Back to Journals » Clinical Interventions in Aging » Volume 10
Cell-based therapies of liver diseases: age-related challenges
Authors Yarygin K ![]() , Lupatov A, Kholodenko I
, Lupatov A, Kholodenko I
Received 9 October 2015
Accepted for publication 20 October 2015
Published 1 December 2015 Volume 2015:10 Pages 1909—1924
DOI https://doi.org/10.2147/CIA.S97926
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Richard Walker
Konstantin N Yarygin, Alexei Y Lupatov, Irina V Kholodenko
Laboratory of Cell Biology, Institute of Biomedical Chemistry, Moscow, Russia
Abstract: The scope of this review is to revise recent advances of the cell-based therapies of liver diseases with an emphasis on cell donor’s and patient’s age. Regenerative medicine with cell-based technologies as its integral part is focused on the structural and functional restoration of tissues impaired by sickness or aging. Unlike drug-based medicine directed primarily at alleviation of symptoms, regenerative medicine offers a more holistic approach to disease and senescence management aimed to achieve restoration of homeostasis. Hepatocyte transplantation and organ engineering are very probable forthcoming options of liver disease treatment in people of different ages and vigorous research and technological innovations in this area are in progress. Accordingly, availability of sufficient amounts of functional human hepatocytes is crucial. Direct isolation of autologous hepatocytes from liver biopsy is problematic due to related discomfort and difficulties with further expansion of cells, particularly those derived from aging people. Allogeneic primary human hepatocytes meeting quality standards are also in short supply. Alternatively, autologous hepatocytes can be produced by reprogramming of differentiated cells through the stage of induced pluripotent stem cells. In addition, fibroblasts and mesenchymal stromal cells can be directly induced to undergo advanced stage hepatogenic differentiation. Reprogramming of cells derived from elderly people is accompanied by the reversal of age-associated changes at the cellular level manifesting itself by telomere elongation and the U-turn of DNA methylation. Cell reprogramming can provide high quality rejuvenated hepatocytes for cell therapy and liver tissue engineering. Further technological advancements and establishment of national and global registries of induced pluripotent stem cell lines homozygous for HLA haplotypes can allow industry-style production of livers for immunosuppression-free transplantation.
Keywords: cell aging, rejuvenation, liver engineering, liver cell therapy
Introduction
No liver pathology occurs specifically at advanced age, but disease progression, its incidence, and the patient’s reaction to drugs and medical manipulations may differ in younger and older people. It should be recognized though, that chronologic age is an arbitrary characteristic of a person. Much of human aging research concerns older individuals. However, senescence does not necessarily start at an advanced age. Recently, it has been shown that differences in the pace of aging can be readily detected in people in their late 30s.1 This study quantitatively evaluated physiological deterioration across multiple organ systems – cardiovascular, renal, hepatic, immune, and others – in 38-year-old people and found that already at this stage a substantial proportion of young individuals were aging more rapidly than their peers and were showing lower physical fitness, cognitive decline, regression of renal, heart and liver function, and self-reported ill health. Accordingly, not just chronological, but biological age clearly is a very important factor to be considered when new methods of treatment of the people of any age are developed. Regenerative medicine based primarily on stem cell biology research is a new approach to disease treatment largely focused on the correction of age- and pathology-related malfunction of organs and systems by the enhancement of tissue regeneration and substitution of dysfunctional and senescent cells, tissues, and organs.
Liver pathology is a serious health problem worldwide because of high morbidity and mortality in end-stage liver disease associated primarily though not solely with growing cirrhosis and cancer incidence in at least half-a-billion patients with persistent hepatitis B and C infection and in tens of millions of alcoholics.2–4 Allogeneic liver transplantation can save lives and improve health, but only in 20%–30% of patients due to donor shortage, medical contraindications, and social and economic reasons.5,6 In general, present day therapeutic techniques do not consistently ensure patient’s recovery or even the upholding of the status quo. Cell-based technologies of regenerative medicine using autologous or allogeneic hepatocytes or cells capable of hepatogenic differentiation offer a novel approach to better management and diagnostics of liver disease. Indeed, cell transplantation is by far less invasive and expensive compared to transplantation of whole liver or its part, while organ engineering has the potential to solve the problem of liver donor shortage. In addition, human hepatocytes are used in extracorporeal liver support systems and their long-term cultures are gradually substituting animal experiments in drug testing and in vitro disease modeling. Accordingly, availability of sufficient amounts of functional human hepatocytes becomes imperative.
Aging presents additional and not yet fully understood challenges in both hepatocyte manufacture and clinical or experimental applications. The response of the recipient body to cell therapy or a tissue engineering construct transplantation depends on his or her age. Biological aging is a universal phenomenon occurring at different levels of organization including cellular level. During the lifetime of multicellular organisms, cells undergo changes and develop signs of senescence. They also find themselves in a distorted microenvironment.
Mechanisms of cellular senescence have been only partly disclosed and include free radical damage to the inner machinery of the cell, loss of telomerase activity resulting in the shortening of telomeres and cell cycle arrest, accumulation of DNA modifications, alterations of mitochondria functions, and others.7–9 The above-listed mechanisms are of course inter-related. For example, oxidative free radical stress causes nuclear DNA and mitochondria damage. In principle, age-related changes can induce altered reactions of cultured cells taken from older donors to differentiation and dedifferentiation stimuli.10 However, some data accumulated so far suggest that is not always the case. Neither replicative aging in vitro nor cell donor’s advanced age prevents adult differentiated cells, for example, skin fibroblasts from dedifferentiation into induced pluripotent stem cells (iPSCs), while redifferentiation of iPSCs can yield fibroblasts with the characteristics of juvenile proliferating cells, not initial senescent fibroblasts, thus demonstrating the reversibility of aging at the cellular level.11,12 Therefore, cell therapy and tissue engineering followed by tissue and organ transplantation may offer not only relief from liver and other diseases but also a method of rejuvenation, at least at the tissue and organ level. Cell transplantation (cell therapy) and tissue and organ engineering present a novel and yet not fully explored and tested approach to disease treatment. However, it seems to have bright prospects because it is based on high quality and well subsidized fundamental and applied research in cell biology, molecular biology, biology of aging, and related fields.
This review is aimed to expose age-related aspects of human hepatocytes preparation by differentiation of iPSCs obtained from cells taken from adult donors or by transdifferentiation of mesenchymal stem cells (MSCs) and fibroblasts. It also depicts some of the prospective problems associated with the application of cell-based therapies to the treatment of patients with a special focus on age-related issues.
Generation of iPSCs from differentiated cells taken from donors of various ages
Reprogramming of somatic cells into iPSCs presents a unique opportunity to obtain autologous pluripotent cells for cell therapy, tissue engineering, drug testing, and disease modeling. Originally, human iPSCs have been generated from skin fibroblasts of an adult person by transfecting them with a set of four genes highly expressed in early embryogenesis – Oct4, Sox2, Klf4 and c-Myc – often called Yamanaka factors and abbreviated as OSKM.13 Earlier, the same battery of genes had been used to induce pluripotency in mouse embryonic and adult fibroblasts14 suggesting fundamental similarities of the mechanisms of pluripotency induction across the species. Significantly, the induction of pluripotency occurs in a stochastic manner and initially the percentage of reprogrammed cells was quite low.
Studies examining the impact of somatic cell donor age upon the efficiency of reprogramming to pluripotency in mice demonstrated lower reprogramming frequency in cells derived from older animals.15–17 Bone marrow cells from 23-month-old mice transfected with Yamanaka factors generated five times less colonies positive for the stem cell marker alkaline phosphatase compared to cells isolated from 2-month-old animals. Moreover, in older mice, reprogramming took twice as long than in younger ones.
Unlike data from mouse experiments, studies with human cells produced conflicting results and did not show clear impact of cell donor’s age on reprogramming efficacy. Remarkably, iPSCs could be obtained from the fibroblasts of ≥100 year-old people.18 These “centenarian” iPSCs expressed pluripotency markers and were actually pluripotent being able to differentiate into the derivatives of the three germ layers – ectoderm, endoderm, and mesoderm. Using four Yamanaka factors, Somers et al19 derived 100 cell lines from fibroblasts donated by people aged 8–64 years. Reprogramming efficacy was 0.1%–1.5% and did not correlate with donor’s age. All the resultant cells expressed pluripotency markers and robustly differentiated along the endoderm lineage route. In contrast, Sharma et al,20 also using OSKM array, found that skin fibroblasts obtained from donors aged 50–85 years reprogrammed with substantially lower efficacy than cells derived from younger 0–18-year-old donors, but not from 20–49-year-old donors. The nature of these inconsistencies is not fully understood. They may be associated with differences in many parameters characterizing the reprogrammed cells including their proliferative potential or culture conditions.
Since the pioneering works of Takahashi et al,13,14 where OSKM cassette was delivered to fibroblasts by retroviral vectors, other combinations of reprogramming factors and different gene delivery vehicles supplemented by specific iRNAs, proteins and biologically active small molecules have been successfully tested to convert somatic cells to pluripotent state.21 All these approaches provide iPSCs and many of them improve the yield of pluripotent cells. On the other hand, iPSCs produced by different methods may be not quite identical hindering the evaluation of the impact of different parameters including age.
Lapasset et al used a cocktail of six factors (traditional OSKM cassette plus Nanog and Lin28) to effectively reprogram aging cells obtained by prolonged in vitro passaging and showing all signs of replicative senescence, as well as cells derived from people of very advanced (92–101 years) age.11 All the resulting iPSC clones were positive for pluripotency markers Tra1-60 and SSEA-4 and were able to differentiate into the ectoderm, endoderm, and mesoderm derivatives. In addition, they were characterized by low expression levels of inhibitors of cyclin-dependent kinases p16INK4A and p21CIP1 blocking cell cycle progression from phase G1 to phase S, elongated telomeres which did not shorten during passaging, and normal mitochondrial metabolism similar to that of the embryonic stem cells. The authors of the paper argue that neither replicative aging in vitro nor cell donor’s advanced age prevent cells from dedifferentiation into iPSCs. Redifferentiation of iPSCs yielded fibroblasts with the characteristics of juvenile proliferating cells, once more demonstrating the reversibility of aging at the cellular level. These data have been confirmed using a more extended panel of age-dependent cell characteristics including telomere length, mitochondrial function, heterochromatin loss, state of the nuclear lamina, DNA repair machinery, and percentage of aging cells in the population to evaluate differences in the reprogramming of fibroblasts obtained from young and old donors.12 Age-related changes of each of the described parameters observed in fibroblasts taken from older people were eliminated after pluripotency induction and redifferentiation.
The reprogramming efficacy and pace depend upon the levels of stem cell-related genes expression in the initial somatic cells. For example, reprogramming of keratinocytes goes two times faster and 100 times more effectively compared to skin fibroblasts.22 Unlike fibroblasts, keratinocytes have stem cell-related genes expression pattern similar to embryonic stem cells. Two independent studies demonstrated that mouse neural stem/progenitor cells expressing high levels of endogenous Sox2 and c-Myc can be effectively reprogrammed by just two (Oct4 + Klf4 or Oct4 + c-Myc)23 or three (Oct4, Klf4, c-Myc)24 factors. These data suggest higher reprogramming efficacy of stem/progenitor cells compared to adult fully differentiated cells.25
This hypothesis is supported by significant experimental evidence, including data on the reprogramming of B lymphocytes.26,27 Forced expression of Yamanaka factors proved to be insufficient to reprogram B cells to iPSCs, even in case of very efficient exogene transfer when all B cells overexpress OSKM. Pluripotency induction occurred only after overexpression of the OSKM cassette was supplemented with overexpression of the CEBPα transcription factor or knockdown of its suppressor Pax5. Unlike mature cells, B lymphocyte progenitors were easily reprogrammed just by introduction of classical Yamanaka battery.
Abramovich et al were the first to suggest as early as in 2008 that reprogramming of adult somatic cells to iPSCs is accompanied by the reversal of the indications of cell senescence resulting in “rejuvenation” at the cellular level.28 Later, this point of view concerning human cells became predominant.11,12,29,30 It was further supported by demonstrating telomere elongation,31 mitochondria rejuvenation up to the state characteristic of the embryonic stem cells,32 and enhanced DNA reparation capacity33,34 in iPSCs, though in different iPSC lines these changes may be pronounced to a varying extent. In the course of reprogramming to iPSCs, somatic cells undergo profound modification allowing them to revert to the state of “stemness”. Fully reprogrammed iPSCs are similar (though not identical) to embryonic stem cells regarding their gene expression profile35,36 with elevated activity of genes responsible for pluripotence and cell renewal (Oct4, Nanog, Sox2, Lin28, Zic3, Fgf4, Tdgf1, and Rex1) and low activity of genes related to tissue specialization. Their DNA methylation profile also reverts to embryonic state. However, iPSCs may retain remnants of the pattern of DNA methylation characteristic of differentiated cells from which they were derived, the so-called “epigenetic memory”, making them and embryonic stem cells somewhat different.37 The impact of the epigenetic memory on the process of somatic cell reprogramming to iPSCs, stability of iPSCs characteristics during passaging, and the results of redifferentiation have been repeatedly discussed in the literature.38–40 Possibly, epigenetic memory is retained merely at early passages, while full reprogramming including its loss may be a continuing process.
Hence, in humans, cell donor’s age is not crucial for the reprogramming of somatic cells to iPSCs and redifferentiation of the latter. These processes are to a greater extent influenced by other factors including tissue origin of donor cells, extent of their differentiation, presence of hereditary and somatic mutations, nongenetic donor pathologies, and number of iPSCs passages before redifferentiation.
Hepatogenic differentiation of iPSC
Adult human liver tissue contains multiple cell types among which hepatocytes are the prevailing species comprising 70%–80% of the total cell number. Hepatocytes and one other type of liver tissue cells, biliary cells, differentiate from bipotent progenitors belonging to the endoderm lineage and are called hepatoblasts.41 Most other liver cells, including endothelium, stellate cells, and resident macrophages usually designated as Kupffer cells are derived from the mesoderm.
Hepatocytes carry out the majority of crucially important liver functions including glucose metabolism, synthesis, storage and degradation of other metabolites, detoxification of poisonous substances and drugs, bile production, and others. For a long time, primary hepatocytes isolated from human liver biopsies or autopsies served as the “golden standard” in drug metabolism research during preclinical drug testing.42 Consequently, they were a very intensely studied cell type and their production was a top priority. As shown in the Cell therapy and tissue engineering in the treatment of liver diseases section of this article, primary hepatocytes were also tested in cell therapy of liver pathology. Unluckily, primary human hepatocytes have very restricted in vitro proliferation capacity making them virtually inaccessible in quantities sufficient for extensive preclinical research and liver tissue reconstruction.43 In addition, in vitro manipulations can deprive primary human hepatocytes from some of their essential properties causing changes as significant as cytochrome P450 inactivation.44 These limitations shaped further efforts to improve the methods of functional human hepatocytes production, iPSCs obviously being one of the most convenient starting materials for the delivery of both autologous and allogeneic human hepatocytes.
The development of the methods of hepatocytes production from iPSCs started soon after the introduction of iPSC technology. Quite logically, the related protocols were based on procedures initially applied to embryonic stem cells and comprised several steps more or less closely reproducing the stages of hepatocyte differentiation throughout the ontogenesis. Each step started with application of a certain set of growth and transcription factors. Song et al were among the first to introduce a four-step differentiation protocol of the hepatogenic differentiation of human iPSCs starting with iPSC differentiation into the definitive endoderm followed by the stages of hepatocytic specification, hepatoblast expansion, and hepatocyte maturation.45 Three-step protocols also including consecutive stimulation of iPSC differentiation into definitive endoderm, immature hepatocytes, and mature hepatocytes induced by three different sets of growth and transcription factors followed soon.46,47
As indicated above, at the first stage of hepatogenic differentiation, embryonic stem cells or iPSCs are converted into the definitive endoderm. Activin A, a member of the activin family of the transforming growth factor beta (TGF-beta) superfamily, is one of the key factors shaping this process. It has been demonstrated that activin A participates in maintaining the “stemness” via SMAD-dependent activation of pluripotency factors such as Oct-4, Nanog, Nodal, and others.48,49 On the other hand, activin A is known to inhibit cell growth and proliferation by stimulating the transcription of the cell cycle inhibitors p15, p21, and p27KIP150,51 and to enhance differentiation through inhibition of c-Myc translation.52 Furthermore, the Nodal/Wnt signal pathway was shown to play a significant role in human embryonic stem cell differentiation along the endodermal route.53 Nodal is an activin analog also belonging to the TGF-beta superfamily. Activin/Nodal signaling regulates the transcription of Smad2/3 factor essential for differentiation to the definitive endoderm.54 It is also capable of activating many other genes in the course of endoderm development during embryogenesis.50
Activin A promotes differentiation into the definitive endoderm in both mouse55 and human56 embryonic stem cells. However, it has been suspected for a long time that the effects of activin A on human embryonic stem cell differentiation strongly depend on its concentration.57 Low (5 ng/mL) activin A concentrations contribute to the maintenance of undifferentiated state of embryonic stem cells58 and iPSCs59 probably due to the induction of Oct-4, Nanog, Nodal, Wnt3, fibroblast growth factor (FGF)-2, and FGF-8 and suppression of bone morphogenic protein (BMP) signaling, while high concentrations (25–100 ng/mL) promote their differentiation into the definitive endoderm in a dose-dependent manner.55,60 Consequently, activin A concentration of about 100 ng/mL is routinely used at the first stage of most newer iPSC hepatogenic differentiation protocols.61,62 In addition to activin A, other factors including Wnt3a combined with hepatocyte growth factor (HGF),61 B27 serum-free supplement initially designed for maintenance of hippocampal and cortical neurons in culture,62 and LY294002, the specific inhibitor of phosphatidylinositol-3-phosphatase,63 are used to enhance the efficacy of differentiation at stage 1.
During the first phase of hepatogenic differentiation, iPSC cultures undergo substantial morphological and molecular transformation.46,61–63 Dissociation of intercellular contacts prompts formation of more loosely packed associations of spiky shaped cells expressing definitive endoderm markers FoxA2, GATA4, and Sox17 instead of compact clusters typical for iPSCs. The efficacy of endodermal differentiation lies within the 60%–80% range as judged by the emergence of cells expressing definitive endoderm markers.45,46
The second stage of the hepatogenic differentiation of human iPSCs embraces the specification of the definitive endoderm and the start of hepatogenic differentiation per se. Stage 2 can be initiated by the addition of a cocktail comprising HGF, FGF-2/-4, and BMP-2/-4.62,64 The first of the cited publications presents data for human, while the second for porcine iPSC differentiation. Earlier, it has been shown that BMP-4 and FGF-2 play a pivotal role in the process of hepatocytic specification in mouse embryos.65,66 HGF is also crucial for normal liver development. HGF knock-out mice failed to acquire normal liver architecture during embryogenesis and suffered from the disintegration of liver parenchyma.67 HGF and its c-MET receptor are essential for cell proliferation, survival, motility, tissue invasion, and morphogenesis.68 Some authors along with the cocktail of growth factors described above induced the second hepatogenic differentiation stage with chemical substances such as dimethyl sulfoxide and/or beta-mercaptoethanol.61,64
During stage 2, cells continued to proliferate and went through further morphological modification acquiring spindle-like or polygonal epithelium-like shape. Expression of the definitive endoderm markers gradually decreased while the expression of hepatoblast markers hepatocyte nuclear factor 4alpha (HNF-4alpha) and alpha-fetoprotein gradually increased.46,61,62 At this point, 80% of cells were positive for HNF-4alpha.46
During the last phase of the hepatogenic differentiation of iPSCs, hepatoblasts are converted to functionally active hepatocyte-like cells. Oncostatin M (OSM), a member of the cytokine IL-6 family, is the key factor used to guide cells through this stage. In fetal liver, OSM is synthesized by the hematopoietic cells. In the middle of the gestation period, OSM together with glucocorticoids, HGF, and Wnt supports the differentiation of hepatocytes.69–71 In vitro OSM induces the maturation of mouse fetal hepatoblasts manifested by the expression of glucose-6-phosphatase and tyrosine aminotransferase and the accumulation of glycogen.69 OSM stimulates the metabolic maturation of hepatocytes through the activation of gp130 receptor and JAK/Stat3 signal pathway.72 It also assists the morphological maturation of hepatocytes through K-ras activation and formation of E-cadherin-based adherence junctions between fetal cells,73 as well as through the expression of claudin-2 which participates in the establishment of tight junctions enhancing the paracellular barrier function and regulating the permeability of the paracellular zones.74 During hepatogenic differentiation of mouse iPSCs, OSM induces hepatocyte maturation by induction of glucocorticoid production.75 The role of glucocorticoids is further stressed by older works reporting that rat hepatocytes can be maintained in functionally active state and switched from the synthesis of alpha-fetoprotein to albumin production by the synthetic glucocorticoid dexamethasone.76,77
In the course of maturation, hepatocytes acquired features characteristic of mature cells: cuboidal form, numerous vacuoles, and vesicles in the cytoplasm, elevated cytoplasm-to-nucleus ratio, and noticeable well-defined nucleolus. Besides, hepatocyte-like cells derived from iPSCs started to express markers of mature hepatocytes, such as albumin, alpha-1-antitripsin, cytokeratin/s including cytokeratine-8, -18, and -19, different enzymes including cytochrome P450 complex and glutathione S-transferase, and transporters P-glycoprotein 3 and multidrug-resistance protein 1. Studies of the functional maturity showed that these cells secrete high quantities of albumin, utilize urea, and accumulate glycogen.46,47,62,64 The main stages of in vitro iPSC hepatic differentiation are summarized in Table 1.
 | Table 1 Hepatogenic differentiation of induced pluripotent stem cells |
To our knowledge, no one has studied the effects of the age of the donor of cells used to produce hepatocytes via iPSCs upon the quality of the ensuing hepatocytes. However, as shown in the Generation of iPSCs from differentiated cells taken from donors of various ages section, in humans, cell donor’s age is not pivotal for redifferentiation of iPSCs to fibroblasts and the quality of the resultant fibroblasts. Hence, age may not be a major factor in hepatocyte production via iPSCs as well, though of course this issue has to be addressed experimentally.
Hepatogenic transdifferentiation of mesenchymal stem cells and skin fibroblasts
Two preceding sections describe a two-step procedure of in vitro hepatocyte derivation via the stage of induced pluripotency. At step one, differentiated cells, for example, fibroblasts are converted to pluripotency and at step two, the ensuing pluripotent cells undergo hepatogenic differentiation. As will be shown in this section, probably there are ways of one-step derivation of hepatocytes from adult tissue cells. Bone marrow and other tissues contain minute quantities of pluripotent stem cells throughout life. Many of those cells are of mesodermal origin. They can be isolated from bone marrow, adipose tissue, skin, and stroma of virtually all internal organs along with multipotent (ie, capable of differentiation to different cell types within mesodermal lineage) mesenchymal cells and differentiated fibroblasts and maintained in MSC cultures where they comprise less than 1% of total cell counts. Tissue pluripotent stem cells can be induced to one-step differentiation into the ectoderm and endoderm derivatives including hepatocytes. It is also possible that some partly or even fully differentiated cells can be induced to transdifferentiate into cells of different lineages without full dedifferentiation to embryonic stem cell-like condition.
Since it has been suggested that blood-borne cells of bone marrow origin may be the source of stem cells for liver regeneration,78–82 different marrow cells have been studied as candidates for this role and proved to be able to undergo at least partial hepatogenic differentiation. These include major cell types, namely, MSC78 and hematopoietic cells,81 as well as rare cell species coisolated with stromal cells comprising the so-called multipotent adult progenitor cells82 and very small embryonic-like stem cells.83 It should be noted that since multipotent adult progenitor cells are able to transdifferentiate across the lineage borders (from mesoderm to endoderm), they of course should be named “pluripotent”, not “multipotent”.
Transdifferentiation of MSCs into ectoderm and endoderm derivatives has been repeatedly demonstrated both in vitro and in vivo. Petersen et al79 experimenting with a rat model of liver injury were the first ones to prove formation of oval cells believed to be hepatic stem/progenitor cells from the bone marrow. Schwartz et al82 showed the ability of bone marrow cells to convert into the functional liver cells. Several research groups demonstrated that nonmarrow MSCs isolated from different tissues also can be induced to acquire hepatocyte-like morphology and express hepatocyte marker genes.84–86
Importantly, only a fraction of cultured MSC undergoes differentiation. This fraction probably includes pluripotent stem cells, preexisting in the culture, such as multipotent adult progenitor cells and/or very small embryonic-like stem cells. The other option is genuine transdifferentiation of progenitor cells committed to differentiation into mesodermal lineage derivatives or even of terminally differentiated fibroblasts, also present in the culture. The capacity of hepatogenic differentiation was revealed in MSCs from such sources as adipose tissue,87–89 Wharton’s jelly,90,91 umbilical cord blood,92,93 tooth pulp,94 and many others.
Though evidence concerning molecular pathways involved in MSC transdifferentiation into hepatocytes has been accumulating for more than a decade, it is still limited. MSC transdifferentiation is a complex process regulated by intracellular and external signals. Exact molecular events involved in the conversion of different cell types constituting heterogenous MSC populations may vary in detail. However, activin A, FGF, BMPs, HGF, and OSM are the main external signals switching and maintaining the conversion of any cell type in MSC cultures.41,95,96
Many protocols of MSC transdifferentiation into functionally active hepatocytes have been suggested. The most effective employ consecutive addition of growth factors and other biologically active molecules to the culture medium to achieve partial in vitro reconstruction of the changes in differentiating cell microenvironment occurring in vivo. The most frequently used protocol includes two stages. At stage 1, hepatogenic differentiation is induced by FGF and HGF, while at stage 2 hepatocyte maturation is promoted by OSM.88,90,97,98 The efficacy of hepatogenic differentiation can be enhanced by the addition of dexamethasone, nicotinamide, or insulin–transferrin–selenium. Differentiation is usually carried out in serum-free conditions,88,99 but some effective protocols utilize low serum media.98 The efficiency of hepatocyte formation is higher if cells are initially at the postmitotic stage of the cell cycle achieved in highly confluent (80%–100%) cultures100 or by inhibition of cell proliferation by the addition of epidermal growth factor (EGF) and FGF-2.98
Hepatogenic differentiation of MSCs can be monitored by a standard set of methods. Differentiation-associated changes include morphological transformation, modification of the pattern of expressed genes, and the onset of functional activities characteristic of hepatocytes. In the course of morphological transformation, MSCs acquire polygonal shape, granulated cytoplasm, and tight intercellular contacts.90,97,101 Emergent hepatocyte-like cells start expressing specific marker proteins such as alpha-fetoprotein, albumin, cytokeratine-18 and -19, HNF-4alpha and HNF-1alpha, and others.88,91,102 Alpha-fetoprotein is expressed at the onset of differentiation, while later its expression goes down. Hepatocyte-like cells also express such enzymes as CYP7A1, CYP1A1, CYP2C9, CYP3A4, and NADPH-cytochrome P450 reductase involved in the metabolism of drugs and xenobiotics and synthesis of stearic acid and bile acids.97 It should be noted that in some cases, hepatocyte marker protein expression may be not a very reliable sign of hepatogenic differentiation of MSC. As shown by Campard et al,90 MSCs isolated from the umbilical cord and maintained in standard culture conditions constitutively express hepatocyte markers albumin, alpha-fetoprotein, connexin 32, and cytokeratine-8, -18, and -19.
Microarray analysis of gene expression profile of hepatocytes derived by transdifferentiation of adipose tissue MSCs demonstrated its similarity to the expression profile of adult human hepatocytes.103 Comparison of full-genome expression profiles of adipose tissue-derived MSCs cultivated for 4 weeks in pro-hepatogenic conditions and naïve cells showed activation of genes associated with liver-specific functions including protein metabolism, regulation of the innate immune response, and toxin biodegradation.104 Moreover, mesenchymal line-specific genes were downregulated and epithelium-specific genes upregulated confirming transformation of mesenchymal cells to epithelial state typical for most cells of internal organ parenchyma including hepatocytes.
Functional activity analysis of hepatocyte-like cells derived from MSCs demonstrated secretion of albumin and urea, accumulation of intracellular glycogen and low density lipoprotein uptake.92–94
MSCs from different tissues may be a very convenient starting material for autologous hepatocyte production because of relative simplicity and low cost of the isolation, expansion, and differentiation procedures. Animal experiments have demonstrated that autologous, allogenic, and even xenogenic hepatocyte-like cells from MSCs are able to integrate into adult liver parenchyma.90,105–107
MSCs reside in the stromal and vascular portion of all organs and tissues and can be easily isolated and maintained in vitro due to their plastic adherence and ability to proliferate in conventional culture media.108–111 Remarkably, fibroblasts, though supposed to be fully differentiated cells, can be isolated and cultured in exactly the same conditions. Analysis of morphology, expression of surface markers, and differentiation potential of human MSCs isolated from different sources and “fibroblasts” from skin and liver stroma demonstrated coincidence of many parameters of MSCs and fibroblast cultures.109,112–115 All studied cultures of MSCs and fibroblasts were heterogeneous and, importantly, contained cell subpopulations of varying size differing by the ability to differentiate within (into mesoderm derivatives) and across (into ectoderm and endoderm derivatives) the borders of their primary germ layer. Not surprisingly, some cells showed signs of mesenchymal–epithelial transition revealing the presence of pluripotent cells.115 In vitro cultures of “MSCs” and “fibroblasts” seem to contain the same cell types, but in different proportions: MSC cultures comprise more stem/progenitor cells and fibroblast cultures are stuffed primarily with differentiated cells. Apparently, fibroblast cultures should be similar to MSC cultures as a feasible source of cells for hepatogenic transdifferentiation.
Despite the diversity of tissues from which MSCs and fibroblast cultures for hepatocyte production can be obtained, skin is probably the most suitable source just because of its accessibility. Cultured skin fibroblast-like cells contain subpopulations capable of differentiation within and out of the boundaries of the mesodermal lineage: into adipocytes, osteoblasts, chondrocytes, smooth muscle cells, neurons, astrocytes, and insulin-producing cells.109,112,116–120 Cultures of skin plastic adherent cells contain cells originating from different structures forming this complex organ including dermis, adipose tissue, hair follicles, sweat and sebaceous glands, and others. It should be noted that fibroblast-like cells derived from skin specimens taken from the skull are partly of ectoderm origin because during embryogenesis in this area dermis is formed from the neural crest. The wide assortment of cell types present in the cultures of skin plastic adherent cells ensures the broad spectrum of their possible differentiation routes. Hepatogenic differentiation of some of those cell species has been actually demonstrated.
For instance, Huang et al121 isolated the so-called foreskin-derived fibroblast-like stromal cells (FDSCs) from human foreskin. FDSCs were able to differentiate into adipocytes, osteoblasts, smooth muscle cells, and Schwann cells. They formed spheroids if maintained in Dulbecco’s Modified Eagle’s Medium (DMEM)-F12 with the addition of EGF and FGF-2, while in the absence of growth factors they were growing as a plastic-adherent culture displaying fibroblast-like morphology. Cells from both suspension and plastic-adherent cultures expressed similar spectra of MSC markers being CD90-, CD105-, CD29-, CD44-, SH3-, SH4-, and CD73-positive and CD45- and CD34-negative. Adherent FDSCs expressed less of such embryonic stem cell markers as Oct-4 and E-cadherin, although two types of cultures expressed comparable levels of other embryonic stem cell markers SSEA-1 and SSEA-4. Unlike adhesive cells, spheroid-forming FDSCs expressed the neural crest stem cell marker neurotrophin receptor p75 NTR and hepatocyte markers alpha-fetoprotein and c-Met growth factor suggesting the presence of pluripotent cells and/or cells committed to differentiate along ectodermal or endodermal route. However, not just suspension culture cells but also adhesive cells could be induced to undergo hepatogenic differentiation.122 In the course of transformation, they expressed hepatocyte markers alpha-fetoprotein, albumin, cytokeratine-18 and -19, and CYP3A4. Both cell types got through morphological changes acquiring epithelium-like shape and became able to accumulate glycogen and low density lipoprotein uptake. FDSCs retained differentiation potential for at least 15 passages and after freeze/thaw procedures.
The so-called skin-derived progenitors are another kind of skin cells with high differentiation potential capable of hepatogenic differentiation.116,123 They display fibroblast- like morphology, are cultivated in the presence of EGF and FGF-2, express MSC markers CD29, CD44, CD90, and CD105, and do not express CD14, CD34, CD45, and CD68.124 Unlike FDSCs, skin-derived progenitors do not express p75 NTR.125 In the course of the in vitro differentiation into hepatocytes, skin-derived progenitors consecutively expressed markers of the early and middle phases of hepatogenic differentiation in vivo: CK18, HNF-4alpha, and HNF-1alpha, while no morphological changes occurred. The latter manifested themselves at a more advanced stage when cells acquired polygonal cuboid shape and started to produce albumin.126
Lysy et al127 compared human skin fibroblasts and bone marrow MSCs by their ability to differentiate into the mesoderm (osteoblasts and adipocytes) and endoderm (hepatocytes) derivatives. Skin fibroblasts expressed the pattern of surface markers typical for MSC, had classical fibroblast morphology, and were able to differentiate into osteoblasts and adipocytes confirming their mesodermal origin. After induction of hepatogenic differentiation, both skin fibroblasts and bone marrow MSC acquired hepatocyte-like morphology, started to express liver-specific genes at the transcriptional and translational levels, and accumulated urea. However, some differences between two cultures were revealed. Fibroblasts accumulated less urea than MSCs. Gene expression analysis showed that after hepatogenic differentiation, fibroblasts still remained in the state of mesenchymal–epithelial transition. Finally, fibroblasts retained the capacity for hepatogenic differentiation during three passages, while MSCs during eight passages.
Cell therapy and tissue engineering in the treatment of liver diseases
Human hepatocytes derived from adult cells by reprogramming via iPSCs or by direct transdifferentiation can be used to treat liver pathology applying one of the two existing practical approaches – transplantation of the suspension of hepatocytes (cell therapy) or ex vivo fabrication of the whole liver or its part followed by total or partial surgical substitution of the patient’s liver (tissue/organ engineering) (Figure 1). Both approaches are still in their infancy, but the development of a much simpler method of cell therapy was initiated earlier and diverse cell types and transplantation routes have already been tested in preclinical animal experiments and clinical trials. Initially, hepatocytes tested in preclinical and clinical studies were primary hepatocytes isolated from liver biopsy or autopsy samples, while trials with in vitro processed cells began later.
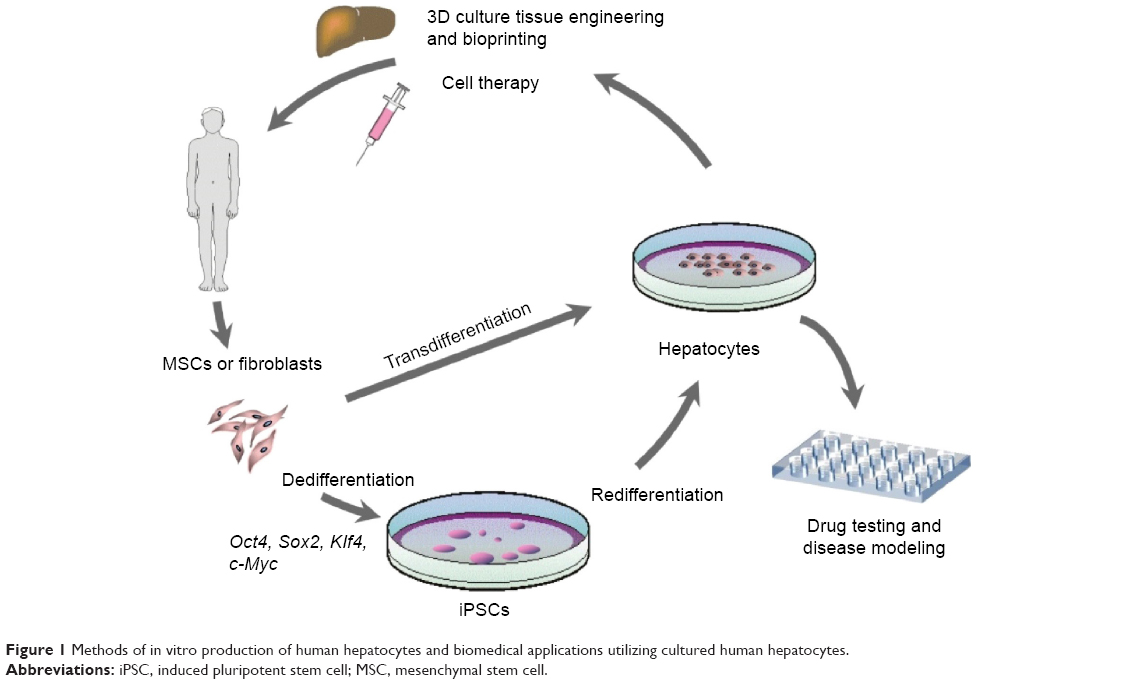 | Figure 1 Methods of in vitro production of human hepatocytes and biomedical applications utilizing cultured human hepatocytes. |
Primary hepatocyte transplantation proved quite effective in the experimental setup128–130 and in the treatment of some liver metabolic disorders, but not acute liver failure or chronic liver disease.131–135 Liver tissue engraftment and participation in its de novo formation is likely to be the major mechanism providing the beneficial effects of primary hepatocytes transplantation. However, other mechanisms, such as paracrine action stimulating tissue regeneration are also involved.
The very limited success of primary hepatocyte transplantation in humans is at least partly related to the inaccessibility of the proper cellular material. Direct isolation of autologous hepatocytes from patient’s liver biopsy is associated with patient’s inconvenience and difficulties obtaining sufficient quantities of viable cells. Therefore, in the clinical context, the only human primary hepatocytes available are poor quality allogeneic cells derived from livers unsuitable for organ transplantation. Lack of reliable sources of primary human hepatocytes remains a major obstacle for their use. Some animal experiments demonstrated effective engraftment of xenogeneic hepatocytes into the liver tissue.136,137 However, the suggested transplantation of animal hepatocytes into humans will hardly be adopted in the near future because of safety concerns.
Further research is underway to improve hepatocyte transplantation methods. Recently, experiments carried out in mice showed very high regeneration-promoting activity of the so-called hybrid periportal hepatocytes residing in portal triads of healthy liver and capable of replenishing the entire chronically damaged parenchyma.138 Importantly, hybrid periportal hepatocytes checked in three disease models exhibited unmatched regeneration-promoting activity, but never originated cancer cells. Now, characterization of analogous human cells and testing their liver regeneration capacity is on the agenda of regenerative hepatology.
Transplantation of MSC by itself produces relief in animal models of liver diseases85,139 and in patients with liver pathology.140–142 Mechanisms underlying the beneficial effects of MSC transplantation may include in situ transdifferentiation of transplanted cells into hepatocytes, paracrine stimulatory action upon resident liver stem cells, or immunomodulation. Liver pathology-related symptoms may also be attenuated by the transplantation of hematopoietic cells143 or their derivatives such as macrophages.144
However, the major part of preclinical and clinical research is conducted with MSCs due to their unique features, such as low immunogenicity, affinity to the sites of ischemia, inflammation or trauma, and ability to modulate immune responses.
Hence, several cell types were tried out in cell therapy of liver diseases. Hepatocytes may well not be the most effective cells, but only autologous hepatocytes fit such applications as liver tissue engineering, underscoring the significance of the development of the methods of their production from adult human cells, either by induction of pluripotency followed by hepatogenic differentiation or direct transdifferentiation of adult stem cells or even terminally differentiated cells.
Liver tissue engineering is still in the initial phase of technological development. The approaches tested so far in animals include obtaining decellularized organ scaffold followed by its repopulation with hepatocytes and other liver cells and bioprinting.135 During decellularization, cells and immunogenic molecules are removed by perfusion of the organ with detergent- and enzyme-containing solutions leaving the extracellular matrix scaffold reproducing organ architecture and detailed 3D microstructure providing adequate framework for blood vessels and biliary ducts.145 The extracellular matrix scaffold can be repopulated by hepatocytes and other liver cells in a bioreactor providing a “neo-organ”. Recellularized liver graft was first successfully transplanted into the rat by Uygun et al.146 The methods were further elaborated147,148 and later included the use of humanized model of porcine liver scaffold and human cells.149
Bioprinting using elements of extracellular matrix and live cells allows precise formation of elaborate 3D tissue and organ structures, including vascular network. This method was successfully applied to make metabolically active 3D hepatic tissue constructs.150,151 At present, 3D bioprinted fragments of liver tissue do not survive for more than a few days. However, this method seems to have great potential for its further development.
New approaches to liver tissue engineering are being developed and important improvements introduced. For example, microencapsulation of hepatocytes before their engraftment into the tissue constructs facilitates long-time survival of functionally intact cells.152 Scaffold-free methods of the assembly of large tissue fragments or whole organs are aimed to totally exclude the use of allogenic biomaterials in organ manufacturing process.153
In addition to donor organ shortage, liver transplantation is restrained by the need to control adverse immune reactions. The use of autologous cells to construct a new organ for transplantation can solve both problems, but has two serious disadvantages. Firstly, building an individual organ for each patient will be very costly. Secondly, it will take at least several months to make a liver or its part from autologous cells and this is not suitable for patients with acute liver failure. Fortunately, recent research developments give hope to produce organs for immunosuppression-free transplantation using allogenic cells. The HLA specificity of an individual is determined by two coexpressed haplotypes, each represented by an HLA-A, HLA-B, and HLA-DR gene. Due to the influence of a number of internal and environmental factors, the combinations of these three genes are not random and there are statistically preferable patterns. It is possible to select homozygous donors with statistically prevalent HLA gene combinations that match substantial numbers of vastly heterozygous potential cell and organ transplantation recipients. Based on these assumptions, Nakatsuji et al calculated that 30 homozygous iPSC lines derived from donors selected from 15,000 Japanese individuals would match 82.2% of potential Japanese recipients, while 50 lines originating from donors selected from 24,000 individuals would raise the score to 90.7%.154 The corresponding figures for UK are the following: 150 selected homozygous HLA-typed volunteers could match 93% of the UK population.155 These findings provided the basis for the idea of a global iPSC lines registry.156 The success of cell therapy of liver diseases and liver tissue engineering depends upon many factors including the age of the cell donor and recipient. As already noted above, biological aging occurs at different levels including cellular level. Throughout lifetime, hepatocytes, fibroblasts, MSCs, hematopoietic cells, macrophages, and other cells mentioned in this review undergo mutually dependent age-related modifications, such as free radical damage to the inner machinery of the cell, shortening of telomeres, accumulation of DNA modifications, alterations of mitochondria functions, and others.7 In stem cells, these changes result in replicative senescence first described in cultivated fibroblasts and widely regarded as a universal tumor-suppressive mechanism.157 Replicative senescence manifests itself by permanent cell cycle arrest, telomere shortening, telomerase-reverse transcriptase dysfunction, irreparable DNA damage, metabolic shift from Krebs cycle toward glycolysis, and finally to cell death. It leads to partial eradication of resident and stem cell populations including those in bone marrow and liver, decline in tissue regeneration capacity, and ultimately causes reduction of parenchymal cell numbers and functional impairment in most organs and tissues including liver.
Biopsies taken from older individuals presumably contain more cells with somatic mutations, chromosome modifications, and fewer stem cells. However, in cell culture, most damaged cells die and cells with short telomeres do not proliferate. This makes the clonal composition of cultured cells population different from clonal composition of cells in originating tissue. Due to elimination of damaged cells, populations of cultured cells seem to become “younger”, at least at first passages. Research in this field is impeded by the lack of an accurate quantitative method of evaluation of the biological age of cells and tissues. The value of the currently prevalent indicator, telomere length, is disputed because it undergoes age-related changes at a different pace in different tissues.158 Recently, Horvath introduced a novel method of evaluation of cell and tissue age based on the studies of cytosine-5 methylation within CpG dinucleotides, also called DNA methylation.159 In this and following studies, DNA methylation showed an excellent correlation with chronological age of cell or tissue donor in most human cell types and tissues, including liver and hepatocytes.159,160 It should be noted that both telomere length and DNA methylation assays show that pluripotency induction in somatic cells is accompanied by the rejuvenation of cells converted into iPSC and shifting of both parameters to the values characteristic of embryonic stem cells.31,159 The situation with rejuvenation of directly transdifferentiated somatic cells is less clear and should be reassessed using more accurate criteria including epigenetic approaches like Horvath’s DNA methylation test.
Conclusion
There is no simple solution to the problems of liver pathology, age-related liver pathology, or age-related diseases in general. Modern medicine still relays primarily on the use of xenobiotic drugs to relieve symptoms. As pointed out by Richard F Walker back in 2006:
[…] a proactive, holistic approach intended to delay onset or avoid development of age-related disease is more logical than a reactive, symptomatic approach.161
Since 2006, there is little change in clinical medicine but huge progress in fundamental research laying the basis for the transformation of the paradigm.
Remarkably, classical donor organ transplantation, including liver transplantation, displays distinct features of a holistic tactic because replacement of heart, kidney, or liver provides coordinated normalization of a number of crucially important homeostasis parameters. Cell-based technologies including cell therapy and tissue/organ engineering offer further advancement of transplantation methodology with much better control over the quality of transplanted material. In addition, research in this field, particularly stem cell and differentiation/dedifferentiation studies, deliver better understanding of aging and age-linked pathology. There are still many questions to be answered and many basic and technical problems to be solved. But it is already clear that cell-based therapies will play an increasingly important role in the development of novel methods of the management of age-related issues. Studies of age-related aspects of the control of liver diseases using cell-based technologies are in their initial phase, but the prospects of cell therapy and organ engineering utilizing rejuvenated hepatocytes produced from somatic cells via iPSC or by direct transdifferentiation seem quite promising.
Production of liver or liver lobes for transplantation is not likely in near future, but achievable in a longer prospective. Autologous or HLA-matched human iPSCs are probably the most convenient source of cell material. Currently, this technological trend attracts investment and in some countries enjoys support from national and local governments. Concerted studies in the field of iPSC technology-based human tissue and organ engineering currently conducted in Japan already deliver results. Masayo Takahashi group of the RIKEN Center for Developmental Biology in Kobe, Japan, was the first to carry out a clinical study of an iPSC-based technology using retinal pigment epithelium cells obtained by differentiation of iPSC derived from autologous dermal fibroblasts.162 Transplantation of retinal pigment epithelium cell sheet into the subretinal space of an elderly woman with age-related moist macular degeneration did not produce serious adverse effects and resulted in partial vision restoration.163 Serious efforts are focused on the engineering of kidneys and methods designed in these studies may be applied to engineering of livers as well.164
At present, California Institute of Regenerative Medicine Human Pluripotent Stem Cell Repository, the largest human iPSC bank, holds just 300 human iPSC lines which is of course very far from quantities needed to provide cellular material for immunosuppression-free therapy. However, several companies started commercial iPSC and derivative production and biobanking. Among these, ReproCELL, Kanagawa, Japan, offers hepatocytes produced from iPSC lines. There are a number of national and international initiatives to create cell repositories and registers big enough to provide starting material to develop immunosuppression-free transplantation technologies.
Transplantation of hepatocytes, MSC, or hematopoietic cells delivered encouraging results in many animal models and some clinical trials. Unfortunately, not enough has been done to reveal the differences in the reactions of younger and older animals or patients with liver pathology to cell transplantation. This is a very important subject for further studies. Taking into account that aging starts early in life and manifests itself at varying chronological age,1 every successful clinical tactic aimed to reverse age-related changes actually contributes to geriatrics. However, to accurately evaluate these approaches, better ways of age assessment at the cellular and tissue levels are needed. The recently introduced method of age estimation at the epigenomic level159 supplements the traditional practice based on telomere length measurements and the combination of those two approaches may provide a more accurate measure of cell and tissue senescence and its reversal. It fully relates to liver pathology management. Liver is an organ with a complex tissue architecture which is difficult to reproduce ex vivo. However, existing approaches including repopulation of decellularized cadaveric human liver scaffolds with hepatocytes and other cells and bioprinting have good chances to be transferred to practical medicine within a decade or two,135,164 while new developments will be arriving.
Acknowledgment
This work was supported by the Russian Science Foundation (grant number 14-15-00648).
Disclosure
The authors report no conflicts of interest in this work.
References
Belsky DW, Caspi A, Houts R, et al. Quantification of biological aging in young adults. Proc Natl Acad Sci U S A. Epub 2015 Jul 6. | ||
Friedman SL. Liver fibrosis – from bench to bedside. J Hepatol. 2003;38(Suppl 1):S38–S53. | ||
Lozano R, Naghavi M, Foreman K, et al. Global and regional mortality from 235 causes of death for 20 age groups in 1990 and 2010: a systematic analysis for the Global Burden of Disease Study 2010. Lancet. 2012;380(9859):2095–2128. | ||
Caldwell S, Park SH. The epidemiology of hepatocellular cancer: from the perspectives of public health problem to tumor biology. J Gastroenterol. 2009;44(Suppl 19):96–101. | ||
D’Amico G, Garcia-Tsao G, Pagliaro L. Natural history and prognostic indicators of survival in cirrhosis: a systematic review of 118 studies. J Hepatol. 2006;44(1):217–231. | ||
Lucey MR, Terrault N, Ojo L, et al. Long-term management of the successful adult liver transplant: 2012 practice guideline by the American Association for the Study of Liver Diseases and the American Society of Transplantation. Liver Transpl. 2013;19(1):3–26. | ||
Fukada S, Ma Y, Uezumi A. Adult stem cell and mesenchymal progenitor theories of aging. Front Cell Dev Biol. 2014;2:10. | ||
Beerman I, Rossi DJ. Epigenetic control of stem cell potential during homeostasis, aging, and disease. Cell Stem Cell. 2015;16(6):613–625. | ||
Nurkovic JS, Volarevic V, Lako M, et al. Aging of stem and progenitor cells: mechanisms, impact on the therapeutic potential and rejuvenation. Rejuvenation Res. Epub 2015 Jun 9. | ||
Studer L, Vera E, Cornacchia D. Programming and reprogramming cellular age in the era of induced pluripotency. Cell Stem Cell. 2015;16(6):591–600. | ||
Lapasset L, Milhavet O, Prieur A, et al. Rejuvenating senescent and centenarian human cells by reprogramming through the pluripotent state. Genes Dev. 2011;25(21):2248–2253. | ||
Miller JD, Ganat YM, Kishinevsky S, et al. Human iPSC-based modeling of late-onset disease via progerin-induced aging. Cell Stem Cell. 2013;13(6):691–705. | ||
Takahashi K, Tanabe K, Ohnuki M, et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131(5):861–872. | ||
Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126(4):663–676. | ||
Li H, Collado M, Villasante A, et al. The Ink4/Arf locus is a barrier for iPS cell reprogramming. Nature. 2009;460(7259):1136–1139. | ||
Wang B, Miyagoe-Suzuki Y, Yada E, et al. Reprogramming efficiency and quality of induced pluripotent stem cells (iPSCs) generated from muscle-derived fibroblasts of mdx mice at different ages. PLoS Curr. 2011;3:RRN1274. | ||
Cheng Z, Ito S, Nishio N, et al. Establishment of induced pluripotent stem cells from aged mice using bone marrow-derived myeloid cells. J Mol Cell Biol. 2011;3(2):91–98. | ||
Yagi T, Kosakai A, Ito D, et al. Establishment of induced pluripotent stem cells from centenarians for neurodegenerative disease research. PLoS One. 2012;7:e41572. | ||
Somers A, Jean JC, Sommer CA, et al. Generation of transgene-free lung disease-specific human induced pluripotent stem cells using a single excisable lentiviral stem cell cassette. Stem Cells. 2010;28(10):1728–1740. | ||
Sharma A, Diecke S, Zhang WY, et al. The role of SIRT6 protein in aging and reprogramming of human induced pluripotent stem cells. J Biol Chem. 2013;288(25):18439–18447. | ||
Seki T, Fukuda K. Methods of induced pluripotent stem cells for clinical application. World J Stem Cells. 2015;7(1):116–125. | ||
Aasen T, Raya A, Barrero MJ, et al. Efficient and rapid generation of induced pluripotent stem cells from human keratinocytes. Nat Biotechnol. 2008;26(11):1276–1284. | ||
Kim JB, Zaehres H, Wu G, et al. Pluripotent stem cells induced from adult neural stem cells by reprogramming with two factors. Nature. 2008;454(7204):646–650. | ||
Eminli S, Utikal J, Arnold K, et al. Reprogramming of neural progenitor cells into induced pluripotent stem cells in the absence of exogenous Sox2 expression. Stem Cells. 2008;26(10):2467–2474. | ||
Liebau S, Mahaddalkar PU, Kestler HA, et al. Hierarchy in reprogramming capacity in different tissue microenvironments: what we know and what we need to know. Stem Cells Dev. 2013;22(5):695–706. | ||
Hanna J, Markoulaki S, Schorderet P, et al. Direct reprogramming of terminally differentiated mature B lymphocytes to pluripotency. Cell. 2008;133(2):250–264. | ||
Maherali N, Ahfeldt T, Rigamonti A, et al. A high-efficiency system for the generation and study of human induced pluripotent stem cells. Cell Stem Cell. 2008;3(3):340–345. | ||
Abramovich A, Muradian KK, Fraifeld VE. Have we reached the point for in vivo rejuvenation? Rejuvenation Res. 2008;11(2):489–492. | ||
Wahlestedt M, Norddahl GL, Sten G, et al. An epigenetic component of hematopoietic stem cell aging amenable to reprogramming into a young state. Blood. 2013;121(21):4257–4264. | ||
Rohani L, Johnson AA, Arnold A, Stolzing A. The aging signature: a hallmark of induced pluripotent stem cells? Aging Cell. 2014;13(1):2–7. | ||
Marion RM, Blasco MA. Telomere rejuvenation during nuclear reprogramming. Curr Opin Genet Dev. 2010;20(2):190–196. | ||
Prigione A, Hossini AM, Lichtner B, et al. Mitochondrial-associated cell death mechanisms are reset to an embryonic-like state in aged donor-derived iPS cells harboring chromosomal aberrations. PLoS One. 2011;6:e27352. | ||
Fan J, Robert C, Jang YY, et al. Human induced pluripotent cells resemble embryonic stem cells demonstrating enhanced levels of DNA repair and efficacy of nonhomologous end-joining. Mutat Res. 2011; 713(1–2):8–17. | ||
Luo LZ, Gopalakrishna-Pillai S, Nay SL, et al. DNA repair in human pluripotent stem cells is distinct from that in nonpluripotent human cells. PLoS One. 2012;7:e30541. | ||
Mikkelsen TS, Hanna J, Zhang X, et al. Dissecting direct reprogramming through integrative genomic analysis. Nature. 2008;454(7200):49–55. | ||
Chin MH, Mason MJ, Xie W, et al. Induced pluripotent stem cells and embryonic stem cells are distinguished by gene expression signatures. Cell Stem Cell. 2009;5(1):111–123. | ||
Deng J, Shoemaker R, Xie B, et al. Targeted bisulfite sequencing reveals changes in DNA methylation associated with nuclear reprogramming. Nat Biotechnol. 2009;27(4):353–360. | ||
De Carvalho DD, You JS, Jones PA. DNA methylation and cellular reprogramming. Trends Cell Biol. 2010;20(10):609–617. | ||
Bar-Nur O, Russ HA, Efrat S, Benvenisty N. Epigenetic memory and preferential lineage-specific differentiation in induced pluripotent stem cells derived from human pancreatic islet beta cells. Cell Stem Cell. 2011;9(1):17–23. | ||
Vaskova EA, Stekleneva AE, Medvedev SP, Zakian SM. “Epigenetic memory” phenomenon in induced pluripotent stem cells. Acta Naturae. 2013;5(4):15–21. | ||
Clotman F, Lemaigre FP. Control of hepatic differentiation by activin/TGFβ signaling. Cell Cycle. 2006;5(2):168–171. | ||
Hewitt NJ, Lechon MJ, Houston JB, et al. Primary hepatocytes: current understanding of the regulation of metabolic enzymes and transporter proteins, and pharmaceutical practice for the use of hepatocytes in metabolism, enzyme induction, transporter, clearance, and hepatotoxicity studies. Drug Metab Rev. 2007;39(1):159–234. | ||
Safinia N, Minger SL. Generation of hepatocytes from human embryonic stem cells. Methods Mol Biol. 2009;481:169–180. | ||
Terry C, Hughes RD. An optimized method for cryopreservation of human hepatocytes. Methods Mol Biol. 2009;481:25–34. | ||
Song Z, Cai J, Liu Y, et al. Efficient generation of hepatocyte-like cells from human induced pluripotent stem cells. Cell Res. 2009;19(11):1233–1242. | ||
Si-Tayeb K, Noto FK, Nagaoka M, et al. Highly efficient generation of human hepatocyte-like cells from induced pluripotent stem cells. Hepatology. 2010;51(1):297–305. | ||
Sullivan GJ, Hay DC, Park IH, et al. Generation of functional human hepatic endoderm from human induced pluripotent stem cells. Hepatology. 2010;51(1):329–335. | ||
James D, Levine AJ, Besser D, Hemmati-Brivanlou A. TGFbeta/activin/nodal signaling is necessary for the maintenance of pluripotency in human embryonic stem cells. Development (Cambridge, England). 2005;132(6):1273–1282. | ||
Vallier L, Alexander M, Pedersen RA. Activin/nodal and FGF pathways cooperate to maintain pluripotency of human embryonic stem cells. J Cell Sci. 2005;118(Pt 19):4495–4509. | ||
Burdette JE, Jeruss JS, Kurley SJ, et al. Activin A mediates growth inhibition and cell cycle arrest through Smads in human breast cancer cells. Cancer Res. 2005;65(17):7968–7975. | ||
Fukuchi Y, Yamato K, Kawamura C, et al. p27KIP1 and GATA-1 are potential downstream molecules in activin A-induced differentiation and apoptosis pathways in CML cells. Oncol Rep. 2006;16(5):1099–1103. | ||
Matsuo SE, Leoni SG, Colquhoun A, Kimura ET. Transforming growth factor-beta1 and activin A generate antiproliferative signaling in thyroid cancer cells. J Endocrinol. 2006;190(1):141–150. | ||
Payne C, King J, Hay D. The role of activin/nodal and Wnt signaling in endoderm formation. Vitam Horm. 2011;85:207–216. | ||
Fei T, Zhu S, Xia K, et al. Smad2 mediates activin/nodal signaling in mesendoderm differentiation of mouse embryonic stem cells. Cell Res. 2010;20(12):1306–1318. | ||
Kubo A, Shinozaki K, Shannon JM, et al. Development of definitive endoderm from embryonic stem cells in culture. Development. 2004;131(7):1651–1662. | ||
Guo S, Mao X, He F, et al. Activin A supplement in the hESCs culture enhances the endoderm differentiation efficiency. Cell Biol Int. 2014; 38(7):849–856. | ||
Xiao L, Yuan X, Sharkis SJ. Activin A maintains self-renewal and regulates fibroblast growth factor, Wnt, and bone morphogenic protein pathways in human embryonic stem cells. Stem Cells. 2006;24(6):1476–1486. | ||
Wang Z, Li W, Chen T, et al. Activin A can induce definitive endoderm differentiation from human parthenogenetic embryonic stem cells. Biotechnol Lett. 2015;37(8):1711–1717. | ||
Tomizawa M, Shinozaki F, Sugiyama T, et al. Activin A maintains pluripotency markers and proliferative potential of human induced pluripotent stem cells. Exp Ther Med. 2011;2(3):405–408. | ||
D’Amour KA, Agulnick AD, Eliazer S, et al. Efficient differentiation of human embryonic stem cells to definitive endoderm. Nat Biotechnol. 2005;23(12):1534–1541. | ||
Chen YF, Tseng CY, Wang HW, et al. Rapid generation of mature hepatocyte-like cells from human induced pluripotent stem cells by an efficient three-step protocol. Hepatology. 2012;55(4):1193–1203. | ||
Mobarra N, Soleimani M, Kouhkan F, et al. Efficient differentiation of human induced pluripotent stem cell (hiPSC) derived hepatocyte-like cells on hMSCs feeder. Int J Hematol Oncol Stem Cell Res. 2014;8(4):20–29. | ||
Sekine K, Takebe T, Suzuki Y, Kamiya A, Nakauchi H, Taniguchi H. Highly efficient generation of definitive endoderm lineage from human induced pluripotent stem cells. Transplant Proc. 2012;44(4):1127–1129. | ||
Ao Y, Mich-Basso JD, Lin B, Yang L. High efficient differentiation of functional hepatocytes from porcine induced pluripotent stem cells. PLoS One. 2014;9(6):e100417. | ||
Jung J, Zheng M, Goldfarb M, Zaret KS. Initiation of mammalian liver development from endoderm by fibroblast growth factors. Science. 1999;284(5422):1998–2003. | ||
Rossi JM, Dunn NR, Hogan BL, Zaret KS. Distinct mesodermal signals, including BMPs from the septum transversum mesenchyme, are required in combination for hepatogenesis from the endoderm. Genes Dev. 2001;15(15):1998–2009. | ||
Schmidt C, Bladt F, Goedecke S, et al. Scatter factor/hepatocyte growth factor is essential for liver development. Nature. 1995;373(6516):699–702. | ||
Liu X, Newton RC, Scherle PA. Developing c-MET pathway inhibitors for cancer therapy: progress and challenges. Trends Mol Med. 2010;16(1):37–45. | ||
Kamiya A, Kinoshita T, Miyajima A. Oncostatin M and hepatocyte growth factor induce hepatic maturation via distinct signaling pathways. FEBS Lett. 2001;492(1–2):90–94. | ||
Michalopoulos GK, Bowen WC, Mule K, Luo J. HGF-, EGF-, and dexamethasone-induced gene expression patterns during formation of tissue in hepatic organoid cultures. Gene Expr. 2003;11(2):55–75. | ||
Suzuki A, Iwama A, Miyashita H, et al. Role for growth factors and extracellular matrix in controlling differentiation of prospectively isolated hepatic stem cells. Development. 2003;130(11):2513–2524. | ||
Ito Y, Matsui T, Kamiya A, et al. Retroviral gene transfer of signaling molecules into murine fetal hepatocytes defines distinct roles for the STAT3 and ras pathways during hepatic development. Hepatology. 2000;32(6):1370–1376. | ||
Matsui T, Kinoshita T, Morikawa Y, et al. K-Ras mediates cytokine-induced formation of E-cadherin-based adherens junctions during liver development. EMBO J. 2002;21(5):1021–1030. | ||
Imamura M, Kojima T, Lan M, et al. Oncostatin M induces upregulation of claudin-2 in rodent hepatocytes coinciding with changes in morphology and function of tight junctions. Exp Cell Res. 2007;313(9):1951–1962. | ||
Zhang Q, Yang Y, Zhang J, et al. Efficient derivation of functional hepatocytes from mouse induced pluripotent stem cells by a combination of cytokines and sodium butyrate. Chin Med J. 2011;124(22):3786–3793. | ||
Nawa K, Nakamura T, Kumatori A, Noda C, Ichihara A. Glucocorticoid-dependent expression of the albumin gene in adult rat hepatocytes. J Biol Chem. 1986;261(36):16883–16888. | ||
de Juan C, Benito M, Fabregat I. Regulation of albumin expression in fetal rat hepatocytes cultured under proliferative conditions: role of epidermal growth factor and hormones. J Cell Physiol. 1992;152(1):95–101. | ||
Prockop DJ. Marrow stromal cells as stem cells for nonhematopoietic tissues. Science. 1997;276(5309):71–74. | ||
Petersen BE, Bowen WC, Patrene KD, et al. Bone marrow as a potential source of hepatic oval cells. Science. 1999;284(5417):1168–1170. | ||
Alison MR, Poulsom R, Jeffery R, et al. Hepatocytes from non-hepatic adult stem cells. Nature. 2000;406(6793):257. | ||
Lagasse E, Connors H, Al-Dhalimy M, et al. Purified hematopoietic stem cells can differentiate into hepatocytes in vivo. Nat Med. 2000;6(11):1229–1234. | ||
Schwartz RE, Reyes M, Koodie L, et al. Multipotent adult progenitor cells from bone marrow differentiate into functional hepatocyte-like cells. J Clin Invest. 2002;109(10):1291–1302. | ||
Kucia M, Reca R, Campbell FR, et al. A population of very small embryonic-like (VSEL) CXCR4(+)SSEA-1(+)Oct-4+ stem cells identified in adult bone marrow. Leukemia. 2006;20(5):857–869. | ||
Pittenger MF, Mackay AM, Beck SC, et al. Multilineage potential of adult human mesenchymal stem cells. Science. 1999;284(5411):143–147. | ||
Phinney DG, Prockop DJ. Concise review: mesenchymal stem/multipotent stromal cells: the state of transdifferentiation and modes of tissue repair – current views. Stem Cells. 2007;25(11):2896–2902. | ||
Jameson E. Cellular transplantation for liver diseases. Gastroen Res. 2008;1(1):8–13. | ||
Zuk PA, Zhu M, Mizuno H, et al. Multilineage cells from human adipose tissue: implications for cell-based therapies. Tissue Eng. 2001;7(2):211–228. | ||
Talens-Visconti R, Bonora A, Jover R, et al. Hepatogenic differentiation of human mesenchymal stem cells from adipose tissue in comparison with bone marrow mesenchymal stem cells. World J Gastroenterol. 2006;12(36):5834–5845. | ||
Zemel R, Bachmetov L, Ad-El D, Abraham A, Tur-Kaspa R. Expression of liver-specific markers in naive adipose-derived mesenchymal stem cells. Liver Int. 2009;29(9):1326–1337. | ||
Campard D, Lysy PA, Najimi M, Sokal EM. Native umbilical cord matrix stem cells express hepatic markers and differentiate into hepatocyte-like cells. Gastroenterology. 2008;134(3):833–848. | ||
Zhang YN, Lie PC, Wei X. Differentiation of mesenchymal stromal cells derived from umbilical cord Wharton’s jelly into hepatocyte-like cells. Cytotherapy. 2009;11(5):548–558. | ||
Hong SH, Gang EJ, Jeong JA, et al. In vitro differentiation of human umbilical cord blood-derived mesenchymal stem cells into hepatocyte-like cells. Biochem Biophys Res Commun. 2005;330(4): 1153–1161. | ||
Waclawczyk S, Buchheiser A, Flogel U, Radke TF, Kögler G. In vitro differentiation of unrestricted somatic stem cells into functional hepatic-like cells displaying a hepatocyte-like glucose metabolism. J Cell Physiol. 2010;225(2):545–554. | ||
Ishkitiev N, Yaegaki K, Calenic B, et al. Deciduous and permanent dental pulp mesenchymal cells acquire hepatic morphologic and functional features in vitro. J Endod. 2010;36(3):469–474. | ||
Kinoshita T, Miyajima A. Cytokine regulation of liver development. Biochim Biophys Acta. 2002;1592(3):303–312. | ||
Zhao R, Duncan SA. Embryonic development of the liver. Hepatology. 2005;41(5):956–967. | ||
Banas A, Teratani T, Yamamoto Y, et al. Adipose tissue-derived mesenchymal stem cells as a source of human hepatocytes. Hepatology. 2007;46(1):219–228. | ||
Saulnier N, Lattanzi W, Puglisi MA, et al. Mesenchymal stromal cells multipotency and plasticity: induction toward the hepatic lineage. Eur Rev Med Pharmacol Sci. 2009;13(Suppl 1):71–78. | ||
Taléns-Visconti R, Bonora A, Jover R, et al. Human mesenchymal stem cells from adipose tissue: differentiation into hepatic lineage. Toxicol In Vitro. 2007;21(2):324–329. | ||
Ong SY, Dai H, Leong KW. Hepatic differentiation potential of commercially available human mesenchymal stem cells. Tissue Eng. 2006;12(12):3477–3485. | ||
Coradeghini R, Guida C, Scanarotti C, et al. A comparative study of proliferation and hepatic differentiation of human adipose-derived stem cells. Cells Tissues Organs. 2010;191(6):466–477. | ||
Snykers S, Vanhaecke T, De Becker A, et al. Chromatin remodeling agent trichostatin A: a key-factor in the hepatic differentiation of human mesenchymal stem cells derived of adult bone marrow. BMC Dev Biol. 2007;7:24. | ||
Yamamoto Y, Banas A, Murata S, et al. A comparative analysis of the transcriptome and signal pathways in hepatic differentiation of human adipose mesenchymal stem cells. FEBS J. 2008;275(6):1260–1273. | ||
Saulnier N, Piscaglia AC, Puglisi MA, et al. Molecular mechanisms underlying human adipose tissue-derived stromal cells differentiation into a hepatocyte-like phenotype. Dig Liver Dis. 2010;42(12):895–901. | ||
Aurich H, Sgodda M, Kaltwaßer P, et al. Hepatocyte differentiation of mesenchymal stem cells from human adipose tissue in vitro promotes hepatic integration in vivo. Gut. 2009;58(4):570–581. | ||
Hwang S, Hong HN, Kim HS, et al. Hepatogenic differentiation of mesenchymal stem cells in a rat model of thioacetamide-induced liver cirrhosis. Cell Biol Int. 2012;36(3):279–288. | ||
Yu J, Cao H, Yang J, et al. In vivo hepatic differentiation of mesenchymal stem cells from human umbilical cord blood after transplantation into mice with liver injury. Biochem Biophys Res Commun. 2012;422(4):539–545. | ||
da Silva Meirelles L, Chagastelles PC, Nardi NB. Mesenchymal stem cells reside in virtually all post-natal organs and tissues. J Cell Sci. 2006;119(Pt 11):2204–2213. | ||
Suzdal’tseva YG, Burunova VV, Vakhrushev IV, Yarygin VN, Yarygin KN. Capability of human mesenchymal cells isolated from different sources to differentiation into tissues of mesodermal origin. Bull Exp Biol Med. 2007;143(1):114–121. | ||
Crisan M, Yap S, Casteilla L, et al. A perivascular origin of mesenchymal stem cells in multiple human organs. Cell Stem Cell. 2008;3(3):301–313. | ||
Bernardo ME, Cometa AM, Pagliara D, et al. Ex vivo expansion of mesenchymal stromal cells. Best Pract Res Clin Haematol. 2011;24(1):73–81. | ||
Yarygin KN, Suzdal’tseva YG, Burunova VV, et al. Comparative study of adult human skin fibroblasts and umbilical fibroblast-like cells. Bull Exp Biol Med. 2006;141(1):161–166. | ||
Lupatov AY, Karalkin PA, Suzdal’tseva YG, et al. Cytofluorometric analysis of phenotypes of human bone marrow and umbilical fibroblast-like cells. Bull Exp Biol Med. 2006;142(4):521–526. | ||
Suzdal’tseva YG, Burunova VV, Petrakova NV, Vakhrushev IV, Yarygin KN, Yarygin VN. Comparative analysis of cytophenotypes of cells of mesenchymal lineage isolated from human tissues. Bull Exp Biol Med. 2007;143(1):114–121. | ||
Lupatov AY, Vdovin AS, Vakhrushev IV, Poltavtseva RA, Yarygin KN. Comparative analysis of the expression of surface markers on fibroblasts and fibroblast-like cells isolated from different human tissues. Bull Exp Biol Med. 2015;158(4):537–543. | ||
Toma JG, McKenzie IA, Bagli D, Miller FD. Isolation and characterization of multipotent skin-derived precursors from human skin. Stem Cells. 2005;23(6):727–737. | ||
Hoogduijn MJ, Gorjup E, Genever PG. Comparative characterization of hair follicle dermal stem cells and bone marrow mesenchymal stem cells. Stem Cells Dev. 2006;15(1):49–60. | ||
Kruse C, Bodo E, Petschnik AE, Danner S, Tiede S, Paus R. Towards the development of a pragmatic technique for isolating and differentiating nestin-positive cells from human scalp skin into neuronal and glial cell populations: generating neurons from human skin? Exp Dermatol. 2006;15(10):794–800. | ||
Tiede S, Kloepper JE, Bodo E, Tiwari S, Kruse C, Paus R. Hair follicle stem cells: walking the maze. Eur J Cell Biol. 2007;86(7):355–376. | ||
Konieva AA, Kholodenko IV, Shragina OA, et al. Functional properties of mesenchymal stem cells labeled with magnetic microparticles in vitro and analysis of their distribution after transplantation. Bull Exp Biol Med. 2010;150(1):131–136. | ||
Huang HI, Chen SK, Ling QD, Chien CC, Liu HT, Chan SH. Multilineage differentiation potential of fibroblast-like stromal cells derived from human skin. Tissue Eng Part A. 2010;16(5):1491–1501. | ||
Huang HI, Chen SK, Wang RYL, Shen CR, Cheng YC. Human foreskin fibroblast-like stromal cells can differentiate into functional hepatocytic cells. Cell Biol Int. 2013;37(12):1308–1319. | ||
Fernandes KJ, McKenzie IA, Mill P. A dermal niche for multipotent adult skin-derived precursor cells. Nat Cell Biol. 2004;6(11):1082–1093. | ||
Buranasinsup S, Sila-Asna M, Bunyaratvej N, Bunyaratvej A. In vitro osteogenesis from human skin-derived precursor cells. Dev Growth Differ. 2006;48(4):263–269. | ||
Toma JG, Akhavan M, Fernandes KJ, et al. Isolation of multipotent adult stem cells from the dermis of mammalian skin. Nat Cell Biol. 2001;3(9):778–784. | ||
De Kock J, Vanhaecke T, Biernaskie J, Rogiers V, Snykers S. Characterization and hepatic differentiation of skin-derived precursors from adult foreskin by sequential exposure to hepatogenic cytokines and growth factors reflecting liver development. Toxicol In Vitro. 2009;23(8):1522–1527. | ||
Lysy PA, Smets F, Sibille C, et al. Human skin fibroblasts: from mesodermal to hepatocyte-like differentiation. Hepatology. 2007;46(5):1574–1585. | ||
Gupta S, Aragona E, Vemuru RP, Bhargava KK, Burk RD, Chowdhury JR. Permanent engraftment and function of hepatocytes delivered to the liver: implications for gene therapy and liver repopulation. Hepatology. 1991;14(1):144–149. | ||
Gupta S, Bhargava KK, Novikoff PM. Mechanisms of cell engraftment during liver repopulation with hepatocyte transplantation. Semin Liver Dis. 1999;19(1):15–26. | ||
Weber A, Groyer-Picard MT, Franco D, Dagher I. Hepatocyte transplantation in animal models. Liver Transpl. 2009;15(1):7–14. | ||
Strom SC, Chowdhury JR, Fox IJ. Hepatocyte transplantation for the treatment of human disease. Semin Liver Dis. 1999;19(1):39–48. | ||
Horslen SP, McCowan TC, Goertzen TC, et al. Isolated hepatocyte transplantation in an infant with a severe urea cycle disorder. Pediatrics. 2003;111(6 pt 1):1262–1267. | ||
Ito M, Nagata H, Miyakawa S, Fox IJ. Review of hepatocyte transplantation. J Hepatobiliary Pancreat Surg. 2009;16(2):97–100. | ||
Dhawan A, Puppi J, Hughes RD, Mitry RR. Human hepatocyte transplantation: current experience and future challenges. Nat Rev Gastroenterol Hepatol. 2010;7(5):288–298. | ||
Lee SY, Kim HJ, Choi D. Cell sources, liver support systems and liver tissue engineering: alternatives to liver transplantation. Int J Stem Cells. 2015;8(1):36–47. | ||
Gupta S. Hog heaven on the road to liver cell therapy. Gastroenterology. 2007;132(1):450–453. | ||
Nagata H, Nishitai R, Shirota C, et al. Prolonged survival of porcine hepatocytes in cynomolgus monkeys. Gastroenterology. 2007;132(1):321–329. | ||
Font-Burgada J, Shalapour S, Ramaswamy S, et al. Hybrid periportal hepatocytes regenerate the injured liver without giving rise to cancer. Cell. 2015;162(4):766–779. | ||
Jameson E. Cellular transplantation for liver diseases. Gastroen Res. 2008;1(1):8–13. | ||
Liu WH, Song FQ, Ren LN, et al. The multiple functional roles of mesenchymal stem cells in participating in treating liver diseases. J Cell Mol Med. 2015;19(3):511–520. | ||
King A, Barton D, Beard HA, et al. REpeated AutoLogous Infusions of STem cells In Cirrhosis (REALISTIC): a multicentre, phase II, open-label, randomised controlled trial of repeated autologous infusions of granulocyte colony-stimulating factor (GCSF) mobilised CD133+ bone marrow stem cells in patients with cirrhosis. A study protocol for a randomised controlled trial. BMJ Open. 2015;5(3):e007700. | ||
Ma XR, Tang YL, Xuan M, Chang Z, Wang XY, Liang XH. Transplantation of autologous mesenchymal stem cells for end-stage liver cirrhosis: a meta-analysis based on seven controlled trials. Gastroenterol Res Pract. 2015;2015:908275. | ||
Zekri AR, Salama H, Medhat E, et al. The impact of repeated autologous infusion of haematopoietic stem cells in patients with liver insufficiency. Stem Cell Res Ther. 2015;6:118. | ||
Forbes SJ, Newsome PN. New horizons for stem cell therapy in liver disease. J Hepatol. 2012;56(2):496–499. | ||
Crapo PM, Gilbert TW, Badylak SF. An overview of tissue and whole organ decellularization processes. Biomaterials. 2011;32(12):3233–3243. | ||
Uygun BE, Soto-Gutierrez A, Yagi H, et al. Organ reengineering through development of a transplantable recellularized liver graft using decellularized liver matrix. Nat Med. 2010;16(7):814–820. | ||
Soto-Gutierrez A, Zhang L, Medberry C, et al. Whole-organ regenerative medicine approach for liver replacement. Tissue Eng Part C Methods. 2011;17(6):677–686. | ||
Baptista PM, Siddiqui MM, Lozier G, Rodriguez SR, Atala A, Soker S. The use of whole organ decellularization for the generation of a vascularized liver organoid. Hepatology. 2011;53(2):604–617. | ||
Barakat O, Abbasi S, Rodriguez G, et al. Use of decellularized porcine liver for engineering humanized liver organ. J Surg Res. 2012;173(1):e11–e25. | ||
Chang R, Emami K, Wu H, Sun W. Biofabrication of a three-dimensional liver micro-organ as an in vitro drug metabolism model. Biofabrication. 2010;2(4):045004. | ||
Ikegami T, Maehara Y. Transplantation: 3D printing of the liver in living donor liver transplantation. Nat Rev Gastroenterol Hepatol. 2013;10(12):697–698. | ||
Li CY, Stevens KR, Schwartz RE, Alejandro BS, Huang JH, Bhatia SN. Micropatterned cell–cell interactions enable functional encapsulation of primary hepatocytes in hydrogel microtissues. Tissue Eng Part A. 2014;20(15–16):2200–2212. | ||
Blakely AM, Manning KL, Tripathi A, Morgan JR. Bio-pick, place, and perfuse: A new instrument for three-dimensional tissue engineering. Tissue Eng Part C Methods. 2015;21(7):737–746. | ||
Nakatsuji N, Nakajima F, Tokunaga K. HLA-haplotype banking and iPS cells. Nat Biotechnol. 2008;26(7):739–740. | ||
Taylor CJ, Peacock S, Chaudhry AN, Bradley JA, Bolton EM. Generating an iPSC bank for HLA-matched tissue transplantation based on known donor and recipient HLA types. Cell Stem Cell. 2012;11(2):147–152. | ||
Turner M, Leslie S, Martin NG, et al. Toward the development of a global induced pluripotent stem cell library. Cell Stem Cell. 2013;13(4):382–384. | ||
Moyse E, Lahousse L, Krantic S. Current research in aging: a report from the 2015 Ageing Summit. Neurodegen Dis Manag. 2015;5(3):187–190. | ||
Sanders J, Boudreau R, Newman A. Understanding the aging process using epidemiologic approaches. In: Newman AB, Cauley JA, editors. The Epidemiology of Aging. Dordrecht/Heidelberg/New York: Springer; 2012:187–214. | ||
Horvath S. DNA methylation age of human tissues and cell types. Genome Biol. 2013;14:R115. | ||
Horvath S, Erhart W, Brosch M, et al. Obesity accelerates epigenetic aging of human liver. Proc Natl Acad Sci U S A. 2014;111(43):15538–15543. | ||
Walker RF. On the evolution of anti-aging medicine. Clin Int Aging. 2006;1(3):201–203. | ||
Kamao H, Mandai M, Okamoto S, et al. Characterization of human induced pluripotent stem cell-derived retinal pigment epithelium cell sheets aiming for clinical application. Stem Cell Reports. 2014;2(2):205–218. | ||
Riardon S, Cyranoski D. Japan stem-cell trial stirs envy. Nature. 2014;513(7518):287–288. | ||
Yamanaka S, Yokoo T. Current bioengineering methods for whole kidney regeneration. Stem Cells Int. 2015;2015:724047. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.