Back to Journals » International Journal of Nanomedicine » Volume 11
Caveolin-1 contributes to realgar nanoparticle therapy in human chronic myelogenous leukemia K562 cells
Authors Shi D, Liu Y, Xi RG, Zou W, Wu LJ, Zhang ZR, Liu ZY, Qu C, Xu BL, Wang XB
Received 16 June 2016
Accepted for publication 1 September 2016
Published 7 November 2016 Volume 2016:11 Pages 5823—5835
DOI https://doi.org/10.2147/IJN.S115158
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Linlin Sun
Dan Shi,1,* Yan Liu,1,* Ronggang Xi,1 Wei Zou,2 Lijun Wu,3 Zhiran Zhang,1 Zhongyang Liu,1 Chao Qu,1 Baoli Xu,1 Xiaobo Wang1
1Department of Pharmacy, The 210th Hospital of People’s Liberation Army, 2College of Life Science, Liaoning Normal University, Dalian, Liaoning, 3Department of Pharmaceutics, College of Pharmacy, Harbin Medical University, Harbin, Heilongjiang, People’s Republic of China
*These authors contributed equally to this work
Abstract: Chronic myelogenous leukemia (CML) is characterized by the t(9;22) (q34;q11)-associated Bcr-Abl fusion gene, which is an essential element of clinical diagnosis. As a traditional Chinese medicine, realgar has been widely used for the treatment of various diseases for >1,500 years. Inspired by nano-drug, realgar nanoparticles (NPs) have been prepared with an average particle size of <100 nm in a previous work. Compared with coarse realgar, the realgar NPs have higher bioavailability. As a principal constituent protein of caveolae, caveolin-1 (Cav-1) participates in regulating various cellular physiological and pathological processes including tumorigenesis and tumor development. In previous studies, it was found that realgar NPs can inhibit several types of tumor cell proliferation. However, the therapeutic effect of realgar NPs on CML has not been fully elucidated. In the present paper, it was demonstrated that realgar NPs can inhibit the proliferation of K562 cells and degrade Bcr-Abl fusion protein effectively. Both apoptosis and autophagy were activated in a dose-dependent manner in realgar NPs treated cells, and the induction of autophagy was associated with class I phosphoinositide 3-kinase/protein kinase B/mammalian target of rapamycin pathway. Morphological analysis indicated that realgar NPs induced differentiation effectively in CML cells. Furthermore, it was identified that Cav-1 might play a crucial role in realgar NP therapy. In order to study the effects of Cav-1 on K562 cells during realgar NP treatment, a Cav-1 overexpression cell model was established by using transient transfection. The results indicated that Cav-1 overexpression inhibited K562 cell proliferation, promoted endogenic autophagy, and increased the sensitivity of K562 cells to realgar NPs. Therefore, the results demonstrated that realgar NPs degraded Bcr-Abl oncoprotein, while the underlying mechanism might be related to apoptosis and autophagy, and Cav-1 might be considered as a potential target for clinical comprehensive therapy of CML.
Keywords: realgar nanoparticles, Bcr-Abl fusion protein, apoptosis, autophagy, caveolin-1
Introduction
Chronic myelogenous leukemia (CML), also called chronic myeloid leukemia, accounts for ~15% of adult leukemia.1 CML is a stem cell driven hematological malignancy, which is characterized by the presence of the Philadelphia chromosome arising from the reciprocal translocation t(9;22) (q34;q11) between breakpoint cluster region (Bcr) and Abelson murine leukemia (Abl) genes.2 The fusion gene, Bcr-Abl, encodes the fusion protein Bcr-Abl, which shows constitutive tyrosine kinase activity.3 Clinical diagnosis of CML is based on the characteristics of splenomegaly, increased peripheral white blood cells, and Bcr-Abl fusion protein expression.4 The fusion protein gives rise to acceleration of cell proliferation, activation of transcription factors, and evasion of apoptosis, which ultimately lead to uncontrolled cell proliferation and immature differentiation.5
Imatinib is a standard first-line therapeutic drug for CML, which is a tyrosine kinase inhibitor (TKI), can interact with Bcr-Abl, and inhibit its activity. Imatinib has been used in clinical application for the treatment of CML since 2001.6 Although the majority of patients with CML have good responses to TKI therapy and live for many years, the quality of life is much lower than normal.7 In addition, over the past decade, some patients still did not respond adequately to imatinib, and ~25% showed primary imatinib resistance.6
Arsenic, a traditional Chinese medicine, has been widely employed for >2,000 years. It has been demonstrated that arsenic has positive effects on cancer therapy.8 In the 1970s, arsenic trioxide (As2O3) was first purified and then used for treating acute promyelocytic leukemia (APL) in China. Complete response rates rose from 52% to 92%.9 In addition, As2S2 inhibited proliferation and induced apoptosis in both K562 and primary CML mononuclear cells.10 Realgar, which contains As4S4 as a main component, has been widely used alone or in combination with other traditional Chinese medicines for >1,500 years in several disease therapies, such as syphilis, psoriasis, malaria, parasitic infections, and especially cancer.11 Research has demonstrated that bioleaching product realgar transforming solution exhibited significant effect against CML and multidrug-resistant CML cell line K562/ADM.12
However, it is insoluble in water and most organic reagents, which leads to poor bioavailability and limits its clinical application. Inspired by nano-drug, the realgar nanoparticles (NPs) have been prepared with an average particle size of <100 nm in a previous work. Compared with coarse realgar, realgar NPs inhibited cell viability more potently and prolonged the presence of arsenic in blood.13 In addition, it has been demonstrated that nano-sized realgar significantly reduced cell viability of human leukemia cell line HL-60 and K562 cells.14 These studies broaden the applicability of realgar NPs greatly.
Cell apoptosis and autophagy, both called programmed cell death, are two catabolic processes, in which cellular components are degraded by proteasome and lysosomal system, respectively. Class I phosphoinositide 3-kinase/protein kinase B/mammalian target of rapamycin (PI3K/Akt/mTOR) signaling pathway is a key pathway in regulating autophagy process.15 It is overactive in many cancers, including hematological malignancy.16 Some studies have demonstrated that PI3K/Akt/mTOR phosphorylation was strengthened after leukemia cell lines and leukemia stem/progenitor cells were treated with As2S2.17 It is worth noting that autophagy participates in the therapy-induced degradation of PML-RARA oncoprotein.18–20
Caveolin-1 (Cav-1), a 22–24 kDa integral membrane protein, is a principal structural component of caveolae membranes. It participates in a variety of cellular physiological and pathological processes by regulating signaling pathways.21 Recently, several studies have shown that autophagy is associated with Cav-1.22–24 Furthermore, we have found that Cav-1 is involved in the autophagy process induced by realgar NPs in human hepatocellular carcinoma cells in our previous work.25
However, the mechanism of realgar NPs in treating CML and whether Cav-1 participates in therapy-induced CML cells death remain unclear. For this purpose, K562 cells (a human Bcr-Abl+ CML cell line) were selected for the research. In the present study, first, the effects of realgar NPs on cellular cytotoxicity, apoptosis, autophagy, and differentiation, as well as the crucial role of Cav-1 in realgar NP therapy, were demonstrated. These data provide an underlying mechanism for therapeutic effects of realgar NPs and suggest a novel potential therapeutic target for CML in clinical treatment.
Materials and methods
Antibodies
Caspase-3 (#9665; Cell Signaling Technology, Danvers, MA, USA), Bcl2 (#2870; Cell Signaling Technology), Bax (#2772; Cell Signaling Technology), P62 (#7695; Cell Signaling Technology), Akt (#9272; Cell Signaling Technology), p-Akt (#4060; Cell Signaling), mTOR (#2983; Cell Signaling Technology), p-mTOR (#5536; Cell Signaling Technology), LC3 (L7543; Sigma-Aldrich, St Louis, MO, USA), Cav-1 (#3267; Cell Signaling Technology), Bcr-Abl (#2862; Cell Signaling Technology), p-Bcr-Abl (#3901; Cell Signaling Technology), CrkL (#3182; Cell Signaling Technology), p-CrkL(Tyr207) (#3181; Cell Signaling Technology), and β-actin (TA-09; ZSGB-BIO, Beijing, China) were used in the present study. Horseradish peroxidase–labeled secondary antibodies anti-mouse IgG (ZB-5305; ZSGB-BIO) and anti-rabbit IgG (ZB-2301; ZSGB-BIO) and fluorescence-labeled secondary antibodies goat anti-mouse IgG (ZF-0512; ZSGB-BIO) and anti-rabbit IgG (ZF-0511; ZSGB-BIO) were also used.
Preparation of realgar NPs
Realgar NPs were prepared by using high-energy ball milling, and the average particle size of 78±8.3 nm was acquired through the optimized preparation process. For detailed preparation methods, refer to Tian et al.13
Cell line and cell culture
Human CML K562 cell line was purchased from the Chinese Academy of Sciences Cell Library. K562 cells were cultured in Roswell Park Memorial Institute (RPMI)-1640 culture medium (C11875500BT; Gibco™, Grand Island, NY, USA) supplemented with 10% new born calf serum (16010-159; Gibco) and incubated in 5% CO2 at 37°C.
Peripheral blood leukocyte separation
Peripheral blood leukocytes were separated from 8- to 10-week-old Sprague Dawley rats purchased from Dalian Medical University Laboratory Animal Center by using peripheral white blood cell separation fluid (P8670; Solarbio, Beijing, China) according to the manufacturer’s instructions. Briefly, heparinized blood was diluted with an equal volume of phosphate-buffered saline (PBS) and layered on an equal volume of leukocyte separation fluid. The solution was centrifuged at 500× g for 25 min to separate leukocytes from red blood cells. The leukocyte layer was collected, washed once with red blood cell lysis buffer, and then washed twice with PBS. The cells were resuspended in RPMI-1640 culture medium. All animal-handling procedures were performed according to the guide for the care and use of laboratory animals of the National Institutes of Health and followed the guidelines of the Animal Welfare Act. All animal experiments were approved by the Experimental Animal Ethical Committee of Dalian Medical University.
Wright–Giemsa staining and hematoxylin–eosin (H&E) staining
Cells were collected by centrifugation and then resuspended with 1× PBS. Cell smears were prepared and dried at room temperature (RT). After the samples were dried, they were fixed in 4% paraformaldehyde at 4°C overnight. Wright–Giemsa dye solution (G1020, Solarbio) and H&E dye solution (G1140, Solarbio; G1100, Solarbio) were used to observe the cell morphologic changes under a light microscope by the following standard protocols.
Cell viability assay
In order to evaluate the cell viability, cell counting kit-8 (CCK-8; C0038; Beyotime, Shanghai, China) assay was performed according to the manufacturer’s instructions. Cells (1×104/well) were seeded into 96-well plates. Later, 10 μL/well of CCK-8 solution was added and incubated for 1 h. The absorbance was measured at 450 nm by using a scanning microplate spectrophotometer. Experiments were repeated in triplicate.
Fluorescence-activated cell sorting analysis
Cell apoptosis was detected by using the Annexin V-fluorescein isothiocyanate (FITC)/propidium iodide (PI) double staining kit (Becton Dickinson Biosciences Franklin Lakes, NJ, USA) according to the manufacturer’s instructions. Briefly, cells were harvested and washed with calcium-free PBS and then resuspended in 1× binding buffer. Subsequently, the cells were double-stained with Annexin V-FITC and PI for 15 min at RT in darkness. After mixed with 1× binding buffer, 5×104 cells per sample were analyzed by using flow cytometry (FCM; Becton Dickinson Biosciences). Data are presented as a percentage of the total cell count.
Transient transfection
The transient transfection was performed by using Lipofectamine® 2000 (Invitrogen Life Technologies, Carlsbad, CA, USA) according to the manufacturer’s protocol with minor modifications; 2×105 cells were seeded in 6-well plates, or 1×104 cells were seeded in 96-well plates. The complete media were replaced with serum-free media before transfection; 4 μg Cav-1 overexpression plasmid was mixed with Lipofectamine 2000. The mixture was vortexed and left for 20 min at RT before adding into wells. Serum was added into each well 4 h after transfection at a final concentration of 10%. After specific time, cells were harvested and subjected to Western blot or CCK-8 analysis.
Western blot
Cells were rinsed twice in PBS and lysed by radio immunoprecipitation assay buffer containing protease inhibitors and phosphatase inhibitors. The protein concentration was determined by using Bradford method. Proteins from total lysates were subjected to 5%–15% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and electrotransferred onto polyvinylidene difluoride membrane. The membrane was blocked with 5% nonfat milk in the mixture of PBS plus 0.1% Tween 20 (PBST) for 1 h and then incubated overnight with primary antibodies. The next day, after three washes in PBST for 10 min, the membrane was incubated with horseradish peroxidase–labeled second antibodies for 1 h at 37°C. After washing, blots were visualized by enhanced chemiluminescence substrate. Quantification of protein bands was performed by using the Gel-Pro analyzer Version 4 software.
Immunofluorescence
After treatment, cells were washed twice with PBS, and then cell smears were prepared. The cells were dried at RT, permeabilized with 0.1% Triton X-100 or 0.5% Triton X-100 for 10 min, and then fixed in 4% paraformaldehyde at 4°C overnight. After washing, the cells were blocked for 1 h with 1% bovine serum albumin (A8020; Solarbio). After blocking, the cells were incubated with primary antibody at 4°C overnight. Then, the cells were washed with PBS three times and incubated with fluorescence-labeled secondary antibody for 2 h at RT. The nuclei were counterstained with Hoechst 33342 (B2261; Sigma-Aldrich) for 5 min at RT. After washing twice, fluorescence signals were analyzed by using a fluorescence microscope (IX73; Olympus, Tokyo, Japan).
Statistical analysis
Data are expressed as mean ± standard deviation. Student’s t-test was used to evaluate the differences between two groups. P<0.05 was considered statistically significant.
Results
Realgar NPs induce cell death and fusion protein degradation in K562 cells
Initial studies were performed to identify the effects of realgar NPs on human myelogenous leukemia K562 cells by using CCK-8 assay. As shown in Figure 1A, the relative survival rate was significantly decreased after treatment with realgar NPs (0–100 μg/mL) in a dose- and time-dependent manner. Bcr-Abl fusion protein degradation is likely to be a determinant of clinical response of CML, which was the reason to investigate whether realgar NPs have an effect on Bcr-Abl fusion protein degradation. In order to address this question, K562 cells were harvested after treatment with specific concentrations of realgar NPs for Western blot analysis. The results showed that the expression of p-Bcr-Abl and Bcr-Abl protein was remarkably reduced in realgar NPs treatment group cells when compared with control group cells (Figure 1B–D). The phosphorylation level of CrkL, a known downstream effector of Bcr-Abl, was also remarkably decreased (Figure 1B and E). These findings suggest that the effect of realgar NPs on CML is probably achieved by degrading Bcr-Abl fusion protein.
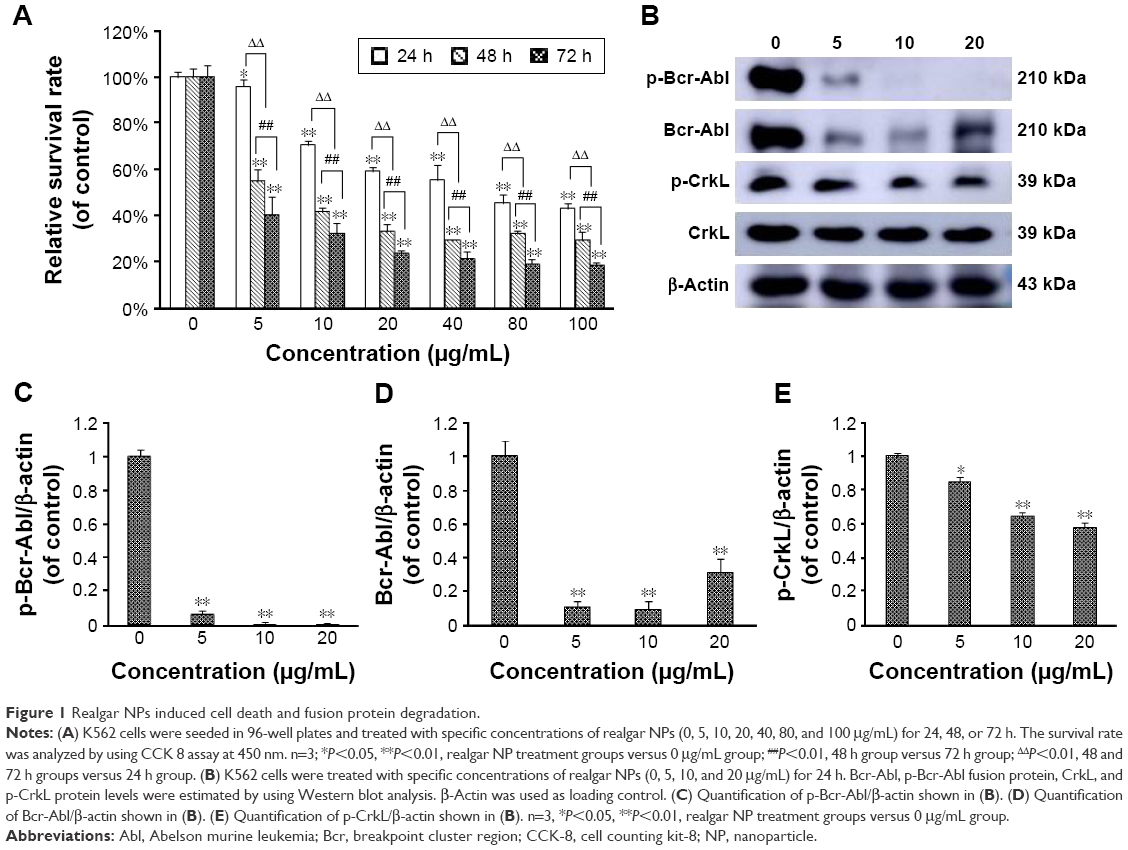 | Figure 1 Realgar NPs induced cell death and fusion protein degradation. |
Apoptosis is activated by realgar NPs
The molecular mechanism of realgar NPs induced cell death was explored. It has been reported that realgar was found to induce apoptosis and differentiation in APL cells.26 Therefore, whether realgar NPs induce apoptosis in CML cells was investigated. In order to achieve this, the apoptosis levels of each group were detected by using FCM. As shown in Figure 2A, after treatment with 10 μg/mL realgar NPs for 48 h, cells expressing Annexin V+/PI− and Annexin V+/PI+ were 24.9%, whereas the control group was 2.5%. In addition, caspase-3 and Bcl2/Bax levels were also used for evaluating apoptosis status. As shown in Figure 2B–F, the expression of cleaved caspase-3 was significantly increased in a dose- and time-dependent manner in realgar NPs treated K562 cells, whereas Bcl2/Bax, which indicates anti-apoptosis, was significantly decreased in the presence of realgar NPs. These results suggest that realgar NPs promote apoptosis significantly in CML cells.
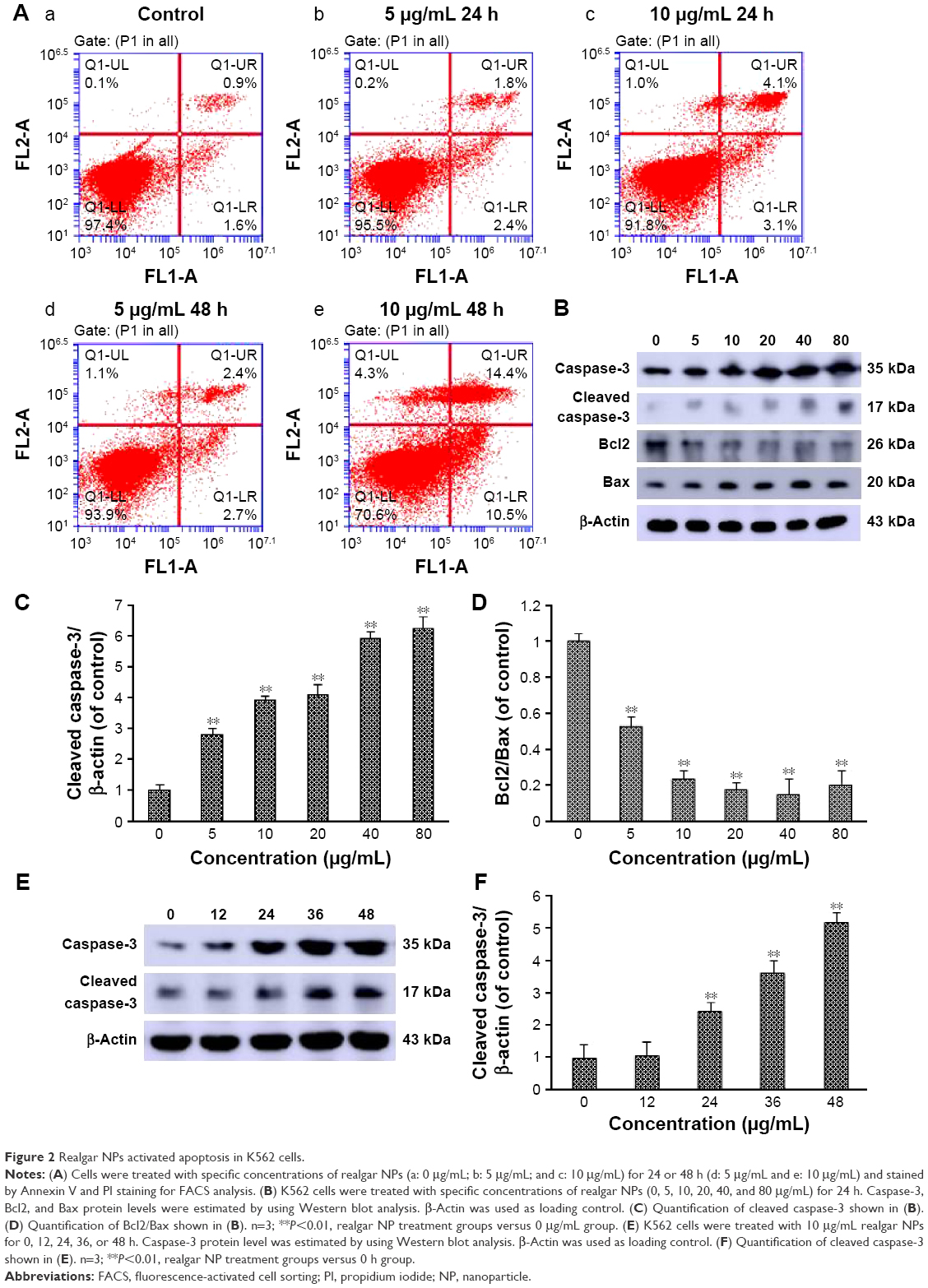 | Figure 2 Realgar NPs activated apoptosis in K562 cells. |
Realgar NPs induce cell autophagy probably through inhibiting PI3K/Akt/mTOR signaling pathway
In addition to the effect of realgar NPs on apoptosis activation, whether realgar NPs induced autophagy was then tested. For this purpose, K562 cells were harvested after treatment with specific concentrations of realgar NPs for 24 h. The mammalian autophagy protein, LC3, was widely regarded as the marker of autophagy. In contrast to the cytoplasmic localization of LC3-I, LC3-II participates in the membrane formation of autophagosome, which can be distinguished from LC3-I by using SDS-PAGE.27 As shown in Figure 3A, the fluorescence intensity of LC3 was enhanced after treatment with realgar NPs. In order to confirm the immunofluorescence results, protein for Western blot analysis was collected. The ratio of LC3-II/LC3-I was also significantly elevated after treatment with realgar NPs (Figure 3B and C).
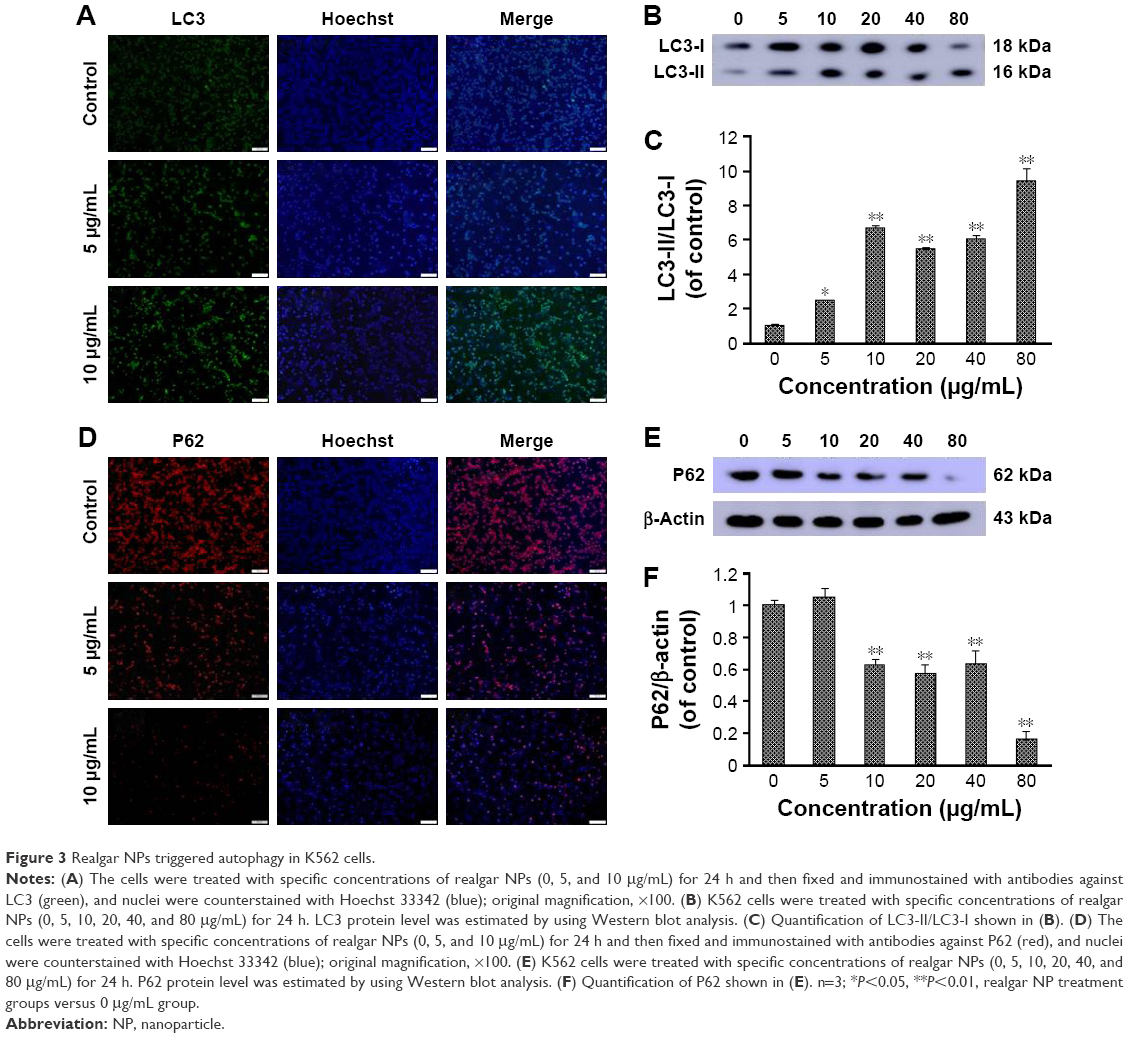 | Figure 3 Realgar NPs triggered autophagy in K562 cells. |
Studies have investigated whether several specific substrates are preferentially degraded through the autophagy–lysosome system, of which the best example is P62.27,28 Therefore, the changes in p62 expression after treatment with realgar NPs were tested by using immunofluorescence and Western blot analysis. As shown in Figure 3D–F, the expression of P62 protein was significantly reduced in realgar NPs treated cells. These data indicate that autophagy is activated by realgar NPs significantly. Notably, previous studies have indicated the crucial role of autophagy in fusion oncoprotein degradation.20 Collectively, these data suggest that autophagy may be involved in realgar NP therapy–induced degradation of Bcr-Abl fusion oncoprotein.
In order to determine whether realgar NPs induced autophagy activation involves the PI3K/Akt/mTOR signaling pathway, the phosphorylation levels of Akt and the downstream signal molecule mTOR after treatment with realgar NPs were detected. The results revealed that Akt and mTOR protein levels remained unchanged, but their phosphorylation status was significantly reduced in a dose-dependent manner in realgar NPs treated cells (Figure 4). The findings suggest that realgar NPs induce cell autophagy possibly by inhibiting PI3K/Akt/mTOR pathway.
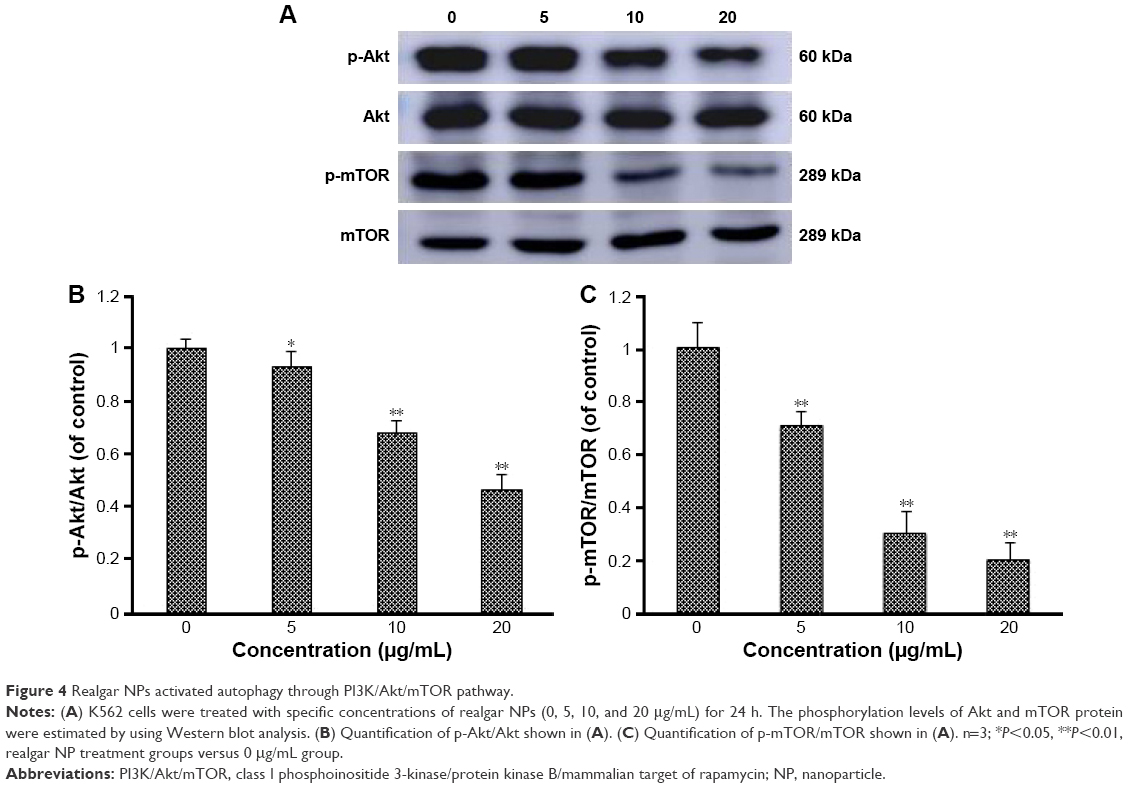 | Figure 4 Realgar NPs activated autophagy through PI3K/Akt/mTOR pathway. |
Realgar NPs induce cells differentiation
To address whether realgar NPs could induce CML cells differentiation, the morphologic changes in K562 cells in the presence or absence of realgar NPs were detected by using Wright–Giemsa staining and H&E staining. The findings showed notable morphologic changes in realgar NP treatment group when compared with control group, as shown in Figure 5. The cytoplasm became more abundant, and the nuclei to cytoplasm ratio decreased after treatment with 5 μg/mL realgar NPs for 48 h. The nucleus of control cells was large and round, whereas the nucleus became semilunar after treatment. These typical morphologic changes indicated a differentiation tendency for K562 cells in the presence of realgar NPs.
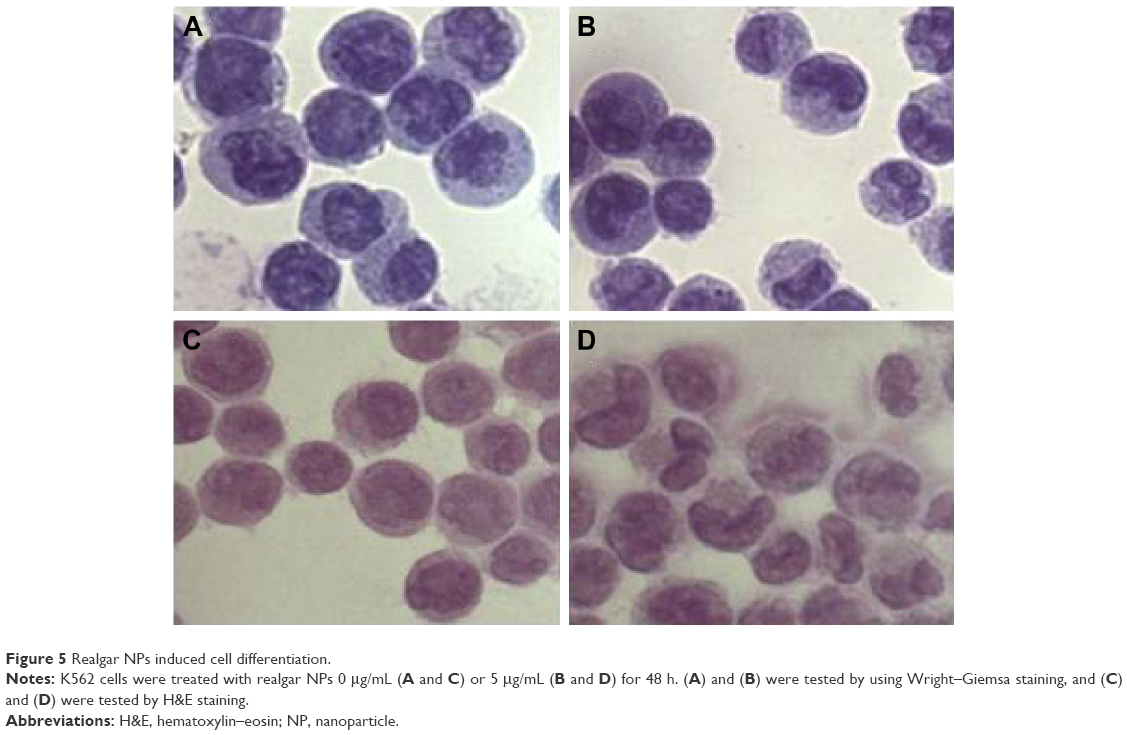 | Figure 5 Realgar NPs induced cell differentiation. |
Cav-1 overexpression increases the sensitivity of K562 cells to realgar NPs
It has been reported that Cav-1 plays a crucial role in the effects of oleanolic acid on APL cells through regulating cell activity, apoptosis, and cell cycle.29 In order to address whether Cav-1 is involved in the treatment process of realgar NPs, first, the level of endogenous Cav-1 in K562 cells was detected by using Western blot with rat peripheral blood leukocytes as positive control. As shown in Figure 6A and B, compared with rat peripheral blood leukocytes, only a small amount of Cav-1 was expressed in K562 cells. Therefore, K562 cells were transiently transfected with Cav-1 overexpression plasmid (Cav-1 OE) or negative control plasmid (Cav-1 NC) for the following research. After being transfected for 24, 48, and 72 h, the cells were harvested, respectively, and subjected to Western blot analysis. Compared with Cav-1 NC group, the level of Cav-1 expression was significantly increased at 24, 48, and 72 h after being transfected with Cav-1 overexpression plasmid (Figure 6C). The endogenous LC3-II protein was increased after Cav-1 overexpression (Figure 6D and E).
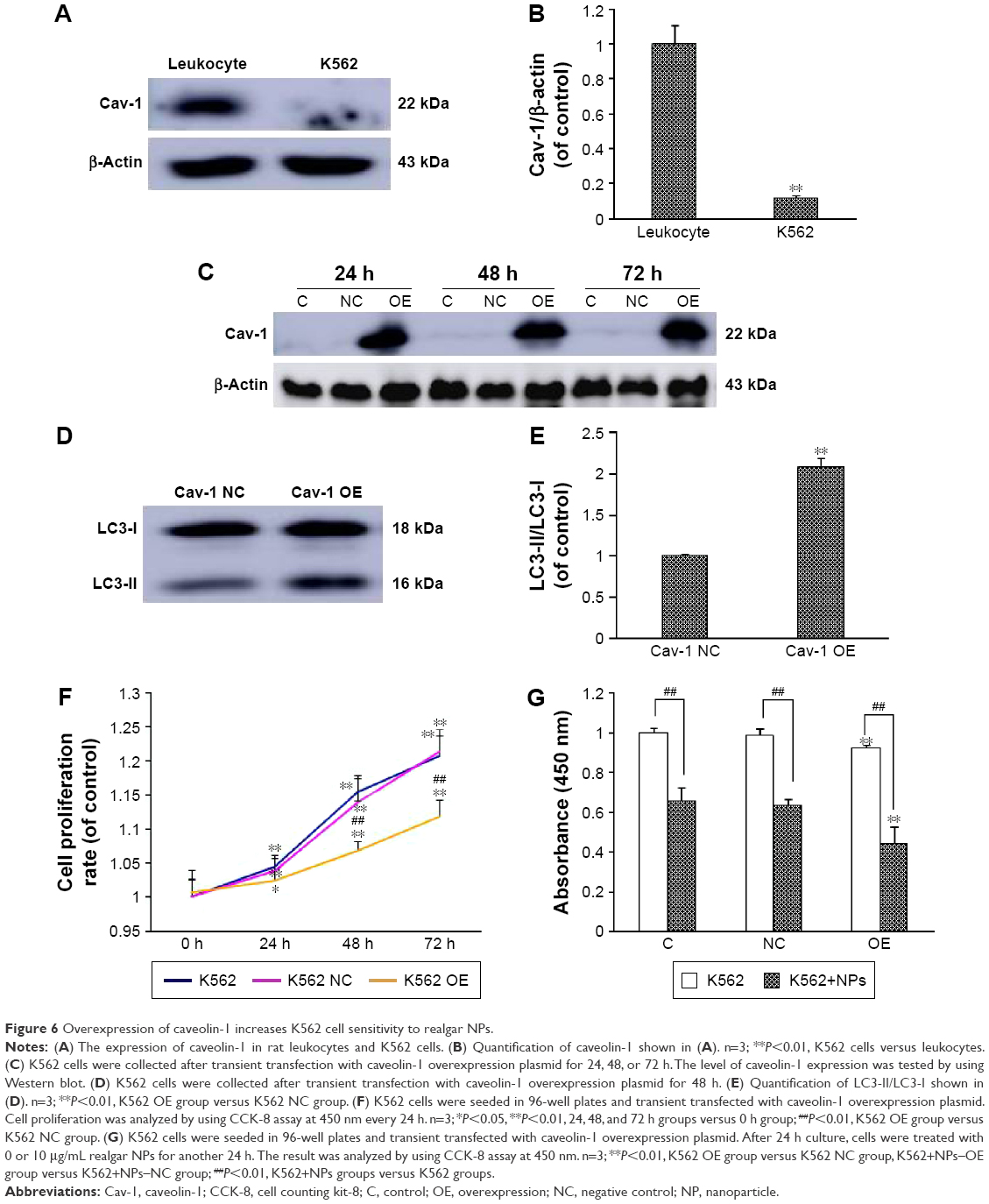 | Figure 6 Overexpression of caveolin-1 increases K562 cell sensitivity to realgar NPs. |
Then, in order to gain further insight into whether Cav-1 participates in regulating realgar NPs induced cell death, the rate of cell proliferation was detected by CCK-8 analysis. The results demonstrated that the rate of cell proliferation was decreased in Cav-1 overexpression cells (Figure 6F), and the cell proliferation inhibition was more significant in Cav-1 OE group than in Cav-1 NC group after treatment with 10 μg/mL realgar NPs for 24 h (Figure 6G). In other words, realgar NPs inhibited both K562 and K562-Cav-1 overexpression cell proliferation, and the inhibitory effect was more significant in K562–Cav-1 overexpression cells. These data indicate that overexpression of Cav-1 not only inhibits K562 cell proliferation, but also increases the sensitivity of K562 cells to realgar NPs.
Discussion
Realgar (As4S4) has been used as a traditional Chinese medicine for >1,500 years. Currently, the clinical application of realgar is not established due to its insolubility.30 Recently, several types of realgar NPs have been developed, and more effective results have been achieved compared with crude realgar in antitumor studies.13,31,32 It has been reported that realgar NP suspension prepared by “solvent relay” strategy induces both apoptosis and necrosis in leukemia cell lines K562 and HL-60.33 In the present study, realgar NP suspension prepared by using high-energy ball milling was used, as reported in a previous study.13 Previous studies have demonstrated that realgar NPs can inhibit the proliferation of human hepatoma carcinoma cells SMMC-7721, HepG2, human breast cancer cells MCF7, and human lung cancer cells A549.13,25 In the present study, it was demonstrated that treatment with realgar NPs (0–100 μg/mL) for 24, 48, or 72 h significantly decreased the relative survival rate of K562 cells in a dose- and time-dependent manner. Notably, Mao et al have investigated that As4S4 exhibits significant beneficial effects on inducing Bcr-Abl degradation in CML, and the mechanism is associated with c-CBL upregulation.34 Therefore, in the present study, the effect of realgar NPs on Bcr-Abl fusion protein degradation in CML cells was also investigated. It was found that both p-Bcr-Abl and Bcr-Abl proteins were remarkably decreased after realgar NPs treatment. In addition, there are several proteins that interact with Bcr-Abl fusion protein. One of the most important substrates and tyrosine-phosphorylated proteins is CrkL protein.35 CrkL can enhance the leukemogenesis mediated by Bcr-Abl and accelerate cell migration.36 The results from the present study demonstrated that realgar NP treatment was able to inhibit the activity of CrkL significantly. These results support the idea that realgar NPs can degrade Bcr-Abl fusion protein and inactivate the Bcr-Abl/CrkL pathway effectively.
Previous studies have investigated whether realgar NPs induce apoptosis in histiocytic lymphoma U937 cells.37,38 Another group reported that nano-realgar could inhibit the proliferation of different cervical carcinoma cell lines and induce cell apoptosis.39 In the present study, the percentage of apoptosis cells increased to 24.9% when treated with 10 μg/mL realgar NPs for 48 h, whereas the percentage in the control group was 2.5%. It is well understood that when apoptosis is activated, it engages a cascade of caspase enzyme activation. During apoptosis, pro-caspase-3 transforms to active enzyme when two cleaved monomers combine to form an active dimer.40 Meanwhile, the balance between prosurvival Bcl2 and proapoptotic protein Bax is crucial for cell survival, and the ratio of Bcl2/Bax is becoming representative of cell apoptosis.41 The present analysis of caspase-3 and Bcl2/Bax expression through Western blot showed that realgar NPs induced apoptosis in a dose- and time-dependent manner in K562 cells. All these data support the hypothesis that realgar NPs trigger apoptosis significantly.
Autophagy, an evolutionarily relatively conserved catabolic process that involves degradation of macromolecular substances or harmful cellular components via the lysosomal system is activated after TKI treatment in CML cells.42 Realgar NPs induced autophagy at lower dosage and apoptosis at higher concentration in melanoma cell lines.43 Consistent with these previous studies, the results revealed that the ratio of LC3-II/LC3-I was significantly increased, whereas the expression of p62 was decreased in K562 cells after realgar NP treatment. PI3K/Akt/mTOR is a key signaling pathway in regulating homeostasis of mammalian cells.44 Recently, studies have revealed that inhibiting PI3K shows a synergistic effect with imatinib on the activation of autophagy in K562 and cultured primary CML cells.45 Imatinib therapy induced autophagy by inhibiting PI3K/Akt/mTOR signaling pathway in CML cells.46 Similarly, in the present study, it was first found that realgar NP treatment reduced the activation of Akt with decreased phosphorylation of its direct downstream substrate of mTOR. Although how realgar NPs inhibit the phosphorylation of Akt and mTOR remains to be further explored, data from the present study testified to the notion that realgar NPs induced autophagy might involve the PI3K/Akt/mTOR signaling pathway. It has been demonstrated that imatinib therapy–induced autophagy degrades the Bcr-Abl oncoprotein in CML cells.47 Taken together, these results indicated that realgar NPs induced Bcr-Abl fusion protein degradation probably via autophagy pathway in CML cells, but the exact mechanism of fusion protein degradation remains to be further investigated.
Cell differentiation was detected by using Wright–Giemsa staining and H&E staining. Morphological changes, including the decreased nuclear to cytoplasm ratio and the nuclear remodeling transforming from rounded to polylobular nuclei, were observed under a light microscope. The results indicated that realgar NPs exerted an effective differentiation effect on K562 cells. Coincidentally, recent research shows that natural drug angelica sinensis polysaccharide is sufficient to induce K562 cell erythroid differentiation.48 In contrast to APL, the development of differentiation therapy for CML is still far from a satisfactory level, and the specific differentiation mechanism still needs to be further investigated.
Cav-1 is a principal constituent protein of caveolae membranes, which regulates multiple physiological and pathological processes, including cell proliferation, survival, metastasis, and migration.49 As a tumor suppressor, Cav-1 is frequently insignificant in various types of malignant cells, such as breast, ovarian carcinomas, and colon cancers.50 It has been reported that compared with peripheral blood leukocytes, only a small amount of Cav-1 is expressed in K562 cells.29 Similarly, only a small amount of Cav-1 was detected in K562 cells in the present study when compared with rat peripheral blood leukocytes. The stable Cav-1 overexpression until 72 h after transfection suggests that the model of Cav-1 overexpression K562 cells is successful. The endogenic autophagy was increased after Cav-1 overexpression, whereas the reason might be associated with inhibiting PI3K/Akt/mTOR signaling pathway. Wang et al have investigated that downregulation of Cav-1 can accelerate the progress of cell cycle in mammary cells via upregulation of cyclin D1.51 Cav-1 overexpression can inhibit HL-60 cell proliferation.29 Consistent with the previous reports, the results from the present study indicate that the proliferation of K562 cells is inhibited after Cav-1 overexpression, and the Cav-1 overexpression K562 cells are more sensitive to realgar NPs when compared with negative control group. Thus, Cav-1 may be considered as a potential target for comprehensive therapy by enhancing the sensitivity of CML treatment with realgar NPs. However, Gilling et al reported that knockdown of Cav-1 expression in chronic lymphocytic leukemia cells resulted in significantly decreased cell proliferation.52 The discrepancy may be caused by differences in cell type, regulatory pathway, and function in and/or out of caveolae.
Conclusion and perspectives
In the present study, it was demonstrated that realgar NPs inhibit the proliferation of CML cells and degrade the Bcr-Abl oncoprotein, while the underlying mechanism may be associated with apoptosis, autophagy, as well as differentiation, and Cav-1 may play a crucial role in realgar NP therapy (Figure 7).
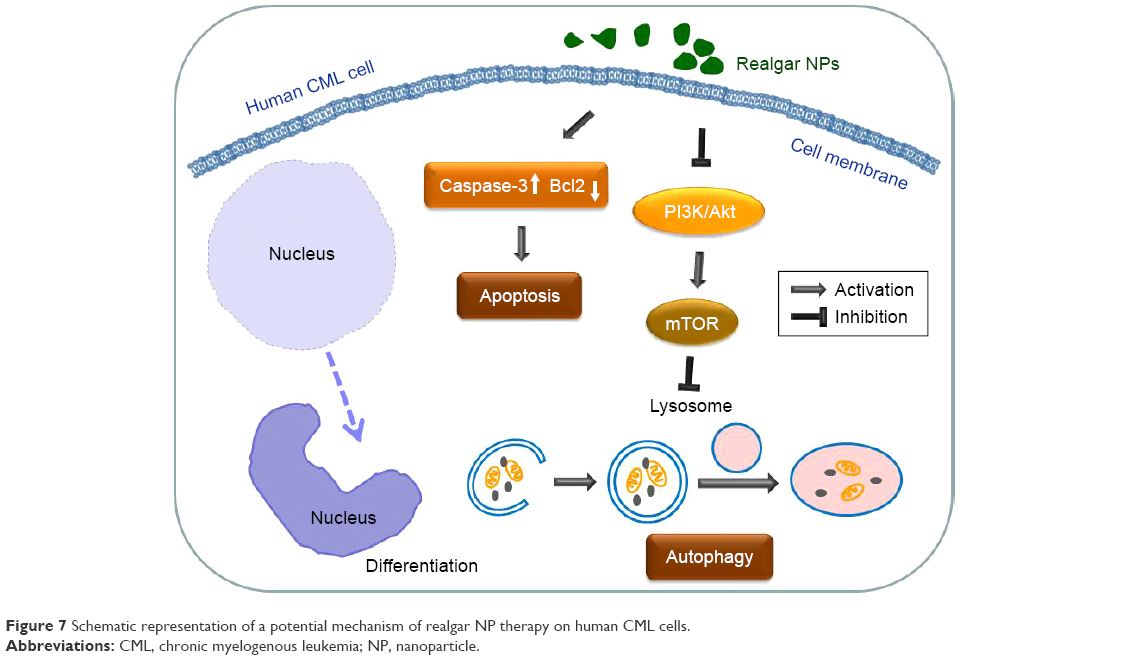 | Figure 7 Schematic representation of a potential mechanism of realgar NP therapy on human CML cells. |
Hence, further studies should be conducted to establish a suitable animal model to explore in vivo anticancer activity of realgar NPs. Moreover, some measures should be taken to overcome the limitation of poor water solubility of realgar NPs and increase the scope for clinical application.
Acknowledgments
The present work was supported and funded by the National Research and Development of Important New Drugs Foundation (No 2009ZXJ 09002-019) and the Key Projects of Shenyang Military Region (CSY13J003). The authors express their special thanks to Dr Wei Zou for generously providing the Cav-1 overexpression plasmid.
Disclosure
The authors report no conflicts of interest in this work.
References
National Comprehensive Cancer Network. NCCN guidelines for patients: chronic myelogenous leukemia. 2016; Version 1. Available from: www.nccn.org/patients/guidelines/cml/index.html#8. Accessed June 17, 2016. | ||
Sawyers CL. Chronic myeloid leukemia. N Engl J Med. 1999;340(17):1330–1340. | ||
Melo JV. The diversity of BCR-ABL fusion proteins and their relationship to leukemia phenotype. Blood. 1996;88(7):2375–2384. | ||
Fu GN, Fan HY, Han XJ, Xin CL. Complex chromosomal rearrangements involving five chromosomes in chronic myelogenous leukemia: a case report. Oncol Lett. 2016;11(4):2651–2653. | ||
Kujak C, Kolesar JM. Treatment of chronic myelogenous leukemia. Am J Health Syst Pharm. 2016;73(3):113–120. | ||
Baccarani M, Deininger MW, Rosti G, et al. European LeukemiaNet recommendations for the management of chronic myeloid leukemia: 2013. Blood. 2013;122(6):872–884. | ||
Phillips KM, Pinilla-Ibarz J, Sotomayor E, et al. Quality of life outcomes in patients with chronic myeloid leukemia treated with tyrosine kinase inhibitors: a controlled comparison. Support Care Cancer. 2013;21(4):1097–1103. | ||
Waxman S, Anderson KC. History of the development of arsenic derivatives in cancer therapy. Oncologist. 2001;6(Suppl 2):3–10. | ||
Chen Z, Chen GQ, Shen ZX, et al. Expanding the use of arsenic trioxide: leukemias and beyond. Semin Hematol. 2002;39(2 Suppl 1): 22–26. | ||
Li JE, Wu WL, Wang ZY, Sun GL. Apoptotic effect of As2S2 on K562 cells and its mechanism. Acta Pharmacol Sin. 2002;23(11):991–996. | ||
Lu DP, Qiu JY, Jiang B, et al. Tetra-arsenic tetra-sulfide for the treatment of acute promyelocytic leukemia: a pilot report. Blood. 2002;99(9):3136–3143. | ||
Wang X, Zhang X, Xu Z, Wang Z, Yue X, Li H. Reversal effect of arsenic sensitivity in human leukemia cell line K562 and K562/ADM using realgar transforming solution. Biol Pharm Bull. 2013;36(4):641–648. | ||
Tian Y, Wang X, Xi R, et al. Enhanced antitumor activity of realgar mediated by milling it to nanosize. Int J Nanomedicine. 2014;9:745–757. | ||
Baláž P, Fabián M, Pastorek M, Sedlák J. Mechanochemical preparation and anticancer effect of realgar As4S4 nanoparticles. Mater Lett. 2009;63(17):1542–1544. | ||
Zhao GX, Pan H, Ouyang DY, He XH. The critical molecular interconnections in regulating apoptosis and autophagy. Ann Med. 2015;47(4):305–315. | ||
Bertacchini J, Heidari N, Mediani L, et al. Targeting PI3K/AKT/mTOR network for treatment of leukemia. Cell Mol Life Sci. 2015;72(12):2337–2347. | ||
Hong Z, Xiao M, Yang Y, et al. Arsenic disulfide synergizes with the phosphoinositide 3-kinase inhibitor PI-103 to eradicate acute myeloid leukemia stem cells by inducing differentiation. Carcinogenesis. 2011;32(10):1550–1558. | ||
Isakson P, Bjørås M, Bøe SO, Simonsen A. Autophagy contributes to therapy-induced degradation of the PML/RARA oncoprotein. Blood. 2010;116(13):2324–2331. | ||
Wang Z, Cao L, Kang R, et al. Autophagy regulates myeloid cell differentiation by p62/SQSTM1-mediated degradation of PML-RARα oncoprotein. Autophagy. 2011;7(4):401–411. | ||
Zeng CW, Chen ZH, Zhang XJ, et al. MIR125B1 represses the degradation of the PML-RARA oncoprotein by an autophagy-lysosomal pathway in acute promyelocytic leukemia. Autophagy. 2014;10(10):1726–1737. | ||
Quest AF, Lobos-González L, Nuñez S, et al. The caveolin-1 connection to cell death and survival. Curr Mol Med. 2013;13(2):266–281. | ||
Martinez-Outschoorn UE, Pavlides S, Whitaker-Menezes D, et al. Tumor cells induce the cancer associated fibroblast phenotype via caveolin-1 degradation: implications for breast cancer and DCIS therapy with autophagy inhibitors. Cell Cycle. 2010;9(12):2423–2433. | ||
Liu Y, Wang Y, Shi D, et al. [Autophagy and caveolin-1 in cancer: a review]. Sheng Wu Gong Cheng Xue Bao. 2012;28(8):912–917. Chinese. | ||
Chen ZH, Cao JF, Zhou JS, et al. Interaction of caveolin-1 with ATG12-ATG5 system suppresses autophagy in lung epithelial cells. Am J Physiol Lung Cell Mol Physiol. 2014;306(11):L1016–L1025. | ||
Yan Liu, Dan Shi, Ronggang Xi, et al. Cav-1 knockdown enhances autophagy induced by realgar nanoparticles in a hepatocellular carcinoma SMMC-7721 cell line. J Comput Theor Nanosci. 2016;13(8): 5262–5268. | ||
Chen S, Fang Y, Ma L, Liu S, Li X. Realgar-induced apoptosis and differentiation in all-trans retinoic acid (ATRA)-sensitive NB4 and ATRA-resistant MR2 cells. Int J Oncol. 2012;40(4):1089–1096. | ||
Mizushima N, Yoshimori T, Levine B. Methods in mammalian autophagy research. Cell. 2010;140(3):313–326. | ||
Pankiv S, Clausen TH, Lamark T, et al. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem. 2007;282(33):24131–24145. | ||
Ma W, Wang DD, Li L, et al. Caveolin-1 plays a key role in the oleanolic acid-induced apoptosis of HL-60 cells. Oncol Rep. 2014;32(1): 293–301. | ||
Wu J, Shao Y, Liu J, Chen G, Ho PC. The medicinal use of realgar (As4S4) and its recent development as an anticancer agent. J Ethnopharmacol. 2011;135(3):595–602. | ||
Xie QJ, Cao XL, Bai L, Wu ZR, Ma YP, Li HY. Anti-tumor effects and apoptosis induction by Realgar bioleaching solution in Sarcoma-180 cells in vitro and transplanted tumors in mice in vivo. Asian Pac J Cancer Prev. 2014;15(6):2883–2888. | ||
Qin YU, Wang H, Liu ZY, Liu J, Wu JZ. Realgar quantum dot induce apoptosis and necrosis in HepG2 cells through endoplasmic reticulum stress. Biomed Rep. 2015;3(5):657–662. | ||
Ning N, Peng ZF, Yuan L, Gou BD, Zhang TL, Wang K. [Realgar nano-particles induce apoptosis and necrosis in leukemia cell lines K562 and HL-60]. Zhongguo Zhong Yao Za Zhi. 2005;30(2):136–140. Chinese. | ||
Mao JH, Sun XY, Liu JX, et al. As4S4 targets RING-type E3 ligase c-CBL to induce degradation of BCR-ABL in chronic myelogenous leukemia. Proc Natl Acad Sci U S A. 2010;107(50):21683–21688. | ||
Salgia R, Pisick E, Sattler M, et al. p130CAS forms a signaling complex with the adapter protein CRKL in hematopoietic cells transformed by the BCR/ABL oncogene. J Biol Chem. 1996;271(41):25198–25203. | ||
Seo JH, Wood LJ, Agarwal A, et al. A specific need for CRKL in p210BCR-ABL-induced transformation of mouse hematopoietic progenitors. Cancer Res. 2010;70(18):7325–7335. | ||
Wang XB, Gao HY, Hou BL, Huang J, Xi RG, Wu LJ. Nanoparticle realgar powders induce apoptosis in U937 cells through caspase MAPK and mitochondrial pathways. Arch Pharm Res. 2007;30(5):653–658. | ||
Xi RG, Huang J, Li D, Wang XB, Wu LJ. Roles of PI3-K/Akt pathways in nanoparticle realgar powders-induced apoptosis in U937 cells. Acta Pharmacol Sin. 2008;29(3):355–363. | ||
Li L, Wang L, Xiao S, Li Y, Cheng C, Xue M. [Effect of nano-realgar on proliferation and apoptosis of human cervical carcinoma cells]. Zhong Nan Da Xue Xue Bao Yi Xue Ban. 2015;40(10):1068–1075. Chinese. | ||
Wall DM, McCormick BA. Bacterial secreted effectors and caspase-3 interactions. Cell Microbiol. 2014;16(12):1746–1756. | ||
Portt L, Norman G, Clapp C, Greenwood M, Greenwood MT. Anti-apoptosis and cell survival: a review. Biochim Biophys Acta. 2011;1813(1):238–259. | ||
Helgason GV, Mukhopadhyay A, Karvela M, Salomoni P, Calabretta B, Holyoake TL. Autophagy in chronic myeloid leukaemia: stem cell survival and implication in therapy. Curr Cancer Drug Targets. 2013;13(7):724–734. | ||
Pastorek M, Gronesova P, Cholujova D, et al. Realgar (As4S4) nanoparticles and arsenic trioxide (As2O3) induced autophagy and apoptosis in human melanoma cells in vitro. Neoplasma. 2014;61(6):700–709. | ||
Cargnello M, Tcherkezian J, Roux PP. The expanding role of mTOR in cancer cell growth and proliferation. Mutagenesis. 2015;30(2):169–176. | ||
Ciarcia R, Damiano S, Montagnaro S, et al. Combined effects of PI3K and SRC kinase inhibitors with imatinib on intracellular calcium levels, autophagy, and apoptosis in CML-PBL cells. Cell Cycle. 2013;12(17):2839–2848. | ||
Sheng Z, Ma L, Sun JE, Zhu LJ, Green MR. BCR-ABL suppresses autophagy through ATF5-mediated regulation of mTOR transcription. Blood. 2011;118(10):2840–2848. | ||
Elzinga BM, Nyhan MJ, Crowley LC, O’Donovan TR, Cahill MR, McKenna SL. Induction of autophagy by imatinib sequesters Bcr-Abl in autophagosomes and down-regulates Bcr-Abl protein. Am J Hematol. 2013;88(6):455–462. | ||
Wang L, Jiang R, Song SD, Hua ZS, Wang JW, Wang YP. Angelica sinensis polysaccharide induces erythroid differentiation of human chronic myelogenous leukemia k562 cells. Asian Pac J Cancer Prev. 2015;16(9):3715–3721. | ||
Boscher C, Nabi IR. Caveolin-1: role in cell signaling. Adv Exp Med Biol. 2012;729:29–50. | ||
Williams TM, Lisanti MP. Caveolin-1 in oncogenic transformation, cancer, and metastasis. Am J Physiol Cell Physiol. 2005;288(3):C494–C506. | ||
Wang X, Feng S, Zhang H, et al. RNA inference-mediated caveolin-1 down-regulation decrease estrogen receptor alpha (ERα) signaling in human mammary epithelial cells. Mol Biol Rep. 2011;38(2):761–768. | ||
Gilling CE, Mittal AK, Chaturvedi NK, et al. Lymph node-induced immune tolerance in chronic lymphocytic leukaemia: a role for caveolin-1. Br J Haematol. 2012;158(2):216–231. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.