Back to Journals » International Journal of Nanomedicine » Volume 11
Cellular uptake mechanism and comparative evaluation of antineoplastic effects of paclitaxel–cholesterol lipid emulsion on triple-negative and non-triple-negative breast cancer cell lines
Authors Ye J ![]() , Xia X
, Xia X ![]() , Dong W, Hao H, Meng L, Yang Y, Wang R, Lyu Y, Liu Y
, Dong W, Hao H, Meng L, Yang Y, Wang R, Lyu Y, Liu Y
Received 26 May 2016
Accepted for publication 23 July 2016
Published 24 August 2016 Volume 2016:11 Pages 4125—4140
DOI https://doi.org/10.2147/IJN.S113638
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Linlin Sun
Jun Ye,1,2 Xuejun Xia,1,2 Wujun Dong,1,2 Huazhen Hao,1,2 Luhua Meng,1,2 Yanfang Yang,1,2 Renyun Wang,1,2 Yuanfeng Lyu,3 Yuling Liu1,2
1State Key Laboratory of Bioactive Substance and Function of Natural Medicines, 2Beijing Key Laboratory of Drug Delivery Technology and Novel Formulation, Institute of Materia Medica, Chinese Academy of Medical Sciences, Peking Union Medical College, Beijing, 3School of Pharmacy, China Pharmaceutical University, Nanjing, People’s Republic of China
Abstract: There is no effective clinical therapy for triple-negative breast cancers (TNBCs), which have high low-density lipoprotein (LDL) requirements and express relatively high levels of LDL receptors (LDLRs) on their membranes. In our previous study, a novel lipid emulsion based on a paclitaxel–cholesterol complex (PTX-CH Emul) was developed, which exhibited improved safety and efficacy for the treatment of TNBC. To date, however, the cellular uptake mechanism and intracellular trafficking of PTX-CH Emul have not been investigated. In order to offer powerful proof for the therapeutic effects of PTX-CH Emul, we systematically studied the cellular uptake mechanism and intracellular trafficking of PTX-CH Emul and made a comparative evaluation of antineoplastic effects on TNBC (MDA-MB-231) and non-TNBC (MCF7) cell lines through in vitro and in vivo experiments. The in vitro antineoplastic effects and in vivo tumor-targeting efficiency of PTX-CH Emul were significantly more enhanced in MDA-MB-231-based models than those in MCF7-based models, which was associated with the more abundant expression profile of LDLR in MDA-MB-231 cells. The results of the cellular uptake mechanism indicated that PTX-CH Emul was internalized into breast cancer cells through the LDLR-mediated internalization pathway via clathrin-coated pits, localized in lysosomes, and then released into the cytoplasm, which was consistent with the internalization pathway and intracellular trafficking of native LDL. The findings of this paper further confirm the therapeutic potential of PTX-CH Emul in clinical applications involving TNBC therapy.
Keywords: paclitaxel, lipid emulsion, triple-negative breast cancers, low-density lipoprotein, tumor targeting
Introduction
Breast cancer (BC) has a high incidence and is the most frequently diagnosed cancer, ranking the second-leading cause of cancer death in women.1,2 Triple-negative BC (TNBC) accounts for about 12%–17% of breast cancer cases, and is characterized by a lack of HER2, estrogen receptor (ER), and progesterone receptor (PR).2,3 Compared with hormonal receptor-positive and HER2-positive BC, TNBC is more aggressive and associated with a worse prognosis, as well as a higher risk of relapse and metastasis and shorter survival time.4,5 Due to lacking both hormone receptors and HER2 expression, TNBC is not susceptible to endocrine therapy or HER2-targeted therapy. The only modality of systemic therapy available for TNBC is chemotherapy with anthracyclines and taxanes,5–8 providing limited options with unavoidable side effects. It is critical to improve the therapies, since the median survival time is only 13.3 months for patients with metastatic TNBC. Virtually all patients with metastatic TNBC ultimately die of the disease, despite receiving systemic treatment.9,10 Therefore, there is a tremendous incentive to refine existing treatment modalities to treat clinically intractable cancers more effectively.
Low density lipoprotein receptor (LDLR), a member of the LDLR family, is overexpressed in various cancer cells,11 including BC,12 but is expressed at low levels in normal cells.13 Therefore, LDLR is a potential receptor target for selective delivery of antineoplastic agents to BC. Interestingly, the expression profile of LDLR is dissimilar across different subtypes of BC. Compared with MCF7 cells (ER-positive), LDLR messenger RNA abundance is three- to fivefold higher in MDA-MB-231 cells (TNBC), and MDA-MB-231 cells exhibit high capacity and high affinity binding of LDL in comparison with MCF7 cells.12,14 LDL accelerates the proliferation of MDA-MB-231 cells, but has little effect on the proliferation of MCF7 cells, which can be attributed to the ability of TNBC cells to take up, store, and utilize exogenous cholesterol (CH; mainly LDL-CH) mediated by LDLR.15 Increased expression of LDLR in TNBC cells is consistent with the aggressive and metastatic nature of TNBC.16 These findings show the potential of LDLR as a targeting receptor for the design of TNBC-targeted chemotherapy-delivery systems. Although many studies have exploited LDLR as a target for tumor diagnosis and treatment of diverse types of cancer, including brain glioma, liver cancer, lung cancer, prostate cancer, and colorectal cancer,13,17–20 few have extensively explored LDLR as a potential receptor for targeted therapy of TNBC.
Over the last decade, several studies reported the utility of a CH-rich emulsion termed “LDE” to deliver therapeutic agents to cancers. LDE, consisting of a cholesteryl ester core coated with a monolayer of phospholipids, resembles the LDL lipid-portion structure and has the ability to bind to LDLR on the cancer cell surface.21–25 Although LDE has been confirmed with tumor-targeting effects mediated by LDLR and shows good drug-loading capacity of paclitaxel (PTX), it is stable for only 8 days at 4°C.21 The instability of LDE-PTX, which might be attributable to the poor lipophilicity of PTX,22 makes it less promising for clinical application. Alternatively, a novel lipid emulsion, which was composed of a PTX-CH complex surrounded by a phospholipid monolayer, was developed in our previous work (Figure 1).26 Compared with the tumor-targeting LDE-PTX loaded with PTX and cholesteryl ester reported previously,21 substitution of the PTX-CH complex for PTX and cholesteryl ester of LDE-PTX has the following advantages: the PTX-CH complex not only greatly improves PTX solubility in the oil phase but can also function as a component of LDL-CH.27 The resulting lipid emulsion (PTX-CH Emul) exhibits an ideal particle size, high drug-loading capability, high drug-encapsulation efficiency, and excellent stability (12 months at 6°C).26 Furthermore, PTX-CH Emul shows superior in vitro antitumor efficacy, higher specificity and efficiency in intratumoral accumulation, better safety profiles, and superior antitumor efficacy in vivo against TNBC compared to the conventional PTX Emul and Taxol.27
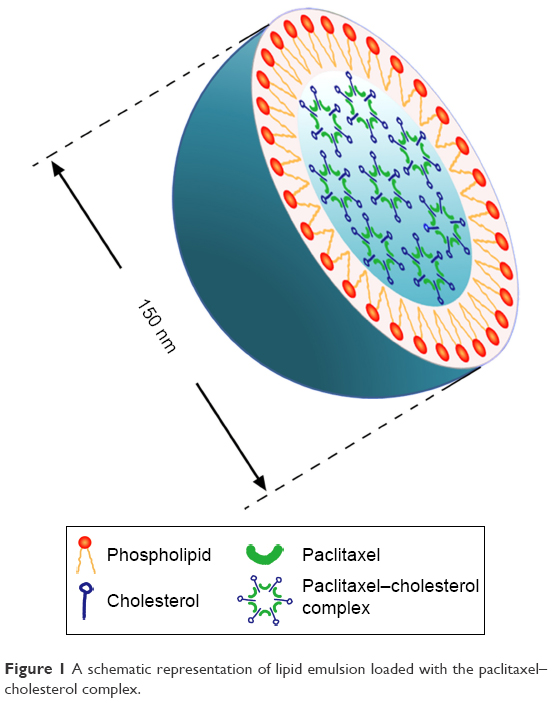 | Figure 1 A schematic representation of lipid emulsion loaded with the paclitaxel–cholesterol complex. |
Although PTX-CH Emul shows great potential as a drug carrier for PTX in clinical applications involving TNBC therapy,26,27 intracellular trafficking and its underlying mechanisms have not been investigated. In order to improve the therapeutic potential of PTX-CH Emul for the treatment of TNBC further, it is important to make a comparative evaluation of the antineoplastic effects of PTX-CH Emul on TNBC and non-TNBC cells. Therefore, the aim of the present study was to elucidate the cellular uptake mechanism and intracellular trafficking of PTX-CH Emul and comparatively evaluate its antineoplastic effects on TNBC and non-TNBC cells. The results of our research provide valuable evidence, which supports the development of PTX-CH Emul for targeted therapy of TNBC.
Materials and methods
Materials
PTX was purchased from Guilin Huiang Biopharmaceutical Co Ltd (Guilin, People’s Republic of China). The Taxol was a kind gift from Beijing Union Pharmaceutical Factory. Cell Counting Kit (CCK)-8 and an annexin V–fluorescein isothiocyanate apoptosis-detection kit were supplied by Dojindo Laboratories (Tokyo, Japan). Fluorescein was obtained from Sigma-Aldrich (St Louis, MO, USA). DiR was obtained from AAT Bioquest Inc. (Sunnyvale, CA, USA). DiI was purchased from Beyotime Institute of Biotechnology (Haimen, People’s Republic of China). A micro-bicinchoninic acid protein-assay kit was purchased from Pierce Biotechnology Inc. (Rockford, IL, USA). LDL and lipoprotein-deficient serum (LPDS) were purchased from Xiesheng Biotechnology Company (Beijing, People’s Republic of China).
Preparation of PTX-CH Emul
The PTX-CH complex, PTX-CH Emul, conventional PTX Emul, fluorescein-labeled (FL)-PTX-CH Emul, FL-PTX Emul, and DiR-PTX-CH Emul were prepared as previously reported.26,27
Cell lines and cell culture
H9c2 (2-1), MDA-MB-231, and MCF7 were supplied by the Cell Resource Center, Peking Union Medical College (Beijing, People’s Republic of China). H9c2 (2-1) and MCF7 cells were cultured in Dulbecco’s Modified Eagle’s Medium supplemented with 10% fetal bovine serum (FBS), 100 U/mL penicillin, and 100 μg/mL streptomycin. MDA-MB-231 cells were cultured in Roswell Park Memorial Institute 1640 growth medium supplemented with 10% FBS, 100 U/mL penicillin, and 100 μg/mL streptomycin. All cells were maintained at 37°C in a humidified chamber with 5% CO2.
Animals and human tumor xenografts
Female BALB/c nude mice (6–8 weeks old, 21–25 g) were obtained from HFK Bioscience Company (Beijing, People’s Republic of China). All animal protocols were approved by the institutional animal care and use committee of Peking Union Medical College. The experiments reported herein were conducted according to the “Guide for the Care and Use of Laboratory Animals” from the National Institutes of Health (8th Edition). Female BALB/c nude mice were inoculated subcutaneously in the right armpit with 0.2 mL (2×106) MDA-MB-231 or 0.2 mL (1×107) MCF7 cell suspension. The tumors were allowed to grow to a median size of 100 mm3 before treatment was initiated.
LDLR-expression level
The LDLR-expression level in H9c2 (2-1), MCF7, or MDA-MB-231 cells was determined using an LDLR enzyme-linked immunosorbent assay kit (Cusabio, Wuhan, People’s Republic of China) according to a method described previously.13
In vitro cellular uptake
MDA-MB-231 or MCF7 cells were seeded into six-well chamber slides and incubated with FL-PTX-CH Emul for 4 hours, washed with phosphate-buffered saline (PBS), fixed with 4% paraformaldehyde (PFA), stained with Hoechst 33258 (Thermo Fisher Scientific, Waltham, MA, USA), and observed with confocal laser-scanning microscopy (CLSM; FV1000; Olympus, Tokyo, Japan). Flow cytometry (Accuri C6; BD Biosciences, San Jose, CA, USA) was used to evaluate the intracellular uptake of FL-PTX-CH Emul in MDA-MB-231 or MCF7 cells quantitatively.
To confirm further the potential of LDLR to mediate the uptake of PTX-CH Emul, MDA-MB-231, or MCF7 cells, which were incubated for 24 hours in Roswell Park Memorial Institute 1640 or Dulbecco’s Modified Eagle’s Medium supplemented with 10% FBS or 10% LPDS, were treated with FL-PTX-CH Emul and excess LDL (200 μg/mL) individually or in combination, followed by CLSM or high-performance liquid chromatography (Agilent Technologies, Santa Clara, CA, USA) analysis.
In vitro cytotoxicity assay
The cytotoxicity of PTX-CH Emul in MDA-MB-231 or MCF7 cells was measured using the CCK-8 kit. Cells were seeded in 96-well plates at a density of 2×103 cells/well and allowed to attach overnight. Afterward, cells were treated with PTX-CH Emul at various concentrations of PTX. After 72 hours of incubation, viability was assessed using the CCK-8 kit according to the manufacturer’s protocol.
In vitro cell-apoptosis assay
MDA-MB-231 or MCF7 cells were seeded in six-well plates and incubated for 24 hours at 37°C. Next, cells were treated for 24 hours with Taxol, PTX Emul, PTX-CH Emul, or culture medium as a control treatment. Afterward, cells were stained with Hoechst 33258 and examined under CLSM. For quantitative analysis of apoptosis, cells were left untreated or treated with various PTX formulations. After 24 hours of treatment, cells were stained with annexin V–fluorescein isothiocyanate and propidium iodide for 15 minutes and analyzed using flow cytometry.
Tumor-spheroid penetration and growth inhibition
Three-dimensional multicellular tumor spheroids of MDA-MB-231 or MCF7 cells were developed using a liquid-overlay system.27,28 MDA-MB-231 or MCF7 cells (2×103 cells/well) were seeded in a 96-well plate precoated with 50 μL agarose (1.5%, w/v). After 7 days, separate groups of MDA-MB-231-tumor spheroids were incubated with FL-dimethyl sulfoxide (DMSO) solution, FL-PTX Emul, and FL-PTX-CH Emul for 8 hours, rinsed with cold PBS thrice, and fixed with 4% PFA overnight at 4°C. The fluorescence intensity of different depths of tumor spheroids was examined under CLSM. In order to make a comparative evaluation of the penetration efficiency of PTX-CH Emul in breast-tumor spheroids, MDA-MB-231- or MCF7-tumor spheroids were incubated with FL-PTX-CH Emul for 8 hours.
To investigate the growth inhibition of tumor spheroids, MDA-MB-231- or MCF7-tumor spheroids were treated with PTX-CH Emul (0.5 μg/mL) and the size of tumor spheroids monitored with an inverted-phase microscope at 0, 1, 3, 5, and 7 days of treatment according to a method described previously.27
In vivo tumor-targeting efficiency
To make a comparative evaluation of tumor-targeting efficiency of PTX-CH Emul in MDA-MB-231 and MCF7 tumor-bearing nude mice, DiR-PTX-CH Emul (0.2 mg/kg) was injected into the mice via the tail vein. Each group had three mice. In vivo imaging was detected at different time points (2, 4, 8, 12, and 24 hours) postinjection using the In Vivo IVIS spectrum-imaging system (PerkinElmer, Waltham, MA, USA). At 24 hours postinjection, the mice were killed and the tumors and other major organs collected, and ex vivo imaging was captured.
Cellular internalization mechanism and intracellular fate
To investigate the intracellular uptake of the PTX and CH components of PTX-CH Emul, MDA-MB-231 cells were incubated with PTX-CH Emul doubly labeled with 14C-CH and 3H-PTX at different PTX concentrations for 2 hours or at 10 μg/mL PTX concentration for different incubation times. After incubation, cells were washed thrice with cold PBS. NaOH (300 μL, 0.5 M) was added to the plates to disrupt the cells before radioactivity measurement.
For binding assays, cells were incubated with FL-DMSO, FL-PTX Emul, or FL-PTX-CH Emul for 2 hours at 4°C. After incubation, cells were washed thrice with cold PBS and incubated with DiI for 15 minutes at room temperature to stain the cell membrane. Then, cells were fixed with 4% PFA, stained with Hoechst 33258, and observed with CLSM. This experiment was performed in triplicate.
In order to investigate whether endocytosis was the main pathway for the cellular uptake of PTX-CH Emul, cells were incubated with PTX-CH Emul at 4°C instead of the regular 37°C. Additionally, cells were preincubated with sucrose (0.45 M) to inhibit the formation of clathrin-coated pits,29 genistein (0.2 mM) to inhibit caveolae-mediated endocytosis,30 or cytochalasin D (30 μM) to inhibit macropinocytosis29 at 37°C for 1 hour. Then, PTX-CH Emul and different inhibitors at the same concentrations as described were added and incubated at 37°C for another 2 hours. After incubation, cells were washed thrice with cold PBS, trypsinized, and centrifuged at 8,000 rpm for 5 minutes to obtain cell pellets, which were subsequently analyzed using high-performance liquid chromatography.
For investigation of the intracellular fate of PTX-CH Emul, cells were seeded onto cover glasses in a six-well plate and incubated for 24 hours at 37°C. Then cells were incubated with FL-PTX-CH Emul for 0.5, 1, 2, and 4 hours at 37°C. At different time points, cells were treated with 75 nM LysoTracker red, fixed, and stained with Hoechst 33258. Confocal images of cells were captured with CLSM. This experiment was performed in triplicate.
Statistical analysis
All data subjected to statistical analysis were obtained from at least three parallel experiments, and the results are expressed as means ± standard deviation. One-way analysis of variance was used for significance analysis, and P<0.05 was considered statistically significant.
Results
LDLR-expression level
The LDLR-expression levels in H9c2 (2-1), MCF7, and MDA-MB-231 cells were 24.1±5.6, 198±13.2, and 462.5±81.5 ng/g of protein, respectively (Figure 2). In contrast with H9c2 (2-1) cardiac myocytes, BC cells, including MDA-MB-231 and MCF7 cells, overexpressed LDLR. Additionally, the expression level of LDLR was much higher in MDA-MB-231, a typical TNBC cell line, than in MCF7, an ER-positive BC cell line. Moreover, incubation of MDA-MB-231 cells with 10% LPDS increased the LDLR-expression level to 768.1±45 ng/g from 462.5±81.5 ng/g, the baseline level obtained with cells incubated with 10% FBS.
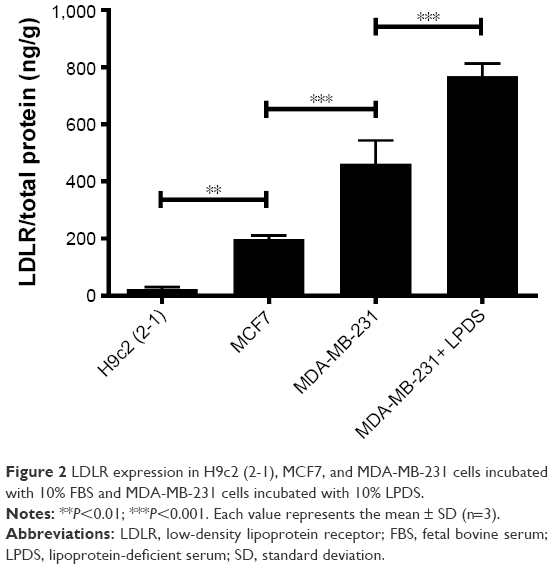 | Figure 2 LDLR expression in H9c2 (2-1), MCF7, and MDA-MB-231 cells incubated with 10% FBS and MDA-MB-231 cells incubated with 10% LPDS. |
In vitro cellular uptake
In comparison with MCF7 cells incubated with FL-PTX-CH Emul, the fluorescence intensity was about fivefold higher in MDA-MB-231 cells (Figure 3A and B), indicating that the expression profile of LDLR modulated the uptake of FL-PTX-CH Emul in BC cells. To confirm that FL-PTX-CH Emul was internalized into MDA-MB-231 or MCF7 cells via LDLR, we firstly incubated cells with 10% LPDS to stimulate the expression of LDLR in MDA-MB-231 or MCF7 cells, and then excess native LDL was added to saturate LDLR. It was found that the uptake of FL-PTX-CH Emul was significantly increased in MDA-MB-231 cells incubated with 10% LPDS compared with that in cells incubated with 10% FBS. In the competition assay, adding excess native LDL reduced the uptake of FL-PTX-CH Emul (Figure 3C and D). Similar results were confirmed in MCF7 cells (Figure S1). These results demonstrated that FL-PTX-CH Emul was recognized by the LDLR of BC cells and internalized through an endocytic pathway.
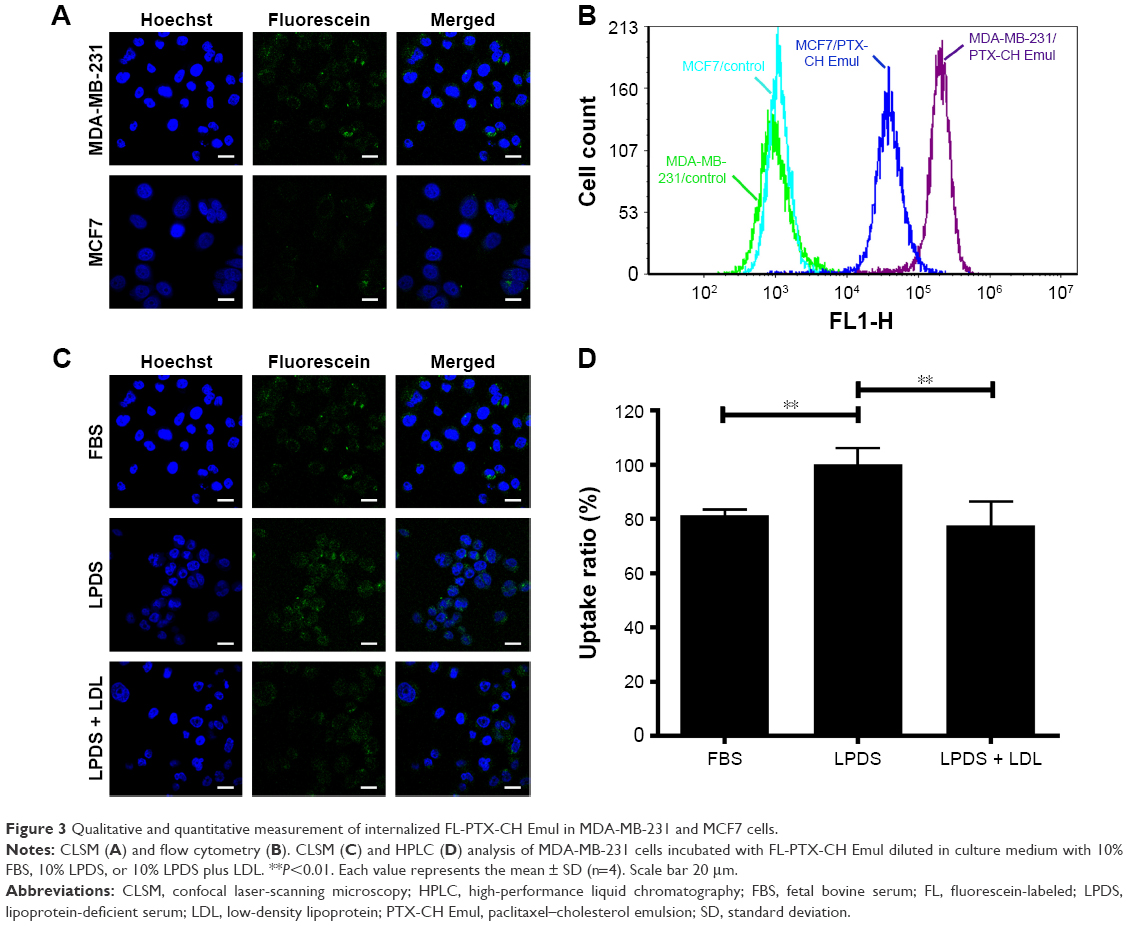 | Figure 3 Qualitative and quantitative measurement of internalized FL-PTX-CH Emul in MDA-MB-231 and MCF7 cells. |
In vitro cytotoxicity assay
To clarify the connection between LDLR expression and cytotoxicity, MCF7 and MDA-MB-231 cells, which have different LDLR-expression profiles, were used to evaluate the cytotoxicity of PTX-CH Emul. In accordance with the results of cellular uptake described earlier, PTX-CH Emul exhibited significantly better cytotoxicity in MDA-MB-231 cells (half-maximal inhibitory concentration [IC50] 2.13 ng/mL)27 than in MCF7 cells (IC50 8.37 ng/mL) (Figure S2).
In vitro cell-apoptosis assay
To test the effect of the encapsulation of PTX in PTX-CH Emul on cell apoptosis, MCF7 and MDA-MB-231 cells were stained with Hoechst 33258 after treatment with different PTX formulations. As shown in Figure 4A and B, the nuclei of untreated MCF7 and MDA-MB-231 cells were homogeneous without segmentation or fragmentation. However, when cells were treated with Taxol or PTX Emul, chromatin condensation and nuclear fragmentation were seen, with the formation of apoptotic bodies containing dense granular particles, and PTX-CH Emul induced even more apparent apoptotic changes in comparison with Taxol and PTX Emul.
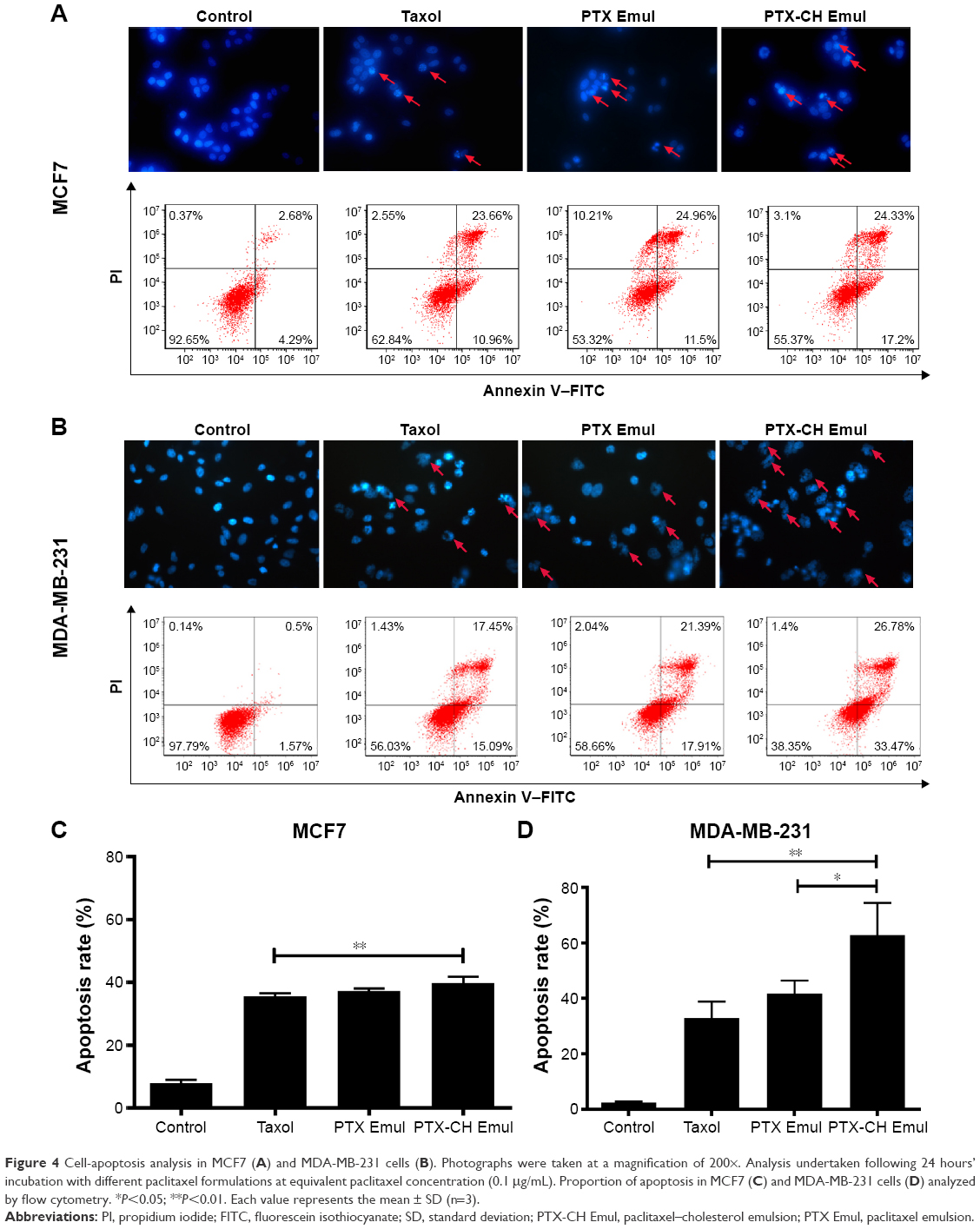 | Figure 4 Cell-apoptosis analysis in MCF7 (A) and MDA-MB-231 cells (B). Photographs were taken at a magnification of 200×. Analysis undertaken following 24 hours’ incubation with different paclitaxel formulations at equivalent paclitaxel concentration (0.1 μg/mL). Proportion of apoptosis in MCF7 (C) and MDA-MB-231 cells (D) analyzed by flow cytometry. *P<0.05; **P<0.01. Each value represents the mean ± SD (n=3). |
For quantitative analysis, the percentage of apoptotic cells treated with the PTX formulations was determined using flow cytometry. The percentages of apoptotic cells in MCF7 cells treated with Taxol, PTX Emul, and PTX-CH Emul were 35.61%±0.89%, 37.22%±0.84%, and 39.8%±2.04%, respectively, while the percentages of apoptotic cells in MDA-MB-231 cells treated with Taxol, PTX Emul, and PTX-CH Emul were 32.95%±5.8%, 41.73%±4.7%, and 62.88%±11.65%, respectively (Figure 4C and D). These results indicated that the percentage of apoptotic cells induced by PTX-CH Emul was much higher in MDA-MB-231 cells (62.88%±11.65%) than in MCF7 cells (39.8%±2.04%). These results were in accord with those obtained with the nuclear staining and in vitro cytotoxicity assay. The significant morphologic modifications and increased percentage of apoptotic cells caused by PTX-CH Emul in comparison with Taxol and PTX Emul may be attributed to the improved cellular uptake of PTX-CH Emul in MDA-MB-231 cells.27 Furthermore, in comparison with its effects in MCF7 cells, PTX-CH Emul induced a significantly greater percentage of apoptotic cells in MDA-MB-231 cells, further supporting the LDLR-specific targeting of PTX-CH Emul.
Tumor-spheroid penetration and growth inhibition
As targeted drug-delivery systems are superior to conventional drug-delivery systems for delivering drugs specifically to tumors, it is of high significance to test the penetration ability of PTX-CH Emul. To this end, tumor spheroids were used to investigate the penetrating efficiency of PTX-CH Emul. As shown in Figure 5A, FL-PTX-CH Emul penetrated more deeply and distributed more extensively in MDA-MB-231-tumor spheroids than FL-DMSO and FL-PTX Emul. In addition, FL-PTX-CH Emul penetrated more deeply into tumor spheroids formed by MDA-MB-231 cells than those formed by MCF7 cells (Figure 5B), indicating that LDLR led to higher penetration efficiency of FL-PTX-CH Emul in MDA-MB-231-tumor spheroids.
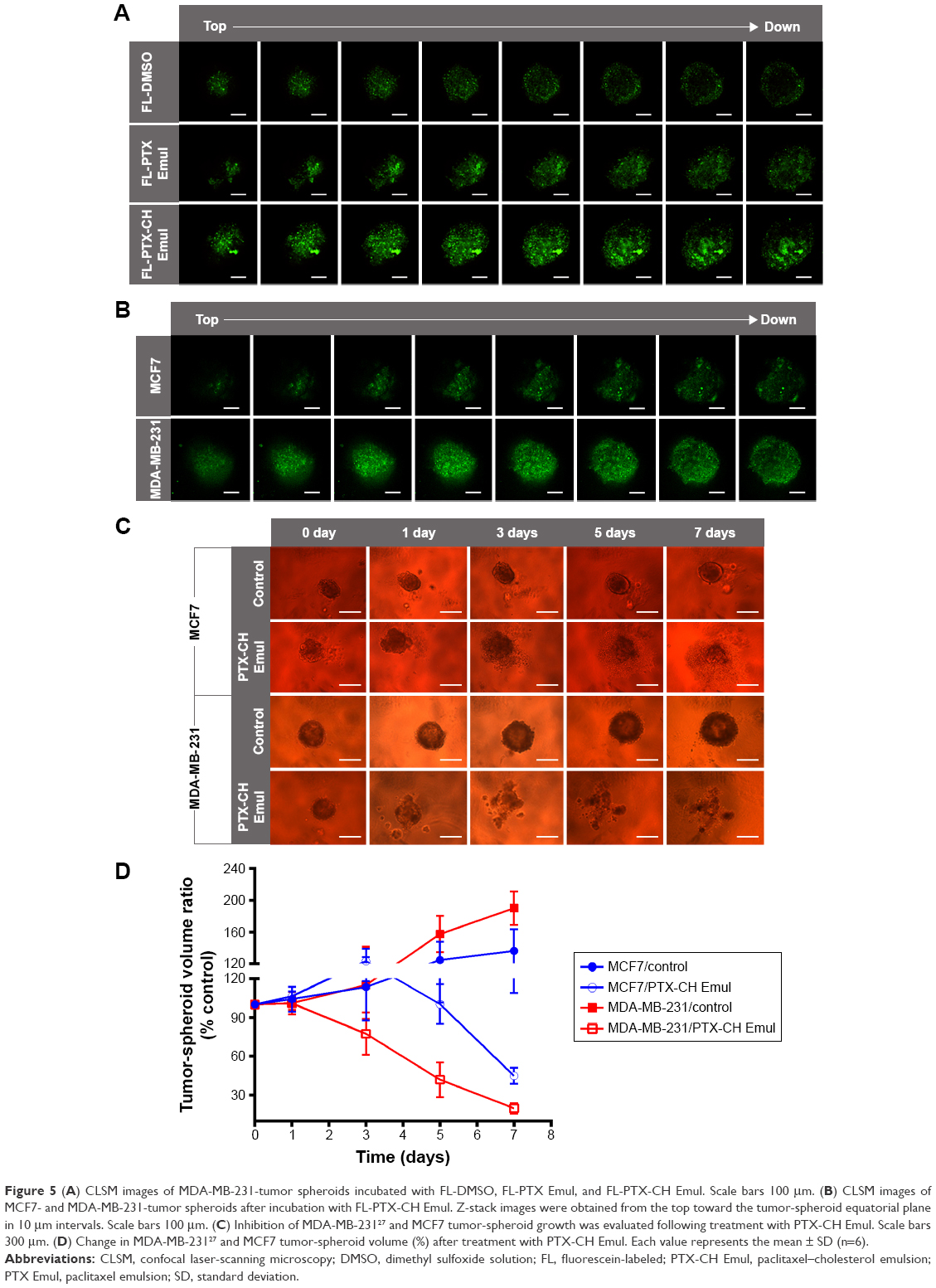 | Figure 5 (A) CLSM images of MDA-MB-231-tumor spheroids incubated with FL-DMSO, FL-PTX Emul, and FL-PTX-CH Emul. Scale bars 100 μm. (B) CLSM images of MCF7- and MDA-MB-231-tumor spheroids after incubation with FL-PTX-CH Emul. Z-stack images were obtained from the top toward the tumor-spheroid equatorial plane in 10 μm intervals. Scale bars 100 μm. (C) Inhibition of MDA-MB-23127 and MCF7 tumor-spheroid growth was evaluated following treatment with PTX-CH Emul. Scale bars 300 μm. (D) Change in MDA-MB-23127 and MCF7 tumor-spheroid volume (%) after treatment with PTX-CH Emul. Each value represents the mean ± SD (n=6). |
As presented in Figure 5C, MCF7- and MDA-MB-231-tumor spheroids grew fast and became compact when treated with the cell-culture medium. In comparison with MCF7-tumor spheroids, MDA-MB-231-tumor spheroids grew much more rapidly, consistent with the growth characteristics of solid tumors in vivo. However, after treatment with PTX-CH Emul, both types of tumor spheroids shrank, with some cell detachment from the tumor spheroids, and almost lost their three-dimensional structure, indicating that PTX-CH Emul effectively inhibited spheroid-cell proliferation. After 7 days of treatment with PTX-CH Emul, the tumor-spheroid volume ratios of MDA-MB-231 and MCF7 cells were 19.9%±4%27 and 45%±6.2%, respectively (Figure 5D). Therefore, it is worth mentioning that PTX-CH Emul exhibited a much more pronounced inhibitory effect on MDA-MB-231-tumor spheroids than that on MCF7-tumor spheroids, which was consistent with the results of in vitro cytotoxicity and cell apoptosis described earlier.
In vivo tumor-targeting efficiency
In our previous study, we found that the fluorescence intensity of DiR-PTX-CH Emul detected in tumor sites was much stronger than that of DiR-PTX Emul at all time points postinjection.27 To confirm the in vivo tumor-targeting efficiency of PTX-CH Emul further, a comparative evaluation was made in MDA-MB-231 and MCF7 tumor-bearing nude mice. As shown in Figure 6A, the fluorescence intensity of tumor sites in MDA-MB-231 tumor-bearing nude mice was much higher than that in MCF7 tumor-bearing nude mice at all observed time points. Additionally, the ex vivo fluorescent image of the excised tumors from MDA-MB-231 tumor-bearing nude mice also clearly showed about threefold-higher fluorescence intensity than that from MCF7 tumor-bearing nude mice (Figure 6B), demonstrating a further piece of evidence of in vivo tumor-targeting efficiency of PTX-CH Emul.
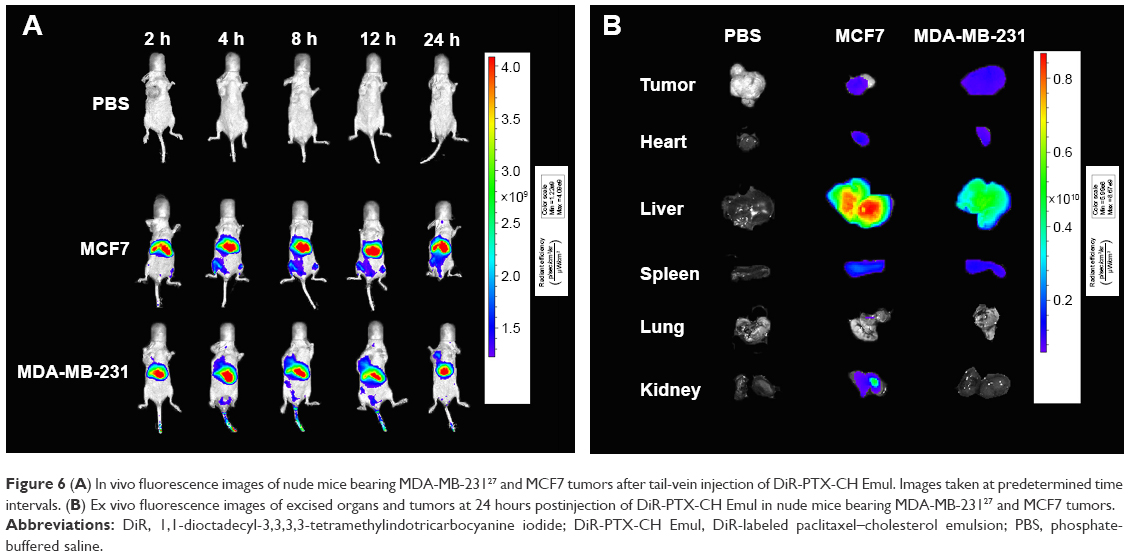 | Figure 6 (A) In vivo fluorescence images of nude mice bearing MDA-MB-23127 and MCF7 tumors after tail-vein injection of DiR-PTX-CH Emul. Images taken at predetermined time intervals. (B) Ex vivo fluorescence images of excised organs and tumors at 24 hours postinjection of DiR-PTX-CH Emul in nude mice bearing MDA-MB-23127 and MCF7 tumors. |
Cellular internalization mechanism and intracellular fate
The LDLR-binding profiles of FL-DMSO, FL-PTX Emul, and FL-PTX-CH Emul in the MDA-MB-231 cells were assessed qualitatively using CLSM. As shown in Figure S3, FL-PTX-CH Emul exhibited significantly greater green fluorescence overlapped with red fluorescence (corresponding to the cell membrane) in comparison with FL-DMSO and FL-PTX Emul in MDA-MB-231 cells, implying that FL-PTX-CH Emul had a higher binding affinity to LDLR than FL-DMSO and FL-PTX Emul did. Additionally, when MDA-MB-231 and MCF7 cells were incubated with FL-PTX-CH Emul, the higher green-fluorescence intensity was observed in MDA-MB-231 cells, indicating that the expression profile of LDLR modulated the binding capacity of FL-PTX-CH Emul to LDLR in BC cells.
Cytochalasin D, sucrose, and genistein were employed to inhibit macropinocytosis-, clathrin-, and caveolin-mediated endocytosis, respectively. We selected 4°C to investigate the energy impact on cellular uptake. With the reduced incubation temperature, the cellular uptake of PTX-CH Emul decreased dramatically in comparison to the control (37°C), suggesting that the endocytosis was an energy-dependent process (Figure 7A). When cells were preincubated with sucrose, cytochalasin D, or genistein, the reduced cellular uptake extent was 57%, 78%, or 81%, respectively, indicating that the internalization of PTX-CH Emul was mediated by the combined clathrin- and caveolae-mediated endocytosis pathways, as well as macropinocytosis, and the clathrin-mediated endocytosis was the main endocytosis pathway.
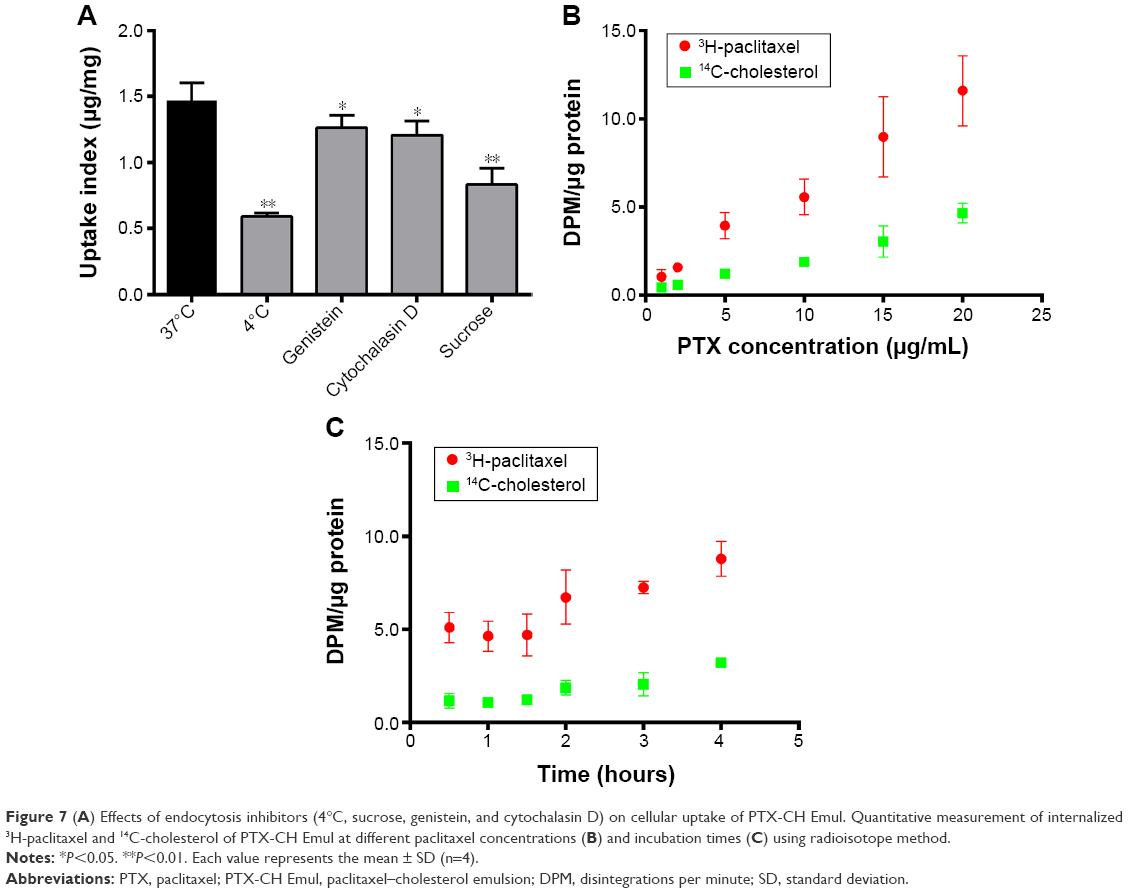 | Figure 7 (A) Effects of endocytosis inhibitors (4°C, sucrose, genistein, and cytochalasin D) on cellular uptake of PTX-CH Emul. Quantitative measurement of internalized 3H-paclitaxel and 14C-cholesterol of PTX-CH Emul at different paclitaxel concentrations (B) and incubation times (C) using radioisotope method. |
When cells were incubated with PTX-CH Emul doubly labeled with 14C-CH and 3H-PTX at increasing PTX concentrations for 2 hours or at a constant PTX concentration for increasing incubation time, the increase in the uptake was proportional to the two labels (Figure 7B and C). This result indicated that both PTX and CH components of PTX-CH Emul were taken up together by MDA-MB-231 cells.
To investigate the intracellular fate of PTX-CH Emul following its uptake, cells were stained with LysoTracker red. In the stained cells, the red fluorescence represented the lysosomes, whereas green fluorescence indicated the FL-PTX-CH Emul. Therefore, the yellow fluorescence demonstrated the colocalization of lysosomes and FL-PTX-CH Emul in merged images. After 30 minutes’ incubation, it was found that FL-PTX-CH Emul colocalized partly with lysosomes in cells (Figure 8). At the longer incubation time points (1 and 2 hours), the density of the cytoplasmic green fluorescence increased significantly, with more aggregates in the lysosomal vesicles. After prolonged incubation (4 hours), the green fluorescence in the lysosomes moved to the perinuclear area, suggesting that FL-PTX-CH Emul was released to the cytoplasm under acidic condition in lysosomes.
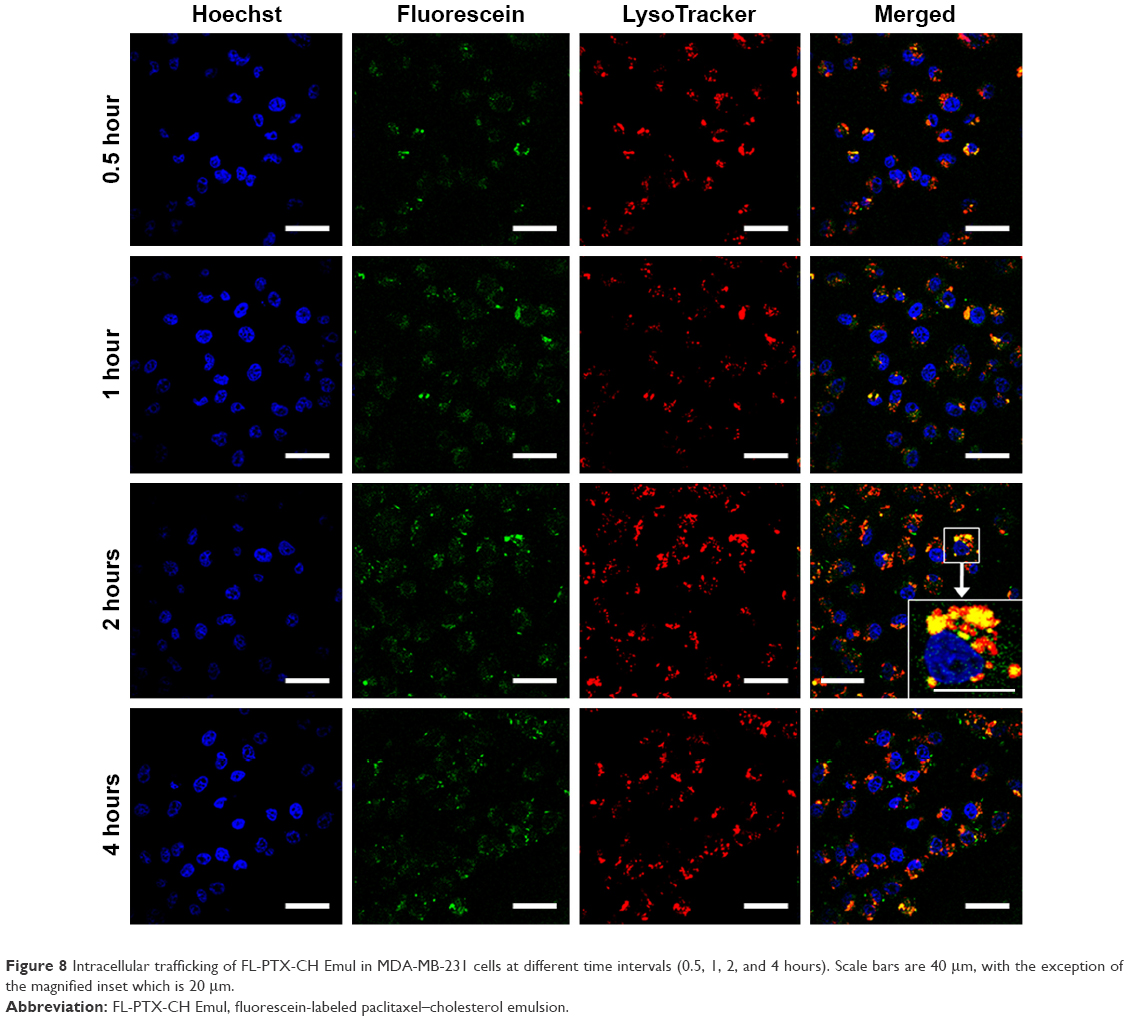 | Figure 8 Intracellular trafficking of FL-PTX-CH Emul in MDA-MB-231 cells at different time intervals (0.5, 1, 2, and 4 hours). Scale bars are 40 μm, with the exception of the magnified inset which is 20 μm. |
Discussion
Much attention has been paid to the design of novel nanocarriers based on LDL,18,19,31,32 due to the overexpression of LDLR in many tumor cells, which offers the advantage of tumor targeting for LDL-based nanocarriers. However, an LDL-based nanocarrier is less than ideal as a targeted drug-delivery system, not only because the quantity of natural LDL extracted from human serum is low but also because it is difficult to isolate the ApoB, which is large and aggregates easily during purification.33,34 Alternatively, LDL-mimicking nanocarriers without protein, including lipid nanoparticles and Emuls, have emerged as promising candidates for the targeted delivery of antineoplastic drugs.17,21,33,35,36 In our previous study, a novel lipid Emul resembling the structure of LDL, which is composed of a PTX-CH complex surrounded by a phospholipid monolayer, was prepared and the in vitro and in vivo antitumor efficacy evaluated in a TNBC model.26,27 The resulting lipid PTX-CH Emul exhibited superior in vitro antitumor efficacy, higher specificity and efficiency in intratumoral accumulation, better safety profiles, and superior antitumor efficacy in vivo against TNBC compared to the conventional PTX Emul and Taxol.27 In this work, the cellular uptake mechanism and intracellular trafficking of PTX-CH Emul were studied to elucidate its tumor-targeting mechanism in TNBC cells. Additionally, a comparative evaluation of antineoplastic effects of PTX-CH Emul on TNBC and non-TNBC cells was performed to investigate further the therapeutic potential of PTX-CH Emul for the treatment of TNBC.
Evidence demonstrates that BC cells have high LDL requirements and express relatively high levels of LDLR on cancer cell membranes, and TNBC cell lines share these characteristics.12,14–16 Moreover, LDLR is expressed at low levels in normal cells, such as cardiac myocytes and breast epithelial cells, suggesting that LDLR could be a potential receptor target for selective delivery of antitumor agents to BC cells. The expression profile of LDLR on H9c2 (2-1), MCF7, and MDA-MB-231 cells was measured by enzyme-linked immunosorbent assay (Figure 2). The results indicated the LDLR-expression level of BC cells was much higher than that of H9c2 (2-1) cardiac myocytes, and LDLR was expressed much more highly in MDA-MB-231, a typical TNBC cell line, in comparison with MCF7, an ER-positive BC cell line, which is in line with the results of previous reports.12,14,37 Apart from H9c2 (2-1) cardiac myocytes, breast epithelial cells should be considered additional control cells in future study, although the comparative analysis of LDLR-expression level on a breast epithelial cell line (MCF10A) and BC cell lines (MDA-MB-231 or MCF7) has been investigated in previous studies.16,37 Additionally, MDA-MB-231 cells incubated with 10% LPDS showed elevated LDLR expression, in contrast to those incubated with 10% FBS. This result was consistent with a previous study, which demonstrated that lipoprotein depletion significantly upregulated LDLR messenger-RNA expression.12
Based on the different expression profiles of LDLR in MCF7 and MDA-MB-231 cells incubated with 10% FBS and 10% LPDS, the cellular uptake level of PTX-CH Emul was evaluated to determine whether endocytosis of PTX-CH Emul was associated with LDLR. The results indicated that the expression profile of LDLR modulated the cellular uptake of FL-PTX-CH Emul in BC cells (Figure 3A and B) and overexpression of LDLR significantly facilitated the cellular uptake of PTX-CH Emul in MDA-MB-231 cells, but this effect was inhibited by excess LDL (Figure 3C and D). Therefore, we can conclude that endocytosis of PTX-CH Emul was mediated by LDLR in BC cells.
To clarify the connection between LDLR expression and in vitro antitumor efficacy, MCF7 and MDA-MB-231 cells, which have different LDLR-expression profiles, were used to evaluate the cytotoxicity and cell apoptosis-inducing activity of PTX-CH Emul. In comparison with MCF7 cells, PTX-CH Emul exhibited significantly superior antiproliferative and cell apoptosis-inducing activity in MDA-MB-231 cells (Figure 4), which could be attributed to the selective accumulation of PTX-CH Emul in MDA-MB-231 cells expressing high levels of LDLR, as presented earlier (Figure 3A and B). It is equally important to mention that PTX-CH Emul possessed much higher cell apoptosis-inducing activity compared to PTX Emul and Taxol in MDA-MB-231 cells (Figure 4B and D), which was consistent with the results of our previous in vitro cytotoxicity study,27 further supporting the improved cellular uptake of PTX-CH Emul mediated by LDLR in MDA-MB-231 cells. Therefore, PTX-CH Emul reported here may serve as a tumor-targeting vehicle for PTX directed against MDA-MB-231 cells with abundant LDLR and then greatly improve the in vitro antitumor efficacy of PTX.
To achieve an effective chemotherapeutic response, nanocarriers must penetrate and accumulate drug within the tumor.38 Compared with the conventional 2-D monolayer model, the 3-D tumor-spheroid model has a closer resemblance to cells growing in the in vivo tissue microenvironment by mimicking many of the physiological characteristics of the native tumor environment.38,39 The results of tumor-spheroid penetration demonstrated that FL-PTX-CH Emul possessed greater penetration efficiency compared to FL-DMSO and FL-PTX Emul in MDA-MB-231-tumor spheroids (Figure 5A). Furthermore, the penetration capacity of FL-PTX-CH Emul was stronger in MDA-MB-231-tumor spheroids than in MCF7-tumor spheroids (Figure 5B), indicating that the enhanced tumor spheroid-penetration efficiency of FL-PTX-CH Emul could have been associated with the elevated expression profile of LDLR in MDA-MB-231 cells. To clarify the correlation between tumor spheroid-penetration efficiency and chemotherapeutic response, tumor-spheroid growth inhibition by PTX-CH Emul was evaluated for 7 days. The results demonstrated that PTX-CH Emul exhibited much more pronounced inhibitory activity in MDA-MB-231-tumor spheroids than in MCF7-tumor spheroids (Figure 5C and D), which is consistent with the results of the in vitro cytotoxicity and apoptosis assays described earlier. The improved growth inhibition of PTX-CH Emul in MDA-MB-231-tumor spheroids may be attributed to the higher 3-D tumor spheroid-penetration efficiency and superior 2-D monolayer cell cytotoxicity.
Our previous results demonstrated that PTX-CH Emul preferentially accumulates in tumors in mice bearing MDA-MB-231 xenografts after intravenous administration compared with PTX Emul, possibly as a result of both enhanced permeability and retention effects and tumor-targeting effects mediated by LDLR.27 The tumor-targeting effect and the real-time biodistribution of PTX-CH Emul were also investigated in mice bearing MCF7 xenografts by noninvasive near-infrared fluorescence optical imaging technology. The results revealed that both the tumor sites in live animals and the excised tumors showed higher fluorescence intensity from MDA-MB-231 tumor-bearing nude mice over those from MCF7 tumor-bearing nude mice (Figure 6). PTX-CH Emul exhibited higher distribution selectively in the tumor regions of MDA-MB-231 tumor-bearing nude mice, which may be attributed to the more abundant expression of LDLR in the MDA-MB-231 cells than in the MCF7 cells.
To internalize a lipoprotein, like LDL, the receptor must undergo endocytosis within a plasma-membrane microdomain.40 Normally, LDL binds specifically to LDLR with high affinity at the cell membrane, the LDL–LDLR complexes cluster into clathrin-coated pits, and are then internalized by clathrin-mediated endocytosis. After being internalized into cytoplasm, LDL–LDLR complexes are transferred into early endosomes, where low pH triggers dissociation of LDL from LDLR. The dissociated LDL fuses with lysosomes, and is eventually degraded to CH, lipids, and free amino acids by acidic hydrolases in the endosomes.19,40,41 In order to investigate whether PTX-CH Emul could mimic the internalization pathway and the fate of LDL, the LDLR-binding assay, endocytosis pathway, and intracellular trafficking of PTX-CH Emul were studied. As shown in the results of the binding assay, FL-PTX-CH had a higher binding affinity to LDLR than FL-DMSO and FL-PTX Emul in MDA-MB-231 cells. In accordance with the cellular uptake results described earlier, overexpression of LDLR significantly facilitated the binding capacity of FL-PTX-CH Emul in MDA-MB-231 cells when compared with MCF7 cells (Figure S3), which is in line with the results of a previous report.14 The results of the endocytosis-inhibition assay indicated that the clathrin-mediated endocytosis was the main endocytosis pathway of PTX-CH Emul (Figure 7A). During the whole endocytosis process, both PTX and CH components of PTX-CH Emul were taken up together by MDA-MB-231 cells (Figure 7B and C). After being internalized into cytoplasm, PTX-CH Emul was first localized in lysosomes and then gradually released into the cytoplasm under acidic conditions in lysosomes (Figure 8). Taken together, from these results we can conclude that PTX-CH Emul may be internalized into BC cells through the LDLR-mediated internalization pathway via clathrin-coated pits, localized in lysosomes, and then released into the cytoplasm, which is consistent with the internalization pathway and intracellular trafficking of native LDL.
Conclusion
Although many studies have developed LDL-mimicking nanocarriers to deliver antineoplastic agents for treatment of diverse types of cancer, few have explored these nanocarriers for targeted therapy of BC. PTX-CH Emul, which was designed to mimic the structure of native LDL, exhibited superior antitumor efficacy against TNBC compared to PTX Emul and Taxol in our previous work. Therefore, it is necessary to elucidate the cellular uptake mechanism and intracellular trafficking of PTX-CH Emul.
PTX-CH Emul dramatically enhanced intracellular delivery of PTX into LDLR-overexpressing TNBC cells via LDLR-mediated endocytosis, resulting in enhanced in vitro and in vivo antitumor efficacy on TNBC models, as previously evaluated. Furthermore, PTX-CH Emul exhibited superior in vitro antitumor efficacy, greater tumor spheroid-penetration efficiency, and higher tumor-region distribution in the TNBC cell line in comparison with the non-TNBC cell line, which may be attributed to the more abundant expression profile of LDLR in the TNBC cell line. The findings of the present study not only provide new strategies to design LDL-mimicking nanocarriers but also provide strong proof for the targeted therapeutic efficacy of PTX-CH Emul in clinical applications involving TNBC therapy.
Acknowledgments
This work was financially supported by the National Mega-project for Innovative Drugs (2012ZX09301002-001) and Beijing Municipal Science and Technology Commission Preclinical Research Projects (500101009). The authors thank Liang-Nian Song for his assistance in language polishing.
Disclosure
The authors report no conflicts of interest in this work.
References
Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin. 2016;66(1):7–30. | ||
DeSantis CE, Fedewa SA, Sauer AG, Kramer JL, Smith RA, Jemal A. Breast cancer statistics, 2015: convergence of incidence rates between black and white women. CA Cancer J Clin. 2016;66(1):31–42. | ||
Foulkes WD, Smith IE, Reis-Filho JS. Triple-negative breast cancer. N Engl J Med. 2010;363(20):1938–1948. | ||
Bauer KR, Brown M, Cress RD, Parise CA, Caggiano V. Descriptive analysis of estrogen receptor (ER)-negative, progesterone receptor (PR)-negative, and HER2-negative invasive breast cancer, the so-called triple-negative phenotype: a population-based study from the California Cancer Registry. Cancer. 2007;109(9):1721–1728. | ||
Liedtke C, Mazouni C, Hess KR, et al. Response to neoadjuvant therapy and long-term survival in patients with triple-negative breast cancer. J Clin Oncol. 2008;26(8):1275–1281. | ||
De Laurentiis M, Cianniello D, Caputo R, et al. Treatment of triple negative breast cancer (TNBC): current options and future perspectives. Cancer Treat Rev. 2010;36 Suppl 3:S80–S86. | ||
Carey LA, Dees EC, Sawyer L, et al. The triple negative paradox: primary tumor chemosensitivity of breast cancer subtypes. Clin Cancer Res. 2007;13(8):2329–2334. | ||
Isakoff SJ. Triple negative breast cancer: role of specific chemotherapy agents. Cancer J. 2010;16(1):53–61. | ||
Kassam F, Enright K, Dent R, et al. Survival outcomes for patients with metastatic triple-negative breast cancer: implications for clinical practice and trial design. Clin Breast Cancer. 2009;9(1):29–33. | ||
Mayer IA, Abramson VG, Lehmann BD, Pietenpol JA. New strategies for triple-negative breast cancer: deciphering the heterogeneity. Clin Cancer Res. 2014;20(4):782–790. | ||
Nikanjam M, Blakely EA, Bjornstad KA, Shu X, Budinger TF, Forte TM. Synthetic nano-low density lipoprotein as targeted drug delivery vehicle for glioblastoma multiforme. Int J Pharm. 2007;328(1):86–94. | ||
Stranzl A, Schmidt H, Winkler R, Kostner GM. Low-density lipoprotein receptor mRNA in human breast cancer cells: influence by PKC modulators. Breast Cancer Res Treat. 1997;42(3):195–205. | ||
Zhang B, Sun X, Mei H, et al. LDLR-mediated peptide-22-conjugated nanoparticles for dual-targeting therapy of brain glioma. Biomaterials. 2013;34(36):9171–9182. | ||
Rotheneder M, Kostner GM. Effects of low- and high-density lipoproteins on the proliferation of human breast cancer cells in vitro: differences between hormone-dependent and hormone-independent cell lines. Int J Cancer. 1989;43(5):875–879. | ||
Antalis CJ, Arnold T, Rasool T, Lee B, Buhman KK, Siddiqui RA. High ACAT1 expression in estrogen receptor negative basal-like breast cancer cells is associated with LDL-induced proliferation. Breast Cancer Res Treat. 2010;122(3):661–670. | ||
Antalis CJ, Uchida A, Buhman KK, Siddiqui RA. Migration of MDA-MB-231 breast cancer cells depends on the availability of exogenous lipids and cholesterol esterification. Clin Exp Metastasis. 2011;28(8):733–741. | ||
Kim JH, Kim Y, Bae KH, Park TG, Lee JH, Park K. Tumor-targeted delivery of paclitaxel using low density lipoprotein-mimetic solid lipid nanoparticles. Mol Pharm. 2015;12(4):1230–1241. | ||
Reynolds L, Mulik RS, Wen X, Dilip A, Corbin IR. Low-density lipoprotein-mediated delivery of docosahexaenoic acid selectively kills murine liver cancer cells. Nanomedicine (Lond). 2014;9(14):2123–2141. | ||
Jain A, Jain K, Kesharwani P, Jain NK. Low density lipoproteins mediated nanoplatforms for cancer targeting. J Nanopart Res. 2013;15(9):1888. | ||
Lee JY, Kim JH, Bae KH, et al. Low-density lipoprotein-mimicking nanoparticles for tumor-targeted theranostic applications. Small. 2015;11(2):222–231. | ||
Rodrigues DG, Covolan CC, Coradi ST, Barboza R, Maranhao RC. Use of a cholesterol-rich emulsion that binds to low-density lipoprotein receptors as a vehicle for paclitaxel. J Pharm Pharmacol. 2002;54(6):765–772. | ||
Rodrigues DG, Maria DA, Fernandes DC, et al. Improvement of paclitaxel therapeutic index by derivatization and association to a cholesterol-rich microemulsion: in vitro and in vivo studies. Cancer Chemother Pharmacol. 2005;55(6):565–576. | ||
Pires LA, Hegg R, Valduga CJ, Graziani SR, Rodrigues DG, Maranhão RC. Use of cholesterol-rich nanoparticles that bind to lipoprotein receptors as a vehicle to paclitaxel in the treatment of breast cancer: pharmacokinetics, tumor uptake and a pilot clinical study. Cancer Chemother Pharmacol. 2009;63(2):281–287. | ||
Teixeira RS, Valduga CJ, Benvenutti LA, Schreier S, Maranhão RC. Delivery of daunorubicin to cancer cells with decreased toxicity by association with a lipidic nanoemulsion that binds to LDL receptors. J Pharm Pharmacol. 2008;60(10):1287–1295. | ||
Kretzer IF, Maria DA, Guido MC, Contente TC, Maranhao RC. Simvastatin increases the antineoplastic actions of paclitaxel carried in lipid nanoemulsions in melanoma-bearing mice. Int J Nanomedicine. 2016;11:885–904. | ||
Xia XJ, Guo RF, Liu YL, et al. Formulation, characterization and hypersensitivity evaluation of an intravenous emulsion loaded with a paclitaxel-cholesterol complex. Chem Pharm Bull (Tokyo). 2011;59(3):321–326. | ||
Ye J, Liu Y, Xia X, et al. Improved safety and efficacy of a lipid emulsion loaded with a paclitaxel-cholesterol complex for the treatment of breast tumors. Oncol Rep. 2016;36(1):399–409. | ||
Nagelkerke A, Bussink J, Sweep FC, Span PN. Generation of multicellular tumor spheroids of breast cancer cells: how to go three-dimensional. Anal Biochem. 2013;437(1):17–19. | ||
Tahara K, Sakai T, Yamamoto H, Takeuchi H, Hirashima N, Kawashima Y. Improved cellular uptake of chitosan-modified PLGA nanospheres by A549 cells. Int J Pharm. 2009;382(1–2):198–204. | ||
Chiu YL, Ho YC, Chen YM, et al. The characteristics, cellular uptake and intracellular trafficking of nanoparticles made of hydrophobically-modified chitosan. J Control Release. 2010;146(1):152–159. | ||
Zhu QL, Zhou Y, Guan M, et al. Low-density lipoprotein-coupled N-succinyl chitosan nanoparticles co-delivering siRNA and doxorubicin for hepatocyte-targeted therapy. Biomaterials. 2014;35(22):5965–5976. | ||
Huang H, Cruz W, Chen J, Zheng G. Learning from biology: synthetic lipoproteins for drug delivery. Wiley Interdiscip Rev Nanomed Nanobiotechnol. 2015;7(3):298–314. | ||
Meng F, Asghar S, Xu Y, et al. Design and evaluation of lipoprotein resembling curcumin-encapsulated protein-free nanostructured lipid carrier for brain targeting. Int J Pharm. 2016;506(1–2):46–56. | ||
Nikanjam M, Gibbs AR, Hunt CA, Budinger TF, Forte TM. Synthetic nano-LDL with paclitaxel oleate as a targeted drug delivery vehicle for glioblastoma multiforme. J Control Release. 2007;124(3):163–171. | ||
Emami J, Rezazadeh M, Varshosaz J. Formulation of LDL targeted nanostructured lipid carriers loaded with paclitaxel: a detailed study of preparation, freeze drying condition, and in vitro cytotoxicity. J Nanomater. 2012;2012:358782. | ||
Kretzer IF, Maria DA, Maranhão RC. Drug-targeting in combined cancer chemotherapy: tumor growth inhibition in mice by association of paclitaxel and etoposide with a cholesterol-rich nanoemulsion. Cell Oncol (Dordr). 2012;35(6):451–460. | ||
Zelenko Z, LeRoith D, Antoniou IM, Rajan L, Gallagher EJ. Silencing the LDLR in breast cancer cells leads to decreased growth of tumor xenografts in a mouse model of hyperlipidemia. Poster presented at: 2016 Endocrine Society Annual Meeting; April 1–4, 2016; Boston, MA. | ||
Lei H, Hofferberth SC, Liu R, et al. Paclitaxel-loaded expansile nanoparticles enhance chemotherapeutic drug delivery in mesothelioma 3-dimensional multicellular spheroids. J Thorac Cardiovasc Surg. 2015;149(5):1417–1425. | ||
Xin H, Sha X, Jiang X, Zhang W, Chen L, Fang X. Anti-glioblastoma efficacy and safety of paclitaxel-loading Angiopep-conjugated dual targeting PEG-PCL nanoparticles. Biomaterials. 2012;33(32):8167–8176. | ||
Ivaturi S, Wooten CJ, Nguyen MD, Ness GC, Lopez D. Distribution of the LDL receptor within clathrin-coated pits and caveolae in rat and human liver. Biochem Biophys Res Commun. 2014;445(2):422–427. | ||
Xu S, Olenyuk BZ, Okamoto CT, Hamm-Alvarez SF. Targeting receptor-mediated endocytotic pathways with nanoparticles: rationale and advances. Adv Drug Deliv Rev. 2013;65(1):121–138. |
Supplementary materials
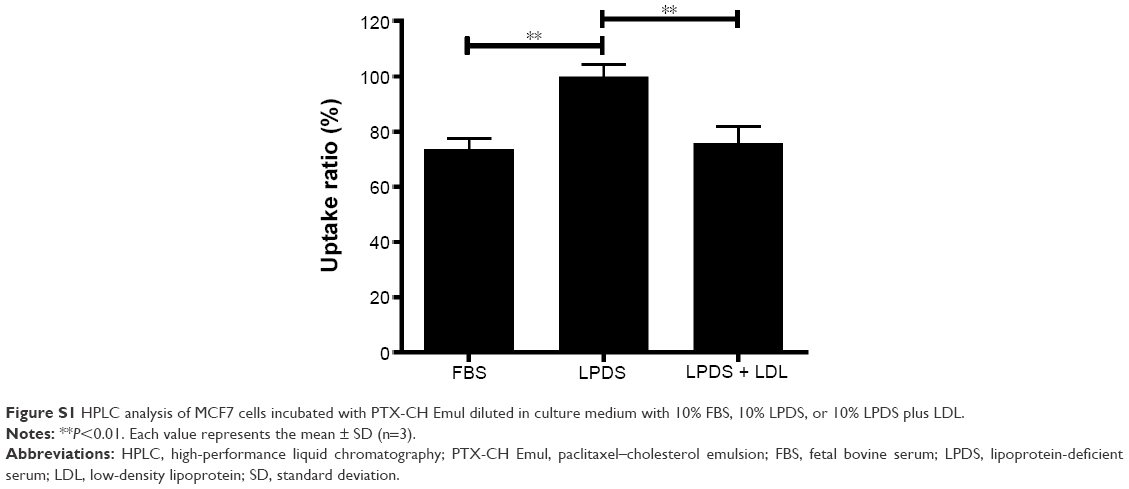 | Figure S1 HPLC analysis of MCF7 cells incubated with PTX-CH Emul diluted in culture medium with 10% FBS, 10% LPDS, or 10% LPDS plus LDL. |
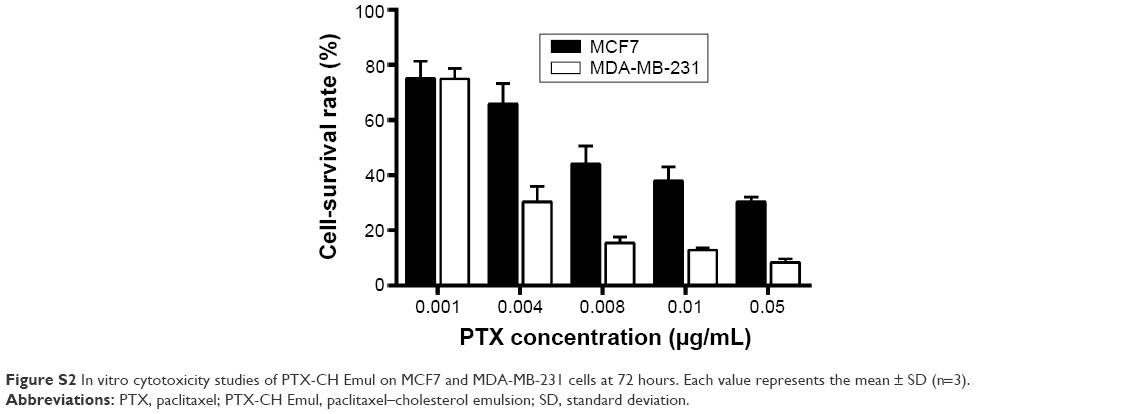 | Figure S2 In vitro cytotoxicity studies of PTX-CH Emul on MCF7 and MDA-MB-231 cells at 72 hours. Each value represents the mean ± SD (n=3). |
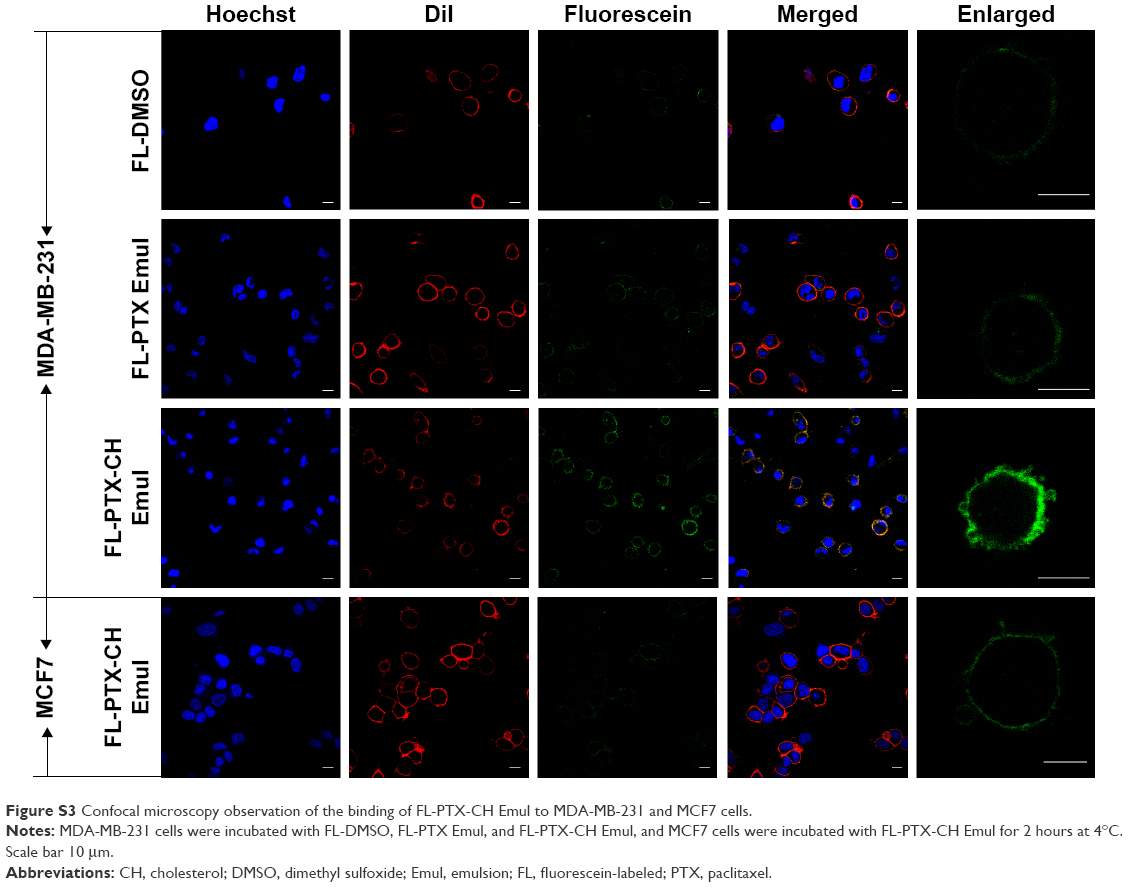 | Figure S3 Confocal microscopy observation of the binding of FL-PTX-CH Emul to MDA-MB-231 and MCF7 cells. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.