Back to Journals » Cancer Management and Research » Volume 12
CFTR Functions as a Tumor Suppressor and Is Regulated by DNA Methylation in Colorectal Cancer
Authors Liu C, Song C, Li J, Sun Q ![]()
Received 5 February 2020
Accepted for publication 13 May 2020
Published 8 June 2020 Volume 2020:12 Pages 4261—4270
DOI https://doi.org/10.2147/CMAR.S248539
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Chien-Feng Li
Can Liu,1 Chao Song,2 Jiaxi Li,3 Qing Sun1,3
1Department of Pathology, Shandong Provincial Qianfoshan Hospital, Cheeloo College of Medicine, Shandong University, Jinan, Shandong Province, People’s Republic of China; 2Department of Pathology, Zibo Central Hospital, Zibo, Shandong Province, People’s Republic of China; 3Department of Pathology, Shandong Provincial Qianfoshan Hospital, The First Hospital Affiliated with Shandong First Medical University, Jinan, Shandong Province, People’s Republic of China
Correspondence: Qing Sun
Department of Pathology, Shandong Provincial Qianfoshan Hospital, Cheeloo College of Medicine, Shandong University, Jingshi Street, Jinan 250014, Shandong Province, People’s Republic of China
Tel +86-531-89269710
Email [email protected]
Purpose: Cystic fibrosis transmembrane conductance regulator (CFTR) was shown to be downregulated or silenced in carcinomas and acts as a candidate tumor suppressor gene. However, the function of CFTR gene in colorectal cancer (CRC) is still unclear. This aim of this study was to investigate the CFTR promoter methylation status and its impact on the expression and functional role of CFTR in CRC development.
Patients and Methods: CFTR expression in CRC tissues and CRC cell lines was detected via quantitative real-time polymerase chain reaction (qRT-PCR) and immunohistochemistry (IHC). The promoter methylation status of CFTR was measured using methylation-specific PCR (MSP). colony formation, transwell, and 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assays were used to evaluate the effect of CFTR overexpression in CRC cell lines.
Results: qRT-PCR and IHC results indicated that CFTR expression was downregulated in the CRC tissues compared to the adjacent normal tissues. The promoter methylation status of CFTR was further analyzed in 70 CRC specimens. MSP validation showed methylation of CFTR promoter in 62.2% (45/70) of CRC tissues. The methylation of CFTR promoter was significantly associated with age (P=0.013) and lymph node metastasis (P=0.026) in CRC tissues. Results of transwell, MTT, and colony formation assays showed that CFTR overexpression inhibited the migration, invasion, and proliferation of CRC cells.
Conclusion: CFTR expression was downregulated in CRC and promoter methylation may be responsible for this downregulation. Overexpression of CFTR may suppress CRC tumor growth by inhibiting the proliferation, migration, and invasion of CRC cells. CFTR promoter methylation was significantly correlated with lymph node metastasis; thus, CFTR may be a potential marker for lymph node metastasis of CRC.
Keywords: CFTR, DNA methylation, colorectal cancer, lymph node metastasis
Introduction
Colorectal cancer (CRC) is the third most common cancer and the fourth leading cause of cancer-related mortality worldwide,1 with nearly 500,000 deaths annually.2 From the perspective of the biological etiology of CRC, the progression of benign tumors (ie polyps) to malignant tumors there is an accumulation of both genetic and epigenetic changes.3 Epigenetics refers to modifications that occur in the expression of heritable genes without involving changes in DNA sequences. Epigenetic regulation of gene expression occurs in normal tissues and plays a significant role in embryonic development, gene imprinting, and cell differentiation. Accordingly, epigenetic mechanisms play an important role in tumor development, including abnormal DNA methylation and post-translational modification of histones, micro-RNA, and non-coding RNA.4 CRC research shows that the occurrence and development of CRC is closely related to abnormal DNA methylation. Particularly, abnormal DNA methylation leads to the invasion of intestinal epithelial cells and carcinogenesis.
DNA methylation is catalyzed by DNA methyltransferase, which takes s-adenosine methionine as the methyl donor. The cytosine 5ʹ carbon covalent bond of CpG dinucleotide in the genome binds to a methyl group, thus affecting gene expression.5 In 1989, Riordan et al first cloned and identified the cDNA fragment of the CFTR gene, which was located in 7q31.2, with a total length of 190 kb and a total number of 27 exons. The messenger RNA (mRNA) sequence is 6132 bp long, encoding CFTR protein, which is composed of 1480 amino acids and has a relative molecular weight of 168 kDa.6 CFTR is an ATP-bound transmembrane protein that is located in the apical membrane of epithelial cells of the exocrine gland and mainly provides selective channels for the transmembrane movement of chloride ions. CFTR protein is widely distributed in several organ systems (respiratory, system, system, and endocrine), sweat glands, and other tissues, maintaining the balance of electrolytes and homeostasis.7 CFTR gene mutation was initially thought to cause cystic fibrosis, and subsequent studies have found that CFTR disorder is associated with many diseases including chronic obstructive pulmonary diseases, pulmonary fibrosis, and cancer.8 CFTR is also highly expressed in various epithelial cells of the intestinal mucosa. CFTR is expressed in the apical membrane of intestinal epithelial cells and is the main ion channel transporter in intestinal crypt epithelial cells.9 Notably, hypermethylation of CFTR gene has been reported to occur frequently in bladder and liver cancers.10–12 However, the function of CFTR in CRC remains unclear. Therefore, this study aimed to investigate the CFTR promoter methylation status and its impact on the expression and function of CFTR in CRC.
Patients and Methods
Tumor Samples and Cell Lines
This study was approved by the Medical Ethics Committee of the First Affiliated Hospital of Shandong First Medical University. A total of 70 formalin-fixed paraffin-embedded (FFPE) specimens and 35 fresh surgical tissue samples were used in this study. All tissue specimens were collected from CRC patients admitted to The First Affiliated Hospital of Shandong First Medical University (Shandong Provincial Qianfoshan Hospital, Shandong University). Specifically, the specimens were CRC tissue and paired adjacent normal tissues (>5 cm from the corresponding tumor edge). No patient received any adjuvant therapy before surgery. Clinicopathological information was also collected. Normal colorectal cells (FHC) and seven human CRC cell lines (ie, HCT116, CaCo-2, HT29, LOVO, SW480, SW620, and SW1463) were used. All cell lines were purchased from GeneChem (Shanghai, China). HCT116, CaCo-2, HT29, and LOVO were cultured in Dulbecco’s modified eagle medium (DMEM) (BasalMedia, Shanghai, China) with 10% fetal bovine serum (FBS) (Gibco, Gaithersburg, MD, USA) and 1% penicillin-streptomycin. SW480, SW620, and SW1463 were cultured in RPMI-1640 medium (BasalMedia, Shanghai, China) with 10% inactivated FBS and 1% penicillin-streptomycin. All cells were cultured in a humidified atmosphere of 5% CO2 at 37°C.
Written informed consent was acquired from all human participants after complete description of the study. All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki Declaration and its later amendments or comparable ethical standards.
Quantitative Real-Time Polymerase Chain Reaction
Quantitative real-time polymerase chain reaction (qRT-PCR) were used to determine the CFTR expression in CRC cells and tissues. TRIzol (Cwbio, Beijing, China) was used to extract total RNA from fresh tissues and cells. cDNA was synthesized using a reverse transcription kit (Takara, Shiga, Japan), according to the manufacturer’s instructions. qRT-PCR was performed with an ABI 7500 real-time PCR system (Thermo Fisher, Waltham, MA, USA), using SYBR® Green PCR Master Mix (Takara, Shiga, Japan) with beta-actin (ACTB) as a control. Each sample was tested in triplicate. The PCR primer sequences for CFTR were as follows: 5′-ACTGGAATCTGAAGGCAGGAGTCC-3′ (forward) and 5′-CGAAGGCACGAAGTGTCCATAGTC-3′ (reverse). The PCR primer sequences for ACTB were as follows: 5′-CCTGGCACCCAGCACAAT-3′ (forward) and 5′-GGGCCGGACTCGTCATAC-3′ (reverse).
Immunohistochemistry
Mouse monoclonal antibody against CFTR (Abcam, St. Louis, MO, USA) was diluted at 1:500. Immunohistochemistry was conducted as follows. The tissue samples were sliced into 4-μm sections; dewaxed in a 60°C incubator for 40 min; and rinsed sequentially in xylene I for 10 min, xylene II for 10 min, and xylene III for 10 min. Thereafter, they were rehydrated using absolute ethyl alcohol for 7 min and 95%, 80%, and 70% ethyl alcohol for 5 min each. Then, the antigen in the measured tissue slices was repaired with citric acid buffer (pH=6) and kept at high temperature for 5 min. After antigen repair, the antigen was cooled to room temperature (approximately 3 h) then washed three times with PBS for 3 min each time. The slides were then incubated in 3% hydrogen peroxide for 15 min and then washed again for three times with PBS for 3 min each. Then, the slides were incubated with the anti-CFTR antibody (1:500) at 4°C overnight. The next day, the slides were placed at room temperature (RT) for 30 min then incubated with a secondary antibody for 30 min at 37°C. Then, the slices were washed three times with PBS for 3 min each and incubated with horseradish peroxidase-labeled streptomycin anti-biotin antibody for 30 min at RT. The slides were washed again three times, and then color development with DAB (15 s) was conducted. The slides were washed with water, the cell nuclei were dyed using hematoxylin (5 s), and the slides were washed again with water (15–30 s). Microscope at 200× light magnification was randomly selected for each slice.
Methylation-Specific Polymerase Chain Reaction
Methylation-specific PCR (MSP) primers were designed in the locations of CpG islands in the promoter region of CFTR gene according to MethyPrimer (Figure 1A) (http://www.urogene.org/cgi-bin/methprimer/methprimer.cgi). Genomic DNA was extracted using TIANamp FFPE DNA kit (Tiangen, Beijing, China) and prepared according to the manufacturer’s instructions. Extracted genomic DNA was treated with EpiTect Bisulfite Kit (Qiagen, Hilden, Germany), following the manufacturer’s instruction. Sulfite-modified DNA samples (2 µL) were amplified using Methylation-specific PCR (MSP) kit (Tiangen). Cycle conditions were as follows: 95°C for 5 min for 1 cycle; 94°C for 20 s, 60°C for 30 sec, and 72°C for 20 s for 35 cycles; and 72°C for 5 min for extension. Then, to prepare 2% agarose gel, 10 µL PCR product was added to each hole, and 120 V vertical water level electrocoagulation for 45 min was performed for observation and to collect images. MSP primers were designed as follows: 5′-TTAAGGAATTCGATTAGGTTAECG-3′ (M-forward) and 5′-TCTCTTTAAATCCAATTAACAACGC-3′ (M-reverse), 5′-TAGGAATTTGATTAGGATTATTGG-3′ (U-forward) and 5′-CTCTTTAAATCCAATTAACAACACT-3′ (U-reverse).
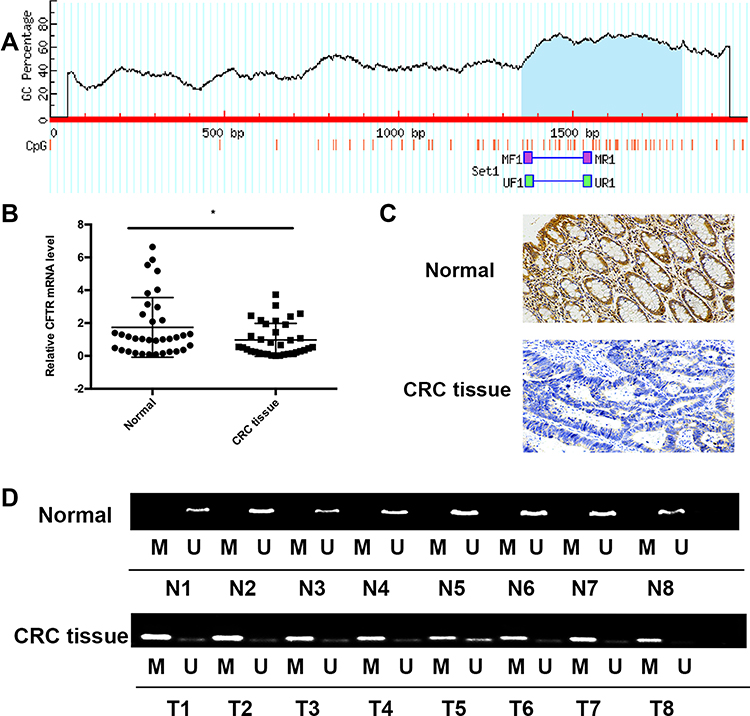 |
Figure 1 Expression and promoter methylation status of CFTR in colorectal tissues and adjacent normal tissues. Notes: (A) Schematic diagram of CpG islands in the CFTR promoter region. (B) CFTR mRNA expression in colorectal cancer tissues and paired adjacent normal tissues (>5 cm from the corresponding tumor edge). The experiment was repeated three times (*P<0.05). (C) Representative images of CFTR IHC in CRC tissues and paired adjacent normal tissues (magnification, ×200). (D) Representative methylation of CFTR promoter in CRC tissues and adjacent normal tissues. “M” indicates methylated CFTR; “U” indicates unmethylated CFTR.Abbreviations: MF, MSP forward primer; MR, MSP reverse primer; UF, unmethylation forward primer; UR, unmethylation reverse primer. |
Demethylation Using 5-Aza-2′-Deoxycytidine Treatment
Cell lines at logarithmic growth stage were inoculated in 6-well plates at 1×105 cells/well and cultured for 24 h. After cell adherence, the degree of fusion reached 40–50%. Medium containing 5-AZA-CDR was replaced, and the final concentration was 5 µM. Culture was continued for 48 h, and cells in the control group were added with equal amount of DMSO. CFTR expression in the experimental and the control cells was detected using qRT-PCR.
Construction of CFTR-Overexpressing HCT116 and CaCo-2 Cell Lines
HCT116 and Caco-2 cells in logarithmic growth stage were inoculated in 6-well plates at 1×105 cells/well and cultured for 24 h. A total of 20–30% confluent HCT116 and CaCo-2 cells were transfected with CFTR lentivirus (LV), according to the manufacturer’s protocols (GeneChem, Shanghai, China). The HCT116 and CaCo-2 cells were also transiently transfected with control empty plasmids (GeneChem). The culture system follows the reagent manufacturer’s instructions. Briefly, serum-free DMEM medium containing 20 µL p-HitransG and CFTR lentivirus (LV) or empty plasmids was mixed and added to the cells and the culture was continued for 12 h. The cells were then cultured in DMEM (BasalMedia) with 10% FBS (Gibco) and 1% penicillin-streptomycin for 72 h. After 42–72 hours from the transfection, plasmid transfection efficiency was evaluated.
Colony Formation Assay
Cells were seeded at 1000 cells/well in 6-well plates and cultured in triplicate. The complete growth medium condition was changed every 48 h. After 2 weeks, the cells were fixed with 4% paraformaldehyde for 30 minutes, stained with 0.2% crystal violet for 10 minutes, and counted. Each experiment was repeated three times.
Cell Viability Detection
Cells were plated into 96-well plates at 6000 cells/well, and cell viability was measured using the MTT kit (Solarbio, Beijing, China) at 0, 24, 48, and 72 h. Absorbance was measured on an Epoch microplate reader (Biotek US, Winooski, VT, USA) at a wavelength of 490 nm. Each experiment was repeated three times.
Transwell Assay for Migration
The effect of CFTR on cell migration was detected using the Transwell Assay System (Corning, High Wycombe, UK). Cells were suspended and harvested in serum free medium and then placed into the upper well at a concentration of 104 cells/200 µL. The complete medium containing 10% FBS was added into the lower well (600 µL). The chamber was incubated for 48 h. The cells remaining on the upper surface were scraped gently and washed with PBS two times. The cells that migrated to the lower surface of the membrane were stained with 0.2% crystal violet and counted. Each experiment was repeated three times.
Transwell Assay for Invasion
Cells were suspended at 104 cells/200μL in serum-free medium (200 μL) and placed into the upper compartment of an invasion chamber. The extracellular matrix (ECM; MatrigelTM, BD Biosciences, Franklin Lakes, NJ) was coated onto the upper well. After 48 h incubation, the invasive cells migrated through the ECM layer to the complete medium in the lower well. The cells were stained with 0.2% crystal violet for 10 minutes and counted. Each experiment was repeated three times.
Statistical Analysis
All statistical analyses were performed using SPSS 18.0. Data were analyzed using Student’s t-test (independent-samples t-test) and the chi-square (also termed ×2) test as appropriate. All tests were bilateral with a significance level of 0.05.
Results
CFTR Expression Was Significantly Decreased in Colorectal Cancer Tissues Compared to Normal Tissues
The mRNA expression of CFTR in 35 pairs of CRC tissues and adjacent normal tissues was measured using qRT-PCR. The results showed that compared with adjacent normal tissues, the mRNA expression of CFTR was significantly decreased in CRC tissues (P=0.034, Figure 1B). Furthermore, protein expression of CFTR was examined in 70 paired CRC tissues and adjacent normal tissues via immunohistochemistry (IHC). CFTR staining was observed mainly in the cytoplasm and cell membranes. The positive expression rates of CFTR protein in adjacent tissue samples and colorectal cancer tissues were 62.9% (44/70) and 45.7% (32/70), respectively, with statistically significant differences (P=0.04) (Figure 1C), indicating that CFTR is downregulated in CRC tissues.
CFTR Promoter Was Frequently Methylated in Colorectal Cancer
We obtained the promoter sequence of CFTR by using UCSC (http://genome.ucsc.edu). The Li Lab (a software for predicting CPG island and designing MSP primers, http://www.urogene.org/cgi-bin/methprimer/methprimer.cgi) analysis showed that CFTR contains a typical CpG island (from 1354 to 1814 bp) (Figure 1A). To explore the methylation status of CFTR promoter, 70 paired samples of FFPE specimens (ie CRC tissues and adjacent normal tissues) were analyzed using MSP (Figure 1D). CFTR promoter methylation was significantly higher in CRC tissues compared to that in adjacent normal tissues [62.2% (45/70) vs 41.4% (29/70), P=0.0068; Figure 1D]. These results indicate that the CFTR promoter was frequently methylated in human CRC.
CFTR Expression Was Inversely Associated with Promoter Methylation in Colorectal Cancer
To determine whether CFTR downregulation in CRC was due to its promoter methylation, 35 samples of CRC tissues and paired adjacent normal tissues were analyzed via MSP. The results of qRT-PCR and MSP showed that CFTR promoter methylation occurred in 16 of the 23 cancer tissues with downregulated CFTR mRNA expression and in 4 of the 12 cancer tissues with upregulated CFTR mRNA expression. Analysis of these data using chi-square test showed that the expression of CFTR mRNA was negatively correlated with the methylation of its promoter (P=0.04) (Table 1). To further verify whether promoter methylation influenced CFTR expression, normal colorectal cell lines (FHC) and seven human CRC cell lines (ie HCT116, CaCo-2, LoVo, HT29, SW480, SW620, and SW1463) were subjected to qRT-PCR and MSP. qRT-PCR results showed that compared with FHC cell lines, the expression of CFTR mRNA was highest in LoVo cell lines, followed by HT29 and SW1463 cell lines, while it was distinctly suppressed in HCT116, CaCo-2, SW480, and SW620 cell lines (Figure 2A). Meanwhile, the results of MSP showed that unmethylation of the CFTR promoter was found in FHC and LoVo cell lines, whereas complete methylation was found in HCT116, CaCo-2, SW480, and SW620 cell lines, and partial methylation was found in HT29 and SW1463 cell lines (Figure 2B). Reduced CFTR expression was associated with promoter methylation in CRC cell lines. To further identify the relationship between the expression and promoter methylation of CFTR, CRC cell lines were treated with 5-AZA-CDR. After 5-AZA-CDR treatment, the expression of CFTR mRNA in HCT116, CaCo-2, SW480, and SW620 cell lines increased significantly, whereas no significant changes were found in LoVo and FHC cell lines. Meanwhile, the expression of CFTR mRNA in HT29 and SW1463 cell lines was slightly increased (Figure 2C). As expected, the expression of CFTR mRNA of CRC cell lines with complete methylation or partial methylation of CFTR promoter was decreased after 5-AZA treatment (Figure 2C). These results indicate that CFTR expression is regulated by promoter methylation in human CRC.
 |
Table 1 Analysis of the Correlation Between CFTR Promoter Methylation and CFTR Expression in Colorectal Cancer |
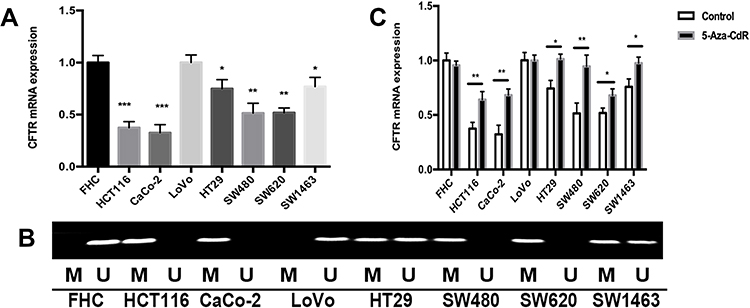 |
Figure 2 CFTR expression is regulated by its promoter methylation in CRC. Notes: (A) CFTR mRNA expression in CRC cell lines (*P<0.05, **P<0.01, ***P<0.001). (B) Representative methylation of CFTR in CRC cell lines. “M” indicates methylated; “U” indicates unmethylated. (C) Changes of CFTR mRNA expression in CRC cell lines after 5-Aza-CdR treatment. The experiment was repeated three times (*P<0.05, **P<0.01). |
Relationship Between the Methylation of CFTR Promoter and Clinicopathologic Characteristics
Subsequently, we evaluated the relationship between the methylation of CFTR promoter and the clinicopathological characteristics of CRC patients using FFPE specimens from 70 CRC patients. Analysis of the CRC FFPE samples showed that there was no correlation between the methylation of CFTR promoter and clinicopathological characteristics such as sex, tumor size, tumor location, T stage, distant metastases or stage (Table 2). However, in CRC specimens, the methylation of the CFTR promoter was associated with age (P=0.013), lymph node metastasis (P=0.026), and regional lymph node involvement (P=0.022) in CRC specimens (Table 2).
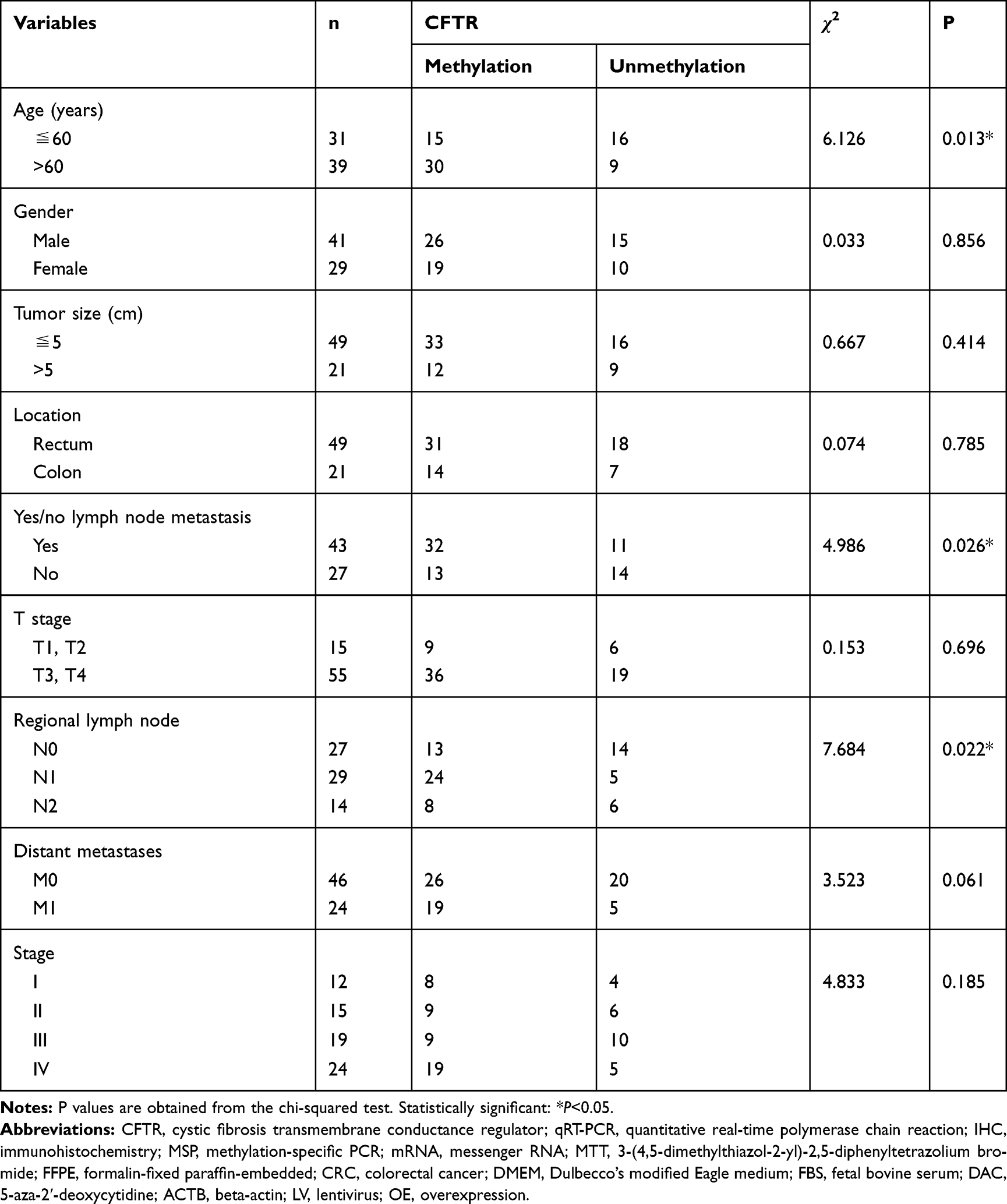 |
Table 2 Relationship Between CFTR Promoter Methylation and Clinicopathological Characteristics of CRC |
CFTR Overexpression Suppresses Colorectal Cancer Cell Proliferation
The results of qRT-PCR and MSP showed that CFTR expression was lower and completely methylated in HCT116 and CaCo-2 cell lines. Therefore, CFTR-overexpressing HCT116 and CaCo-2 cell lines were used to investigate the function of CFTR in CRC. Lentivirus transfection efficiency in HCT116 and CaCo-2 cell lines was evaluated via qRT-PCR (Figure 3A). The effect of CFTR on cell proliferation was studied using MTT and colony formation assay. In HCT116 cell lines with CFTR overexpression, the OD values were 1.16 ± 0.04 vs 0.69 ± 0.05 before and after CFTR overexpression, respectively (Figure 3B). In CaCo-2 cell lines with CFTR overexpression, the OD values were 1.69 ± 0.08 vs 0.86 ± 0.05 before and after CFTR overexpression, respectively (Figure 3B). OD values significantly decreased in both cell lines (both P<0.01) with overexpression of CFTR.
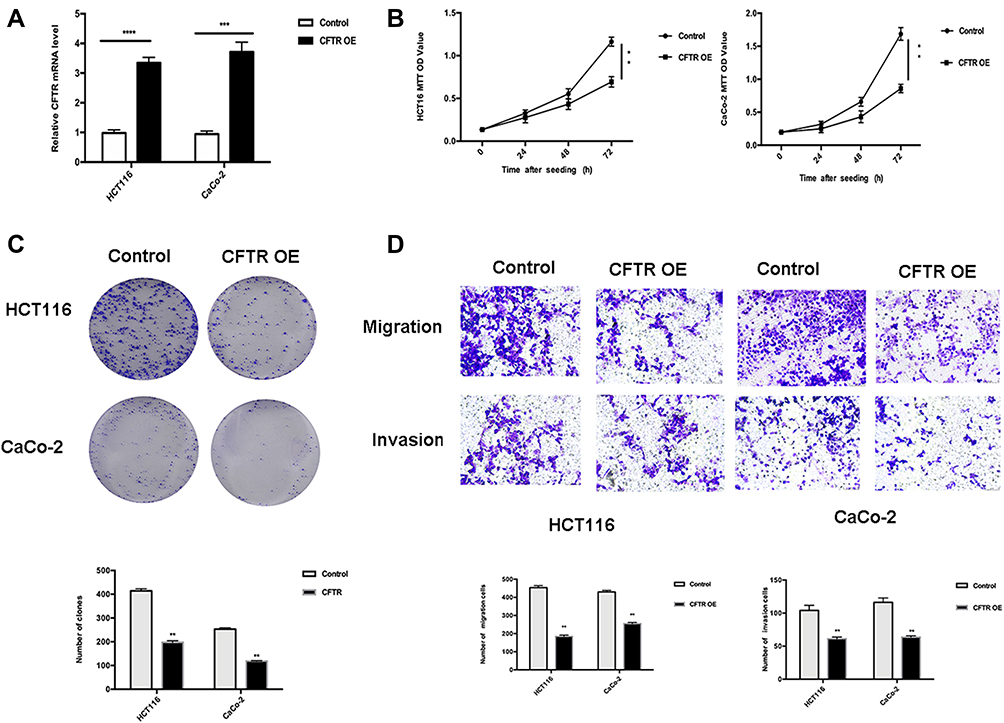 |
Figure 3 CFTR overexpression inhibits the proliferation and migration of CRC cells in HCT116 and CaCo-2 cell lines. Notes: (A) CFTR mRNA expression in lentivirus transfection cell lines. The experiment was repeated three times (***P<0.001, ****P<0.0001). (B) Growth curve showing the effect of CFTR on cell proliferation in HCT116 and CaCo-2 cell lines before and after CFTR overexpression as detected via MTT for 72 hours. The experiment was repeated three times. **P<0.01 compared to control. (C) Effects of CFTR overexpression on colony formation of HCT116 and CaCo-2 cell lines. The experiment was repeated three times. **P<0.01 compared to control. (D) CFTR overexpression inhibits cell migration and invasion in HCT116 and CaCo-2 cell lines. The transwell assay showed that HCT116 cells and CaCo-2 cell lines migrated and invaded after 48 hours of culture (×200, magnification). The results were plotted as the average number of migrated and invaded cells from six random microscope fields. All data are representative of at least three independent experiments. **P<0.01 compared to controls. |
The colony numbers were 414.33 ± 7.04 vs 197.67 ± 5.44 in HCT116 cell lines and 253.00 ± 4.55 vs 118.00 ± 1.70 in CaCo-2 cell lines before and after CFTR overexpression, respectively (Figure 3C).
Colony numbers were significantly reduced after overexpression of CFTR (both P<0.01). These results suggest that overexpression of CFTR inhibits cell proliferation in CRC.
CFTR Overexpression Inhibits Cell Migration and Invasion in HCT116 and CaCo-2 Cell Lines
As CFTR promoter methylation was associated with lymph node metastasis in human CRC, we investigated the effects of CFTR overexpression on cell migration in CRC. mRNA levels of CFTR in HCT116 and CaCo-2 cell lines were detected via qRT-PCR to determine whether CFTR was overexpressed (Figure 3A). The number of migrated cells were 453.67 ± 9.03 vs 184.67 ± 5.73 in HCT116 and 429.67 ± 7.13 vs 255.00 ± 5.72 in CaCo-2 cell lines before and after CFTR overexpression. The number of invasive cells were 104.21 ± 4.51 vs 61.33 ± 1.45 in HCT116 cells, and 116.32 ± 3.84 vs 63.67 ± 1.21 in CaCo-2 cells, before and after overexpression of CFTR. The number of migrated and invasive cells decreased significantly after overexpression of CFTR in CRC cells (both P<0.01).The results of the transwell assay suggest that CFTR overexpression inhibits cell migration and invasion in HCT116 and CaCo-2 cell lines (Figure 3D).
Discussion
Several studies have recently suggested that abnormal expression or function of CFTR may be associated with cancer incidence.12–14 A large cohort study has shown that patients with cystic fibrosis (CF) in the United States have a significantly increased risk of developing cancers of the digestive tract.14 In addition, another study also found increased risks of renal cancer, thyroid lymphoma, and skin and prostate malignancies in CF patients.12,15,16 This relationship between CFTR and cancer risk suggests that CFTR plays a key role in cancer progression.
Dietary and lifestyle changes have increased the incidence of CRC. In Asia, CRC has become the third most common malignancy,17 with the incidence increased by 2 to 4 times in China, Singapore, Japan, South Korea, and other Asian countries.18 However, the function of CFTR in CRC remains unclear. In the present study, the results of qRT-PCR and IHC indicated that compared to the adjacent normal tissues, CFTR expression was downregulated in the CRC tissues. The inactivation of tumor suppressor genes is mainly caused by gene mutations and loss of chromatin. Recent studies that analyzed CpG island methylation of gene promoter has confirmed that methylation is the third mechanism of tumor suppressor gene inactivation, and in some cases, the only mechanism of tumor suppressor gene inactivation.19 The occurrence and development of CRC is a complex and slow process involving multiple factors and processes. In addition to genetic changes, such as gene mutations, epigenetic changes are also involved. For example, DNA methylation plays a significant role in tumor formation and progression.20 In the normal genome, 70–80% of CpG can be methylated, and there is a CPG-rich region on the promoter (ie CpG island) that usually does not undergo methylation.21–23 In tumor tissues, genome-wide hypomethylation and hypermethylation in promoter regions occur, particularly in tumor suppressor gene promoters.24 Some sites of CRC often show obvious hypermethylation, such as the CpG island of some tumor suppressor genes, and can be stably inherited.25 Notably, hypermethylation of CFTR gene has been reported to occur frequently in bladder and liver cancers.10–12 CFTR contains a typical CpG island (from 1354 to 1814 bp) (Figure 1A). Thus, we explored the CFTR promoter methylation status and its impact on CFTR expression in CRC. As expected, our results suggest that CFTR expression is regulated by and inversely associated with its promoter methylation in human CRC. We then further investigated the relationship between CFTR promoter methylation and clinicopathological characteristics in 70 CRC specimens. The data showed that CFTR promoter methylation was significantly associated with age (P=0.013) and lymph node metastasis (P=0.026) in CRC specimens. We also found that the methylation probability of CFTR in colorectal cancer patients with stage IV with distant metastasis was 79.2% (19/24), which was higher than that in patients without distant metastasis 56.5% (26/46); however, there was no statistical significance between the two groups (P=0.061). Therefore, expanding the sample size to further verify the significance of CFTR methylation in patients with stage IV with metastasis may be needed. In addition, the qRT-PCR, MSP and demethylation treatment assays of CRC cell lines (ie FHC, HCT116, CaCo-2, LoVo, HT29, SW480, SW620, and SW1463) also showed that CFTR overexpression is negatively correlated with the methylation of its promoter. Consequently, we investigated the biological functions of CFTR overexpression using colony formation, transwell, and MTT assays in HCT116 and CaCo-2 cell lines. The results showed that CFTR overexpression suppresses cell proliferation, migration and invasion in CRC. Epithelial–mesenchymal transition (EMT) can inhibit the proliferation and migration ability of endometrial tumor cells,26,27 and some studies found that EMT can be promoted by inhibiting the expression of CFTR.16 These results further suggest that CFTR may be a candidate tumor suppressor gene for CRC.
This study has some limitations. The number of CRC samples used in this study was insufficient, and thus we were unable to determine the possible relationship between CFTR promoter methylation and survival prognosis of CRC. Although our analysis shows that CFTR promoter methylation is associated with lymph node metastasis, further studies are needed.
Conclusion
CFTR expression is downregulated in CRC tissues and CFTR promoter methylation may be responsible for the downregulation of CFTR. Overexpression of CFTR inhibits the proliferation, migration, and invasion of CRC cells, and this may suppress CRC tumor growth. CFTR promoter methylation is significantly correlated with lymph node metastasis and thus may be a potential marker for lymph node metastasis of CRC.
Data Sharing Statement
Some or all of the data used during the current study are available upon request from the corresponding author.
Acknowledgments
We thank the Pathology Department of the First Affiliated Hospital of Shandong First Medical University (Shandong Provincial Qianfoshan Hospital, Shandong University). This work was funded by the National Science Foundation of China (NSFC NO:81272420) and Shandong Province's Major Scientific and Technological Innovation Project (NO:2017CXGC1201 and 2019JZZY010108).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Jemal A, Siegel R, Xu J, Ward E. Cancer statistics, 2010. CA Cancer J Clin. 2010;60(5):277–300. doi:10.3322/caac.20073
2. Weitz J, Koch M, Debus J, Hohler T, Galle PR, Buchler MW. Colorectal cancer. Lancet. 2005;365(9454):153–165. doi:10.1016/S0140-6736(05)17706-X
3. Kinzler KW, Vogelstein B. Lessons from hereditary colorectal cancer. Cell. 1996;87(2):159–170. doi:10.1016/S0092-8674(00)81333-1
4. Choong MK, Tsafnat G. Genetic and epigenetic biomarkers of colorectal cancer. Clin Gastroenterol Hepatol. 2012;10(1):9–15. doi:10.1016/j.cgh.2011.04.020
5. Bestor TH. The DNA methyltransferases of mammals. Hum Mol Genet. 2000;9(16):2395–2402. doi:10.1093/hmg/9.16.2395
6. Riordan JR, Rommens JM, Kerem B. et al. Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Science. 1989;245(4922):1066–1073. doi:10.1126/science.2475911
7. Linsdell P. Cystic fibrosis transmembrane conductance regulator chloride channel blockers: pharmacological, biophysical and physiological relevance. World J Biol Chem. 2014;5:26–39. doi:10.4331/wjbc.v5.i1.26
8. Raju SV, Tate JH, Peacock SK, et al. Impact of heterozygote CFTR mutations in COPD patients with chronic bronchitis. Respir Res. 2014;15(1):18. doi:10.1186/1465-9921-15-18
9. De Lisle RC, Borowitz D. The cystic fibrosis intestine. Cold Spring Harb Perspect Med. 2013;3(9):a009753. doi:10.1101/cshperspect.a009753
10. Yu J, Zhu T, Wang Z, et al. A novel set of DNA methylation markers in urine sediments for sensitive/specific detection of bladder cancer. Clin Cancer Res. 2007;13(24):7296–7304. doi:10.1158/1078-0432.CCR-07-0861
11. Moribe T, Iizuka N, Miura T, et al. Methylation of multiple genes as molecular markers for diagnosis of a small, well-differentiated hepatocellular carcinoma. Int J Cancer. 2009;125(2):388–397. doi:10.1002/ijc.24394
12. Ding S, Gong BD, Yu J, et al. Methylation profile of the promoter CpG islands of 14 “drug-resistance” genes in hepatocellular carcinoma. World J Gastroenterol. 2004;10:3433–3440. doi:10.3748/wjg.v10.i23.3433
13. Son JW, Kim YJ, Cho HM, et al. Promoter hypermethylation of the CFTR gene and clinical/pathological features associated with non-small cell lung cancer. Respirology. 2011;16:1203–1209. doi:10.1111/j.1440-1843.2011.01994.x
14. Maisonneuve P, Marshall BC, Knapp EA, Lowenfels AB. Cancer risk in cystic fibrosis: a 20-year nationwide study from the United States. J Natl Cancer Inst. 2013;105(2):122–129. doi:10.1093/jnci/djs481
15. Xie C, Jiang XH, Zhang JT, et al. CFTR suppresses tumor progression through miR-193b targeting urokinase plasminogen activator (uPA) in prostate cancer. Oncogene. 2013;32(18):
16. Zhang JT, Jiang XH, Xie C, et al. Downregulation of CFTR promotes epithelial-to-mesenchymal transition and is associated with poor prognosis of breast cancer. Biochim Biophys Acta. 2013;1833(12):2961–2969. doi:10.1016/j.bbamcr.2013.07.021
17. Pourhoseingholi MA, Vahedi M, Baghestani AR. Burden of gastrointestinal cancer in Asia; an overview. Gastroenterol Hepatol Bed Bench. 2015;8:19–27.
18. Sung JJ, Lau JY, Goh KL, Leung WK. Asia Pacific Working Group on Colorectal C. Increasing incidence of colorectal cancer in Asia: implications for screening. Lancet Oncol. 2005;6(11):871–876. doi:10.1016/S1470-2045(05)70422-8
19. Bhattacharya SK, Ramchandani S, Cervoni N, Szyf M. A mammalian protein with specific demethylase activity for mCpG DNA. Nature. 1999;397(6720):579–583. doi:10.1038/17533
20. Yan W, Guo M. Epigenetics of colorectal cancer. Methods Mol Biol. 2015;1238:405–424.
21. Jabbari K, Bernardi G. Cytosine methylation and CpG, TpG (CpA) and TpA frequencies. Gene. 2004;333:143–149. doi:10.1016/j.gene.2004.02.043
22. Herman JG, Baylin SB. Gene silencing in cancer in association with promoter hypermethylation. N Engl J Med. 2003;349(21):2042–2054. doi:10.1056/NEJMra023075
23. Weber M, Hellmann I, Stadler MB, et al. Distribution, silencing potential and evolutionary impact of promoter DNA methylation in the human genome. Nat Genet. 2007;39(4):457–466. doi:10.1038/ng1990
24. Grady WM, Carethers JM. Genomic and epigenetic instability in colorectal cancer pathogenesis. Gastroenterology. 2008;135(4):1079–1099. doi:10.1053/j.gastro.2008.07.076
25. Zhao Y, Zhang S, Fu B, Xiao C. Abnormalities of tumor suppressor genes P16 and P15 in primary maxillofacial squamous cell carcinomas. Cancer Genet Cytogenet. 1999;112(1):26–33. doi:10.1016/S0165-4608(98)00259-3
26. Xia X, Wang J, Liu Y, Yue M. Lower cystic fibrosis transmembrane conductance regulator (CFTR) promotes the proliferation and migration of endometrial carcinoma. Med Sci Monit. 2017;23:966–974. doi:10.12659/MSM.899341
27. Huang W, Jin A, Zhang J, et al. Upregulation of CFTR in patients with endometriosis and its involvement in NFkappaB-uPAR dependent cell migration. Oncotarget. 2017;8(40):66951–66959. doi:10.18632/oncotarget.16441
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.