Back to Journals » Drug Design, Development and Therapy » Volume 20
cGAS-STING and PANoptosis: Interplay, Underlying Mechanisms, and Therapeutic Targets
Authors Wang Y ![]() , Chen J, Feng W, Li N, Zhang X, Zhao S, Shi K, Wang E, Jin Y
, Chen J, Feng W, Li N, Zhang X, Zhao S, Shi K, Wang E, Jin Y
Received 3 January 2026
Accepted for publication 8 April 2026
Published 16 April 2026 Volume 2026:20 593380
DOI https://doi.org/10.2147/DDDT.S593380
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Tuo Deng
Yumin Wang,1,* Jinxia Chen,2,* Wenxin Feng,3,* Ning Li,4 Xiu Zhang,5 Shuang Zhao,6 Kerui Shi,7 Erdan Wang,7 Yuzi Jin7
1Department of Respiratory and Critical Care Medicine, Aerospace Center Hospital, Peking University Aerospace School of Clinical Medicine, Beijing, People’s Republic of China; 2Department of Blood Transfusion, The Fourth Hospital of Hebei Medical University, Shijiazhuang, People’s Republic of China; 3Department of Anesthesiology, Basic Medical College, Guangxi Medical University, Nanning, People’s Republic of China; 4Department of Biochemistry and Molecular Biology, Shenyang Medical College, Liaoning Province Key Laboratory for Phenomics of Human Ethnic Specificity and Critical Illness(LPKL-PHESCI), Shenyang Key Laboratory for Phenomics, Shenyang, People’s Republic of China; 5Department of Stomatology, Shenyang Medical College, Liaoning Province Key Laboratory for Phenomics of Human Ethnic Specificity and Critical Illness (LPKL -PHESCI), Shenyang Key Laboratory for Phenomics, Shenyang, People’s Republic of China; 6Laboratory Animal Center, Affiliated Hospital of Chengde Medical University, Chengde, People’s Republic of China; 7Department of Pediatrics, Central Hospital Affiliated to Shenyang Medical College, Shenyang, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Yuzi Jin, Email [email protected]
Abstract: The cGAS-STING pathway is an essential cytosolic DNA sensing mechanism that activates innate immune responses upon detection of microbial or aberrant self-DNA. This evolutionarily conserved signaling axis plays critical roles in autoimmune diseases, sterile inflammation, and cellular senescence. While its transient activation provides protective immunity, dysregulated cGAS-STING signaling contributes to the pathogenesis of various inflammatory and autoimmune conditions. Growing evidence indicates its functional convergence with multiple cell death pathways—particularly PANoptosis, a distinct inflammatory programmed cell death (PCD) pathway that integrates key features of pyroptosis, apoptosis, and necroptosis. The dynamic interplay between cGAS-STING signaling and PANoptosis has emerged as an important pathogenic mechanism across multiple diseases, revealing new therapeutic opportunities. In this review, we propose a unifying conceptual framework in which cGAS-STING activation functions as a predominant upstream driver of PANoptosis across diverse pathological contexts, orchestrated through a convergent molecular axis involving cytosolic DNA sensing, ZBP1-PANoptosome assembly, and coordinated inflammatory cell death. We begin by outlining the core molecular architecture of the cGAS-STING pathway and its implications in disease. We then examine the mechanisms and pathophysiological consequences of cGAS-STING –PANoptosis crosstalk in various disorders, followed by recent advances in therapeutic strategies specifically targeting this interface. Finally, we discuss translational challenges, such as the poor bioavailability and systemic toxicity of conventional STING agonists, and highlight innovative solutions including nanomedicine-based delivery systems that enable tumor-specific activation while minimizing off-target effects. By highlighting the therapeutic potential of pharmacological modulation at this junction, we identify promising strategies for treating inflammation-associated diseases. The center shows two overlapping sections labeled PANoptosis and cGAS-STING signaling, connected by two opposite-direction arrows. Text near the overlap lists: cytosolic DNA accumulation; activating STING; activating cGAS; upregulation of ZBP1, cleaved caspases, pMLKL, GSDMD. At the top is a label: ZBP1 agonist CBL0137. At the bottom are therapy labels: STING agonist DMXAA, diABZI, cGAMP, STBF plus ADU-S100 plus anti-LAG3; and cGAS/cGAS-STING inhibitor RU.521, SN-011. Around the center are disease callouts with illustrations: Cancer (diffuse large B-cell lymphoma; triple-negative breast cancer) with a tumor-cell cluster; Metabolic inflammation (diabetes; obesity); Digestion system (hepatic injure) with a liver; CNS inflammation (Alzheimer; Parkinson) with a brain; Infection (bacteria; virus; fungus); Respiratory system (acute lung injure; asthma) with lungs.Infographic on PANoptosis and cGAS-STING signaling, showing related diseases and drug targets.
Keywords: cGAS-STING, PANoptosis, Programmed cell death, crosstalk
Introduction
The innate immune system serves as the body’s first line of defense against infection and cellular stress, relying on a sophisticated network of sensors and signaling pathways to detect danger and orchestrate appropriate responses.1 Among these, the cyclic GMP-AMP synthase–stimulator of interferon genes (cGAS-STING) pathway has emerged as a pivotal cytosolic DNA-sensing mechanism that triggers robust type I interferon and pro-inflammatory cytokine responses upon detecting aberrantly localized double-stranded DNA.2,3 Although its transient activation is essential for host defense, chronic or dysregulated signaling drives pathology across a broad spectrum of diseases, establishing cGAS-STING as a central amplifier of sterile inflammation.4–9
Parallel to advances in understanding innate immune sensing, the field of programmed cell death (PCD) has evolved beyond the classical paradigms of apoptosis, pyroptosis, and necroptosis. The recent conceptualization of PANoptosis represents a significant paradigm shift. PANoptosis is defined as an integral inflammatory PCD pathway that incorporates key molecular features and effectors from pyroptosis, apoptosis, and necroptosis, yet cannot be fully explained by any one of these alone. It is driven by the assembly of sophisticated multi-protein complexes termed PANoptosomes, which are nucleated by sensors like Z-DNA-binding protein 1 (ZBP1), absent in melanoma 2 (AIM2), and receptor-interacting serine/threonine -protein kinase 1 (RIPK1).10 This coordinated cell death process acts as a powerful defense mechanism against pathogens and malignant cells but, when uncontrolled, contributes significantly to immunopathology and tissue damage in conditions such as viral infections, sepsis, ischemic injury, and cancer.11–22
Emerging evidence now underscores a critical and dynamic interplay between the cGAS-STING pathway and PANoptosis.23 However, the term “interplay” encompasses several distinct types of relationships that must be carefully delineated to avoid conceptual ambiguity. In the context of this review, we consider three principal patterns. First, coexistence or parallel activation refers to scenarios in which both pathways are activated in the same pathological setting without a demonstrated mechanistic link. Second, parallel pathways triggered by shared upstream damage describes situations where a common upstream insult (such as mitochondrial DNA release) independently activates both cGAS-STING signaling and PANoptosis, but the two processes operate in parallel without a direct hierarchical connection. Third, upstream–downstream relationships with demonstrated mechanistic coupling denotes a causal link in which activation of the cGAS-STING axis directly promotes PANoptosome assembly and the execution of PANoptosis, typically through intermediate effectors such as ZBP1, type I interferons, or TNF-α. Distinguishing among these patterns is critical for both mechanistic understanding and therapeutic targeting.
In numerous disease contexts, activation of the cGAS-STING axis serves as a crucial upstream trigger for PANoptosome assembly and the subsequent execution of PANoptosis. This sequence is often initiated by the cytosolic release of DNA (eg, mitochondrial DNA), which activates cGAS-STING signaling, leading to the production of cytokines like type I IFN and tumor necrosis factor-alpha (TNF-α). These cytokines, in synergy with the initial DNA damage signal, create a permissive environment for the activation of sensors such as ZBP1, culminating in PANoptosis. This interplay has been implicated in the pathogenesis of severe infections (eg, SARS-CoV-2, influenza),24 acute lung and liver injury,25,26 septic immune dysfunction,27 neurodegenerative processes,28 and cancer progression.29 Conversely, in some contexts, PANoptosis may influence or coincide with cGAS-STING activation, suggesting a complex, context-dependent relationship. Targeting this intricate interplay holds immense therapeutic promise. In oncology, inducing immunogenic PANoptosis via cGAS-STING activation presents a novel strategy to overcome immunosuppressive tumor microenvironments and enhance the efficacy of immunotherapies. For inflammatory and degenerative diseases, inhibiting this axis may mitigate excessive cell death and tissue destruction.
This review aims to provide a comprehensive synthesis of the current understanding of the cGAS-STING pathway and PANoptosis. We will first delineate the core molecular architecture and physiological functions of the cGAS-STING axis, followed by an exploration of the mechanisms and regulators defining PANoptosis. The central focus will be on examining their multifaceted interplay across various disease models, with explicit attention to distinguishing among the three patterns of relationship described above. We will highlight how cGAS-STING activation can orchestrate PANoptosis and the consequent pathological outcomes, while also acknowledging cases in which the two processes operate in parallel. Furthermore, we will discuss the latest advances in therapeutic strategies, particularly focusing on nanomedicine approaches designed to precisely modulate this interface. Finally, we will address the prevailing translational challenges and future directions. By elucidating the mechanisms and therapeutic potential of the cGAS-STING -PANoptosis nexus, this review seeks to illuminate new avenues for treating a wide array of intractable human diseases.
cGAS-STING Signaling Pathway
Overview of the cGAS-STING Signaling Pathway
The cGAS-STING signaling axis consists of cyclic GMP-AMP synthase (cGAS) and the stimulator of interferon genes (STING) (Figure 1). Functioning upstream of STING,2,3 cGAS acts as a cytosolic DNA sensor that recognizes pathogenic or misplaced double-stranded DNA in a sequence-independent manner, thereby initiating a type I interferon (IFN)-dependent innate immune response essential for host defense against infections.30 In addition to microbial DNA, cGAS can be activated by endogenous DNA—such as mitochondrial DNA or nuclear chromatin released upon genomic stress—linking this pathway to autoinflammatory diseases, sterile inflammation, and cellular senescence.30 Upon binding dsDNA, cGAS undergoes a conformational shift that enhances its catalytic activity,31–35 leading to the synthesis of 2′,3′-cyclic GMP-AMP (cGAMP) from ATP and GTP [26]. This secondary messenger binds to and activates STING, a ∼40 kDa transmembrane adaptor protein residing in the endoplasmic reticulum (ER),3,36,37 prompting its oligomerization into dimers and higher-order complexes.38,39 Activated STING then translocates to the ER-Golgi intermediate compartment, where it recruits TBK1 and IKK kinases. TBK1 mediates phosphorylation of itself and STING, resulting in IRF3 phosphorylation and activation, while IKK promotes NF-κB signaling via IκBα phosphorylation.30 These events facilitate nuclear translocation of IRF3 and NF-κB, driving the expression of type I interferons (eg, IFN-β) and proinflammatory cytokines such as TNF and IL-6.40 Together, these responses coordinate antimicrobial and antiviral defense mechanisms.30 Following activation, STING is targeted for degradation through endolysosomal pathways.30 Beyond its classical role in infection, the cGAS-TING pathway also senses tissue damage and cellular stress, and participates in regulating autophagy, metabolism, senescence, and programmed cell death. It is crucial for maintaining tissue homeostasis, and its dysregulation has been associated with a spectrum of disorders, including inflammatory, autoimmune, degenerative, and malignant diseases.30
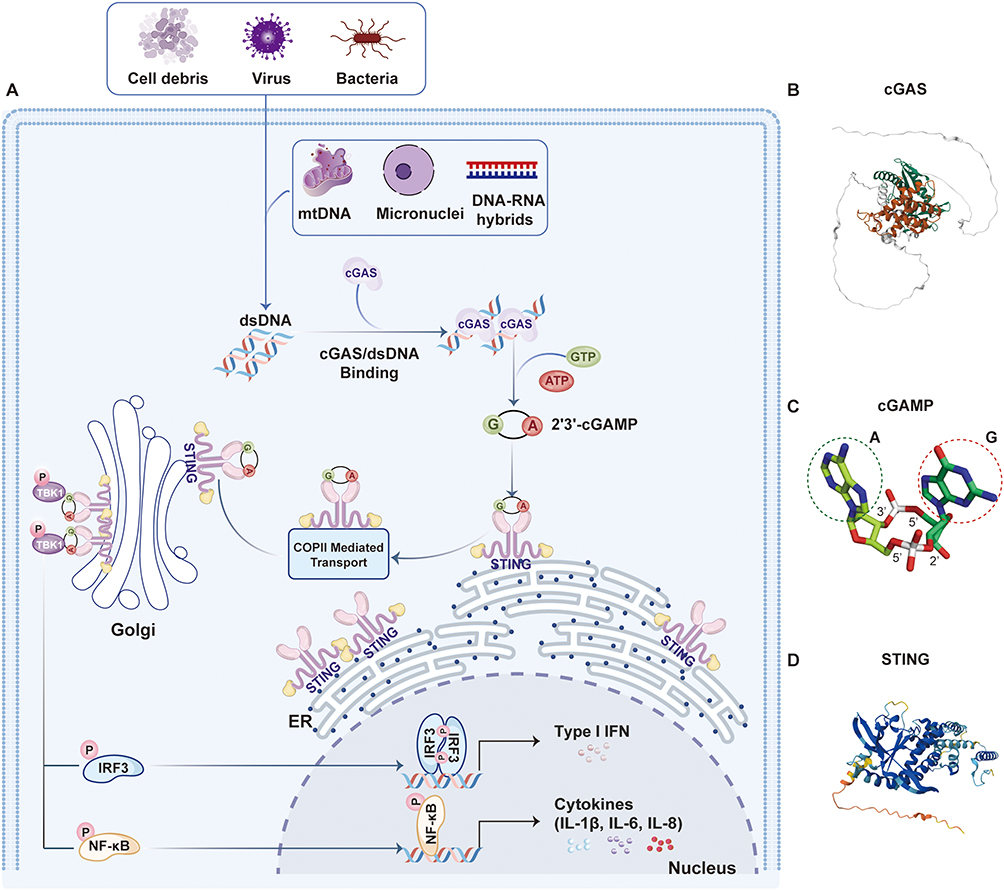 |
Figure 1 The cGAS-STING Signaling Cascade. (A) The cGAS-STING pathway is initiated by the accumulation of cytosol double-stranded DNA(dsDNA), which are introduced by virus, bacteria, dead cells, mitochondria and cancer cells et al cGAS recognizes dsDNA exposed during pathogen infection or cellular stress, leading to produce the second messenger, 2′3′ cyclic GMP-AMP (cGAMP). Upon accumulation, cGAMP binds to STING located on the endoplasmic reticulum (ER) membrane and facilitates further pathway activation. The binding of cGAMP to STING mediates STING dimerization and translocation from the ER to the Golgi apparatus, where STING forms a complex with TANK-binding kinase 1 (TBK1), which, through auto-phosphorylation and STING phosphorylation, mediates recruitment. (B) Structural overview of full-length cGAS. The structured catalytic domains (green/brown) are flanked by flexible terminal regions (white loops). (C) Stick representation of 2′,3′-cGAMP. Key moieties include the Adenine (A, green circle) and Guanine (G, red circle) bases, with annotated ribose sugar positions and phosphate linkages. (D) Ribbon diagram of STING bound to cGAMP. The protein forms a dimeric complex (dark/light blue), illustrating the ligand-induced conformational changes essential for downstream signaling. |
The Role of cGAS-STING in Non-Cancer Diseases: A Unified Mechanism Linking Tissue Injury to Inflammation
The cGAS-STING pathway, initially characterized as a sentinel for microbial DNA, has emerged as a central mediator of sterile inflammation across diverse organ systems. Its activation in non-malignant diseases is almost uniformly triggered by the accumulation of cytosolic self-DNA, most frequently mitochondrial DNA (mtDNA) released from stressed or damaged cells. This common initiating event drives a stereotyped type I interferon and pro-inflammatory cytokine response that, depending on context, can contribute to either tissue repair or pathological chronic inflammation.41–43
In the central nervous system, mtDNA released from damaged neurons activates cGAS in microglia and astrocytes, driving sustained type I IFN production that accelerates tau hyperphosphorylation and α-synuclein aggregation in Alzheimer’s and Parkinson’s diseases, respectively.42,44 Similarly, in the lung, mtDNA from injured epithelial cells activates cGAS-STING in resident macrophages and fibroblasts, promoting the persistent inflammation and fibroblast-to-myofibroblast differentiation characteristic of chronic obstructive pulmonary disease and idiopathic pulmonary fibrosis.45–49 In the cardiovascular system, DNA released from dying cells within atherosclerotic plaques or infarcted myocardium triggers STING-dependent inflammation, driving plaque instability, maladaptive cardiac remodeling, and heart failure.6,43,50 A parallel cascade occurs in the liver, where lipotoxicity and oxidative stress in non-alcoholic steatohepatitis cause hepatocyte mtDNA release, activating STING in Kupffer cells and hepatic stellate cells to perpetuate injury and fibrosis.51–53 In the kidney, ischemic or toxic insults to tubular epithelial cells similarly release cytosolic DNA, triggering STING-mediated production of inflammatory and profibrotic mediators that exacerbate acute injury and accelerate progression to chronic kidney disease.54–56 Finally, in metabolic tissues, nutrient excess and endoplasmic reticulum stress cause mtDNA release from adipocytes, activating STING in adipose tissue macrophages and driving the chronic low-grade inflammation that underlies insulin resistance in obesity and type 2 diabetes.57 Thus, while the organ-specific pathologies differ, the underlying mechanism is remarkably uniform: cellular stress leads to cytosolic DNA accumulation, cGAS-STING activation, and downstream inflammatory signaling. This recurring theme positions the cGAS-STING pathway as a central amplifier of sterile inflammation, explaining its broad involvement across seemingly disparate diseases and highlighting its therapeutic potential.
The Role of cGAS-STING in Cancer
Epigenetic silencing of the cGAS-STING pathway represents a common immune evasion mechanism in cancer, with its downregulation observed across numerous human malignancies.58,59 This suppression occurs through diverse processes —including epigenetic alterations, post-translational modifications (PTMs), and dysregulated intracellular metabolic pathways—collectively contributing to therapy resistance. Key regulatory factors include DNA and histone modifiers, non-coding RNAs (ncRNAs), and RNA modification writers such as those mediating 5-methylcytosine (m5C). These aberrant epigenetic changes promote oncogenesis by disrupting normal gene expression, altering protein dynamics, and facilitating malignant transformation.60 Specifically, DNA methylation,61 histone methylation and demethylation,62–67 m5C modification,68 and ncRNA-mediated regulation69,70 have been implicated in the suppression of cGAS and STING. Additionally, PTMs—including ubiquitination,71–74 phosphorylation,75–77 methylation,78,79 and lactylation,80 palmitoylation81—modulate the expression and function of cGAS and STING through multiple mechanisms. Metabolic pathways such as serine,82 purine synthesis,83 glucose,68 fatty acid,84 and ATP metabolism85 further regulate this axis. A deeper understanding of these metabolic reprogramming mechanisms may reveal therapeutic vulnerabilities to reactivate cGAS-STING signaling and enhance antitumor immunity.
In oncology, cGAS-TING activation exerts antitumor effects by stimulating innate immunity, promoting senescence in pre-malignant cells, enhancing conventional therapy responses, and inducing regulated cell death (RCD) via both interferon (IFN)-dependent and -independent mechanisms.86–88 These functions underscore its role as a tumor-suppressive pathway with therapeutic promise. However, monotherapy with STING agonists has shown limited clinical efficacy in patients with advanced cancer. Recent preclinical studies using in vitro and in vivo models have begun to uncover how cancer cells autonomously and non-autonomously evade cGAS-STING-mediated immune surveillance.61,62,64,68,69
PANoptosis
Core Mechanism of PANoptosis
PANoptosis, a term first conceptualized by Kanneganti’s research team in 2019, describes the simultaneous occurrence of pyroptosis, apoptosis, and necroptosis within cells.10 Functioning as a distinct inflammatory programmed cell death (PCD) pathway, it integrates key features of these three modes yet cannot be fully explained by any one alone.89 PANoptosis serves as a defensive response against external stimuli and pathogens, thereby helping to maintain cellular homeostasis and organismal stability. This phenomenon was initially identified in macrophages exposed to influenza A virus.15 It is an innate immune, lytic cell death process driven by caspases and receptor-interacting serine/threonine-protein kinases (RIPKs), and is centrally regulated by large multi-protein complexes known as PANoptosomes.10,90 These multifaceted complexes assemble by integrating molecular components from the machinery of other cell death modalities, such as the inflammasome for pyroptosis, the apoptosome for apoptosis, and the necrosome for necroptosis.10 The assembly of PANoptosomes and the activation of PANoptosis are triggered by specific cues, making it crucial to distinguish this process from other cell death pathways for precise therapeutic targeting10 (Figure 2). To date, several specific PANoptosomes have been molecularly characterized, including those nucleated by Z-DNA-binding protein 1 (ZBP1), AIM2, RIPK1, and NLR family pyrin domain-containing 12 (NLRP12).13,14,90–93 This process is physiologically critical for clearing pathogens, mediating cancer treatment responses, and maintaining immune homeostasis.11,12 Consequently, PANoptosis has been implicated in a wide array of pathological conditions, including infectious diseases, autoinflammatory disorders, neurodegenerative diseases, and cancer.13–22 PANoptosis is critical not only for host defense against pathogens and cancer but also for maintaining immune homeostasis and preventing disease development.94,95
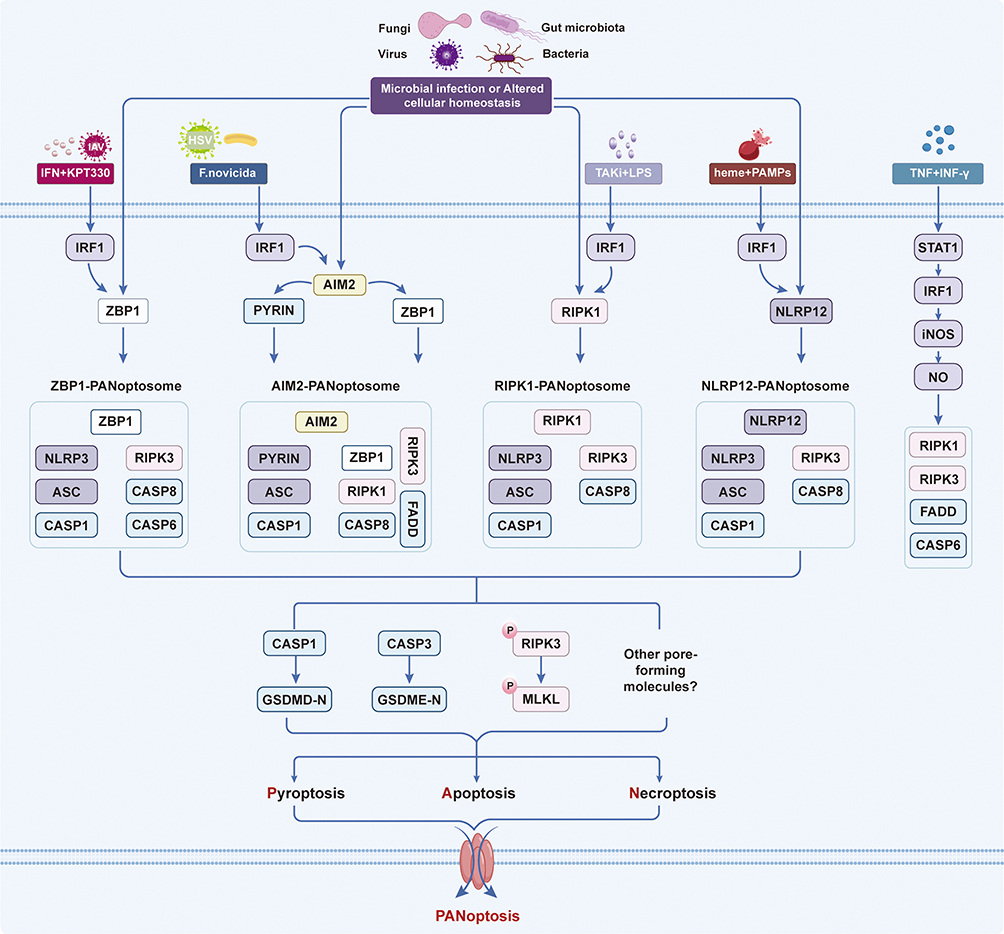 |
Figure 2 Core Mechanisms of PANoptosis. PANoptosis is driven by the assembly of distinct PANoptosome complexes in response to microbial infections and cellular homeostatic disturbances. Key sensors including ZBP1, AIM2, RIPK1, and NLRP12 recruit downstream molecules to form four PANoptosomes: ZBP1-PANoptosome, AIM2-PANoptosome, RIPK1-PANoptosome, and NLRP12-PANoptosome. These complexes act as molecular platforms to activate caspase-3/7, cleave GSDMD/GSDME, and phosphorylate MLKL, ultimately triggering membrane pore formation and PANoptotic cell death. Interferon regulatory factor 1 (IRF1) critically regulates the assembly and activation of all four PANoptosomes under distinct stimulatory conditions, such as viral infection, pathogen challenge, and cytokine or drug treatment. |
While the PANoptosis paradigm has gained considerable traction, it is important to acknowledge that its conceptual framework remains a subject of active debate within the cell death field. Critics have noted that the concurrent activation of pyroptosis, apoptosis, and necroptosis in response to certain stimuli may reflect parallel engagement of independent death pathways rather than the operation of a unified, evolutionarily conserved program orchestrated by a dedicated molecular platform.96,97 Some investigators argue that distinguishing PANoptosis as a separate cell death modality requires definitive evidence of unique regulatory nodes that are indispensable for the coordinated activation of all three constituent pathways—criteria that are still being refined and validated.98,99 Furthermore, the specificity and universality of PANoptosome complexes across different cell types and physiological contexts remain incompletely characterized. This ongoing discourse underscores the importance of continued mechanistic dissection to establish whether PANoptosis represents a genuinely discrete cell death mechanism or, alternatively, a frequently observed co-activation of parallel death cascades under specific inflammatory conditions.
Role of PANoptosis in Diseases
PANoptosis has been implicated in a variety of diseases. Given the central theme of this review, we focus on pathological conditions where PANoptosis is known to intersect with the cGAS-STING pathway. A detailed discussion of the mechanistic crosstalk is provided in this section, while this section briefly outlines the disease contexts in which PANoptosis operates.
Respiratory Diseases
In viral respiratory infections, PANoptosis serves as a critical host defense mechanism but can also drive immunopathology. During influenza A virus (IAV) infection, ZBP1 senses viral RNA and assembles the ZBP1-PANoptosome, activating caspase-1, caspase-8, caspase-3, and MLKL to induce inflammatory cell death that limits viral spread.100 In SARS-CoV-2 infection, synergistic action of TNF-α and IFN-γ triggers PANoptosis, contributing to hyperinflammation and tissue damage.20,21,101 Importantly, the cGAS-STING pathway has been identified as an upstream activator of ZBP1-dependent PANoptosis in bystander cells during SARS-CoV-2 infection.24 In bacterial pneumonia, PANoptosis helps clear infected cells but may exacerbate tissue injury and cytokine storms.102,103
Cardiovascular Diseases
PANoptosis contributes to the pathogenesis of atherosclerosis, myocardial infarction (MI), ischemia/reperfusion (I/R) injury, and doxorubicin-induced cardiotoxicity. In atherosclerosis, core PANoptosome components (AIM2, NLRP3, RIPK1/RIPK3) drive inflammation, foam cell formation, and plaque instability.104–112 During MI and I/R injury, pyroptosis, apoptosis, and necroptosis are concurrently activated, and emerging evidence suggests that ZBP1-mediated PANoptosis plays a detrimental role.113–121 In doxorubicin-induced cardiomyopathy, mitochondrial DNA (mtDNA) release triggers PANoptosis, and the protective protein FUNDC1 stabilizes mtDNA to inhibit this process.122 The cGAS-STING pathway is increasingly recognized as a key upstream regulator in these cardiovascular contexts.25,112
Kidney Diseases
PANoptosis is involved in acute kidney injury (AKI), chronic kidney disease (CKD), and diabetic kidney disease (DKD). In sepsis-induced AKI, EIF2AK2 upregulates AIM2, promoting PANoptosome formation and exacerbating kidney injury.123 In ischemia-reperfusion injury, the NLRP3 inhibitor MNS reduces PANoptosis and alleviates renal damage.124 Cisplatin-induced nephrotoxicity involves PANoptosis, which can be mitigated by activating the Nrf2 pathway. In DKD, the TRAIL/DR5 axis mediates podocyte PANoptosis, and inhibition of this pathway reduces proteinuria.125 Notably, cGAS-STING activation downstream of mtDNA release has been shown to drive PANoptosis in models of atrazine-induced nephrotoxicity and aristolochic acid nephropathy.126,127
Liver Diseases
PANoptosis plays a pivotal role in metabolic dysfunction-associated steatotic liver disease (MASLD), acute liver failure (ALF), drug-induced liver injury, and cholestatic fibrosis. In MASLD, mitochondrial dysfunction and mtDNA release stimulate PANoptosis in hepatocytes.128,129 In ALF, intestinal endotoxemia promotes hepatocyte PANoptosis via the TLR4/TAK1 axis.130 Acetaminophen overdose aggravates liver injury through AIM2-mediated oxidative stress and PANoptosis.131,132 Importantly, the cGAS-STING pathway is activated by mtDNA during hepatic ischemia-reperfusion injury and drives PANoptosis in macrophages,25 and environmental toxin-induced AIM2-PANoptosome formation has been linked to hepatic steatosis.133
Viral Infections
PANoptosis serves as a critical host defense mechanism against diverse viruses. IAV, HSV-1, and Francisella novicida infections activate PANoptosis via ZBP1- or AIM2-PANoptosomes.13,15 In coronavirus infections, impaired NLRP3 inflammasome activation leads to alternative PANoptosis via caspase-8 and RIPK3.134 The cGAS-STING pathway acts upstream to induce ZBP1-mediated PANoptosis in bystander cells during SARS-CoV-2 infection24 and in HSV-1-induced acute retinal necrosis.28
Cancer
PANoptosis exerts dual roles in cancer, acting both as a tumor suppressor and a potential therapeutic target. In colorectal cancer, IRF1 promotes PANoptosis, and its deficiency increases tumor burden.135 TNF-α and IFN-γ synergistically induce PANoptosis in multiple human cancer cell lines, suggesting a broad antitumor mechanism.136 In melanoma, combination therapy with IFN and nuclear export inhibitors upregulates ZBP1-mediated PANoptosis, leading to tumor regression.19 Importantly, activation of the cGAS-STING pathway by STING agonists or by SAMHD1 deficiency promotes PANoptosis in diffuse large B-cell lymphoma and enhances the efficacy of PD-L1 blockade.29
The Interplay of cGAS-STING and PANoptosis in Diseases
Activation of cGAS-STING Induces PANoptosis
A unifying mechanistic principle has emerged from studies across diverse pathological conditions: the cGAS-STING pathway functions as a critical upstream trigger that, upon sensing cytosolic double-stranded DNA (dsDNA), orchestrates the assembly of PANoptosome complexes and the subsequent execution of PANoptosis. This recurring axis is defined by three core mechanistic steps: (1) upstream events that generate cytosolic dsDNA, (2) signal integration via the cGAS-STING cascade leading to ZBP1-dependent PANoptosome nucleation, and (3) the coordinated activation of pyroptosis, apoptosis, and necroptosis (Figure 3).
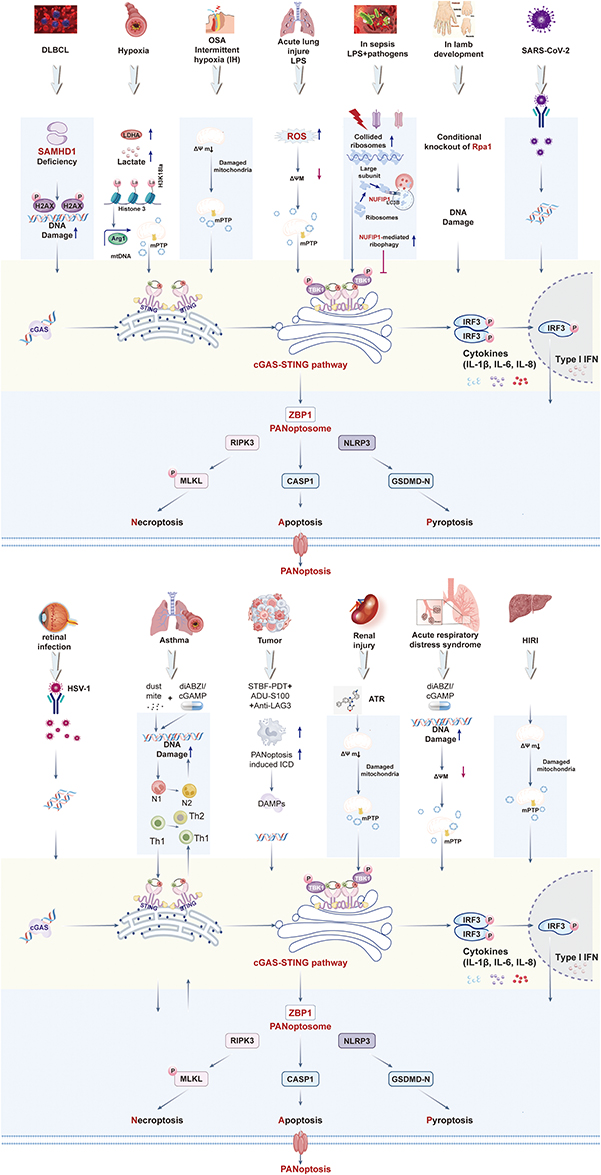 |
Figure 3 Activation of cGAS-STING induces PANoptosis. Illustration of a central and converging molecular pathway where the sensing of dsDNA by cGAS initiates a robust innate immune response that culminates in a coordinated inflammatory cell death program known as PANoptosis. Diverse stressors—including microbial infection (e.g, SARS-CoV-2, HSV-1), mitochondrial dysfunction (e.g, hypoxia, sepsis, ischemia-reperfusion), genomic DNA damage (e.g, SAMHD1/RPA1 deficiency), and sterile inflammation—trigger the release of cytosolic dsDNA. Diverse stressors—including microbial infection (e.g, SARS-CoV-2, HSV-1), mitochondrial dysfunction (e.g, hypoxia, sepsis, ischemia-reperfusion), genomic DNA damage (e.g, SAMHD1/RPA1 deficiency), and sterile inflammation—trigger the release of cytosolic dsDNA. This dsDNA is sensed by cGAS, which synthesizes 2’3’-cGAMP to activate STING. Activated STING drives TBK1-IRF3-mediated type I interferon production and NF-κB-dependent pro-inflammatory cytokine release. The synergistic action of these signals promotes ZBP1-dependent PANoptosome assembly, which coordinates the activation of necroptosis (via RIPK3-MLKL), apoptosis (via caspase-8-caspase-1), and pyroptosis (via NLRP3-GSDMD), collectively executing PANoptosis. This pathway is negatively regulated by NUFIP1-mediated ribophagy and amplified by a self-DNA positive feedback loop, contributing to the pathophysiology of infections, inflammatory diseases, and tissue injury. |
The initiating step—cytosolic dsDNA accumulation—can arise from multiple sources depending on the disease context. In models of acute lung injury, sepsis, and hepatic ischemia-reperfusion, mitochondrial dysfunction leads to mitochondrial DNA (mtDNA) release into the cytosol, a process driven by oxidative stress, mitochondrial permeability transition, or GSDMD-mediated pore formation.23,25–27 In diffuse large B-cell lymphoma and developmental defects caused by RPA1 deficiency, genomic instability results in nuclear DNA leakage.29,137 Viral infections such as SARS-CoV-2 and HSV-1 directly introduce foreign DNA or trigger the accumulation of Z-form nucleic acids.24,28 Notably, even direct pharmacological activation of STING by synthetic agonists induces a feed-forward loop wherein initial cell death releases self-DNA, further amplifying the signal.23,138
Once dsDNA accumulates, the cGAS-STING signaling cascade serves as a central signal integrator. cGAS binds dsDNA and synthesizes 2’3’-cGAMP, which activates STING, leading to TBK1-IRF3-mediated type I interferon production and NF-κB-driven inflammatory cytokine release.24,27,139 This inflammatory milieu —particularly the synergistic actions of IFN-β and TNF-α—creates a permissive environment for ZBP1 upregulation and activation.24,28,137 In multiple systems, ZBP1 has emerged as the critical PANoptosome scaffold. Activated ZBP1 nucleates a multi-protein complex that recruits RIPK3, caspase-8, ASC, and NLRP3, forming the ZBP1-PANoptosome.24–28,137 This complex serves as a molecular hub that simultaneously engages the downstream effectors of all three cell death pathways: caspase-3/7 for apoptosis, MLKL phosphorylation for necroptosis, and gasdermin D/E cleavage for pyroptosis.25–27,29,139 The coordinated activation of these effectors defines PANoptosis as an integrated inflammatory cell death program rather than the parallel engagement of independent pathways.
This mechanistic framework is consistent across a wide range of conditions. In SARS-CoV-2 infection, cGAMP transferred from infected to bystander cells activates STING, driving autophagic degradation of ADAR1 and Z-nucleic acid accumulation, which together enable ZBP1-mediated PANoptosis.24 In hepatic ischemia-reperfusion injury, STING upregulates ZBP1 expression, and genetic or pharmacological inhibition of STING suppresses PANoptosis and ameliorates tissue damage.25 Similarly, in sepsis, ribosomal collision-induced cGAS activation drives STING -dependent ZBP1-PANoptosome formation in CD4+ T cells, leading to immune dysfunction that can be reversed by cGAS-STING inhibition.27 In limb development defects caused by RPA1 deficiency, DNA damage activates cGAS-STING, which transcriptionally upregulates ZBP1; the accumulated ZBP1 then binds cytoplasmic Z-DNA to assemble the PANoptosome and trigger mesenchymal stem cell death.137
Notably, this axis can be amplified by positive feedback loops. STING activation induces PANoptosis and lytic cell death, releasing additional dsDNA (including self-DNA and mitochondrial DNA) that further activates cGAS, perpetuating a cycle of inflammation and cell death.23,138 In the context of obstructive sleep apnea, intermittent hypoxia triggers mtDNA release, which activates cGAS-STING and induces neuronal PANoptosis via ER stress modulation;139 blockade of mtDNA release or cGAS-STING signaling breaks this cycle and confers neuroprotection.
Collectively, these findings establish a conserved mechanistic architecture: diverse pathological insults converge on cytosolic dsDNA accumulation, which activates the cGAS-STING pathway; STING-dependent type I interferon and TNF-α signaling then licenses ZBP1 to assemble the PANoptosome; and the resultant coordinated engagement of pyroptosis, apoptosis, and necroptosis executes inflammatory cell death. This core logic operates across infectious, inflammatory, ischemic, and malignant diseases, positioning the cGAS-STING-PANoptosis axis as a central pathogenic mechanism and a promising therapeutic node.
Induction of PANoptosis Activates cGAS-STING
The relationship between PANoptosis and cGAS-STING is not uniformly unidirectional. While Activation of cGAS-STING Induces PANoptosis establishes cGAS-STING as an upstream trigger of PANoptosis, emerging evidence reveals that PANoptosis can also activate or amplify cGAS-STING signaling, depending on the pathological context and the nature of the initial insult. These studies fall into three mechanistic categories.
First, parallel activation without hierarchical coupling is exemplified in a model of heme-induced sepsis. Li et al demonstrated that in Kupffer cells, elevated heme synergizes with bacterial components to trigger simultaneous PANoptosis and cGAS-STING activation as parallel consequences of mitochondrial damage rather than through a linear cascade.140 Mechanistically, heme activates phospholipase C gamma (PLC-γ), promoting the translocation of cleaved gasdermin D (c-GSDMD) to mitochondria. This results in mitochondrial pore formation, dysfunction, and the release of mitochondrial DNA (mtDNA), which concurrently drives PANoptosis —through coordinated pyroptosis, apoptosis, and necroptosis—and activates cGAS-STING signaling, ultimately leading to cellular senescence.140 Critically, pharmacological inhibition of PLC-γ or scavenging of heme attenuates both outcomes, confirming that mitochondrial damage serves as a shared upstream node. This parallel relationship underscores that cGAS-STING and PANoptosis can operate as co-effectors of a common trigger rather than in a strict hierarchical order.140
Second, a sequential model in which PANoptosis serves as the upstream executor that generates immunostimulatory signals to activate the cGAS-STING pathway has been established in several cancer immunotherapy studies. In this context, PANoptosis acts as the initial event, releasing damage-associated molecular patterns (DAMPs) and cytosolic double-stranded DNA (dsDNA), which subsequently engage cGAS-STING to amplify adaptive immunity. For instance, Yin et al developed an ultrasound-responsive Mn/Se nanozyme that induces mitochondrial reactive oxygen species (ROS) overload and coordinated PANoptosis in bladder cancer cells.141 The resulting immunogenic cell death releases dsDNA, which, in conjunction with manganese ions, potentiates cGAS binding and amplifies type I interferon signaling, dendritic cell maturation, and CD8⁺ T cell infiltration, effectively converting immunologically “cold” tumors into “hot” ones.141 Similarly, Lin et al constructed a TME-responsive nanozyme (Ava@HM/Trop2) that triggers PANoptosis through oxidative stress and mitochondrial damage, leading to the release of tumor-associated antigens and DAMPs that robustly activate the cGAS-STING pathway.142 This activation further reinforces dendritic cell maturation and T-cell cytotoxicity, creating a positive feedback loop that enhances antitumor immunity and synergizes with PD-1 inhibitors.142 In another study, ultrasound-activated engineered extracellular vesicles were shown to induce immunogenic PANoptosis in triple-negative breast cancer cells, with the resulting cytosolic DNA release activating cGAS-STING in antigen-presenting cells.143 Genetic ablation of STING completely abolished the therapeutic efficacy, confirming that the PANoptosis-to-cGAS-STING axis is essential for the observed immune activation.143
Ultrasound-activated engineered extracellular vesicles were found to induce immunogenic PANoptosis in cancer cells, which is critically mediated by the cGAS-STING signaling pathway.143 The nanotherapy causes DNA damage and cytosolic DNA release, activating the cGAS-STING axis in antigen-presenting cells. This activation enhances dendritic cell maturation, macrophage repolarization, and type I interferon production, leading to potent T-cell priming and antitumor immunity.143 Genetic ablation of STING abolished the PANoptosis-driven immune activation and therapeutic efficacy, confirming the pathway’s essential role. Thus, the cGAS-STING pathway acts upstream to initiate and amplify PANoptosis-triggered adaptive immunity, establishing a novel immunotherapeutic strategy against triple-negative breast cancer (Figure 4).143
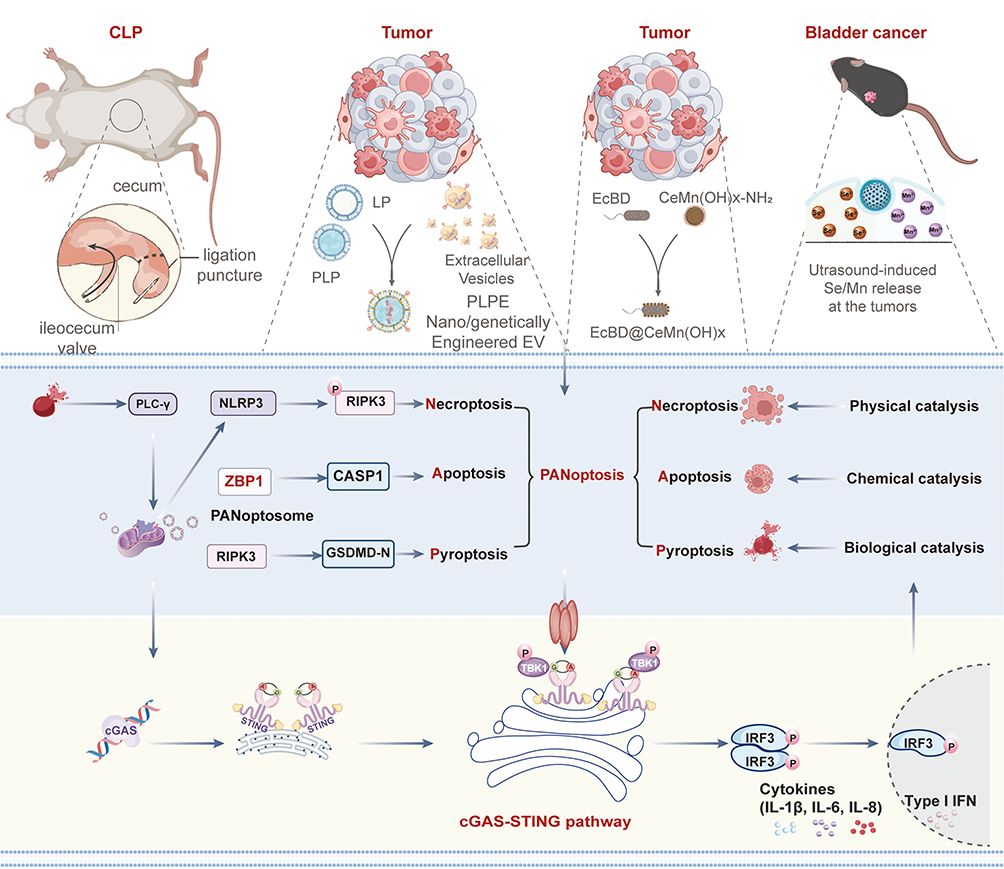 |
Figure 4 CLP and Nano/genetically-engineered extracellular vesicles orchestrate PANoptosis and cGAS-STING innate immune activation across inflammatory and oncological disease models. |
Likewise, Zhu et al developed a nano-biocomposite (EcBD@CeMn(OH)x) in which Ce3⁺ ions induce DNA damage and organelle dysfunction to drive PANoptosis, while Mn2⁺ ions bind to released DNA fragments to stimulate cGAS-STING-mediated innate immunity, thereby establishing a coordinated PANoptosis-to-cGAS-STING cascade that reverses the immunosuppressive tumor microenvironment.144 Collectively, these studies illustrate a recurring theme in oncology: PANoptosis functions as an upstream immunogenic cell death mechanism that provides the critical dsDNA ligand for cGAS-STING activation, which in turn amplifies antitumor immunity.
In summary, the mechanisms by which PANoptosis regulates cGAS-STING are context-dependent and can be categorized into three distinct patterns. The first pattern involves parallel activation, as seen in heme-induced sepsis, where mitochondrial damage simultaneously triggers both pathways without a hierarchical relationship.140 The second pattern, predominantly observed in cancer immunotherapy, positions PANoptosis as an upstream executor that releases dsDNA to activate cGAS-STING, thereby amplifying antitumor immune responses.142–144 The third pattern, exemplified by studies using bavachinin or quercetin, demonstrates a hierarchical relationship in which cGAS-STING acts as an upstream mediator of PANoptosis, with reciprocal amplification loops reinforcing the cascade.145,146 Together, these findings underscore the complexity of the cGAS-STING–PANoptosis interplay and highlight the necessity of considering both the directionality and the mechanistic context when designing therapeutic interventions.
Molecular Interaction Nodes, Spatiotemporal Regulation, and PANoptosome Subtype Specificity in cGAS-STING-Driven PANoptosis
Beyond the established upstream–downstream relationship, a more nuanced understanding requires dissecting the direct and indirect molecular bridges that connect the cGAS-STING cascade to the PANoptosome machinery. These interactions are not monolithic; instead, they exhibit context-dependent spatiotemporal features and differential regulation of specific PANoptosome subtypes.
Direct molecular interactions between STING and core PANoptotic effectors have been identified, suggesting physical platforms for signal integration. For instance, STING has been shown to interact with RIPK1 and RIPK3, particularly following DNA damage or herpesvirus infection, facilitating the assembly of a RIPK1-RIPK3 -caspase-8 complex that can drive concurrent apoptosis and necroptosis.28 Similarly, STING can associate with NLRP3 in the Golgi apparatus following its translocation from the ER, promoting inflammasome activation and subsequent pyroptosis.147 These physical interactions imply that STING may serve as a scaffold that nucleates or stabilizes PANoptosome components, blurring the line between upstream signaling and the death-executing complex. Furthermore, cGAS itself can interact with ZBP1 in the cytosol under conditions of mitochondrial stress, promoting the formation of a ZBP1-cGAS-RIPK3 complex that amplifies PANoptosis in a manner independent of type I interferon signaling.24
Indirect molecular crosstalk is largely mediated by the transcriptional and post-translational effects of cGAS-STING downstream products. The most prominent indirect mechanism involves type I interferons (IFN-α/β) and TNF-α, which are potent inducers of ZBP1 and RIPK1 expression.16 This transcriptional upregulation primes cells for PANoptosome assembly. Additionally, STING-driven NF-κB activation leads to the production of pro-IL-1β and pro-IL-18, providing the substrates for caspase-1-mediated pyroptosis within the PANoptosome. Another critical indirect node is the metabolic crosstalk: STING activation can induce mitochondrial dysfunction and reactive oxygen species (ROS) production, which in turn facilitate the oligomerization of AIM2 and NLRP3, lowering the threshold for PANoptosome activation.112 These indirect pathways create a permissive inflammatory environment that amplifies the core death-inducing signals.
The spatiotemporal regulation of this crosstalk is critical for determining cellular outcomes. Following DNA sensing, STING traffics from the ER to the ER-Golgi intermediate compartment (ERGIC) and then to the Golgi apparatus. PANoptosome assembly appears to be spatially coupled to STING’s itinerary. For instance, the interaction between STING and NLRP3 predominantly occurs in the Golgi, where potassium efflux and other ionic changes facilitate NLRP3 oligomerization.147 Conversely, STING-mediated autophagic degradation of ADAR1, which relieves repression on ZBP1, is thought to occur at the ERGIC, highlighting how STING’s subcellular localization dictates which PANoptosome pathway is engaged.24 Temporally, the cascade is often bifurcated: an early phase of STING-driven interferon production creates a “primed” state, while a subsequent, sustained phase of STING signaling or accumulation of secondary DAMPs (eg, mtDNA) triggers the actual PANoptosome assembly. This temporal separation allows for signal amplification but also provides checkpoints for regulation, such as the NUFIP1-mediated ribophagy that clears stress signals and abrogates the late-phase PANoptosis.27
Importantly, the cGAS-STING pathway exerts divergent regulatory effects on distinct PANoptosome subtypes. For the ZBP1-PANoptosome, cGAS-STING signaling is a dominant upstream activator, primarily through two mechanisms: (1) induction of type I IFNs that transcriptionally upregulate ZBP1, and (2) creation of a Z-nucleic acid-rich environment, either via direct viral sensing or via ADAR1 degradation, which provides the ligand for ZBP1 activation.24,137 In contrast, the AIM2-PANoptosome is more directly engaged by DNA released as a consequence of STING-driven cell death, creating a positive feedback loop where STING activation leads to DNA release, which in turn activates AIM2.23 However, in some contexts, STING can also transcriptionally upregulate AIM2 via IRF3, providing a parallel priming mechanism.123 For the RIPK1-PANoptosome, the relationship is complex: STING can directly interact with and activate RIPK1, but STING-induced TNF-α also engages TNFR1 to drive RIPK1 activation, leading to pathway convergence.29 Lastly, for the NLRP12-PANoptosome, the link to cGAS-STING appears less direct and more context-specific, often involving heme or other DAMPs that simultaneously trigger mitochondrial damage (activating cGAS-STING) and NLRP12 oligomerization, with the two pathways operating in parallel rather than in a strict linear hierarchy.90 These distinctions underscore that while cGAS-STING is a recurrent upstream node, the specific molecular logic and dependency vary significantly depending on the initiating stimulus and the PANoptosome subtype engaged.
Cross-Disease Comparative Analysis of the cGAS-STING-PANoptosis Axis
Beyond the individual disease descriptions, a cross-disease comparison of the cGAS-STING–PANoptosis axis reveals both unifying principles and context-specific adaptations. Across all conditions examined—including respiratory infections, sterile inflammatory diseases, ischemic injuries, and cancer—the axis consistently operates as a two-step cascade: an initial cytosolic DNA sensing event (often involving mitochondrial or genomic DNA) activates STING, which then coordinates the assembly of a PANoptosome, most frequently the ZBP1-containing complex, leading to the simultaneous engagement of pyroptosis, apoptosis, and necroptosis.23–29 This core architecture appears evolutionarily conserved and serves as a rapid, fail-safe mechanism to eliminate stressed or infected cells.
However, the functional outcome of this interplay diverges sharply depending on disease context. In infectious diseases (eg, SARS-CoV-2, influenza, HSV-1), cGAS-STING-driven PANoptosis acts as a protective host defense by limiting pathogen replication, yet when excessive it drives immunopathology and cytokine storm.24,28,100 In sterile inflammatory conditions such as acute lung injury, hepatic ischemia-reperfusion, or obstructive sleep apnea, the same axis becomes a purely maladaptive driver of tissue damage, amplifying inflammation through a self-sustaining loop where initial cell death releases more self-DNA, further activating cGAS-STING.23,25,26,139 Conversely, in oncology, the therapeutic activation of this cascade is being harnessed to convert immunologically “cold” tumors into “hot” ones: STING agonists or nanoparticle-based delivery systems induce immunogenic PANoptosis, enhancing cross-priming of antitumor T cells and synergizing with immune checkpoint inhibitors.29,143,148
Another point of divergence lies in the dominant upstream sensors. While ZBP1 is the most frequently reported PANoptosome initiator downstream of STING in viral infections, sepsis, and ischemia models,24,27,119,137 the AIM2-PANoptosome predominates in contexts involving bacterial DNA or environmental toxins, as seen in hepatic co-exposure to β-hexachlorocyclohexane and nanoplastics,133 and in some forms of acute kidney injury.123,132 Moreover, the hierarchical relationship between cGAS-STING and PANoptosis is not always unidirectional. In heme-induced sepsis, both pathways are co-activated in Kupffer cells as parallel consequences of mitochondrial damage, without a strict upstream-downstream relationship,140 illustrating that the axis can be engaged in distinct regulatory modes depending on the primary insult.
Finally, the comparative analysis underscores a striking uniformity in therapeutic vulnerability. Across diseases, pharmacological inhibition of cGAS or STING consistently suppresses PANoptosis and ameliorates pathology in models of acute lung injury, sepsis, hepatic ischemia-reperfusion, and atrazine-induced nephrotoxicity,24,26,27,126 whereas STING agonists or agents that promote mitochondrial DNA release exert antitumor effects by unleashing immunogenic PANoptosis in lymphoma, breast cancer, and cutaneous squamous cell carcinoma.29,143,148 This convergence suggests that the cGAS-STING–PANoptosis node represents a tractable therapeutic target whose direction of modulation—inhibition in inflammatory diseases versus activation in cancer—can be rationally selected based on disease context.
Therapeutic Potential of cGAS-STING-PANoptosis in Diseases
The pharmacological modulation of the cGAS-STING-PANoptosis axis represents a promising but complex therapeutic frontier. While numerous compounds have demonstrated efficacy in preclinical models (summarized in Table 1), the context-dependent nature of this axis necessitates a critical evaluation of its therapeutic potential, translational challenges, and the dual risks of over-activation or inappropriate inhibition.
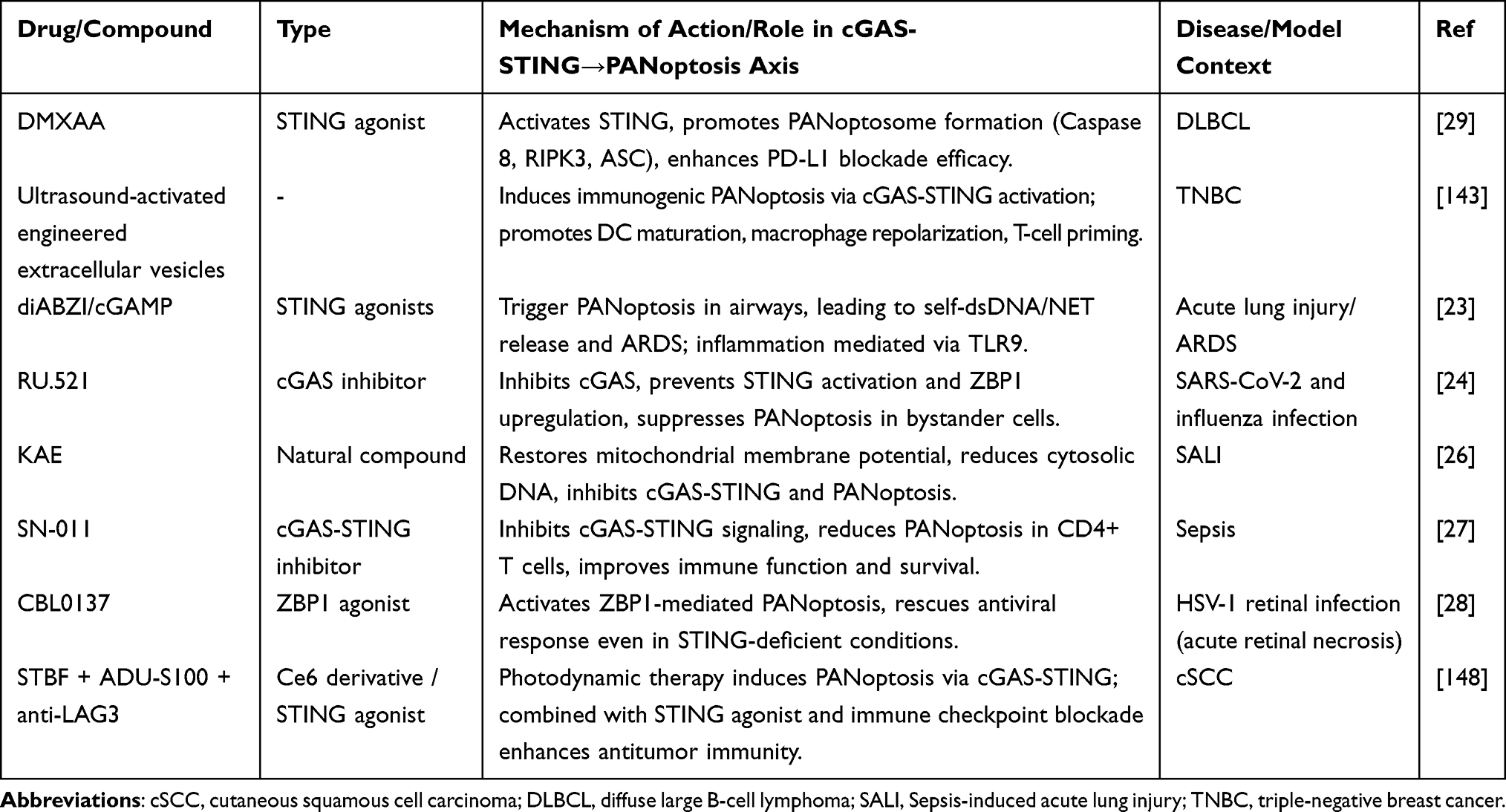 |
Table 1 Summary of Drug/Compound That Modulate cGAS-STING-Mediated PANoptosis Induction in Diseases Therapy |
The Double-Edged Sword: Context-Dependent Therapeutic Strategies
The pharmacological modulation of the cGAS-STING–PANoptosis axis is inherently context-dependent, reflecting its opposing roles in oncology versus inflammatory and infectious diseases. In oncology, the goal is to activate this axis to harness immunogenic cell death, whereas in inflammatory, infectious, and ischemic conditions, the objective shifts to its inhibition to prevent excessive tissue damage.
In cancer contexts, several strategies have been developed to activate the cGAS-STING-PANoptosis cascade. For instance, STING agonists such as DMXAA and diABZI are designed to directly engage STING, thereby promoting PANoptosome assembly and triggering immunogenic PANoptosis, a strategy that has shown promise in models of diffuse large B-cell lymphoma (DLBCL).29,137,138 Complementing this, ultrasound-activated engineered extracellular vesicles have been employed to achieve tumor-specific delivery of STING agonists, inducing immunogenic PANoptosis while minimizing systemic toxicity, as demonstrated in triple-negative breast cancer models.143 Further expanding the toolkit, CBL0137, a ZBP1 agonist, has been shown to activate ZBP1-mediated PANoptosis and can even rescue antiviral responses in STING-deficient conditions, highlighting a parallel route to engage cell death independently of STING.28
Conversely, in inflammatory and infectious diseases, the emphasis is on restraining this axis to prevent pathology. For example, RU.521, a specific cGAS inhibitor, has been reported to suppress STING activation and ZBP1 upregulation, thereby preventing bystander cell PANoptosis during SARS-CoV-2 and influenza infections and reducing associated lung injury.24 In the context of sepsis, SN-011, an inhibitor targeting both cGAS and STING, effectively attenuates PANoptosis in CD4+ T lymphocytes, leading to restored immune function and improved survival.27 In parallel, natural compounds have also shown efficacy in inhibiting this pathway. KAE, for instance, ameliorates LPS-induced acute lung injury by restoring mitochondrial membrane potential and reducing cytosolic DNA, thus inhibiting the cGAS-STING-PANoptosis axis.26 Similarly, lycopene exerts protective effects in atrazine-induced nephrotoxicity by stabilizing mitochondrial DNA (mtDNA) and preventing STING-dependent PANoptosis.126
Finally, the therapeutic landscape is further enriched by combination and emerging strategies that integrate these principles. Photodynamic therapy using a Ce6 derivative (STBF) has been shown to induce PANoptosis via cGAS-STING activation, and when combined with the STING agonist ADU-S100 and immune checkpoint blockade (anti-LAG3), it produces synergistic antitumor immunity in models of cutaneous squamous cell carcinoma (cSCC).146 Bavachinin (BVC) triggers ZBP1 -mediated PANoptosis in endometrial cancer (EC) cells, and the cGAS-STING pathway acts as a critical intermediate signaling axis in this process.145 BVC first induces mitochondrial ROS accumulation and DNA damage, which promotes the release of cytoplasmic double-stranded DNA and activates the cGAS-STING -TBK1-IRF3 signaling cascade. Activated cGAS-STING signaling further upregulates ZBP1 expression, driving the assembly of the PANoptosome complex and initiating integrated pyroptosis, apoptosis and necroptosis.145 Moreover, BVC directly targets TLR4 to boost mitochondrial ROS production, forming a positive feedback loop that amplifies cGAS-STING activation and subsequent PANoptosis.145 Inhibition of STING markedly suppresses ZBP1 expression and reverses BVC-induced PANoptosis, confirming the regulatory link between cGAS -STING and PANoptosis. Additionally, BVC enhances cisplatin chemosensitivity by aggravating DNA damage and strengthening cGAS-STING -mediated PANoptosis, and these anti-tumor effects are verified in vivo.145 Collectively, these studies underscore that the direction of therapeutic modulation—activation versus inhibition—must be carefully tailored to the specific disease context to harness the full potential of the cGAS-STING -PANoptosis axis.
Together, these studies underscore the context-dependent duality of targeting the cGAS-STING-PANoptosis axis. While STING agonists and PANoptosis inducers hold promise for converting immunologically “cold” tumors into “hot” ones, inhibitors of this axis are critical for curbing excessive inflammation and tissue damage in non-malignant diseases. The successful translation of these strategies will likely depend on precise spatiotemporal control, cell-type-specific delivery, and combination with existing therapies.
Translational Challenges and Potential Adverse Effects
Despite promising preclinical data, several significant hurdles impede clinical translation. First, achieving cell-type-specific modulation remains a major challenge. The cGAS-STING pathway is ubiquitously expressed, and its downstream effects vary dramatically between cell types. For example, STING activation in antigen -presenting cells is desirable for anti-tumor immunity, while its activation in cardiomyocytes or neurons can be detrimental in ischemic injury.25,139 Systemic administration of STING agonists or inhibitors risks unintended consequences, including autoimmunity, cytokine storms, or increased susceptibility to infections.
Second, the complex feedback loops and crosstalk with other pathways complicate therapeutic outcomes. As highlighted in Section 4, cGAS-STING activation can lead to PANoptosis, which in turn releases self-DNA that further activates cGAS-STING, creating a self-amplifying inflammatory loop.23,24 While this can be targeted for therapeutic benefit, it also increases the risk of off-target inflammation. Furthermore, the relationship between cGAS-STING and PANoptosis is not always unidirectional; they can act as parallel downstream effectors of a common upstream trigger, as seen in heme-induced sepsis.140 This suggests that targeting a single node (eg, STING) may not fully abrogate the pathological cell death if parallel pathways are still active.
Third, the lack of robust predictive biomarkers for patient stratification remains a critical gap. Preclinical studies often use acute models with clear temporal dynamics, but human diseases are typically chronic with heterogeneous etiologies. Identifying which patients are most likely to benefit from STING agonism (eg, those with “cold” tumors) versus STING inhibition (eg, those with overactive interferon signatures) is essential for clinical success. Currently, there are no validated biomarkers to guide such patient selection.
Emerging Strategies to Overcome Translational Barriers
To address these challenges, innovative delivery systems are being developed. Nanomedicine platforms, such as ultrasound-activated engineered extracellular vesicles, enable tumor-specific delivery of STING agonists, inducing immunogenic PANoptosis while minimizing systemic toxicity.143 Similarly, stimuli-responsive nanoparticles can be designed to release cGAS-STING modulators only in response to disease-specific microenvironments (eg, low pH, specific enzymes), enhancing precision. Additionally, targeting downstream effectors like ZBP1 may offer more context-specific intervention. For example, the ZBP1 agonist CBL0137 can rescue antiviral PANoptosis even in STING-deficient conditions, while ZBP1 inhibition might be a more selective strategy to block PANoptosis without fully abrogating all cGAS-STING functions.28
Finally, combination therapies represent a promising avenue. In cancer, STING agonists are being combined with immune checkpoint inhibitors (eg, anti-PD-L1, anti-LAG3) to synergistically enhance T-cell-mediated antitumor immunity.29,148 In inflammatory diseases, combining cGAS-STING inhibitors with agents that stabilize mitochondria (eg, lycopene) or restore cellular homeostasis may provide additive or synergistic benefits.126
In summary, the therapeutic modulation of the cGAS-STING-PANoptosis axis holds immense promise but requires a nuanced approach. Future success will depend on the development of precision delivery systems, identification of predictive biomarkers, and a deeper mechanistic understanding of the cell-type- and context-specific roles of this axis in human disease.
Conclusions and Perspectives
The intricate interplay between the cGAS-STING pathway and PANoptosis represents a rapidly evolving frontier in immunology and cell biology, with profound implications for understanding disease pathogenesis and developing novel therapeutic strategies. This review has systematically delineated the molecular architecture of the cGAS-STING signaling cascade and the core mechanisms of PANoptosis (as detailed in Section 3.1), highlighting their individual and synergistic roles across a spectrum of diseases, particularly in cancer. The cGAS-STING axis, initially recognized as a sentinel of cytosolic DNA and a cornerstone of innate immunity, is now appreciated as a central regulator of tissue homeostasis, inflammation, and cell fate. Concurrently, PANoptosis has emerged as a unique, coordinated cell death pathway that integrates the key features of pyroptosis, apoptosis, and necroptosis, governed by multifaceted PANoptosome complexes. The convergence of these two powerful biological processes forms a critical regulatory node that significantly influences disease outcomes, from cancer progression and therapy resistance to acute tissue injury and chronic inflammatory disorders.
A paramount theme elucidated throughout this review is the predominant role of the cGAS-STING pathway as a crucial upstream activator of PANoptosis in numerous pathological contexts. Evidence from diverse models—including cancer, respiratory diseases, neurological conditions, hepatic ischemia-reperfusion injury, and septic immune dysfunction—consistently demonstrates that cGAS-STING activation, often triggered by cytosolic DNA released under conditions of cellular stress, genomic instability, or infection, serves as a potent initiator of PANoptosome assembly and subsequent PANoptosis. This recurring motif underscores a fundamental biological principle: the sensing of intracellular nucleic acid threats by cGAS-STING can escalate into a robust, inflammatory cell death program designed to eliminate damaged or infected cells and alert the immune system.
However, the relationship is not universally unidirectional. As noted in specific contexts like heme-induced sepsis, cGAS-STING activation and PANoptosis can be parallel, co-incident events both downstream of a common upstream trigger (eg, mitochondrial damage), without a strict hierarchical relationship. This nuance highlights the context-dependent nature of this interplay and cautions against oversimplification. The dysregulation of either pathway can lead to severe pathology. Uncontrolled cGAS-STING signaling can drive excessive inflammation and tissue damage, while aberrant PANoptosis can contribute to the destruction of healthy tissues in ischemic injuries, neurodegenerative diseases, and organ failure. Therefore, a precise understanding of the spatiotemporal regulation of this crosstalk is essential for therapeutic targeting.
The therapeutic potential of leveraging the cGAS-STING-PANoptosis axis is immense, particularly in oncology. The limitations of conventional STING agonists—such as poor bioavailability, rapid clearance, and systemic toxicity—have spurred the development of innovative nanomedicine approaches. Engineered nanoparticles and extracellular vesicles show remarkable promise in achieving tumor-specific delivery of STING agonists, thereby locally activating the pathway, inducing immunogenic PANoptosis, and reshaping the tumor microenvironment to be more permissive to immune attack. These strategies can effectively turn immunologically “cold” tumors “hot” and synergize with existing immunotherapies. Furthermore, natural compounds and small-molecule inhibitors that modulate specific nodes within this axis (eg, Lycopene stabilizing mtDNA, Oridonin targeting SIRT2/NLRP3) offer additional avenues for intervention, demonstrating efficacy in mitigating injury in models of kidney, liver, and lung diseases.
Despite significant progress in elucidating the cGAS-STING-PANoptosis axis, several critical questions remain unresolved, presenting key avenues for future investigation. A primary challenge is the identification of novel molecular regulators that govern the crosstalk between these two pathways. While the central roles of cGAS, STING, ZBP1, and core PANoptosome components are established, it is highly likely that additional sensors, adaptors, and post-translational modifications modulate the intensity, duration, and context-specific outcomes of this interplay. For instance, how distinct DNA damage signatures or mitochondrial stress signals differentially engage the PANoptosome machinery remains poorly understood. Unbiased proteomic and genetic screens will be essential to map the full interactome and identify new nodes of regulation. Second, achieving cell-type- and context -specific modulation of this axis is a major therapeutic hurdle. The ubiquitous expression of cGAS-STING and PANoptosis components means that systemic activation or inhibition could lead to unintended on-target off-tissue effects, such as autoimmunity, chronic inflammation, or impaired host defense. Developing strategies to precisely target this axis in disease-relevant cell populations—for example, tumor cells versus immune cells in the tumor microenvironment, or specific neuronal subsets in neurodegeneration—is a pressing need. Advances in nanomedicine, cell-type -specific delivery vehicles (eg, engineered extracellular vesicles), and the use of disease-responsive prodrugs offer promising avenues, but their translational potential requires rigorous validation. Third, the lack of robust, clinically applicable biomarkers for pathway activation represents a significant barrier to patient stratification and therapeutic monitoring. Measuring pathway activity in patient tissues or biofluids is challenging due to the transient nature of these signaling events and the limited accessibility of affected tissues. Identifying and validating stable, easily detectable biomarkers—such as circulating cGAMP levels, cell death-associated breakdown products, or specific transcriptional signatures in peripheral blood mononuclear cells—will be crucial for selecting patients most likely to benefit from agonists or inhibitors and for assessing pharmacodynamic responses in clinical trials. Fourth, the precise mechanistic relationship between cGAS-STING signaling and PANoptosis in chronic, low-grade inflammatory conditions remains underexplored. Most evidence derives from acute disease models, whereas the dynamics of this crosstalk in chronic settings such as metabolic dysfunction -associated steatotic liver disease, atherosclerosis, or age-related neurodegeneration may involve distinct molecular mechanisms and feedback loops. Longitudinal studies using sophisticated in vivo models that better recapitulate chronic disease are needed to dissect the temporal sequence and causal contributions of this axis over disease progression. Finally, how the cGAS-STING-PANoptosis interplay integrates with other stress responses —including autophagy, ferroptosis, mitochondrial dynamics, and the gut microbiome —constitutes a vast and largely uncharted territory. Understanding these complex networks will be critical for predicting therapeutic outcomes and avoiding unexpected resistance or compensatory mechanisms.
Another critical frontier is the translation of preclinical findings into human applications. Most current evidence derives from animal models, which may not fully recapitulate the complexity of human diseases. Robust biomarker development is needed to identify patient populations most likely to benefit from therapies targeting the cGAS-STING-PANoptosis axis. Moreover, investigating the potential feedback loops and adaptive resistance mechanisms that tumors or diseased tissues might employ to evade this form of immunogenic cell death will be vital for designing effective combination therapies and preventing relapse.
A critical dimension of the cGAS-STING-PANoptosis axis that warrants further exploration is its intersection with autophagy and cellular senescence. Although introduced as key downstream processes in the Introduction, these pathways are intimately linked to the core mechanisms discussed throughout this review. Autophagy, for instance, can both positively and negatively regulate cGAS-STING signaling: while autophagic degradation of damaged mitochondria (mitophagy) limits mtDNA release and thus suppresses STING activation, certain autophagy components can also facilitate STING trafficking or, as shown in the context of sepsis, ribophagy-mediated clearance of ribosomal RNA can curtail cGAS activation and subsequent PANoptosis.27 Conversely, chronic or dysregulated cGAS-STING activation is a well-established driver of cellular senescence, contributing to the inflammatory secretome characteristic of aging and age-related diseases.30 The interplay between senescence and PANoptosis remains incompletely understood, but emerging evidence suggests they may operate as parallel or sequential responses to mitochondrial damage, as observed in heme-induced Kupffer cell dysfunction during sepsis.140 From a therapeutic perspective, these connections offer additional nodes for intervention: modulating autophagy may fine-tune cGAS-STING activation thresholds, while targeting senescence-associated inflammation could complement strategies aimed at limiting PANoptosis. Future studies integrating these interconnected processes will be essential for developing more precise and context -dependent therapeutic approaches.
Another critical frontier is the translation of preclinical findings into human applications. Most current evidence derives from animal models, which may not fully recapitulate the complexity of human diseases. A key caveat lies in the marked species differences between mouse and human cGAS-STING pathway components. Notably, murine STING (mSTING) exhibits higher basal activity and stronger responsiveness to cyclic dinucleotides compared with human STING (hSTING), and the two orthologs differ substantially in their ligand-binding affinities, oligomerization dynamics, and downstream signaling outcomes. Moreover, human STING displays considerable genetic polymorphism (eg, the common R232H variant) that profoundly influences agonist responsiveness and disease susceptibility, features not captured in standard inbred mouse strains. Similarly, although key PANoptosome components such as ZBP1, AIM2, and RIPK1 are evolutionarily conserved, their regulatory networks and the relative contribution of pyroptosis, apoptosis, and necroptosis to the integrated PANoptotic response may differ between species due to variations in inflammasome architecture, caspase redundancy, and interferon signaling strength. These interspecies disparities underscore that findings from mouse models and in vitro cell lines should be interpreted with caution and may not directly predict human pathophysiology. Therefore, future studies should prioritize human-relevant experimental systems, including humanized mouse models, organoids, and primary human tissue validation, alongside careful consideration of genetic background when evaluating the translational potential of targeting the cGAS-STING-PANoptosis axis.
In conclusion, the dialogue between the cGAS-STING pathway and PANoptosis constitutes a fundamental regulatory layer in health and disease. It embodies the complex interplay between innate immune sensing and inflammatory cell death, a relationship that can be either protective or pathogenic depending on the context. The strategic manipulation of this axis, particularly through the sophisticated toolkit of nanomedicine, opens up a new paradigm for treating a wide array of intractable diseases, with cancer at the forefront. As we continue to unravel the molecular intricacies of this crosstalk and overcome the existing pharmacological hurdles, the prospect of developing targeted, effective, and safer therapeutics that harness the power of this potent biological duo moves from a compelling possibility to an attainable reality, promising to make a significant impact on future medical practice.
Abbreviations
AIM2, Absent in melanoma 2; AKI, Acute kidney injury; ALF, Acute liver failure; ALI, Acute lung injury; ARDS, Acute respiratory distress syndrome; ASC, Apoptosis-associated speck-like protein containing a CARD; cGAMP, 2′,3′-cyclic GMP-AMP; cGAS, Cyclic GMP-AMP synthase; CKD, Chronic kidney disease; CNS, Central nervous system; COPD, Chronic obstructive pulmonary disease; DAMP, Damage-associated molecular pattern; DKD, Diabetic kidney disease; DLBCL, Diffuse large B-cell lymphoma; dsDNA, Double-stranded DNA; ER, Endoplasmic reticulum; GSDMD, Gasdermin D; GSDME, Gasdermin E; HCC, Hepatocellular carcinoma; HIRI, Hepatic ischemia-reperfusion injury; HSC, Hepatic stellate cell; IAV, Influenza A virus; IFN, Interferon; IL, Interleukin; IPF, Idiopathic pulmonary fibrosis; IRF, Interferon regulatory factor; I/R, Ischemia/reperfusion; LPS, Lipopolysaccharide; MASLD, Metabolic dysfunction-associated steatotic liver disease; MI, Myocardial infarction; MLKL, Mixed lineage kinase domain-like pseudokinase; mtDNA, Mitochondrial DNA; NAFLD, Non-alcoholic fatty liver disease; NASH, Non-alcoholic steatohepatitis; NET, Neutrophil extracellular trap; NF-κB, Nuclear factor kappa B; NLRP, NLR family pyrin domain containing; NSCLC, Non-small cell lung cancer; PANoptosis, Pyroptosis, apoptosis, and necroptosis; PCD, Programmed cell death; RCC, Renal cell carcinoma; RIPK, Receptor-interacting serine/threonine-protein kinase; SARS-CoV-2, Severe acute respiratory syndrome coronavirus 2; STING, Stimulator of interferon genes; TBK1, TANK-binding kinase 1; TLR, Toll-like receptor; TNF, Tumor necrosis factor; ZBP1, Z-DNA-binding protein 1.
Ethics Approval
Not applicable. This review does not involve new data collection or experimental procedures on human participants or animals.
Funding
This work was supported in part by the Beijing Natural Science Foundation (No. 7252174), Wu Jieping Medical Foundation (320.6750.2024-13-59), Team Construction Project of Liaoning Province Education Department (LJ222410164013), General project of Liaoning Province Education Department (LJ212410164019; LJ212510164017), Liaoning Provincial Science and Technology Joint Fund Project (2025JH2/101800065),Talent development plan for the future in Medical-Engineering Integration by BRA-CDCHE and ZTA (MBRC0012025029), Science Foundation of ASCH (YN202305; YN202402; YN202423), and the Science Foundation of AMHT (2024YK04; 2025YK10).
Disclosure
The authors have no competing financial or non-financial interests to disclose.
References
1. Paiva SL. Tapping the therapeutic potential of the innate immune system. Nat Rev Drug Discov. 2020;19(4):236. doi:10.1038/d41573-020-00040-0
2. Sun L, Wu J, Du F, Chen X, Chen ZJ. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science. 2013;339:786–24. doi:10.1126/science.1232458
3. Wu J, Sun L, Chen X, et al. Cyclic GMP-AMP is an endogenous second messenger in innate immune signaling by cytosolic DNA. Science. 2013;339:826–830. doi:10.1126/science.1229963
4. Mou J, Chen Y, Zhu X, et al. Emerging role of the cGAS-STING pathway in cardiovascular diseases: biologic function, mechanisms and targeted therapy. Mol Med. 2025;31:218. doi:10.1186/s10020-025-01273-8
5. Wang H, Fleishman JS, Wu S, et al. cGAS-STING targeting offers novel therapeutic opportunities in neurological diseases. Ageing Res Rev. 2025;105:102691. doi:10.1016/j.arr.2025.102691
6. Wang Y, Wang W, Zhang Y, Gao P, Fleishman JS, Wang H. cGAS-STING targeting offers a novel therapeutic paradigm in cardiovascular diseases. Eur J Pharm Sci. 2025;211:107137. doi:10.1016/j.ejps.2025.107137
7. Wang Y, Zhang X, Wang W, Zhang Y, Fleishman JS, Wang H. cGAS-STING targeting offers therapy choice in lung diseases. Biol Direct. 2025;20(1):20. doi:10.1186/s13062-025-00611-4
8. Zhang B, Xu P, Ablasser A, et al. Regulation of the cGAS-STING pathway. Annu Rev Immunol. 2025;43:667–692. doi:10.1146/annurev-immunol-101721-032910
9. Zhou Q, Luo J, Chai X, et al. Therapeutic targeting the cGAS-STING pathway associated with protein and gene: An emerging and promising novel strategy for aging-related neurodegenerative disease. Int Immunopharmacol. 2025;156:114679. doi:10.1016/j.intimp.2025.114679
10. Malireddi R, Kesavardhana S, Kanneganti TD. ZBP1 and TAK1: Master regulators of NLRP3 inflammasome/pyroptosis, apoptosis, and necroptosis (PAN-optosis). Front Cell Infect Microbiol. 2019;9:406. doi:10.3389/fcimb.2019.00406
11. Chen W, Gullett JM, Tweedell RE, Kanneganti TD. Innate immune inflammatory cell death: pANoptosis and PANoptosomes in host defense and disease. Eur J Immunol. 2023;53(11):e2250235. doi:10.1002/eji.202250235
12. Sun X, Yang Y, Meng X, Li J, Liu X, Liu H. PANoptosis: Mechanisms, biology, and role in disease. Immunol Rev. 2024;321(1):246–262. doi:10.1111/imr.13279
13. Lee S, Karki R, Wang Y, Nguyen LN, Kalathur RC, Kanneganti TD. AIM2 forms a complex with pyrin and ZBP1 to drive PANoptosis and host defence. Nature. 2021;597(7876):415–419. doi:10.1038/s41586-021-03875-8
14. Malireddi R, Kesavardhana S, Karki R, Kancharana B, Burton AR, Kanneganti TD. RIPK1 distinctly regulates yersinia-induced inflammatory cell death, PANoptosis. Immunohorizons. 2020;4(12):789–796. doi:10.4049/immunohorizons.2000097
15. Kuriakose T, Man SM, Malireddi RK, et al. ZBP1/DAI is an innate sensor of influenza virus triggering the NLRP3 inflammasome and programmed cell death pathways. Sci Immunol. 2016;1(2):
16. Karki R, Lee S, Mall R, et al. ZBP1-dependent inflammatory cell death, PANoptosis, and cytokine storm disrupt IFN therapeutic efficacy during coronavirus infection. Sci Immunol. 2022;7(74):eabo6294. doi:10.1126/sciimmunol.abo6294
17. Malireddi R, Tweedell RE, Kanneganti TD. PANoptosis components, regulation, and implications. Aging. 2020;12(12):11163–11164. doi:10.18632/aging.103528
18. Mall R, Bynigeri RR, Karki R, Malireddi R, Sharma BR, Kanneganti TD. Pancancer transcriptomic profiling identifies key PANoptosis markers as therapeutic targets for oncology. NAR Cancer. 2022;4(4):zcac033. doi:10.1093/narcan/zcac033
19. Karki R, Sundaram B, Sharma BR, et al. ADAR1 restricts ZBP1-mediated immune response and PANoptosis to promote tumorigenesis. Cell Rep. 2021;37(3):109858. doi:10.1016/j.celrep.2021.109858
20. Banoth B, Tuladhar S, Karki R, et al. ZBP1 promotes fungi-induced inflammasome activation and pyroptosis, apoptosis, and necroptosis (PANoptosis). J Biol Chem. 2020;295(52):18276–18283. doi:10.1074/jbc.RA120.015924
21. Karki R, Sharma BR, Tuladhar S, et al. Synergism of TNF-α and IFN-γ triggers inflammatory cell death, tissue damage, and mortality in SARS-CoV-2 infection and cytokine shock syndromes. Cell. 2021;184(1):149–68.e17. doi:10.1016/j.cell.2020.11.025
22. Hou K, Pan W, Liu L, et al. Molecular mechanism of PANoptosis and programmed cell death in neurological diseases. Neurobiol Dis. 2025;209:106907. doi:10.1016/j.nbd.2025.106907
23. Messaoud-Nacer Y, Culerier E, Rose S, et al. STING agonist diABZI induces PANoptosis and DNA mediated acute respiratory distress syndrome (ARDS). Cell Death Dis. 2022;13(3):269. doi:10.1038/s41419-022-04664-5
24. Yang B, Hu A, Wang T, et al. SARS-CoV-2 infection induces ZBP1-dependent PANoptosis in bystander cells. Proc Natl Acad Sci U S A. 2025;122(28):e2500208122. doi:10.1073/pnas.2500208122
25. Wu C, Miao H, Yi Z, et al. STING-mediated mitochondrial DNA release exacerbates PANoptosis in liver ischemia reperfusion injury. Int Immunopharmacol. 2025;157:114778. doi:10.1016/j.intimp.2025.114778
26. Chen Y, Wu X, Jiang Z, Li X. KAE ameliorates LPS-mediated acute lung injury by inhibiting PANoptosis through the intracellular DNA-cGAS-STING axis. Front Pharmacol. 2024;15:1461931. doi:10.3389/fphar.2024.1461931
27. Zhao P, Li J, He P, et al. NUFIP1-mediated ribophagy alleviates PANoptosis of CD4(+) T lymphocytes in sepsis via the cGAS-STING pathway. Research. 2025;8:0895. doi:10.34133/research.0895
28. Liu W, Cui H, Wang L, et al. STING activates ZBP1-mediated PANoptosis to defend against HSV-1 retinal infection. J Neuroinflamm. 2025;22(1):243. doi:10.1186/s12974-025-03595-0
29. Cai Y, Chen X, Lu T, et al. Activation of STING by SAMHD1 deficiency promotes PANoptosis and enhances efficacy of PD-L1 blockade in diffuse large B-cell lymphoma. Int J Biol Sci. 2023;19(14):4627–4643. doi:10.7150/ijbs.85236
30. Gall A, Treuting P, Elkon KB, et al. Autoimmunity initiates in nonhematopoietic cells and progresses via lymphocytes in an interferon-dependent autoimmune disease. Immunity. 2012;36:120–131. doi:10.1016/j.immuni.2011.11.018
31. Civril F, Deimling T, de Oliveira Mann CC, et al. Structural mechanism of cytosolic DNA sensing by cGAS. Nature. 2013;498:332–337. doi:10.1038/nature12305
32. Gao P, Ascano M, Wu Y, et al. Cyclic [G(2’,5’)pA(3’,5’)p] is the metazoan second messenger produced by DNA-activated cyclic GMP-AMP synthase. Cell. 2013;153:1094–1107. doi:10.1016/j.cell.2013.04.046
33. Kranzusch PJ, Lee AS, Berger JM, Doudna JA. Structure of human cGAS reveals a conserved family of second-messenger enzymes in innate immunity. Cell Rep. 2013;3:1362–1368. doi:10.1016/j.celrep.2013.05.008
34. Li X, Shu C, Yi G, et al. Cyclic GMP-AMP synthase is activated by double-stranded DNA-induced oligomerization. Immunity. 2013;39:1019–1031. doi:10.1016/j.immuni.2013.10.019
35. Zhang X, Shi H, Wu J, et al. Cyclic GMP-AMP containing mixed phosphodiester linkages is an endogenous high-affinity ligand for STING. Mol Cell. 2013;51:226–235. doi:10.1016/j.molcel.2013.05.022
36. Ishikawa H, Barber GN. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature. 2008;455:674–678. doi:10.1038/nature07317
37. Ishikawa H, Ma Z, Barber GN. STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature. 2009;461:788–792. doi:10.1038/nature08476
38. Shang G, Zhang C, Chen ZJ, Bai XC, Zhang X. Cryo-EM structures of STING reveal its mechanism of activation by cyclic GMP-AMP. Nature. 2019;567:389–393. doi:10.1038/s41586-019-0998-5
39. Shu C, Yi G, Watts T, Kao CC, Li P. Structure of STING bound to cyclic di-GMP reveals the mechanism of cyclic dinucleotide recognition by the immune system. Nat Struct Mol Biol. 2012;19:722–724. doi:10.1038/nsmb.2331
40. Motwani M, Pesiridis S, Fitzgerald KA. DNA sensing by the cGAS-STING pathway in health and disease. Nat Rev Genet. 2019;20:657–674. doi:10.1038/s41576-019-0151-1
41. Liu L, Mao Y, Xu B, et al. Induction of neutrophil extracellular traps during tissue injury: involvement of STING and Toll-like receptor 9 pathways. Cell Prolif. 2019;52(3):e12579. doi:10.1111/cpr.12579
42. Decout A, Katz JD, Venkatraman S, Ablasser A. The cGAS-STING pathway as a therapeutic target in inflammatory diseases. Nat Rev Immunol. 2021;21(9):548–569. doi:10.1038/s41577-021-00524-z
43. Briard B, Place DE, Kanneganti TD. DNA sensing in the innate immune response. Physiology. 2020;35(2):112–124. doi:10.1152/physiol.00022.2019
44. Huang Y, Liu B, Sinha SC, Amin S, Gan L. Mechanism and therapeutic potential of targeting cGAS-STING signaling in neurological disorders. Mol Neurodegener. 2023;18(1):79. doi:10.1186/s13024-023-00672-x
45. Tian M, Li F, Pei H, Liu X, Nie H. The role of the cGAS-STING pathway in chronic pulmonary inflammatory diseases. Front Med Lausanne. 2024;11:1436091. doi:10.3389/fmed.2024.1436091
46. Zhang J, Zhang L, Chen Y, Fang X, Li B, Mo C. The role of cGAS-STING signaling in pulmonary fibrosis and its therapeutic potential. Front Immunol. 2023;14:1273248. doi:10.3389/fimmu.2023.1273248
47. De moura Rodrigues D, Lacerda-Queiroz N, Couillin I, Riteau N. STING targeting in lung diseases. Cells. 2022;11(21):3483. doi:10.3390/cells11213483
48. Feng L, Zhang F, Cao J, Xiong W. Role of cGAS-STING pathway in fibrotic disease. Pharmacol Res. 2025;221:107971. doi:10.1016/j.phrs.2025.107971
49. Wang J, Luo J, Rotili D, et al. SIRT6 protects against lipopolysaccharide-induced inflammation in human pulmonary lung microvascular endothelial cells. Inflammation. 2024;47(1):323–332. doi:10.1007/s10753-023-01911-5
50. Li H, Wang X, Luo X, Shi H, Li J. cGAS/STING/NLRP3 signaling pathway-mediated pyroptosis in hypertrophic cardiomyopathy radiotherapy. Front Biosci. 2025;30(3):26084. doi:10.31083/FBL26084
51. Wang Y, Yang R, Cao Y, et al. cGAS-STING targeting offers novel therapeutic opportunities in liver diseases. Drug Des Devel Ther. 2025;19:5835–5853. doi:10.2147/DDDT.S521397
52. Donne R, Saroul-Ainama M, Cordier P, et al. Replication stress triggered by nucleotide pool imbalance drives DNA damage and cGAS-STING pathway activation in NAFLD. Dev Cell. 2022;57(14):1728–41.e6. doi:10.1016/j.devcel.2022.06.003
53. Wang J, Guo Y, Hu J, Peng J. STING activation in various cell types in metabolic dysfunction-associated steatotic liver disease. Liver Int. 2025;45(4):e70063. doi:10.1111/liv.70063
54. Li J, Liu X, Feng H. The cGAS-STING axis in kidney disease: mechanisms and therapeutic potential. Eur J Pharmacol. 2025;1006:178155. doi:10.1016/j.ejphar.2025.178155
55. Wang Y, Zhu Y, Dou L, et al. Pharmacologically targeting cGAS-STING: a novel therapeutic opportunities in kidney diseases. Eur J Pharmacol. 2025;1007:178296. doi:10.1016/j.ejphar.2025.178296
56. Chung KW, Dhillon P, Huang S, et al. Mitochondrial damage and activation of the STING pathway lead to renal inflammation and fibrosis. Cell Metab. 2019;30(4):784–99.e5. doi:10.1016/j.cmet.2019.08.003
57. Bai J, Liu F. The cGAS-cGAMP-STING pathway: a molecular link between immunity and metabolism. Diabetes. 2019;68(6):1099–1108. doi:10.2337/dbi18-0052
58. Liu W, Kim GB, Krump NA, Zhou Y, Riley JL, You J. Selective reactivation of STING signaling to target Merkel cell carcinoma. Proc Natl Acad Sci U S A. 2020;117:13730–13739. doi:10.1073/pnas.1919690117
59. Xia T, Konno H, Barber GN. Recurrent loss of STING signaling in melanoma correlates with susceptibility to viral oncolysis. Cancer Res. 2016;76:6747–6759. doi:10.1158/0008-5472.CAN-16-1404
60. Wang Y, Hu J, Wu S, et al. Targeting epigenetic and posttranslational modifications regulating ferroptosis for the treatment of diseases. Signal Transduct Target Ther. 2023;8(1):449. doi:10.1038/s41392-023-01720-0
61. Cheng G, Wu J, Ji M, Hu W, Wu C, Jiang J. TET2 inhibits the proliferation and metastasis of lung adenocarcinoma cells via activation of the cGAS-STING signalling pathway. BMC Cancer. 2023;23:825. doi:10.1186/s12885-023-11343-x
62. Cai X, Yu X, Tang T, Xu Y, Wu T. JMJD2A promotes the development of castration-resistant prostate cancer by activating androgen receptor enhancer and inhibiting the cGAS-STING pathway. Mol Carcinog. 2024;63:1682–1696. doi:10.1002/mc.23753
63. Lin J, Guo D, Liu H, et al. The SETDB1-TRIM28 complex suppresses antitumor immunity. Cancer Immunol Res. 2021;9(12):1413–1424. doi:10.1158/2326-6066.CIR-21-0754
64. Sun M, Han X, Li J, et al. Targeting KDM4 family epigenetically triggers antitumour immunity via enhancing tumour-intrinsic innate sensing and immunogenicity. Clin Transl Med. 2024;14:e1598. doi:10.1002/ctm2.1598
65. Tang M, Chen G, Tu B, et al. SMYD2 inhibition-mediated hypomethylation of Ku70 contributes to impaired nonhomologous end joining repair and antitumor immunity. Sci Adv. 2023;9(24):eade6624. doi:10.1126/sciadv.ade6624
66. Wu L, Cao J, Cai WL, et al. KDM5 histone demethylases repress immune response via suppression of STING. PLoS Biol. 2018;16(8):e2006134. doi:10.1371/journal.pbio.2006134
67. Zhang W, Liu W, Jia L, et al. Targeting KDM4A epigenetically activates tumor-cell-intrinsic immunity by inducing DNA replication stress. Mol Cell. 2021;81(10):2148–2165.e9. doi:10.1016/j.molcel.2021.02.038
68. Chen T, Xu ZG, Luo J, et al. NSUN2 is a glucose sensor suppressing cGAS/STING to maintain tumorigenesis and immunotherapy resistance. Cell Metab. 2023;35:1782–1798.e8. doi:10.1016/j.cmet.2023.07.009
69. Gao Y, Zhang N, Zeng Z, et al. LncRNA PCAT1 activates SOX2 and suppresses radioimmune responses via regulating cGAS/STING signalling in non-small cell lung cancer. Clin Transl Med. 2022;12:e792. doi:10.1002/ctm2.792
70. Ma F, Lei YY, Ding MG, Luo LH, Xie YC, Liu XL. LncRNA NEAT1 interacted with DNMT1 to regulate malignant phenotype of cancer cell and cytotoxic t cell infiltration via epigenetic inhibition of p53, cGAS, and STING in lung cancer. Front Genet. 2020;11:250. doi:10.3389/fgene.2020.00250
71. Chen YH, Chen HH, Wang WJ, et al. TRABID inhibition activates cGAS/STING-mediated anti-tumor immunity through mitosis and autophagy dysregulation. Nat Commun. 2023;14(1):3050. doi:10.1038/s41467-023-38784-z
72. Fu L, Ding H, Bai Y, Cheng L, Hu S, Guo Q. IDI1 inhibits the cGAS-Sting signaling pathway in hepatocellular carcinoma. Heliyon. 2024;10(5):e27205. doi:10.1016/j.heliyon.2024.e27205
73. Li JY, Zhao Y, Gong S, et al. TRIM21 inhibits irradiation-induced mitochondrial DNA release and impairs antitumour immunity in nasopharyngeal carcinoma tumour models. Nat Commun. 2023;14(1):865. doi:10.1038/s41467-023-36523-y
74. Liu X, Cen X, Wu R, et al. ARIH1 activates STING-mediated T-cell activation and sensitizes tumors to immune checkpoint blockade. Nat Commun. 2023;14(1):4066. doi:10.1038/s41467-023-39920-5
75. Man CH, Lam W, Dang CC, et al. Inhibition of PLK4 remodels histone methylation and activates the immune response via the cGAS-STING pathway in TP53-mutated AML. Blood. 2023;142(23):2002–2015. doi:10.1182/blood.2023019782
76. Mondal I, Das O, Sun R, et al. PP2Ac deficiency enhances tumor immunogenicity by activating STING-type I interferon signaling in glioblastoma. Cancer Res. 2023;83(15):2527–2542. doi:10.1158/0008-5472.CAN-22-3382
77. Zhang KM, Zhao DC, Li ZY, et al. Inactivated cGAS-STING signaling facilitates endocrine resistance by forming a positive feedback loop with AKT kinase in ER+HER2- breast cancer. Adv Sci. 2024;11:e2403592. doi:10.1002/advs.202403592
78. Kim H, Kim H, Feng Y, et al. PRMT5 control of cGAS/STING and NLRC5 pathways defines melanoma response to antitumor immunity. Sci Transl Med. 2020;12(551):eaaz5683. doi:10.1126/scitranslmed.aaz5683
79. Liu J, Bu X, Chu C, et al. PRMT1 mediated methylation of cGAS suppresses anti-tumor immunity. Nat Commun. 2023;14(1):2806. doi:10.1038/s41467-023-38443-3
80. Rao K, Zhang X, Luo Y, et al. Lactylation orchestrates ubiquitin-independent degradation of cGAS and promotes tumor growth. Cell Rep. 2025;44(4):115441. doi:10.1016/j.celrep.2025.115441
81. Fan Y, Gao Y, Nie L, et al. Targeting LYPLAL1-mediated cGAS depalmitoylation enhances the response to anti-tumor immunotherapy. Mol Cell. 2023;83(19):3520–3532.e7. doi:10.1016/j.molcel.2023.09.007
82. Saha S, Ghosh M, Li J, et al. Serine depletion promotes antitumor immunity by activating mitochondrial DNA-mediated cGAS-STING signaling. Cancer Res. 2024;84(16):2645–2659. doi:10.1158/0008-5472.CAN-23-1788
83. Duan Y, Hu Z, Han P, et al. ADSL-generated fumarate binds and inhibits STING to promote tumour immune evasion. Nat Cell Biol. 2025;27(4):668–682. doi:10.1038/s41556-025-01627-8
84. Xiang W, Lv H, Xing F, et al. Inhibition of ACLY overcomes cancer immunotherapy resistance via polyunsaturated fatty acids peroxidation and cGAS-STING activation. Sci Adv. 2023;9(49):eadi2465. doi:10.1126/sciadv.adi2465
85. Jacoberger-Foissac C, Cousineau I, Bareche Y, et al. CD73 inhibits cGAS-STING and cooperates with CD39 to promote pancreatic cancer. Cancer Immunol Res. 2023;11(1):56–71. doi:10.1158/2326-6066.CIR-22-0260
86. Guo J, Huang L. Nanodelivery of cGAS-STING activators for tumor immunotherapy. Trends Pharmacol Sci. 2022;43:957–972. doi:10.1016/j.tips.2022.08.006
87. Wang Y, Zhu Y, Cao Y, et al. The activation of cGAS-STING pathway offers novel therapeutic opportunities in cancers. Front Immunol. 2025;16:1579832. doi:10.3389/fimmu.2025.1579832
88. Zhang Z, Zhang C. Regulation of cGAS-STING signalling and its diversity of cellular outcomes. Nat Rev Immunol. 2025;25:425–444. doi:10.1038/s41577-024-01112-7
89. Pan H, Pan J, Li P, Gao J. Characterization of PANoptosis patterns predicts survival and immunotherapy response in gastric cancer. Clin Immunol. 2022;238:109019. doi:10.1016/j.clim.2022.109019
90. Sundaram B, Pandian N, Mall R, et al. NLRP12-PANoptosome activates PANoptosis and pathology in response to heme and PAMPs. Cell. 2023;186(13):2783–801.e20. doi:10.1016/j.cell.2023.05.005
91. Christgen S, Zheng M, Kesavardhana S, et al. Identification of the PANoptosome: a molecular platform triggering pyroptosis, apoptosis, and necroptosis (PANoptosis). Front Cell Infect Microbiol. 2020;10:237. doi:10.3389/fcimb.2020.00237
92. Zheng M, Karki R, Vogel P, Kanneganti TD. Caspase-6 is a key regulator of innate immunity, inflammasome activation, and host defense. Cell. 2020;181(3):674–87.e13. doi:10.1016/j.cell.2020.03.040
93. Malireddi R, Gurung P, Kesavardhana S, et al. Innate immune priming in the absence of TAK1 drives RIPK1 kinase activity-independent pyroptosis, apoptosis, necroptosis, and inflammatory disease. J Exp Med. 2020;217(3):
94. Pandeya A, Kanneganti TD. Therapeutic potential of PANoptosis: innate sensors, inflammasomes, and RIPKs in PANoptosomes. Trends Mol Med. 2024;30(1):74–88. doi:10.1016/j.molmed.2023.10.001
95. Wang L, Zhu Y, Zhang L, et al. Mechanisms of PANoptosis and relevant small-molecule compounds for fighting diseases. Cell Death Dis. 2023;14(12):851. doi:10.1038/s41419-023-06370-2
96. Kesavardhana S, Malireddi R, Kanneganti TD. Caspases in cell death, inflammation, and pyroptosis. Annu Rev Immunol. 2020;38:567–595. doi:10.1146/annurev-immunol-073119-095439
97. newton K, Strasser A, Kayagaki N, et al. Cell death. Cell. 2024;187(2):235–256. doi:10.1016/j.cell.2023.11.044
98. Enuganti D, Bandharam N, Narra B, et al. Manipulating the PANoptosome: baseless hype or a newfound hope for human immunity. Apoptosis. 2025;30(9–10):2008–2018. doi:10.1007/s10495-025-02152-7
99. Li P, Gao Y, Tao Z, et al. PANoptosis: cross-talk among apoptosis, necroptosis, and pyroptosis in neurological disorders. J Inflamm Res. 2025;18:8131–8140. doi:10.2147/JIR.S526158
100. Zheng M, Kanneganti TD. The regulation of the ZBP1-NLRP3 inflammasome and its implications in pyroptosis, apoptosis, and necroptosis (PANoptosis). Immunol Rev. 2020;297(1):26–38. doi:10.1111/imr.12909
101. Katal S, Eibschutz LS, Radmard AR, et al. Black Fungus and beyond: COVID-19 associated infections. Clin Imaging. 2022;90:97–109. doi:10.1016/j.clinimag.2022.07.005
102. FitzGerald ES, Luz NF, Jamieson AM. Competitive cell death interactions in pulmonary infection: host modulation versus pathogen manipulation. Front Immunol. 2020;11:814. doi:10.3389/fimmu.2020.00814
103. Xiao H, Zhao Q, Yuan J, et al. IFN-γ promotes PANoptosis in Pasteurella multocida toxin-induced pneumonia in mice. Vet Microbiol. 2023;285:109848. doi:10.1016/j.vetmic.2023.109848
104. Pan J, Han L, Guo J, et al. AIM2 accelerates the atherosclerotic plaque progressions in ApoE-/- mice. Biochem Biophys Res Commun. 2018;498(3):487–494. doi:10.1016/j.bbrc.2018.03.005
105. Wang X, Zhang Y, Du L, et al. TUDCA alleviates atherosclerosis by inhibiting AIM2 inflammasome and enhancing cholesterol efflux capacity in macrophage. iScience. 2024;27(6):109849. doi:10.1016/j.isci.2024.109849
106. Zhao Y, Liang B, Sheng S, et al. AIM2 inflammasome regulated by the IFN-γ/JAK2/STAT1 pathway promotes activation and pyroptosis of monocytes in coronary artery disease. Immun Inflamm Dis. 2024;12(6):e1317. doi:10.1002/iid3.1317
107. Zhuo S, Song S, Wang C, et al. Inflammatory corpuscle AIM2 facilitates macrophage foam cell formation by inhibiting cholesterol efflux protein ABCA1. Sci Rep. 2024;14(1):10782. doi:10.1038/s41598-024-61495-4
108. Karunakaran D, Nguyen MA, Geoffrion M, et al. RIPK1 expression associates with inflammation in early atherosclerosis in humans and can be therapeutically silenced to reduce NF-κB activation and atherogenesis in mice. Circulation. 2021;143(2):163–177. doi:10.1161/CIRCULATIONAHA.118.038379
109. Tian F, Yao J, Yan M, et al. 5-Aminolevulinic acid-mediated sonodynamic therapy inhibits RIPK1/RIPK3-Dependent necroptosis In THP-1-derived foam cells. Sci Rep. 2016;6:21992. doi:10.1038/srep21992
110. Zhang Y, Li H, Huang Y, et al. Stage-dependent impact of RIPK1 inhibition on atherogenesis: dual effects on inflammation and foam cell dynamics. Front Cardiovasc Med. 2021;8:715337. doi:10.3389/fcvm.2021.715337
111. Fan X, Han J, Zhong L, et al. Macrophage-derived GSDMD plays an essential role in atherosclerosis and cross talk between macrophages via the mitochondria-STING-IRF3/NF-κB axis. Arterioscler Thromb Vasc Biol. 2024;44(6):1365–1378. doi:10.1161/ATVBAHA.123.320612
112. She H, Zheng J, Zhao G, et al. Arginase 1 drives mitochondrial cristae remodeling and PANoptosis in ischemia/hypoxia-induced vascular dysfunction. Signal Transduct Target Ther. 2025;10(1):167. doi:10.1038/s41392-025-02255-2
113. Gao X, Ma C, Liang S, Chen M, He Y, Lei W. PANoptosis: novel insight into regulated cell death and its potential role in cardiovascular diseases (Review). Int J Mol Med. 2024;54(3):
114. Do Carmo H, Arjun S, Petrucci O, Yellon DM, Davidson SM. The Caspase 1 inhibitor VX-765 protects the isolated rat heart via the RISK pathway. Cardiovasc Drugs Ther. 2018;32(2):165–168. doi:10.1007/s10557-018-6781-2
115. Luedde M, Lutz M, Carter N, et al. RIP3, a kinase promoting necroptotic cell death, mediates adverse remodelling after myocardial infarction. Cardiovasc Res. 2014;103(2):206–216. doi:10.1093/cvr/cvu146
116. Szobi A, Gonçalvesová E, Varga ZV, et al. Analysis of necroptotic proteins in failing human hearts. J Transl Med. 2017;15(1):86. doi:10.1186/s12967-017-1189-5
117. Yang Z, Li C, Wang Y, et al. Melatonin attenuates chronic pain related myocardial ischemic susceptibility through inhibiting RIP3-MLKL/CaMKII dependent necroptosis. J Mol Cell Cardiol. 2018;125:185–194. doi:10.1016/j.yjmcc.2018.10.018
118. Li PB, Bai JQ, Jiang WX, Li HH, Li CM. The mechanosensitive Piezo1 channel exacerbates myocardial ischaemia/reperfusion injury by activating caspase-8-mediated PANoptosis. Int Immunopharmacol. 2024;139:112664. doi:10.1016/j.intimp.2024.112664
119. Cui B, Qi Z, Liu W, Zhang G, Lin D. ZBP1-mediated PANoptosis: a possible novel mechanism underlying the therapeutic effects of penehyclidine hydrochloride on myocardial ischemia-reperfusion injury. Int Immunopharmacol. 2024;137:112373. doi:10.1016/j.intimp.2024.112373
120. Li H, Yang DH, Zhang Y, et al. Geniposide suppresses NLRP3 inflammasome-mediated pyroptosis via the AMPK signaling pathway to mitigate myocardial ischemia/reperfusion injury. Chin Med. 2022;17(1):73. doi:10.1186/s13020-022-00616-5
121. Li Y, Xing N, Yuan J, Yang J. Sevoflurane attenuates cardiomyocyte apoptosis by mediating the miR-219a/AIM2/TLR4/MyD88 axis in myocardial ischemia/reperfusion injury in mice. Cell Cycle. 2020;19(13):1665–1676. doi:10.1080/15384101.2020.1765512
122. Bi Y, Xu H, Wang X, et al. FUNDC1 protects against doxorubicin-induced cardiomyocyte PANoptosis through stabilizing mtDNA via interaction with TUFM. Cell Death Dis. 2022;13(12):1020. doi:10.1038/s41419-022-05460-x
123. Wei S, Wu L, Xiang Z, et al. EIF2AK2 protein targeted activation of AIM2-mediated PANoptosis promotes sepsis-induced acute kidney injury. Ren Fail. 2024;46(2):2403649. doi:10.1080/0886022X.2024.2403649
124. Uysal E, Dokur M, Kucukdurmaz F, et al. Targeting the PANoptosome with 3,4-methylenedioxy-β-nitrostyrene, reduces PANoptosis and protects the kidney against renal ischemia-reperfusion injury. J Invest Surg. 2022;35(11–12):1824–1835. doi:10.1080/08941939.2022.2128117
125. Lv Z, Hu J, Su H, et al. TRAIL induces podocyte PANoptosis via death receptor 5 in diabetic kidney disease. Kidney Int. 2025;107(2):317–331. doi:10.1016/j.kint.2024.10.026
126. Yi BJ, Wang CC, Li XW, et al. Lycopene protects against atrazine-induced kidney STING-dependent PANoptosis through stabilizing mtDNA via interaction with Sam50/PHB1. J Agric Food Chem. 2024;72(26):14956–14966. doi:10.1021/acs.jafc.4c02820
127. Xu C, Wang Q, Du C, et al. Histone deacetylase-mediated silencing of PSTPIP2 expression contributes to aristolochic acid nephropathy-induced PANoptosis. Br J Pharmacol. 2024;181(9):1452–1473. doi:10.1111/bph.16299
128. Tong J, Lan XT, Zhang Z, et al. Ferroptosis inhibitor liproxstatin-1 alleviates metabolic dysfunction-associated fatty liver disease in mice: potential involvement of PANoptosis. Acta Pharmacol Sin. 2023;44:1014–1028. doi:10.1038/s41401-022-01010-5
129. Ma Z, Xie K, Xue X, et al. Si-Wu-Tang attenuates hepatocyte PANoptosis and M1 polarization of macrophages in non-alcoholic fatty liver disease by influencing the intercellular transfer of mtDNA. J Ethnopharmacol. 2024;328:118057. doi:10.1016/j.jep.2024.118057
130. Li W, Zhang W, Zhang D, Shi C, Wang Y. Effect of lipopolysaccharide on TAK1-mediated hepatocyte PANoptosis through Toll-like receptor 4 during acute liver failure. Int Immunopharmacol. 2024;129:111612. doi:10.1016/j.intimp.2024.111612
131. Hu C, Li M, Chen Y, et al. AIM2 regulates autophagy to mitigate oxidative stress in aged mice with acute liver injury. Cell Death Discov. 2024;10(1):107. doi:10.1038/s41420-024-01870-2
132. Zeng FL, Zhang Y, Wang ZH, et al. Neutrophil extracellular traps promote Acetaminophen-induced acute liver injury in mice via AIM2. Acta Pharmacol Sin. 2024;45(8):1660–1672. doi:10.1038/s41401-024-01239-2
133. Lin Q, Cai H, Yu F, et al. AIM2-PANoptosome-driven PANoptosis in hepatic lipid dysregulation induced by β-HCH and nanoplastics co-exposure. Apoptosis. 2025;30(9–10):2340–2357. doi:10.1007/s10495-025-02147-4
134. Zheng M, Williams EP, Malireddi R, et al. Impaired NLRP3 inflammasome activation/pyroptosis leads to robust inflammatory cell death via caspase-8/RIPK3 during coronavirus infection. J Biol Chem. 2020;295(41):14040–14052. doi:10.1074/jbc.RA120.015036
135. Karki R, Sharma BR, Lee E, et al. Interferon regulatory factor 1 regulates PANoptosis to prevent colorectal cancer. JCI Insight. 2020;5(12):e136720. doi:10.1172/jci.insight.136720
136. Malireddi R, Karki R, Sundaram B, et al. Inflammatory cell death, PANoptosis, mediated by cytokines in diverse cancer lineages inhibits tumor growth. Immunohorizons. 2021;5(7):568–580. doi:10.4049/immunohorizons.2100059
137. Yin Q, Peng S, Zhang Z, et al. RPA1 protects DNA damage-induced PANoptosis in limb development. Sci Adv. 2025;11(34):eadw2756. doi:10.1126/sciadv.adw2756
138. Messaoud-Nacer Y, Culerier E, Rose S, et al. STING-dependent induction of neutrophilic asthma exacerbation in response to house dust mite. Allergy. 2025;80(3):715–737. doi:10.1111/all.16369
139. Wang S, Tan J, Zhang Q. Cytosolic escape of mitochondrial DNA triggers cGAS-STING pathway-dependent neuronal panoptosis in response to intermittent hypoxia. Neurochem Res. 2024;49(8):2228–2248. doi:10.1007/s11064-024-04151-7
140. Li T, Adams J, Zhu P, et al. The role of heme in sepsis induced Kupffer cell PANoptosis and senescence. Cell Death Dis. 2025;16(1):284. doi:10.1038/s41419-025-07637-6
141. Yin Y, Qin Z, Zhou H, et al. Ultrasound responsive Mn/Se-nanozyme as PANoptosis initiators for bladder cancer immunotherapy. Adv Healthc Mater. 2026:e04808. 10.1002/adhm.202504808.
142. Lin F, Huang H, Long J, et al. A multifunctional nanozyme integrating panoptosis induction and T-cell metabolic reprogramming to augment the efficacy of PD-1 inhibitors. Colloids Surf B Biointerfaces. 2026;260:115389. doi:10.1016/j.colsurfb.2025.115389
143. Zhou L, Lyu J, Liu F, Su Y, Feng L, Zhang X. Immunogenic PANoptosis-initiated cancer sono-immune reediting nanotherapy by iteratively boosting cancer immunity cycle. Adv Mater. 2024;36(2):e2305361. doi:10.1002/adma.202305361
144. Zhu G, Qian Y, Gao M, et al. A novel nano-biocomposite synergistically activates PANoptosis and cGAS-STING for precise cancer immunotherapy. Biomaterials. 2026;327:123758. doi:10.1016/j.biomaterials.2025.123758
145. Shu W, Chen G, Zhang J, et al. Bavachinin exerts anti-tumor effects by activating TLR4/STING axis-dependent PANoptosis and synergistically enhances chemosensitivity in endometrial cancer. Biochem Pharmacol. 2025;242(Pt 2):117321. doi:10.1016/j.bcp.2025.117321
146. Zhang Y, Jiang Q, Hu H, et al. Quercetin mitigated LPS-induced PANoptosis in the bursa of Fabricius via the cGAS/STING signaling pathway in broilers. Poult Sci. 2026;105(2):106179. doi:10.1016/j.psj.2025.106179
147. Zeng QQ, Wang J, Yue RC, et al. Gelsevirine ameliorates sepsis-associated encephalopathy by inhibiting the STING signalling-mediated pyroptosis pathway in microglia. Phytomedicine. 2024;135:156071. doi:10.1016/j.phymed.2024.156071
148. Chen D, Wang B, Li C, et al. Ce6 derivative photodynamic therapy triggers PANoptosis and enhances antitumor immunity with LAG3 blockade in cutaneous squamous cell carcinoma. Cell Rep Med. 2025;6(7):102239. doi:10.1016/j.xcrm.2025.102239
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.