses. Neurology. 2012;79(2):183–191. doi:10.1212/WNL.0b013e31825f0547
5. Teixeira C, Guimarães A, Bessa C, et al. Clinicopathological and molecular characterization of neuronal ceroid lipofuscinosis in the Portuguese population. J Neurol. 2003;250(6):661–667. doi:10.1007/s00415-003-1050-z
6. Claussen M, Heim P, Knispel J, Goebel HH, Kohlschütter A. Incidence of neuronal ceroid-lipofuscinoses in West Germany: variation of a method for studying autosomal recessive disorders. Am J Med Genet. 1992;42(4):536–538. doi:10.1002/(ISSN)1096-8628
7. Moore SJ, Buckley DJ, MacMillan A, et al. The clinical and genetic epidemiology of neuronal ceroid lipofuscinosis in Newfoundland. Clin Genet. 2008;74(3):213–222. doi:10.1111/j.1399-0004.2008.01054.x
8. Golabek AA, Kida E, Walus M, Wujek P, Mehta P, Wisniewski KE. Biosynthesis, glycosylation, and enzymatic processing in vivo of human tripeptidyl-peptidase I. J Biol Chem. 2003;278(9):7135–7145. doi:10.1074/jbc.M211872200
9. Wlodawer A, Durell SR, Li M, Oyama H, Oda K, Dunn BM. A model of tripeptidyl-peptidase I (CLN2), a ubiquitous and highly conserved member of the sedolisin family of serine-carboxyl peptidases. BMC Struct Biol. 2003;3(1):8. doi:10.1186/1472-6807-3-8
10. Haltia M, Goebel HH. The neuronal ceroid-lipofuscinoses: a historical introduction. Biochim Biophys Acta. 2013;1832(11):1795–1800. doi:10.1016/j.bbadis.2012.08.012
11. Phillips JE, Gomer RH. Partial genetic suppression of a loss-of-function mutant of the neuronal ceroid lipofuscinosis-associated protease TPP1 in Dictyostelium discoideum. Dis Model Mech. 2015;8(2):147–156. doi:10.1242/dmm.018820
12. Zatyka M, Sarkar S, Barrett T. Autophagy in rare (non-lysosomal) neurodegenerative diseases. J Mol Biol. 2020. doi:10.1016/j.jmb.2020.02.012
13. Stumpf M, Muller R, Gassen B, et al. A tripeptidyl peptidase 1 is a binding partner of the Golgi pH regulator (GPHR) in Dictyostelium. Dis Model Mech. 2017;10(7):897–907. doi:10.1242/dmm.029280
14. Smith PK, Sen MG, Fisher PR, Annesley SJ. Modelling of neuronal ceroid lipofuscinosis type 2 in Dictyostelium discoideum suggests that cytopathological outcomes result from altered TOR signalling. Cells. 2019;8(5):469. doi:10.3390/cells8050469
15. Mahmood F, Fu S, Cooke J, Wilson SW, Cooper JD, Russell C. A zebrafish model of CLN2 disease is deficient in tripeptidyl peptidase 1 and displays progressive neurodegeneration accompanied by a reduction in proliferation. Brain. 2013;136(Pt 5):1488–1507. doi:10.1093/brain/awt043
16. Sleat DE, Wiseman JA, El-Banna M, et al. A mouse model of classical late-infantile neuronal ceroid lipofuscinosis based on targeted disruption of the CLN2 gene results in a loss of tripeptidyl-peptidase I activity and progressive neurodegeneration. J Neurosci. 2004;24(41):9117–9126. doi:10.1523/JNEUROSCI.2729-04.2004
17. Geraets RD, Langin LM, Cain JT, et al. A tailored mouse model of CLN2 disease: a nonsense mutant for testing personalized therapies. PLoS One. 2017;12(5):e0176526. doi:10.1371/journal.pone.0176526
18. Awano T, Katz ML, O’Brien DP, et al. A frame shift mutation in canine TPP1 (the ortholog of human CLN2) in a juvenile Dachshund with neuronal ceroid lipofuscinosis. Mol Genet Metab. 2006;89(3):254–260. doi:10.1016/j.ymgme.2006.02.016
19. Gardner E, Bailey M, Schulz A, Aristorena M, Miller N, Mole SE. Mutation update: review of TPP1 gene variants associated with neuronal ceroid lipofuscinosis CLN2 disease. Hum Mutat. 2019;40(11):1924–1938. doi:10.1002/humu.v40.11
20. Nickel M, Simonati A, Jacoby D, et al. Disease characteristics and progression in patients with late-infantile neuronal ceroid lipofuscinosis type 2 (CLN2) disease: an observational cohort study. Lancet Child Adolesc Health. 2018;2(8):582–590. doi:10.1016/S2352-4642(18)30179-2
21. Kohlschütter A, Schulz A. CLN2 disease (classic late infantile neuronal ceroid lipofuscinosis). Pediatr Endocrinol Rev. 2016;13(Suppl 1):682–688.
22. Kohlschütter A, Schulz A, Bartsch U, Storch S. Current and emerging treatment strategies for neuronal ceroid lipofuscinoses. CNS Drugs. 2019;33(4):315–325. doi:10.1007/s40263-019-00620-8
23. Worgall S, Kekatpure MV, Heier L, Ballon D, Dyke JP, Shungu D. Neurological deterioration in late infantile neuronal ceroid lipofuscinosis. Neurology. 2007;69(6):521–535. doi:10.1212/01.wnl.0000267885.47092.40
24. Schulz A, Kohlschütter A, Mink J, Simonati A, Williams R. NCL diseases — clinical perspectives. Biochim Biophys Acta. 2013;1832(11):1801–1806. doi:10.1016/j.bbadis.2013.04.008
25. Steinfeld R, Heim P, von Gregory H, et al. Late infantile neuronal ceroid lipofuscinosis: quantitative description of the clinical course in patients with CLN2 mutations. Am J Med Genet. 2002;112(4):347–354. doi:10.1002/(ISSN)1096-8628
26. Pérez-Poyato MS, Marfa MP, Abizanda IF, Rodriguez-Revenga L, Sánchez VC, González MJ. Late infantile neuronal ceroid lipofuscinosis: mutations in the CLN2 gene and clinical course in Spanish patients. J Child Neurol. 2013;28(4):470–478. doi:10.1177/0883073812448459
27. Orlin A, Sondhi D, Witmer MT, et al. Spectrum of ocular manifestations in CLN2-associated Batten (Jansky-Bielschowsky) disease correlate with advancing age and deteriorating neurological function. PLoS One. 2013;8(8):e73128. doi:10.1371/journal.pone.0073128
28. Quagliato EMAB, Rocha DM, Sacai PY, Watanabe SS, Salomão SR, Berezovsky A. Retinal function in patients with the neuronal ceroid lipofuscinosis phenotype. Arq Bras Oftalmol. 2017;80(4):215–219. doi:10.5935/0004-2749.20170053
29. Fietz M, AlSayed M, Burke D, et al. Diagnosis of neuronal ceroid lipofuscinosis type 2 (CLN2 disease): expert recommendations for early detection and laboratory diagnosis. Mol Genet Metab. 2016;119(1–2):160–167. doi:10.1016/j.ymgme.2016.07.011
30. Wyrwich KW, Schulz A, Nickel M, et al. An adapted clinical measurement tool for the key symptoms of CLN2 disease. J Inborn Errors Metab Screen. 2018;6:1–7. doi:10.1177/2326409818788382
31. Specchio N, Bellusci M, Pietrafusa N, Trivisano M, de Palma L, Vigevano F. Photosensitivity is an early marker of neuronal ceroid lipofuscinosis type 2 disease. Epilepsia. 2017;58(8):1380–1388. doi:10.1111/epi.2017.58.issue-8
32. Löbel U, Sedlacik J, Nickel M, et al. Volumetric description of brain atrophy in neuronal ceroid lipofuscinosis 2: supratentorial gray matter shows uniform disease progression. AJNR Am J Neuroradiol. 2016;37(10):1938–1943. doi:10.3174/ajnr.A4816
33. Dyke JP, Sondhi D, Voss HU, et al. Brain region-specific degeneration with disease progression in late infantile neuronal ceroid lipofuscinosis (CLN2 disease). AJNR Am J Neuroradiol. 2016;37(6):1160–1169. doi:10.3174/ajnr.A4669
34. Albert DV, Yin H, De Los Reyes EC, Vidaurre J. Unique characteristics of the photoparoxysmal response in patients with neuronal ceroid lipofuscinosis type 2: can EEG be a biomarker? J Child Neurol. 2016;31(13):1475–1482. doi:10.1177/0883073816658659
35. Jadav RH, Sinha S, Yasha TC, et al. Clinical, electrophysiological, imaging, and ultrastructural description in 68 patients with neuronal ceroid lipofuscinoses and its subtypes. Pediatr Neurol. 2014;50(1):85–95. doi:10.1016/j.pediatrneurol.2013.08.008
36. Dozières-Puyravel B, Nasser H, Elmaleh-Berges M, et al. Paediatric-onset neuronal ceroid lipofuscinosis: first symptoms and presentation at diagnosis. Dev Med Child Neurol. 2019;62(4):528–530.
37. ClinicalTrials.gov. Trials in patients with CLN2 disease. Available from: https://clinicaltrials.gov/ct2/results?cond=CLN2+disease&term=&cntry=&state=&city=&dist=.
38. Katz ML, Tecedor L, Chen Y, et al. AAV gene transfer delays disease onset in a TPP1-deficient canine model of the late infantile form of Batten disease. Sci Transl Med. 2015;7(313):313ra180. doi:10.1126/scitranslmed.aac6191
39. Passini MA, Dodge JC, Bu J, et al. Intracranial delivery of CLN2 reduces brain pathology in a mouse model of classical late infantile neuronal ceroid lipofuscinosis. J Neurosci. 2006;26(5):1334–1342. doi:10.1523/JNEUROSCI.2676-05.2006
40. Worgall S, Sondhi D, Hackett NR, et al. Treatment of late infantile neuronal ceroid lipofuscinosis by CNS administration of a serotype 2 adeno-associated virus expressing CLN2 cDNA. Hum Gene Ther. 2008;19(5):463–474. doi:10.1089/hum.2008.022
41. Selden NR, Al-Uzri A, Huhn SL, et al. Central nervous system stem cell transplantation for children with neuronal ceroid lipofuscinosis. J Neurosurg Pediatr. 2013;11(6):643–652. doi:10.3171/2013.3.PEDS12397
42. Lojewski X, Staropoli JF, Biswas-Legrand S, et al. Human iPSC models of neuronal ceroid lipofuscinosis capture distinct effects of TPP1 and CLN3 mutations on the endocytic pathway. Hum Mol Genet. 2014;23(8):2005–2022. doi:10.1093/hmg/ddt596
43. Markham A. Cerliponase alfa: first global approval. Drugs. 2017;77(11):1247–1249. doi:10.1007/s40265-017-0771-8
44. Cerliponase Alfa (Brineura) [Summary of Product Characteristics]. Cork, Ireland: BioMarin International Limited; 2017. Available from: https://www.ema.europa.eu/en/documents/product-information/brineura-epar-product-information_en.pdf. Accessed March 23, 2020.
45. Cerliponase Alfa (Brineura) [Prescribing Information]. Novato, CA: BioMarin Pharmaceutical Inc; 2017. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/761052lbl.pdf. Accessed March 23, 2020.
46. Sanders DN, Kanazono S, Wininger FA, et al. A reversal learning task detects cognitive deficits in a Dachshund model of late-infantile neuronal ceroid lipofuscinosis. Genes Brain Behav. 2011;10(7):798–804. doi:10.1111/j.1601-183X.2011.00718.x
47. Katz ML, Coates JR, Cooper JJ, O’Brien DP, Jeong M, Narfstrom K. Retinal pathology in a canine model of late infantile neuronal ceroid lipofuscinosis. Invest Ophthalmol Vis Sci. 2008;49(6):2686–2695. doi:10.1167/iovs.08-1712
48. Chang M, Cooper JD, Sleat DE, et al. Intraventricular enzyme replacement improves disease phenotypes in a mouse model of late infantile neuronal ceroid lipofuscinosis. Mol Ther. 2008;16(4):649–656. doi:10.1038/mt.2008.9
49. Vuillemenot BR, Katz ML, Coates JR, et al. Intrathecal tripeptidyl-peptidase 1 reduces lysosomal storage in a canine model of late infantile neuronal ceroid lipofuscinosis. Mol Genet Metab. 2011;104(3):325–337. doi:10.1016/j.ymgme.2011.06.018
50. Vuillemenot BR, Kennedy D, Cooper JD, et al. Nonclinical evaluation of CNS-administered TPP1 enzyme replacement in canine CLN2 neuronal ceroid lipofuscinosis. Mol Genet Metab. 2015;114(2):281–293. doi:10.1016/j.ymgme.2014.09.004
51. Katz ML, Coates JR, Sibigtroth CM, et al. Enzyme replacement therapy attenuates disease progression in a canine model of late-infantile neuronal ceroid lipofuscinosis (CLN2 disease). J Neurosci Res. 2014;92(11):1591–1598. doi:10.1002/jnr.23423
52. Vuillemenot BR, Kennedy D, Reed RP, et al. Recombinant human tripeptidyl peptidase-1 infusion to the monkey CNS: safety, pharmacokinetics, and distribution. Toxicol Appl Pharmacol. 2014;277(1):49–57. doi:10.1016/j.taap.2014.03.005
53. Schulz A, Ajayi T, Specchio N, et al. Study of intraventricular cerliponase alfa for CLN2 disease. N Engl J Med. 2018;378(20):1898–1907. doi:10.1056/NEJMoa1712649
54. Schulz A, De Los Reyes E, Specchio N, et al. Cerliponase alfa for the treatment of CLN2 disease in an expanded patient cohort including children younger than three years.
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.