Back to Journals » Clinical Ophthalmology » Volume 19
Cataract Induced by Glucocorticoids
Authors Jian YF, Zhang JS ![]() , Wan XH
, Wan XH
Received 6 May 2025
Accepted for publication 12 September 2025
Published 6 October 2025 Volume 2025:19 Pages 3703—3712
DOI https://doi.org/10.2147/OPTH.S537700
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Scott Fraser
Yu-Feng Jian, Jing-Shang Zhang, Xiu-Hua Wan
Beijing Tongren Eye Center, Beijing Tongren Hospital of Capital Medical University, Beijing Key Laboratory of Ophthalmology and Vision Science, Beijing, 100730, People’s Republic of China
Correspondence: Xiu-Hua Wan, Email [email protected] Jing-Shang Zhang, Email [email protected]
Abstract: Glucocorticoids (GCs) remain a cornerstone therapy for noninfectious uveitis and autoimmune disorders; however, chronic administration is strongly associated with sight-threatening complications, particularly glucocorticoid-induced cataracts (GIC). This comprehensive review synthesizes current evidence on the molecular pathogenesis, epidemiological patterns, and clinical management of GIC. Epidemiological analyses indicate that over 50% of patients receiving systemic corticosteroids for > 60 days develop ocular complications, with cataract formation (36%) and glaucoma (16%) representing the predominant sequelae. Histopathologically, GIC manifests as posterior subcapsular opacities, mechanistically linked to oxidative stress, epithelial-mesenchymal transition (EMT), vimentin dysregulation, Na+/K+-ATPase inhibition, apoptosis, and endoplasmic reticulum (ER) stress. Risk stratification models identify cumulative GC dose (> 20,000 mg/m² prednisolone equivalents), treatment duration (> 6 months), and administration route (oral > topical > intravitreal) as critical determinants of cataractogenesis. Although early-stage GIC is clinically silent, progressive opacification leads to debilitating visual acuity loss, photophobia, and impaired quality of life. Current interventions encompass antioxidants, molecular targeting strategies, advanced drug delivery systems, and glucocorticoid-sparing agents. Through systematic integration of epidemiology, pathogenesis, and therapeutic advances, we aim to resolve the GC therapeutic paradox and provide robust frameworks for future clinical management.
Keywords: glucocorticoid, glucocorticoid induced cataract, intervention
Introduction: Glucocorticoid Induced Cataract(GIC)
Glucocorticoid Induced Cataract(GIC)
Glucocorticoid-induced cataract (GIC) represents a pervasive therapeutic paradox: while glucocorticoids (GCs) remain indispensable for managing sight-threatening inflammatory disorders, their chronic use induces blinding posterior subcapsular opacities in 36% of patients, highlighting the critical trade-off between anti-inflammatory efficacy and lens toxicity. This iatrogenic complication imposes substantial global burden. Contemporary evidence reveals alarming risk heterogeneity: pediatric nephrotic syndrome cohorts exhibit 18.1% GIC incidence after 4.3 years of oral prednisolo28,669 mg/m²), while myeloma patients show 36% incidence within six dexamethasone cycles—a 21-fold higher risk than localized sub-Tenon administration (1.7%). Crucially, this dose-route-risk continuum positions targeted delivery as the cornerstone of GIC mitigation.
Unlike prior reviews focusing solely on epidemiology or isolated mechanisms, three critical gaps persist despite six decades of research. First, molecular pathogenesis remains fragmented—oxidative stress, EMT, and vimentin dysregulation operate in isolation rather than as interconnected effectors of glucocorticoid-induced mitochondrial-ER stress crosstalk. Second, clinical risk models ignore pharmacogenomic variables like GR-α polymorphisms—known modifiers of GC sensitivity—despite GR signaling’s role in Na+/K+-ATPase suppression. Third, emerging solutions lack translational frameworks: while polymeric nanoparticles reduce lens GR-α activation, clinical adoption remains limited. This review therefore establishes a tripartite framework addressing: (1) Mechanistic consolidation via ROS-ER-mitochondria cascade; (2) Risk model refinement with GR-α genotypes; (3) Ocular targeting optimization through nanocarriers.
Structurally, we begin by decoding epidemiological determinants through dose-route-susceptibility correlations. We then integrate molecular mechanisms—from oxidative crystallin aggregation to CSPG5-mediated EMT—into a unified pathophysiology. Subsequent sections evaluate targeted interventions including antioxidant combotherapy, CRISPR-based editing, and nanoscale delivery systems. Validation of glucocorticoid-sparing protocols and establishment of precision algorithms incorporating CRISPR-screening and TRB3 biomarkers complete this progression from mechanism to clinical translation, resolving GCs’ therapeutic dilemma.
Epidemiology
The risk profile of GIC exhibits marked heterogeneity across patient populations, reflecting differential susceptibility to glucocorticoid toxicity [Table 1]. In pediatric cohorts receiving long-term oral corticosteroids for idiopathic nephrotic syndrome, GIC incidence reached 18.1% (20/110 cases), with elevated cumulative doses (median: 28,669 mg/m² vs 14,995 mg/m² in non-affected controls) and extended treatment duration (4.3 vs 2.25 years) demonstrating significant dose-response relationships.1 Similarly, children with systemic autoimmune diseases (SADs) treated with oral glucocorticoids exhibited progressive cataract development, with incidence rising from 17.6% at 6 months to 41.2% at 18 months, frequently accompanied by steroid-induced ocular hypertension (SIOH).2
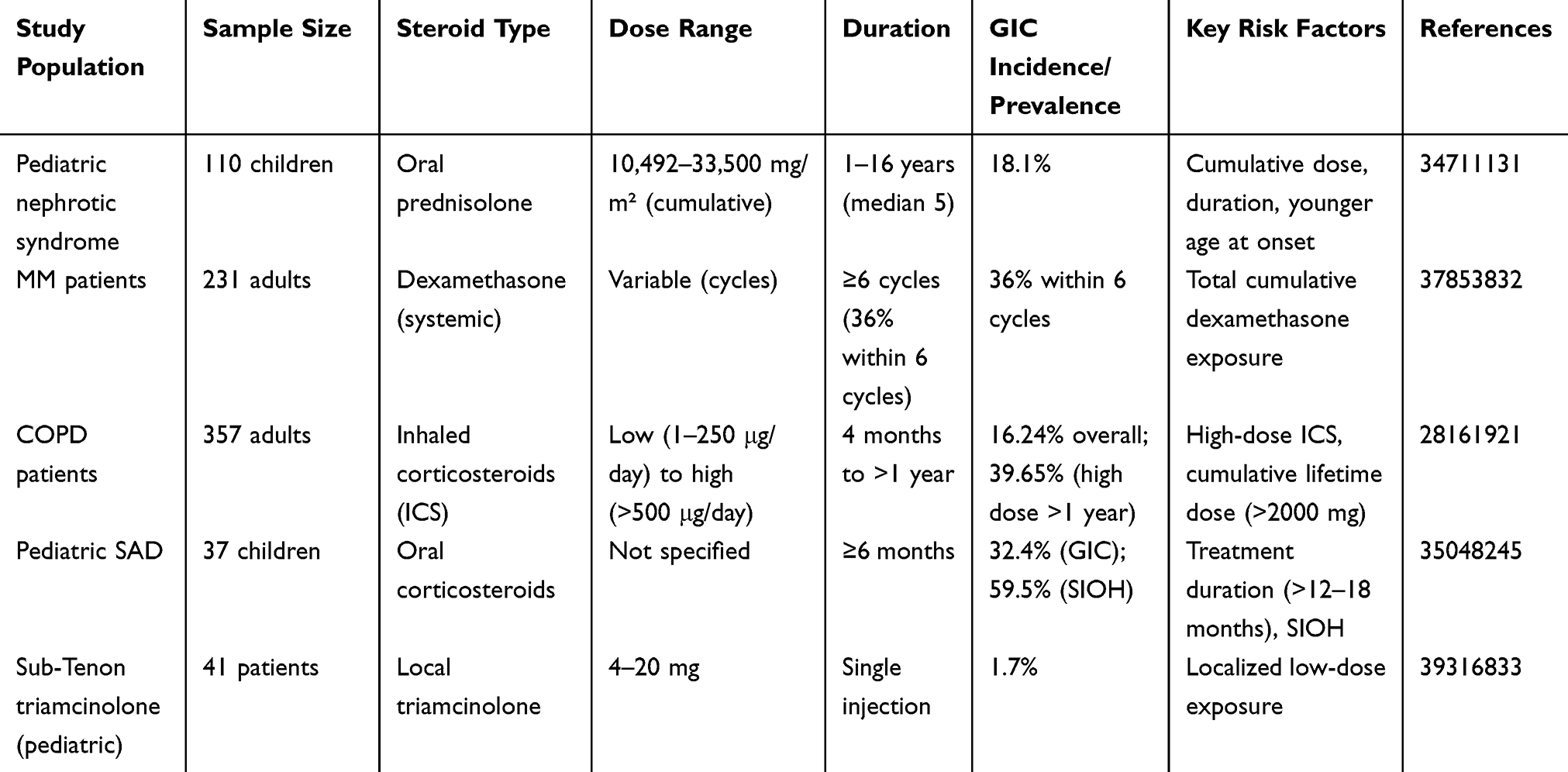 |
Table 1 The Prevalence and Risk Factors of Glucocorticoid -Induced Cataracts (GIC) in Different Patient Groups |
Adult populations display distinct risk patterns: multiple myeloma (MM) patients on dexamethasone-containing regimens developed cataracts with notable rapidity (36% incidence within six treatment cycles), where total cumulative dexamethasone dose directly correlated with cataract severity.3 COPD cohorts using inhaled corticosteroids (ICS) manifested dose- and duration-dependent risks, with high-dose ICS (>500 μg/day fluticasone equivalents) conferring 39.65% cataract prevalence after >12 months of exposure, contrasting sharply with low-dose ICS (<250 μg/day) showing no significant risk.4 Notably, localized administration via sub-Tenon triamcinolone injections (4–20 mg) demonstrated minimal cataractogenicity (1.7% incidence) in pediatric retinal photocoagulation cases, highlighting administration route as a key risk modifier.5
Methods
PubMed searches were performed using the terms “GIC”, or “SIC”, AND “glucocorticoid induced cataract”, or “steroid induced cataract”. The search was performed in July of 2025, with an additional search in May 2025. All original English language articles were considered for this review.
Mechanism
Oxidative Stress
Oxidative stress constitutes a pivotal pathogenic mechanism in GIC, characterized by disrupted equilibrium between reactive oxygen species (ROS) generation and antioxidant defense capacity. Within the lens microenvironment, excessive ROS accumulation or impaired clearance mechanisms destabilize redox homeostasis, precipitating crystallin aggregation, epithelial apoptosis, and progressive opacification.6,7 Lens epithelial cells (LECs) - essential guardians of transparency - undergo programmed cell death when ROS-mediated inactivation of Na+/K+-ATPase triggers intracellular sodium/water overload, a critical pathway to lens opacification.6
Aging potentiates redox imbalance through dual mechanisms: diminished synthesis of endogenous antioxidants (eg, glutathione, GSH) and impaired recycling pathways, which collectively promote disulfide crosslinking in crystallins.7 Experimental evidence identifies GSH depletion as a hallmark of GIC pathogenesis, with hydrocortisone (HC)-treated models demonstrating inverse correlations between lens/liver GSH levels and oxidative markers: 50% hepatic GSH reduction within 24 hours precedes 8–10-fold elevations in lipid peroxides (LPO), culminating in detectable lens opacities by 48 hours.8,9 Insulin administration reverses these metabolic perturbations, confirming the systemic dimension of oxidative injury in cataractogenesis.9
Endoplasmic reticulum (ER) stress amplifies oxidative damage through calcium dyshomeostasis and sustained activation of the unfolded protein response (UPR). Notably, selenium toxicity exacerbates ER stress via microtubule destabilization, creating a permissive environment for accelerated cataract formation.10
Synthesis of these pathways reveals a multimodal oxidative assault: ROS/RNS-mediated protein oxidation, lipid peroxidation chain reactions, and DNA damage collectively compromise lens structural integrity. This mechanistic framework underscores the interdependence of localized oxidative injury and systemic redox dysregulation in GIC progression.10 These temporal metabolic shifts underscore the critical window for antioxidant intervention.
Epithelial-Mesenchymal Transition
Epithelial-mesenchymal transition (EMT) in LECs emerges as a pivotal pathogenic driver of GIC, mediating lens fibrosis and posterior subcapsular opacification through coordinated molecular reprogramming.11 This phenotypic transformation is defined by the transcriptional silencing of epithelial markers (E-cadherin, zonula occludens-1) and concomitant upregulation of mesenchymal effectors (N-cadherin, vimentin, fibronectin, α-smooth muscle actin [α-SMA]), accompanied by cytoskeletal destabilization and pathological extracellular matrix (ECM) deposition.12
The lens microenvironment facilitates EMT activation through TGF-β2-dominated signaling, wherein the canonical Smad2/3 pathway induces nuclear translocation of EMT transcription factors, driving spindle-shaped morphological transition and invasive LEC migration.12 Complementing this mechanism, endoplasmic reticulum (ER) stress activates unfolded protein response (UPR) sensors (GRP78, ATF6, IRE1α) in human LECs (HLECs), as evidenced by tunicamycin/thapsigargin models showing E-cadherin suppression and mesenchymal marker induction.13
GCs exert dual disruptive effects: dexamethasone (Dex) potentiates EMT through coordinated downregulation of N-cadherin, α/β-catenin complexes, and glucocorticoid receptor (GR) expression, thereby destabilizing intercellular junctions and promoting LEC motility.14 Counterregulatory pathways exist, as nerve growth factor (NGF) reverses Dex-induced EMT by restoring p38 MAPK/Akt phosphorylation dynamics, effectively suppressing α-SMA and fibronectin overexpression.15
Notably, chondroitin sulfate proteoglycan 5 (CSPG5) – overexpressed in GIC patients – orchestrates EMT via EZH2/B-Myb nuclear translocation. CRISPR-mediated CSPG5 knockdown abrogates Dex-triggered fibronectin expression, F-actin remodeling, and migratory phenotypes, validating its therapeutic targeting potential.16
Integrative analysis reveals an interconnected EMT network in GIC pathogenesis, encompassing four core pathways: 1.TGF-β/Smad-mediated transcriptional reprogramming; 2.ER stress/UPR-driven proteostatic imbalance; 3.NGF/TrkA signaling axis dysregulation; 4.CSPG5/EZH2 epigenetic modulation. These convergent mechanisms synergistically promote fibrotic plaque formation through ECM overproduction and lens architecture disruption, ultimately compromising optical clarity. These convergent mechanisms synergistically promote fibrotic plaque formation, though their relative contributions remain incompletely stratified in clinical cohorts.
Vimentin
Vimentin, a type III intermediate filament protein (57 kDa, 466 residues), constitutes an essential cytoskeletal regulator in LECs, orchestrating structural stabilization and modulating fundamental processes including apoptotic signaling, mechanotransduction, and focal adhesion dynamics.17 Mammalian lenses demonstrate spatial-temporal regulation of vimentin expression, with prominent immunoreactivity in epithelial and elongating cortical fiber cells that progressively diminishes in mature nuclear fibers, mirroring its stage-specific roles in lens morphogenesis.
Experimental models establish vimentin dysregulation as a direct mediator of cataractogenesis. Dexamethasone (Dex), a synthetic glucocorticoid (GC), induces post-transcriptional downregulation of vimentin protein in LECs independent of mRNA modulation, correlating with lens opacification and cytoskeletal destabilization.18 Mechanistic studies reveal vimentin-glucocorticoid receptor (GR) physical interaction, with GR transcriptional activity governing vimentin expression. Pharmacological GR antagonism using RU486 preserves lens transparency through vimentin level restoration, confirming GR-dependent regulatory circuitry.18 This GR-vimentin axis represents a targetable pathway, though clinical translation requires further validation.
The redox-sensitive molecular architecture of vimentin positions it as a critical oxidative stress sensor. Cysteine residue oxidation induces pathogenic disulfide bridging, triggering filament disassembly and forming cytoplasmic aggregates characteristic of cataractous lenses.17 These structural alterations impair vimentin’s essential functions in organelle spatial coordination, signaling hub assembly, and biomechanical resistance, ultimately disrupting optical clarity.17,19
Transgenic evidence delineates vimentin’s dosage-dependent homeostatic control: overexpression disrupts fiber cell differentiation through impaired signaling coordination, while knockout models compromise epithelial integrity via disrupted intercellular communication. This dual regulatory paradigm establishes vimentin as both a structural stabilizer and redox rheostat.
Glucocorticoid-induced cataract pathogenesis converges on vimentin dysregulation through two synergistic mechanisms: 1. GR-mediated transcriptional suppression; 2. Oxidative modification-induced filament destabilization. These pathways collectively disrupt lens cytoarchitecture through microtubule-actin network decoupling and organelle mispositioning.17,18
Na/K-ATPase
The sodium-potassium adenosine triphosphatase (Na+/K+-ATPase), a pivotal ion transporter in lens physiology, sustains electrolyte homeostasis through precise regulation of intracellular sodium (Na+) and potassium (K+) gradients, thereby maintaining lens transparency via osmotic equilibrium.20 Both hypoactive and hyperactive states of Na+/K+-ATPase disrupt ionic balance, resulting in intracellular Na+ accumulation, osmotic water influx, fiber cell swelling, and subsequent opacification.21 Glucocorticoids (GCs), exemplified by dexamethasone (Dex), directly inhibit Na+/K+-ATPase function through glucocorticoid receptor (GR)-mediated transcriptional regulation. Experimental evidence identifies glucocorticoid response elements (GREs) within the promoter region of the Na+/K+-ATPase α1 subunit, enabling Dex to suppress α1 subunit expression at both mRNA and protein levels in LECs. This suppression demonstrates temporal correlation with progressive enzyme activity reduction and lens opacity development, as validated in ex vivo rat lens models. Concurrent administration of the GR antagonist RU486 abrogates Dex-induced α1 subunit downregulation, preserving enzymatic function and optical clarity, thereby confirming GR-dependent transcriptional control.22
Pathological analyses delineate a dual-phase mechanism: GC-mediated Na+/K+-ATPase inhibition impairs Na+ extrusion, elevating intracellular Na+ concentrations and osmotic pressure. This initiates fiber cell vacuolation, membrane integrity loss, and disordered fiber packing—cardinal features of GIC.22 Paradoxically, Na+/K+-ATPase overexpression similarly destabilizes ionic gradients, inducing comparable osmotic stress and opacification,21 underscoring the necessity for strict enzymatic activity regulation.
Na+/K+-ATPase dysfunction extends beyond ionic imbalance, reducing water-soluble crystallin content while increasing insoluble protein aggregates—key contributors to light scattering and cataract severity.21 Collectively, GCs perturb Na+/K+-ATPase homeostasis via GR signaling cascades, disrupting ionic equilibrium and osmotic regulation. This mechanism constitutes a central pathway in glucocorticoid-induced cataractogenesis.21,22
Apoptosis
Apoptosis—a genetically programmed cell death process marked by cellular shrinkage, caspase activation, and DNA fragmentation—serves as a central mechanism in glucocorticoid (GC)-induced cataractogenesis.23 In LECs, GCs such as dexamethasone (Dex) induce apoptosis through dual-pathway mechanisms: (1) mitochondrial dysfunction via Bcl-2 family protein dysregulation and (2) endoplasmic reticulum (ER) stress-mediated caspase activation. Dex elevates pro-apoptotic Bax expression while suppressing anti-apoptotic Bcl-2, leading to mitochondrial membrane depolarization and cytochrome c release. This cascade sequentially activates caspase-9 and effector caspase-3.24,25 Concurrently, GC-triggered ER stress activates the unfolded protein response (UPR), upregulating CHOP and TRB3 expression, which synergistically enhance caspase-3 cleavage and LEC apoptosis.26
Glucocorticoid receptor (GR)-α signaling orchestrates these apoptotic pathways. Dex binding to GR-α increases GRE-dependent transcriptional activity and GR protein levels, correlating with elevated Bax/caspase-3 activation and reduced LEC survival.27 Intriguingly, low-dose Dex (0.1 μM) transiently stimulates LEC proliferation (via Ki-67 upregulation) but induces apoptosis at higher doses (1–100 μM), demonstrating a biphasic dose-response relationship.25 Pharmacological inhibition of ER stress (4-PBA, TUDCA) or TRB3 knockdown attenuates mitochondrial dysfunction and caspase-3 activation, validating ER-mitochondrial crosstalk in GC-induced apoptosis.26
Clinical observations link LEC calcium (Ca²+) overload to ER stress amplification, which exacerbates apoptosis via oxidative damage and epithelial-mesenchymal transition (EMT). Dex downregulates EphA2—a receptor critical for lens transparency—impairing cell adhesion and promoting EMT. This process coincides with compensatory upregulation of HSP27 and CRYAB as endogenous anti-apoptotic responses.28 Notably, GR antagonism with RU486 incompletely rescues LEC apoptosis and paradoxically amplifies caspase-3 activity at high Dex concentrations, revealing the multifaceted nature of GR signaling.25,27
In summary, GCs drive LEC apoptosis through mitochondrial-ER stress axis dysregulation, caspase cascade activation, and EMT induction, positioning apoptotic modulation as a strategic target in steroid-induced cataract prevention.23,28
Interventions and Drug Delivery Strategies for Glucocorticoid Induced Cataract
Glucocorticoid-induced cataracts (GICs), a significant ocular complication of prolonged glucocorticoid (GC) therapy, demand innovative strategies to counteract oxidative stress and pathological signaling. Current interventions focus on antioxidant supplementation, advanced drug delivery systems, and molecular target modulation [Table 2]. Vitamin E (VE) demonstrates dose-dependent efficacy in chick embryo models, where 50 mg/kg supplementation reduced lens opacification by normalizing lipid peroxide (LPO) levels, preserving Na+/K+-ATPase activity, and restoring glutathione (GSH) content. Treated lenses exhibited elevated total antioxidant status (TAS) and glutathione peroxidase (GPx) activity alongside reduced oxidative markers in ocular and hepatic tissues, with high-dose VE (50 mg/kg) completely preventing nuclear cataracts.29–31 Similarly, astaxanthin (AST; 100 mg/kg) mitigated hydrocortisone-induced opacity in chick embryos by maintaining GSH homeostasis and reducing oxidative stress.32 Beyond antioxidants, molecular targeting of chondroitin sulfate proteoglycan 5 (CSPG5) and nerve growth factor (NGF) shows therapeutic promise. CSPG5 knockdown in dexamethasone-treated lens epithelial cells (LECs) suppressed epithelial-mesenchymal transition (EMT), fibronectin expression, and cell migration, while its overexpression in GIC patients correlated with EZH2/B-Myb-mediated EMT enhancement.16 NGF treatment restored p38 MAPK/Akt phosphorylation dynamics, downregulated α-smooth muscle actin (α-SMA), and inhibited EMT via TrkA receptor signaling, offering protection against GC-induced damage.15
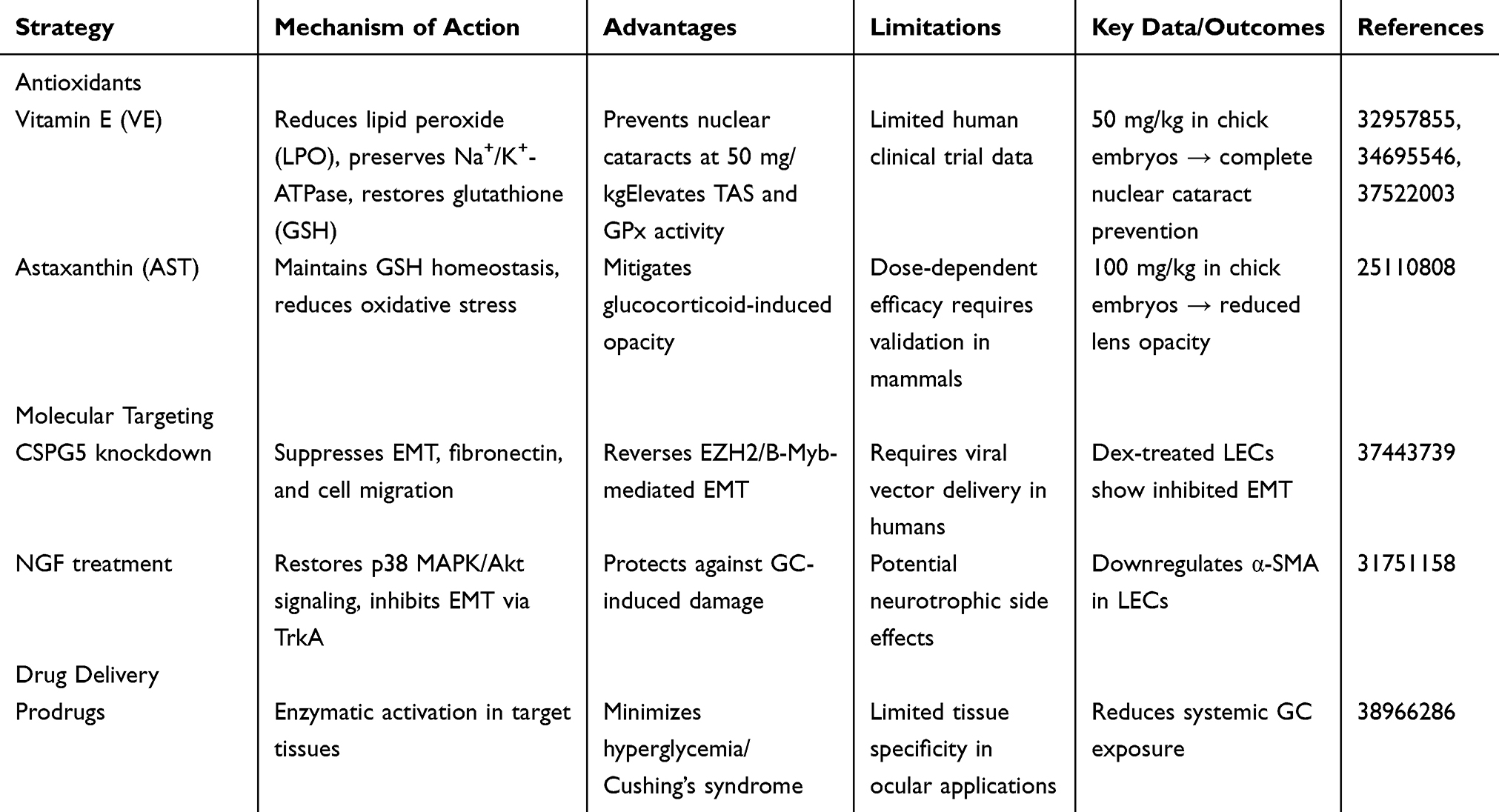 |
Table 2 Comparative Analysis of Interventions for Glucocorticoid-Induced Cataract Prevention |
Advanced drug delivery systems address GC toxicity through precision targeting. Prodrugs—enzymatically activated in target tissues—minimize systemic complications like hyperglycemia and Cushing’s syndrome by reducing off-target GC exposure33 [Figure 1].
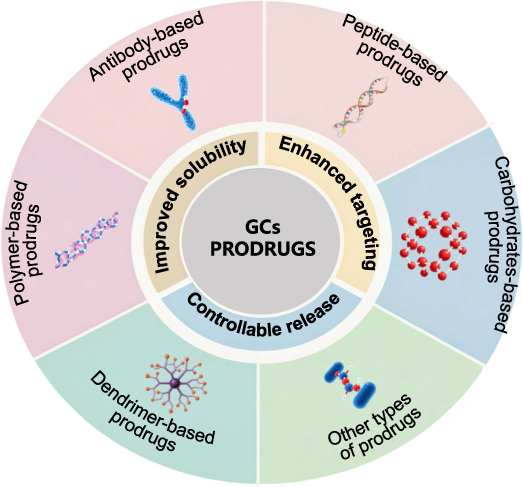 |
Figure 1 Approaches and Methods for Mitigating Adverse Reactions of Glucocorticoid Medications. Reprinted from Liu, H.et al, Glucocorticoids-based prodrug design: Current strategies and research progress. Asian J Pharm Sci. 2024. 19(3): p. 100922. Creative Commons.33 |
Polymeric core-shell nanoparticles (NPs) encapsulating triamcinolone acetonide (TA) demonstrated controlled ocular distribution in diabetic models, maintaining therapeutic efficacy while preventing posterior subcapsular cataracts (PSCs). These NPs, composed of polycaprolactone (PCL) and Pluronic F68 (PF68), avoided glucocorticoid receptor-α (GR-α) activation in LECs, thereby reducing lens accumulation and PSC risk. This targeted approach exemplifies how nanotechnology can decouple GC efficacy from cataractogenic side effects.34
Collectively, while a multi-modal strategy integrating antioxidants, targeted delivery, and molecular interventions provides a robust preclinical framework for GIC prevention, clinical applicability remains constrained by bioavailability challenges, unvalidated safety profiles of nanoparticles, and unresolved CSPG5/NGF modulation protocols. VE and AST counteract oxidative damage, while NPs and prodrugs enhance therapeutic specificity. Molecular modulation of CSPG5 and NGF disrupts EMT-driven pathology, synergizing with redox balance restoration to preserve lens transparency. These approaches collectively reduce epithelial apoptosis, protein aggregation, and fibrotic remodeling, addressing both upstream oxidative triggers and downstream GC-specific pathways. Future clinical translation requires optimizing bioavailability of antioxidant formulations, validating nanoparticle safety profiles, and establishing CSPG5/NGF modulation protocols—critical steps toward transforming these preclinical successes into therapeutic realities.
This table systematically compares preventive interventions for GIC, categorized into three main approaches: (1) Antioxidants (Vitamin E [VE], Astaxanthin [AST]) mitigating lens oxidative stress (reducing lipid peroxide [LPD], preserving glutathione [GSH] homeostasis); (2) Molecular targeting (CSPG5 knockdown suppressing epithelial-mesenchymal transition [EMT]/fibrosis; Nerve Growth Factor [NGF] restoring p38 MAPK/Akt signaling and inhibiting EMT via TrkA); and (3) Drug delivery optimization (Prodrugs activated by target tissue enzymes to minimize systemic glucocorticoid exposure).
Glucocorticoid-Sparing Therapies and Alternative Strategies for Preventing Glucocorticoid Induced Cataracts
Glucocorticoid-sparing strategies are critical for mitigating cataract risk in patients requiring long-term glucocorticoid (GC) therapy for chronic inflammatory conditions like juvenile idiopathic arthritis (JIA), uveitis, and Vogt-Koyanagi-Harada (VKH) disease [Table 3]. Methotrexate (MTX), a dihydrofolate reductase inhibitor, serves as a cornerstone therapy due to its dual anti-inflammatory and immunomodulatory effects. In pediatric uveitis associated with JIA, MTX (0.35–0.65 mg/kg/week) reduces GC dependency, lowering cataract incidence from 23% in GC-treated cohorts while alleviating intraocular pressure (IOP) elevation and growth retardation.35,36 MTX modulates inflammation through JAK-STAT pathway inhibition, adenosine-mediated suppression, and T-cell/monocyte regulation.37,38 Its systemic use with GCs effectively manages VKH syndrome, while intravitreal MTX (400 μg/0.1 mL) achieves therapeutic efficacy in refractory uveitis with minimal ocular toxicity (eg, transient vitreous haze).39 Complementing MTX, TNF-α inhibitors like adalimumab provide alternatives for GC-resistant uveitis, FDA-approved for pediatric use with reduced cataract risk compared to GCs.40 However, 30% of patients experience paradoxical inflammation recurrence, necessitating rigorous monitoring.41,42
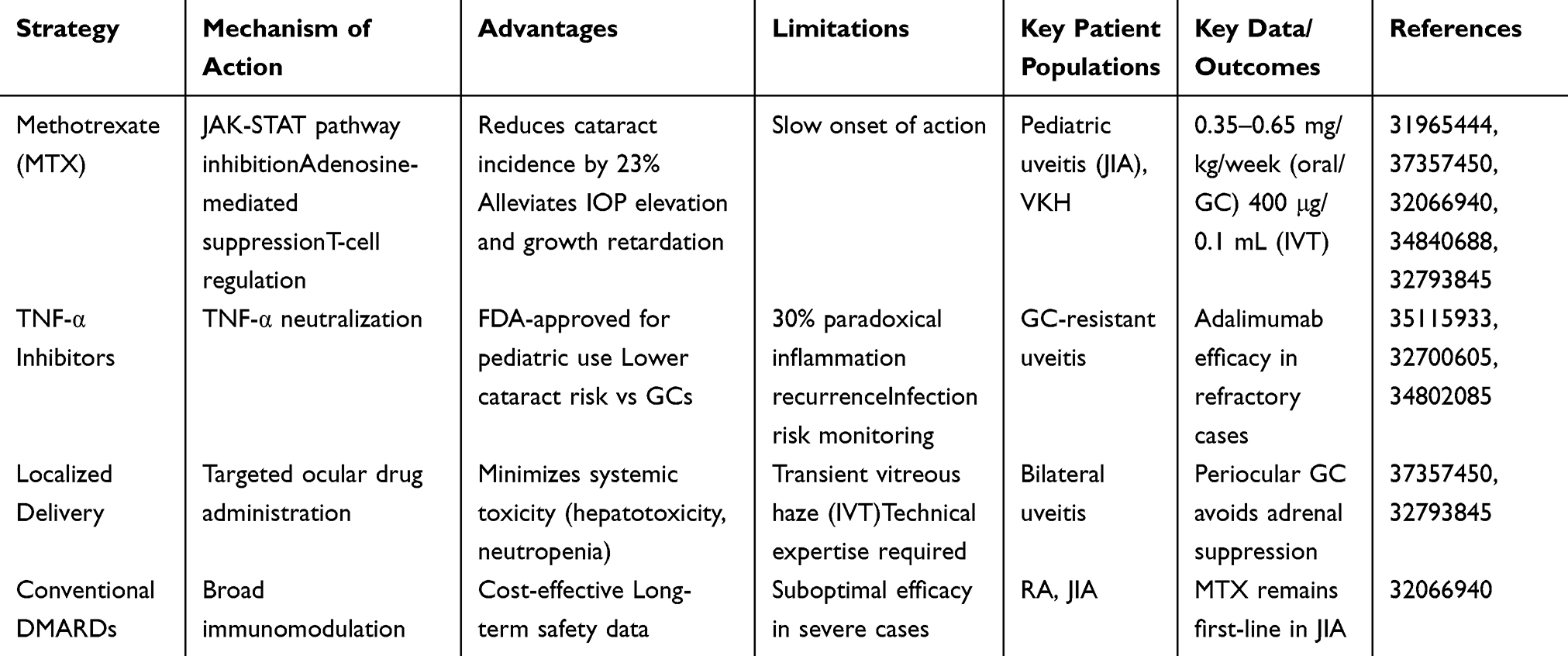 |
Table 3 Comparative Analysis of Glucocorticoid-Sparing Strategies in Cataract Prevention |
Localized drug delivery systems minimize systemic GC exposure while maintaining therapeutic efficacy. Intravitreal MTX achieves targeted ocular concentrations, circumventing systemic complications like hepatotoxicity and neutropenia.39 Periocular GC injections are prioritized in bilateral uveitis to avoid adrenal suppression and growth impairment in pediatric populations.36 These approaches exemplify precision medicine principles, balancing anti-inflammatory benefits with reduced off-target toxicity. Despite these advances, challenges persist: MTX may rarely accelerate preexisting cataracts (causality unconfirmed), while TNF inhibitors require vigilance for infections and inflammatory rebound.38 Conventional disease-modifying antirheumatic drugs (DMARDs) like MTX remain first-line in rheumatoid arthritis (RA) and JIA due to proven cost-effectiveness and long-term safety, even as biologics gain traction.37
Future strategies aim to optimize GC-sparing efficacy through combination therapies and advanced delivery systems. Trials investigating MTX-TNF inhibitor combinations show promise in enhancing anti-inflammatory synergy while reducing treatment frequency. Sustained-release intravitreal implants may further improve compliance by minimizing injection burden. Personalized regimens guided by inflammatory biomarkers (eg, IL-6, TNF-α levels) could refine patient stratification, tailoring interventions to individual risk profiles. However, economic barriers may limit biologics accessibility in resource-limited settings, reinforcing the relevance of conventional DMARDs.These innovations must address residual challenges—validating long-term ocular safety of biologics, clarifying MTX’s cataractogenic potential, and ensuring equitable access to advanced therapies. By integrating pharmacological advances with precision delivery, clinicians can preserve vision while mitigating the irreversible lens damage characteristic of glucocorticoid-induced cataracts.
This table compares four strategies to reduce or replace systemic glucocorticoids (GCs) for GIC prevention: Methotrexate (MTX) inhibits JAK-STAT signaling and T-cell regulation; reduces cataract risk by 32%, alleviates intraocular pressure elevation and growth retardation; slow onset; indicated for pediatric uveitis in juvenile idiopathic arthritis (JIA) (0.35–0.65 mg/kg/week, oral/subcutaneous); TNF-α inhibitors neutralize TNF-α; approved for pediatric use with lower cataract risk vs GCs; 20% paradoxical inflammation recurrence risk; used in GC-resistant uveitis; adalimumab effective in refractory cases; Localized delivery (eg, IVT) enables targeted ocular administration; minimizes systemic toxicity (hepatotoxicity, neutropenia); may cause transient vitreous haze and requires technical expertise; indicated for bilateral uveitis; periocular GC avoids adrenal suppression; Conventional DMARDs provide broad immunosuppression; cost-effective with established long-term safety; suboptimal efficacy in severe cases; used in Behçet’s disease (BA)/JIA; MTX remains first-line in JIA.
Conclusion
Glucocorticoid-induced cataracts (GIC) represent a significant therapeutic challenge in clinical practice, where the well-established anti-inflammatory benefits of glucocorticoids (GCs) may conflict with their potential to induce sight-threatening complications, including posterior subcapsular opacities reported in up to 36% of long-term users. To address this complex balance, our analysis suggests that effective management could require coordinated targeting of three interconnected pathological processes: (1) a proposed mitochondrial-ER stress interaction, wherein GC-associated oxidative stress (eg, ROS accumulation, GSH depletion) and endoplasmic reticulum disturbances (UPR activation, calcium dysregulation) may contribute to TRB3-Bax-caspase-9 apoptotic pathways; (2) EMT-related fibrotic changes, potentially involving TGF-β/Smad-mediated E-cadherin suppression and CSPG5/EZH2 epigenetic modifications in lens epithelial cells; and (3) disruptions in ionic equilibrium, possibly through GR-mediated modulation of Na+/K+-ATPase expression, which could lead to osmotic stress and protein aggregation.
Current targeted strategies show promise but with limitations: GC-sparing agents such as methotrexate (at studied doses of 0.35–0.65 mg/kg/week) and TNF-α inhibitors are associated with approximately 23% lower cataract incidence in some uveitis studies, while core-shell nanoparticle systems have demonstrated reduced GR-α activation in preclinical models by localizing drug release. However, these approaches appear to address isolated mechanisms—antioxidants may alleviate oxidative damage without necessarily preventing EMT, and CRISPR-based CSPG5 modulation might not resolve ionic imbalance. A proposed integrative strategy could explore co-delivery systems for agents like astaxanthin and ER-stress modulators, combined with targeted gene-editing techniques. Preliminary investigations might examine whether pharmacogenomic assessment of GR-α variants and TRB3 levels could inform dosing strategies, potentially benefiting pediatric populations where susceptibility rates have been observed near 18.1% in limited cohorts.
Translational considerations include reports of inflammatory rebound following biologic therapies and biocompatibility challenges with intraocular delivery systems. Emerging approaches such as mitochondrial protein profiling, refined gene-editing platforms, and novel ocular implantation methods offer promising research directions. Through prioritized development of mechanism-informed combination therapies and advanced delivery platforms, this evolving framework could potentially enhance the safety profile of GC therapy while maintaining anti-inflammatory efficacy—progressing toward optimized visual outcomes alongside disease management.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
There is no funding to report.
Disclosure
The authors declare no competing interests in this work.
References
1. Hariharan R, Krishnamurthy S, Kaliaperumal S, et al. Incidence and predictive risk factors for ophthalmological complications in children with nephrotic syndrome receiving long-term oral corticosteroids: a cohort study. Paediatr Int Child Health. 2021;41(3):199–205. doi:10.1080/20469047.2021.1983315
2. Yan H, Tan X, Yu J, et al. The occurrence timeline of steroid-induced ocular hypertension and cataract in children with systemic autoimmune diseases. Int Ophthalmol. 2022;42(7):2175–2184. doi:10.1007/s10792-022-02217-5
3. Banerjee R, Martínez JA, Pérez PA, et al. Association between dexamethasone exposure and visually significant cataracts in multiple myeloma. Am J Hematol. 2024;99(1):E12–E14. doi:10.1002/ajh.27133
4. Nath T, Roy SS, Kumar H, et al. Prevalence of steroid-induced cataract and glaucoma in chronic obstructive pulmonary disease patients attending a tertiary care center in India. Asia Pac J Ophthalmol (Phila). 2017;6(1):28–32. doi:10.22608/APO.201616
5. Patel NA, Hoyek S, López-Font FJ, et al. Incidence of steroid-related ocular hypertension and cataract formation after sub-tenon triamcinolone in nonuveitic pediatric patients. Retina. 2025;45(1):141–146. doi:10.1097/IAE.0000000000004272
6. Hsueh YJ, Chen Y-N, Tsao Y-T, et al. The pathomechanism, antioxidant biomarkers, and treatment of oxidative stress-related eye diseases. Int J Mol Sci. 2022;23(3):1255. doi:10.3390/ijms23031255
7. Honisch C, Rodella U, Gatto C, et al. oxidative stress and antioxidant-based interventional medicine in ophthalmology. Pharmaceuticals. 2023;16(8):1146. doi:10.3390/ph16081146
8. Duman R, Vurmaz A. Role of innate immunity and oxidative stress in steroid-induced cataracts in developing chick embryos. Cutan Ocul Toxicol. 2018;37(3):281–285. doi:10.1080/15569527.2018.1452929
9. Watanabe H, Kosano H, Nishigori H. Steroid-induced short term diabetes in chick embryo: reversible effects of insulin on metabolic changes and cataract formation. Invest Ophthalmol Vis Sci. 2000;41(7):1846–1852.
10. Li J, Buonfiglio F, Zeng Y, et al. Oxidative stress in cataract formation: is there a treatment approach on the horizon? Antioxidants. 2024;13(10):1249. doi:10.3390/antiox13101249
11. Cicinelli MV, Buchan JC, Nicholson M, et al. Cataracts. Lancet. 2023;401(10374):377–389. doi:10.1016/S0140-6736(22)01839-6
12. Li H, Ji L, Shen H, et al. The long noncoding RNA H19 promotes fibrotic processes in lens epithelial cells. Invest Ophthalmol Vis Sci. 2023;64(7):21. doi:10.1167/iovs.64.7.21
13. Zhou S, Yang J, Wang M, et al. Endoplasmic reticulum stress regulates epithelial‑mesenchymal transition in human lens epithelial cells. Mol Med Rep. 2020;21(1):173–180. doi:10.3892/mmr.2019.10814
14. Celojevic D, Carlsson T, Johansson BR, Nannmark U, Petersen A, et al. Cell adhesion molecule expression in human lens epithelial cells after corticosteroid exposure. Open Ophthalmol J. 2012;6:42–48. doi:10.2174/1874364101206010042
15. Hah YS, Yoo W-S, Seo S-W, et al. Reduced NGF level promotes epithelial-mesenchymal transition in human lens epithelial cells exposed to high dexamethasone concentrations. Curr Eye Res. 2020;45(6):686–695. doi:10.1080/02713683.2019.1695844
16. Yoo WS, Seong H, Song C, et al. Role of chondroitin sulfate proteoglycan 5 in steroid-induced cataract. Cells. 2023;12(13):1705. doi:10.3390/cells12131705
17. Viedma-Poyatos A, Pajares MA, Perez-Sala D. Type III intermediate filaments as targets and effectors of electrophiles and oxidants. Redox Biol. 2020;36:101582. doi:10.1016/j.redox.2020.101582
18. Xie GL, Yan H, Lu ZF. Inhibition of glucocorticoid-induced alteration of vimentin by a glucocorticoid receptor antagonist RU486 in the organ-cultured rat lens. Mol Vis. 2011;17:32–40.
19. Alieva IB, Shakhov AS, Dayal AA, et al. Unique role of vimentin in the intermediate filament proteins family. Biochemistry. 2024;89(4):726–736. doi:10.1134/S0006297924040114
20. Giannone AA, Li L, Sellitto C, et al. Physiological mechanisms regulating lens transport. Front Physiol. 2021;12:818649. doi:10.3389/fphys.2021.818649
21. Kim JY, Park JH, Kang -S-S, et al. Topical nerve growth factor attenuates streptozotocin-induced diabetic cataracts via polyol pathway inhibition and Na(+)/K(+)-ATPase upregulation. Exp Eye Res. 2021;202:108319. doi:10.1016/j.exer.2020.108319
22. Xie GL, Yan H, Lu ZF. Inhibition of glucocorticoid-induced changes of Na(+), K(+)-ATPase in rat lens by a glucocorticoid receptor antagonist RU486. Exp Eye Res. 2010;91(4):544–549. doi:10.1016/j.exer.2010.07.005
23. Kupsco A, Schlenk D. Oxidative stress, unfolded protein response, and apoptosis in developmental toxicity. Int Rev Cell Mol Biol. 2015;317:1–66.
24. Feng L, Wei Y, Sun Y, et al. MIR34A modulates lens epithelial cell apoptosis and cataract development via the HK1/caspase 3 signaling pathway. Aging. 2023;15(13):6331–6345. doi:10.18632/aging.204854
25. Petersen A, Carlsson T, Karlsson JO, Jonhede S, Zetterberg M, et al. Effects of dexamethasone on human lens epithelial cells in culture. Mol Vis. 2008;14:1344–1352.
26. Liu J, Tang Y, Li J, et al. TRB3 promotes cataract progression through endoplasmic reticulum stress-mediated mitochondrial dysfunction and cell apoptosis. Cell Biochem Biophys. 2024;83;(1):391–402
27. Wang L, Zhao W, Leng F, et al. Glucocorticoid receptors take part in the apoptotic process of human lens epithelial cells, but the glucocorticoid receptor antagonist RU486 does not rescue the cells fully. Mol Biosyst. 2011;7(6):1926–1937. doi:10.1039/c1mb05045a
28. Pluss CJ, Kustermann S. A human three-dimensional in vitro model of lens epithelial cells as a model to study mechanisms of drug-induced posterior subcapsular cataracts. J Ocul Pharmacol Ther. 2020;36(1):56–64. doi:10.1089/jop.2019.0010
29. Vurmaz A, Ertekin A, Sabaner MC, et al. Effects of vitamin E in a glucocorticoid induced cataract model in chicken embryos. Biotech Histochem. 2021;96(6):431–438. doi:10.1080/10520295.2020.1818284
30. Tanito M. Reported evidence of vitamin E protection against cataract and glaucoma. Free Radic Biol Med. 2021;177:100–119. doi:10.1016/j.freeradbiomed.2021.10.027
31. Zhang T, Yi X, Li J, et al. Vitamin E intake and multiple health outcomes: an umbrella review. Front Public Health. 2023;11:1035674. doi:10.3389/fpubh.2023.1035674
32. Ishikawa S, Hashizume K, Nishigori H, et al. Effect of astaxanthin on cataract formation induced by glucocorticoids in the chick embryo. Curr Eye Res. 2015;40(5):535–540. doi:10.3109/02713683.2014.935445
33. Liu H, Ji M, Xiao P, et al. Glucocorticoids-based prodrug design: current strategies and research progress. Asian J Pharm Sci. 2024;19(3):100922. doi:10.1016/j.ajps.2024.100922
34. Srinivasarao DA, Reddy SS, Reddy GB, et al. Spatio-temporal control on the delivery of triamcinolone acetonide using polymeric nanoparticles reduces steroid induced cataract. Int J Pharm. 2019;568:118474. doi:10.1016/j.ijpharm.2019.118474
35. Petty RE, Zheng Q. Uveitis in juvenile idiopathic arthritis. World J Pediatr. 2020;16(6):562–565. doi:10.1007/s12519-019-00331-6
36. Tugal-Tutkun I. An overview of pediatric uveitis. Turk Arch Pediatr. 2023;58(4):363–370. doi:10.5152/TurkArchPediatr.2023.23086
37. Cronstein BN, Aune TM. Methotrexate and its mechanisms of action in inflammatory arthritis. Nat Rev Rheumatol. 2020;16(3):145–154. doi:10.1038/s41584-020-0373-9
38. Abdi F, Mohammadi SS, Falavarjani KG. Intravitreal Methotrexate. J Ophthalmic Vis Res. 2021;16(4):657–669. doi:10.18502/jovr.v16i4.9756
39. Park JG, Callaway NF, Ludwig CA, et al. Intravitreal methotrexate and fluocinolone acetonide implantation for Vogt-Koyanagi-Harada uveitis. Am J Ophthalmol Case Rep. 2020;19:100859. doi:10.1016/j.ajoc.2020.100859
40. Liu XB, Tang LS, Chen JW, Lin CS, Liu QH, Xu Q, et al. Case report: a promising treatment strategy for noninfectious uveitis. Front Pharmacol. 2021;12:784860. doi:10.3389/fphar.2021.784860
41. Gaggiano C, Sota J, Gentileschi S, et al. The current status of biological treatment for uveitis. Expert Rev Clin Immunol. 2020;16(8):787–811. doi:10.1080/1744666X.2020.1798230
42. Dammacco R, Guerriero S, Alessio G, et al. Natural and iatrogenic ocular manifestations of rheumatoid arthritis: a systematic review. Int Ophthalmol. 2022;42(2):689–711. doi:10.1007/s10792-021-02058-8
 © 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.